

Abstract
To identify suitable lipophilic compounds having high potency and selectivity for vesicular acetylcholine transporter (VAChT), a heteroaromatic ring or a phenyl group was introduced into the carbonyl-containing scaffold for VAChT ligands. Twenty new compounds with ALog D values between 0.53-3.2 were synthesized, and their in vitro binding affinities were assayed. Six of them (19a, 19e, 19g, 19k and 24a-b) displayed high affinity for VAChT (Ki = 0.93 – 18 nM for racemates) and moderate to high selectivity for VAChT over σ1 and σ2 receptors (Ki = 44 – 4400-fold). These compounds have a methyl or a fluoro substitution that provides the position for incorporating PET radioisotopes C-11 or F-18. Compound (-)-[11C]24b (Ki = 0.78 for VAChT, 900-fold over σ receptors) was successfully synthesized and evaluated in vivo in rats and nonhuman primates. The data revealed that (-)-[11C]24b has highest binding in striatum and has favorable pharmacokinetics in the brain.
Keywords: Blood-brain-barrier (BBB), lipophilicity, sigma-1 and sigma-2 receptors, structure-activity relationship (SAR), vesicular acetylcholine transporter (VAChT)
Introduction
Dementia is a clinical syndrome characteristic of loss or decline in memory and other cognitive abilities that result in functional impairment in the elderly. It is a global public health problem.1, 2 The prevalence in the USA has been recently estimated at 5.4 million individuals, including 5.2 million over the age of 65, and 200,000 individuals under the age of 65.1 Increasing lifespan of people will lead to increased number of patients with dementia. The severity of dementia in neurodegenerative diseases is linked to loss of cholinergic neurons and synapses in the central nervous system (CNS). The use of 11C-labeled Pittsburgh Compound-B ([11C]PIB) to assess β-amyloid plaques and diagnose Alzheimer’s disease (AD) was a significant breakthrough.3 However, [11C]PIB cannot specifically assess the loss of cholinergic neurons and synapses in the brain, which correlates to severity of cognitive dysfunction in AD.4-9 For Parkinson’s disease associated with dementia (PDD), the cholinergic deficit in cortical regions is more severe than for non-demented patients with Parkinson’s Disease (PD).10 A PET tracer that can assess the loss of cholinergic synapses would provide a useful tool for assessing the severity of cognitive dysfunction and monitoring the efficacy of cholinergic therapies for dementia in neurodegenerative disorders.
Vesicular acetylcholine transporter (VAChT) and choline acetyltransferase (ChAT) are essential for a cholinergic neuron.11 VAChT is localized in cholinergic terminals, and it transports acetylcholine (ACh) from the cytoplasm into the synaptic vesicles. Anti-ChAT preferentially stains cell bodies, whereas anti-VAChT preferentially stains nerve terminals.4, 12 Anti-ChAT is more useful for monitoring the death of cholinergic cells, whereas anti-VAChT is more useful for monitoring changes in the density of cholinergic terminals. It is widely accepted that VAChT is a reliable biomarker to study cholinergic function in the brain. Currently, (-)-5-[123I]iodobenzovesamicol ([123I]IBVM) is the only radiotracer used for imaging VAChT levels in living human brain using single-photon emission computed tomography (SPECT). The relative distribution for specific binding of [123I]IBVM in human brain corresponds well with postmortem results reported for ChAT.13, 14 When [123I]IBVM was used to assess cholinergic deficiency in patients with dementia, it was found that PDD and AD patients have globally reduced cortical binding.15 In addition, a significant decrease in [123I]IBVM binding (47-62%) in cingulate cortex and parahippocampalamygdaloid in AD subjects compared to control patients has been observed.16 However, the slow binding kinetics of [123I]IBVM requires scanning for approximately 6 hours post-injection, which can be stressful for patients. Positron emission tomography (PET) imaging will be able to carry out scans with higher sensitivity and spatial resolution (3-5 mm) compared to SPECT (10 mm).17, 18 The demand to provide higher accuracy in clinical imaging of VAChT levels in humans makes the identification of a PET tracer for VAChT very important. To date, this goal has not been achieved because of the lack of suitable PET radiotracers.
In efforts to develop a PET tracer for VAChT, investigators have put tremendous effort into optimizing the structure of vesamicol analogues with the goal of identifying highly potent and selective ligands.4, 19-27 Among various radioligands developed, only a small number have been evaluated in vivo in non-human primates and humans.19, 27-29 Despite promising in vitro, ex vivo and initial in vivo studies, most of the ligands are unsuitable for clinical use due to poor selectivity over σ receptors in brain, low extraction from the blood, slow brain kinetics or fast metabolism. Among the physicochemical properties of ligands, lipophilicity is a one of the key properties that plays a pivotal role in absorption, distribution, metabolism, and elimination of ligands.30 For central nervous system drugs, it was found that the blood-brain-barrier (BBB) penetration is optimal with the ALog D values in the range of 1.5 – 3.0, with a mean value of 2.5.31-33 Although other properties of compounds affect the BBB penetration, those ligands with moderate lipophilicity often exhibit highest brain uptake.30 Highly lipophilic radiotracers are usually cleared slowly from the brain.
Our group has reported a new class of VAChT inhibitors containing a carbonyl group attached to the 4 position of the piperidine ring and discussed the structure-activity relationship (SAR) of this new class of compounds.4, 19, 34-36 Among them (as shown in Figure 1), compounds 5, 7 and 8 displayed high potencies and good selectivity for VAChT in vitro.4, 19 In particular, a fluorine-18 labeled version of (-)-2-hydroxy-3-(4-(4-fluorobenzoyl)piperidino)tetralin, (-)-[18F]7 was successfully radiosynthesized and used to conduct in vivo evaluation in rats and monkeys; the initial results were very promising.19 The possibility that (-)-[18F]7 can serve as a clinical PET tracer for quantifying the level of VAChT in vivo is under investigation. The current manuscript focuses on 1) optimizing the structures of this new class of VAChT ligand to identify highly potent ligands having lipophilicity suitable to efficiently cross the BBB. 2) Separate the enantiomers of the optimal compound, and radiosynthesize with carbon-11 PET isotope. 3) In vivo evaluate the new C-11 radiotracer in rodent and non-human primate. The strategies to achieve optimization include: (1) replacing the thiophenyl group in (1-((2S,3S)-3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(thiophen-2-yl)methanone 9 with N-methyl pyrrole4 (the methyl group provides a position for conveniently labeling with carbon-11 isotope), (2) replacing the 4-fluorophenyl group in 74 with pyridine, substituted pyridines and pyrroles and optimizing the substitution so that radiolabeling with carbon-11 or fluorine-18 can be achieved, and (3) converting the primary amino group of compound 6 to monomethyl amino or dimethyl amino, which will provide access for radiolabeling with carbon-11. This investigation was inspired by (1) the observation that a series of carbonyl group containing analogues have high affinity for VAChT and low affinity for σ receptors;4, 19, 34 (2) our in vivo validation of (-)-[18F]719 and its analogue (-)-[11C]8,35 which showed high binding in the striatum, the VAChT enriched regions in the brain; and (3) the high demand for a clinically suitable PET probe for investigating the correlation between loss of cognitive function and loss of cholinergic synapses, which will help improve the early diagnosis of dementia and monitor the therapeutic efficacy of treating Alzheimer’s and other neurodegenerative diseases.
Figure 1.
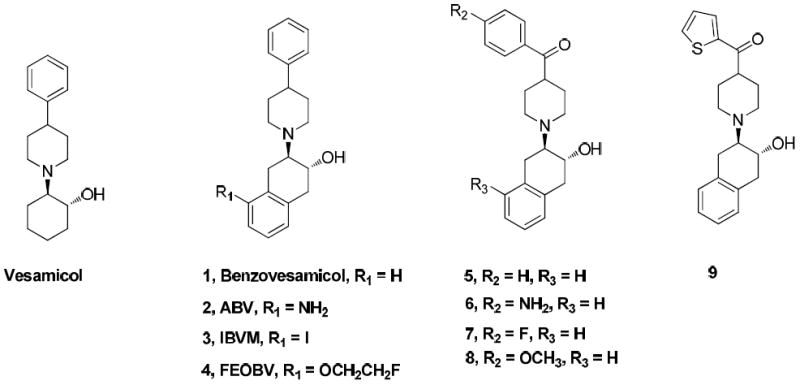
Strcutres of VAChT compounds
Results and Discussion
Chemistry
The synthesis of a new series of vesicular acetylcholine transporter inhibitors was accomplished according to Schemes 1-5. t-Boc-protected piperidine-4-carboxylic acid (10) was treated with 1,1’-carbonyldiimidazole (CDI), and N,O-dimethylhydroxylamine to afford Weinreb amide 11, which served as a versatile intermediate for synthesizing piperidines bearing a substituted heteroaromatic carbonyl group at the 4-position. Bromo substituted heteroaromatic compounds were lithiated with n-butyllithium (n–BuLi) and reacted with Weinreb amide to afford intermediates 16a-i. When synthesizing N-methyl pyrrole derivative 16a, via treatment of N-methylpyrrole with n-BuLi36 followed by compound 11, it was observed that the yield was much higher if the reaction vessel was pre-cooled to -78 °C while adding n-BuLi. To synthesize N-methyl pyridine derivatives 16h and 16i, commercially available 4 or 6-bromine substituted pyridin-2-amines 12h and 12i were first converted to 2-hydroxypyridines 13h and 13i or 2-pyridones 14h and 14i with sodium nitrite in sulfuric acid37, 38 followed by methylation using iodomethane in acetone to form bromo substituted N-methyl-2-pyridones 15h and 15i as major products.39 These pyridones reacted with 11 to afford the corresponding carbonyl group containing intermediates 16h and 16i. Trifluoroacetic acid (TFA) was used to remove the t-Boc group in 16a-i to afford the key intermediates 17a-i. Treatment of 17a-i with epoxide 1a,2,7,7a-tetrahydronaphtho[2,3-b]oxirene (18)4 afforded the target compounds 19a-i as shown in Scheme 1. To confirm whether the methyl group was on the N- or O-atom of 19h and 19i, the X-ray crystal structure of the oxalate salt of 19i was obtained as shown in Figure 2. The X-ray crystal structure revealed that the methyl group is on the nitrogen.
Scheme 1a.
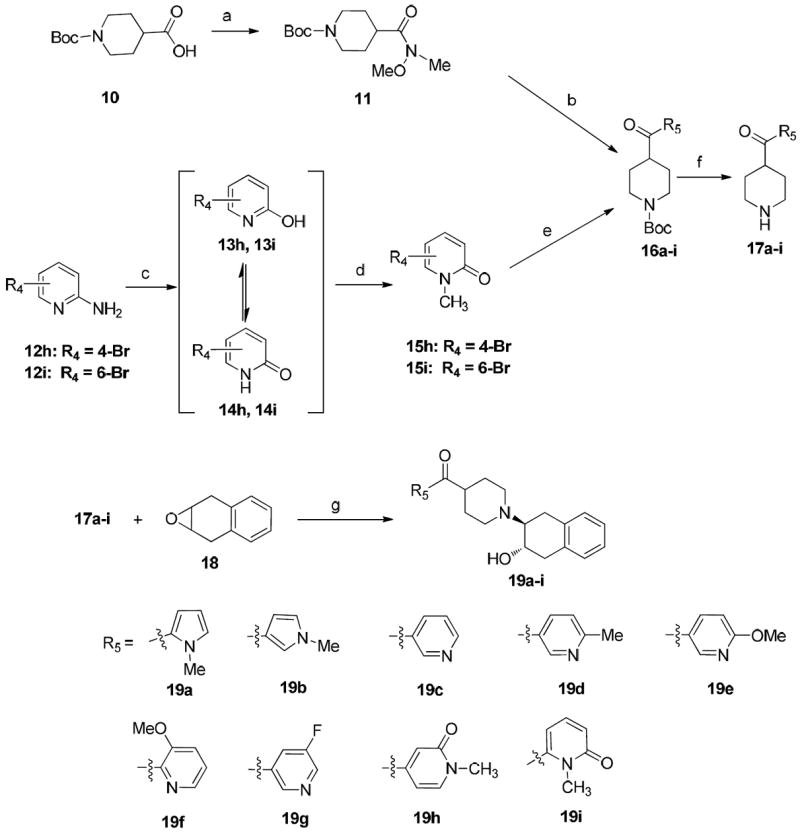
aReagents and conditions:
(a) CDl, N,O-Dimethylhydroxylamine hydrochloride, NEt3, CH2Cl2, rt, overnight; (b) ArLi, THF; (c) H2SO4/NaNO2, 0-5 °C, 2 h; (d) CH3I, K2CO3, acetone, 80 °C, 4 h; (e) 11, BuLi, THF, -78 °C; (f) TFA, CH2Cl2, rt, 4 h; (g) NEt3, EtOH, 75 °C, 36 h.
Scheme 5a.
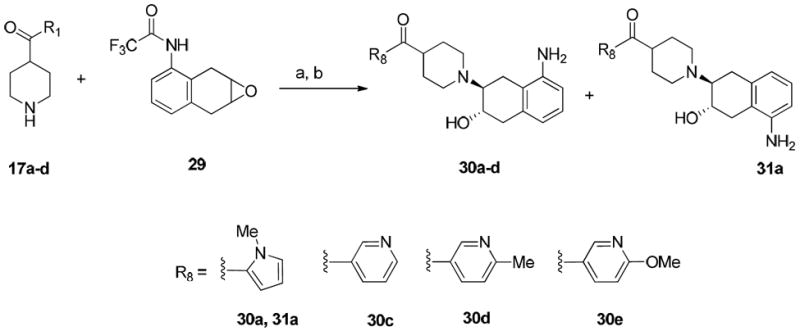
aReagents and conditions:
(a) NEt3, EtOH, 60 °C, 48 h; (b) 1N NaOH, EtOH, 60 °C, overnight.
Figure 2.
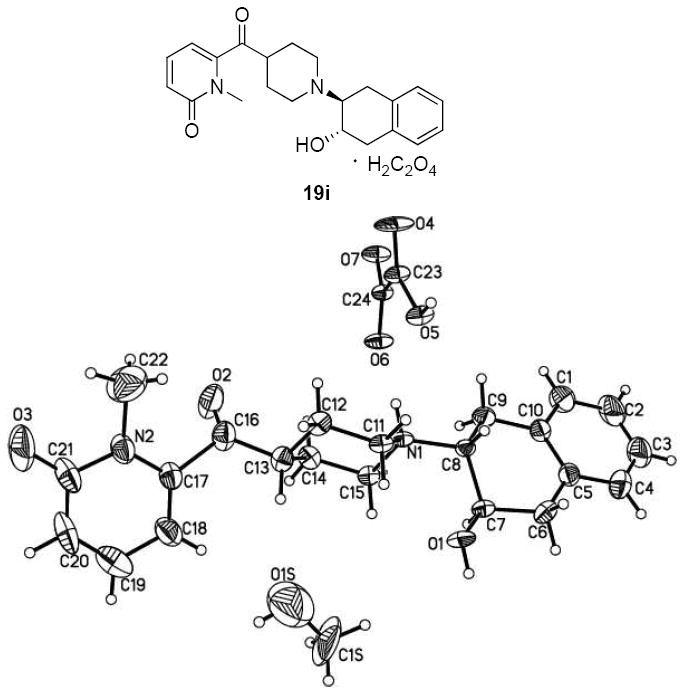
Chemical structure and X-ray crystal structure of 19i.
Although direct condensation of substituted piperidines and epoxide 18 was a common procedure for synthesizing vesicular acetylcholine transporter inhibitors,19, 40 it proved challenging when synthesizing 19j and 19k due to the low solubility of substituted heteroaromatic piperidines in commonly used solvents such as methanol, ethanol, dichloromethane, DMF and DMSO. Thus, an alternative approach was followed in which the t-Boc group in compound 11 was removed first, and the resulting piperidine was treated with 18 to afford compound 20. The free hydroxyl group of compound 20 was protected with the tert-butyldimethylsilyl group by reacting with tert-butyldimethylsilyl chloride to afford compound 21. Commercially available 5-bromo-2-fluoropyridine (17j) or 3-bromo-2-fluoropyridine (17k) was treated with n-BuLi in THF followed by compound 21, and further removal of TBDMS group afforded target compounds 19j and 19k as shown in Scheme 2.
Scheme 2a.
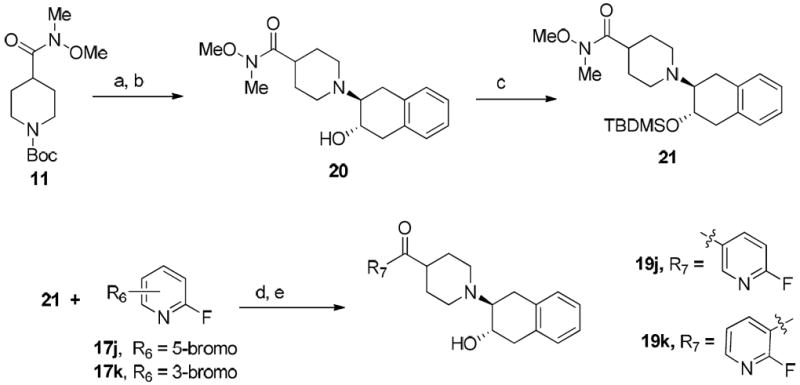
aReagents and conditions:
(a) TFA, CH2Cl2, rt, 4 h; (b) 1,4-dihydronaphthalene oxide, NEt3, EtOH, 75 °C, 36 h; (c) TBDMSCI, imidazole, CH2Cl2, rt, overnight; (d) n-BuLi, THF, -78 °C, 4h; (e) 12N HCl, THF, rt.
Compounds 24a and 24b were synthesized as depicted in Scheme 3. Compound 6 was synthesized following the reported procedure.19 Direct dimethylation of the aromatic amine in compound 6 with iodomethane in the presence of NaH afforded compound 24a very easily. However, the synthesis of the N-monomethylaniline analogue 24b was more complicated. The free hydroxyl group and the primary aniline group of 6 were protected by treating with acetic anhydride to form diacetylated compound 22.41 N-methylation of aromatic amide 22 gave intermediate 23, which upon hydrolysis afforded the target compound 24b.
Scheme 3a.
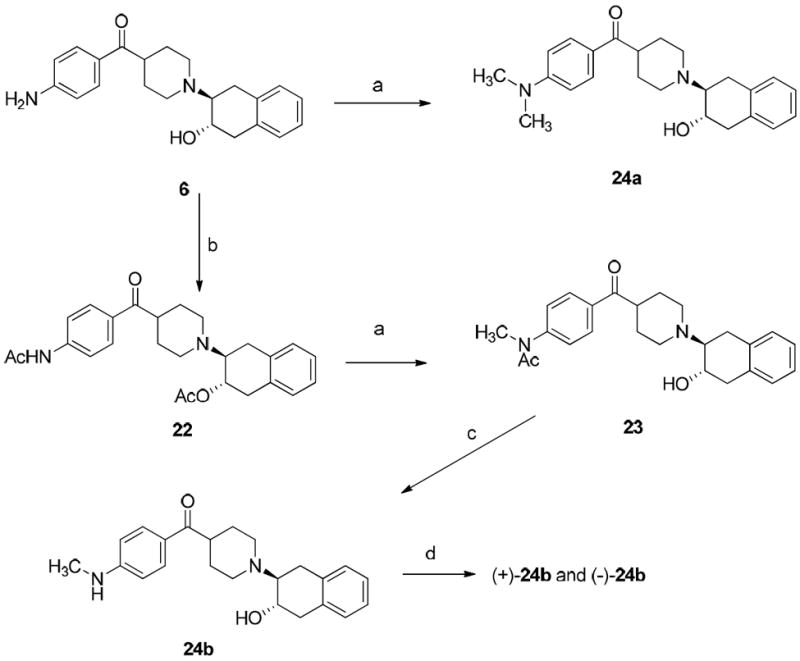
aReagents and conditions:
(a) CH3I, NaH, THF, rt, 0.5 h; (b) (CH3CO)2O, Et3N, CH2Cl2, rt; (c) Concentrated HCl, ethylene glycol, refluxed; d) Separation of enantiomers of 24b on HPLC: column: Chiralcel OD; mobile phase: 35% isopropanol in hexane; flow rate: 4.0 mL/min; (+)-enantiomer at 15 min and (-)-enantiomer at 30 min;
Synthesis of fluoroethoxy analogues 26a and 26b followed two different approaches as shown in Scheme 4. The first approach was to demethylate 19e in the presence of boron tribromide (BBr3) or trimethylsilyl iodide (TMSI) to generate the corresponding alcohol 25, which was reacted with 1-bromo-2-fluoroethane to afford the target O-alkylated compound 26a. However, use of a similar approach to synthesize compound 26b was not successful with either BBr3 or TMSI to remove the methyl group in 19f. To overcome this challenge, an alternative approach was used. Commercially available 2-bromopyridin-3-ol (27) was reacted with 1-bromo-2-fluoroethane via O-alkylation to afford 2-bromo-3-(2-fluoroethoxy)pyridine (28). This compound was lithiated and treated with compounds 11, followed by 18 to afford target compound 26b following the procedure described for synthesis of compounds 19j and 19k.
Scheme 4a.
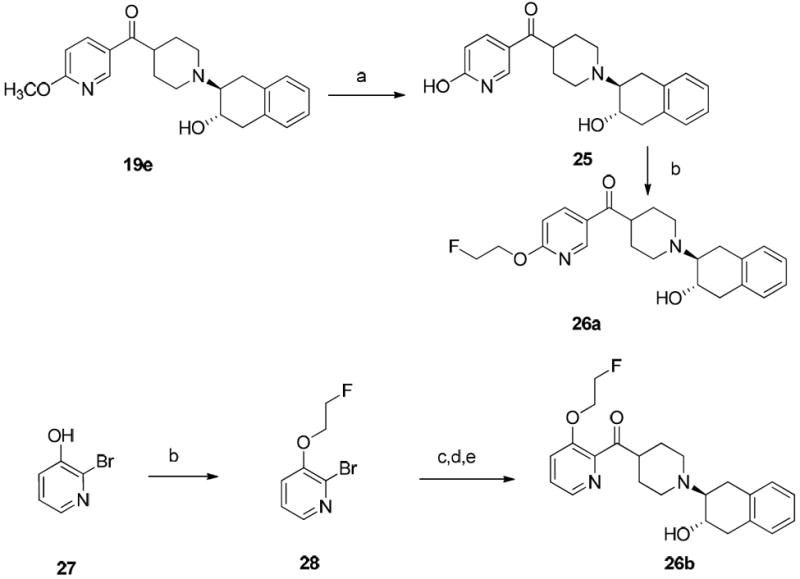
aReagents and conditions:
(a) BBr3 or TMSI; (b) Br(CH2)2F, K2CO3, DMF, rt, 72 h; (c) tert-butyl 4-(methoxy(methyl)carbamoyl) piperidine-1-carboxylate (11), BuLi, THF, -78 °C; (d) TFA, CH2Cl2, rt, 4 h; (e) 1,4-dihydronaphthalene oxide, NEt3, EtOH, 75 °C, 36 h.
To make aminobenzovesamicol (ABV) analogues containing substituted heteroaromatic carbonyl groups, namely 30a-d and 31a, the corresponding piperidine precursors 17a-d containing substituted heteroaromatic carbonyl groups were reacted with 2,2,2-trifluoro-N-(1a,2,7,7atetrahydronaphtho[ 2,3-b]oxiren-3-yl)acetamide (29), which was synthesized by following the literature protocol.4 The trifluoroacetyl protection group was then removed via hydrolysis in the presence of sodium hydroxide to afford the regioisomers 30a-d and 31a as shown in Scheme 5.
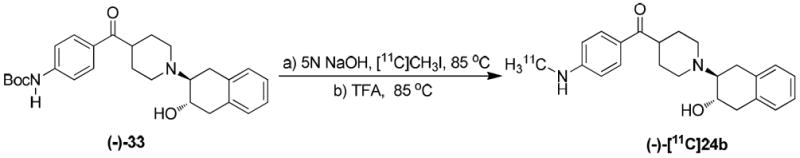
In vitro binding studies revealed that 24b was highly potent. Therefore, the (-)-24b and (+)-24b were obtained by separating the enantiomers on HPLC using chiralcel OD column. The precursor (-)-33 for the radiolabeling of (-)-[11C]24b was synthesized as shown in Scheme 6. Briefly, the enantiomeric separation of 6 was performed on chiral HPLC using Chiralcel OD column to give (+)-6 and (-)-6. The (-)-6 isomer was treated with Boc anhydride in the presence of triethylamine to give the tri-Boc protected intermediate (-)-32. 4-Dimethylaminopyridine (DMAP) was used in stoichiometric amount in this reaction. The tri-Boc protected compound upon treatment with potassium carbonate in methanol under reflux selectively deprotected one Boc group on the aniline nitrogen to give the precursor (-)-33, which was used for radiosynthesis of (-)-[11C]24b.
Scheme 6a. Synthesis of the precusor (-)-33.
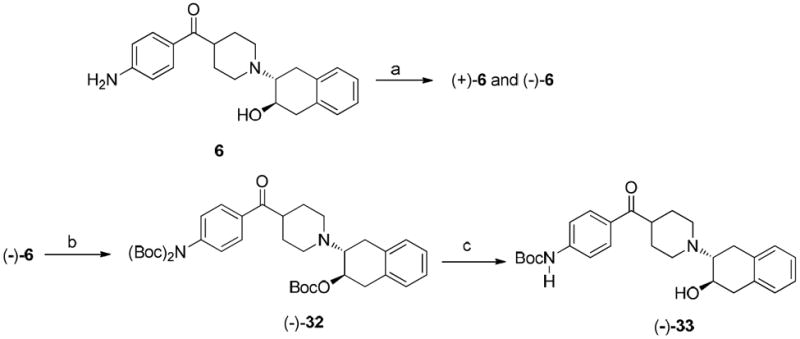
aReagents and conditions:
(a) Separation of enantiomers of 6 on HPLC: column: Chiralcel OD; mobile phase: 34% isopropanol in hexane; flow rate: 4.0 mL/min; (+)-enantiomer at 20.8 min and (-)-enantiomer at 33 min; (b) (Boc)2O, DMAP, Et3N, THF, rt., 1.5 h; (c) K2CO3, MeOH, reflux, overnight.
The radiosynthesis of (-)-[11C]24b was successfully accomplished in two steps from the precursor as shown in Scheme 7. The precursor was first reacted with [11C]CH3I to obtained the intermediate [11C]methylated Boc protected compound. This intermediate was then treated with trifluoroacetic acid in situ to obtain the Boc deprotected product (-)-[11C]24b in high radiochemical yield (40-50%). The new PET radiotracer obtained was subjected to biodistribution evaluations in Sprague–Dawley rats and microPET imaging studies in nonhuman primates.
Scheme 7. Radiosynthesis of (-)-[11C]24b.
In Vitro Binding Studies
VAChT binding was assayed using highly expressed human VAChT with gently homogenized and partially clarified PC12123.7 cells by displacement of bound 5 nM (-)-[3H]vesamicol. Apparent equilibrium dissociation constants (Ki, nM) are reported in Table 1. The σ1 and σ2 binding affinities were assayed in rat brain and in guinea pig membranes, respectively. Apparent equilibrium dissociation constants (Ki, nM) are reported in Table 1. The ligand selectivity is defined in terms of an index that is the Ki for σ1 or σ2 receptors divided by Ki for VAChT (Ki σ1/Ki VAChT or Ki σ2/Ki VAChT). A larger number represents good selectivity for binding to VAChT.
Table 1.
Binding affinities of new analogues for VAChT, σ1 receptor, and σ2 receptor
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
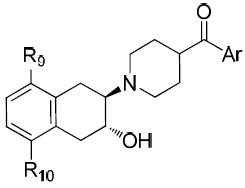
| Compd | Ar | R9 | R10 | Ki (nM) ± SEMa | VAChT selectivity | ALog De | |||
| VAChTb | σ1c | σ2d |
|
|
|||||
| 5 |
|
H | H | 4.30 ± 1.00 | 220 ± 17 | 320 ± 7 | 51 | 74 | 2.78 |
| 6 |
|
H | H | 1.68 ± 0.14 | NA | NA | NA | NA | 1.56 |
| 7 |
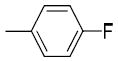
|
H | H | 2.70 ± 0.40 | 191 ± 58 | 251 ± 39 | 71 | 93 | 2.99 |
| 19a |
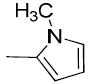
|
H | H | 18.4 ± 2.5 | 1300 ± 93 | 2550 ± 340 | 71 | 140 | 1.42 |
| 19b |
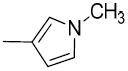
|
H | H | 26.6 ± 3.8 | 3040 ± 190 | 7710 ± 1060 | 110 | 290 | 1.84 |
| 19c |
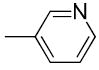
|
H | H | 24.1 ± 2.8 | 12900 ± 3900 | 3030 ± 70 | 540 | 130 | 1.83 |
| 19d |
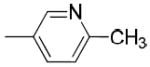
|
H | H | 27.9 ± 8.0 | 2120 ± 290 | 3770 ± 890 | 76 | 140 | 2.29 |
| 19e |
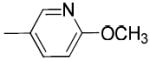
|
H | H | 8.36 ± 0.68 | 986 ± 160 | 2570 ± 140 | 120 | 310 | 2.64 |
| 19f |
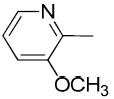
|
H | H | 121 ± 29 | 21000 ± 3800 | 1560 ± 60 | 170 | 13 | 2.96 |
| 19g |
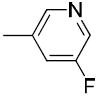
|
H | H | 10.1 ± 1.5 | 709 ± 79 | 1690 ± 60 | 70 | 170 | 1.99 |
| 19h |
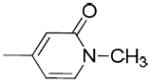
|
H | H | 66.2 ± 10.2 | 3350 ± 50 | 2120 ± 250 | 51 | 32 | 0.60 |
| 19i |
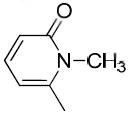
|
H | H | 182 ± 65 | 2160 ± 180 | 3340 ± 130 | 12 | 18 | 0.64 |
| 19j |
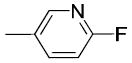
|
H | H | 26.1 ± 2.4 | 682 ± 27 | 1750 ± 90 | 26 | 67 | 2.02 |
| 19k |
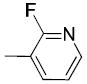
|
H | H | 12.7 ± 1.1 | 1300 ± 50 | 3530 ± 330 | 100 | 280 | 1.65 |
| 24a |
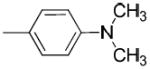
|
H | H | 0.93 ± 0.09 | 40.9 ± 8.2 | 4080 ± 100 | 44 | 4400 | 3.24 |
| 24b |
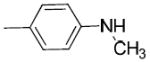
|
H | H | 3.03 ± 0.48 | 305 ± 22 | 6180 ± 540 | 100 | 2000 | 2.61 |
| 26a |
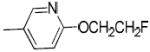
|
H | H | 38.0 ± 3.8 | 2120 ± 50 | 1860 ± 140 | 56 | 49 | 2.87 |
| 26b |
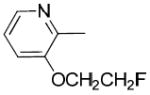
|
H | H | 76.4 ± 8.3 | 20000 ± 6800 | 303 ± 130 | 260 | 4.0 | 3.18 |
| 30a |
|
NH2 | H | 38.7 ± 7.0 | 7660 ± 230 | 6990 ± 1480 | 200 | 180 | 0.53 |
| 30b |
|
NH2 | H | 118 ± 24 | 20300 ± 8100 | 5080 ± 220 | 170 | 43 | 0.87 |
| 30c |
|
NH2 | H | 48.0 ± 11.4 | 11400 ± 2000 | 27500 ± 6900 | 240 | 570 | 1.33 |
| 30d |
|
NH2 | H | 23.3 ± 3.4 | 5680 ± 30 | 10800 ± 1300 | 120 | 220 | 1.66 |
| 31a |
|
H | NH2 | 2310 ± 390 | 9240 ± 790 | 11400 ± 2500 | 4.0 | 4.9 | 0.53 |
| (-)-24b |
|
H | H | 0.78 ± 0.07 | 992 ± 21 | 11443 ± 690 | 1271 | 14670 | 2.61 |
| (+)-24b |
|
H | H | 19.0 ± 1.4 | 1170 ± 66 | 7765 ± 807 | 61.5 | 408 | 2.61 |
Ki values (mean ± SEM) were determined in at least three experiments for VAChT, σ1 and σ2 binding sites.
The VAChT binding assay used expressed human VAChT.
The σ1 binding assay used membrane preparations of guinea pig brain.
The σ2 binding assay used homogenates of rat liver.
Calculated value at pH 7.4 by ACD/Labs, version 7.0 (Advanced Chemistry Development, Inc., Canada).
In the previous studies, we had reported that a new class of carbonyl group containing analogues displayed high potency and high selectivity for VAChT;4 particularly, compound (1-(3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(thiophen-2-yl)methanone (9), which replaces the phenyl group in the structure of compound 5 with a 2-thiophene moiety, displays comparable affinity for VAChT, with a Ki value of 5.00 ± 1.20 nM. In the current study, to explore the structure-activity relationship, we first replaced the thiophen-2-yl group in compound 9 with 1-methyl-1H-pyrrol-2-yl or 1-methyl-1H-pyrrol-3-yl group to obtain compounds 19a and 19b. Both of these two compounds displayed moderately potent binding toward VAChT with the Ki values of 18.4 ± 2.5 and 26.6 ± 3.8 nM for compounds 19a and 19b, respectively (Table 1) for binding to VAChT. Compared to 9, compounds 19a and 19b displayed about 4-5-fold decrease in affinity for VAChT. However, the selectivity of 19a and 19b for VAChT versus σ1 receptor reached at least 71-fold and 114-fold respectively. More importantly, these modified structures (with a methyl group on the N-atom of the pyrrole) provide a site for labeling with carbon-11 via [11C]methyl iodide.
When the benzoyl group was replaced with pyridin-3-carbonyl, 6-methyl-pyridin-3-carbonyl, 6-methoxy-pyridin-3-carbonyl, or 6-fluoro-pyridin-3-carbonyl, compounds 19c, 19d, 19e, and 19j were obtained. The pyridine-3-carbonyl analogue 19c (Ki = 24.1 ± 2.8 nM) displayed 5-fold lower affinity for VAChT compared to its benzoyl counterpart 5 (Ki = 4.30 ± 1.00 nM). When the hydrogen on the 6-position of the pyridine-3-carbonyl group is replaced with a methyl group, methoxy group, or fluoride group, the order of the binding potency for VAChT is -OCH3> -F ≈ -CH3, with Ki values of 8.36 ± 0.68, 26.1 ± 2.4, and 27.9 ± 8.0 nM for compounds 19e, 19j, and 19d, respectively. This demonstrates that an electron donating group, -OCH3, at the 6-position of pyridin-3-carbonyl results in increased affinity for VAChT. When further replacing the methoxy in 19e (Ki value of 8.36 ± 0.68 nM) with fluoroethoxy to afford compound 26a (Ki value of 38.0 ± 3.8 nM), the affinity for VAChT decreased 4.5-fold.
When the substitution pattern in the pyridine ring of 19e is changed from the 6-methoxy-pyridin-3-carbonyl to that of 3-methoxy-pyridin-2-carbonyl (19f), binding affinity (Ki value) for VAChT dropped 15-fold from 8.36 ± 0.68 nM to 121 ± 29 nM. Replacing the methoxy group in 19f with a fluoroethoxy group to obtain compound 26b resulted in slight improvement in VAChT binding (Ki value of 76.4 ± 8.3 nM). The affinities of compounds 19f and 26b are lower than those of the structural isomers 19e and 26a.
The only difference among compounds 19g, 19j and 19k was the position of fluoro substitution in the pyridin-3-carbonyl group. The order of binding affinity toward VAChT is 19g ≈ 19k >19j, in which the fluorine atom is at 5-, 2- and 6- positions of the pyridine-3-carbonyl ring and the dissociation constants for VAChT are 10.1 ± 1.5, 12.7 ± 1.1 and 26.1 ± 2.4 nM, respectively. Compounds 19h and 19i are N-methyl-2-pyridone derivatives. The only difference between them is the position of the bridging carbonyl group located at the 4-position in 19h and at the 6-position in 19i. The binding affinities for VAChT are 66.2 ± 10.2 nM for 19h and 182 ± 65 nM for 19i.
Compound (4-aminophenyl)(1-(3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)methanone (6) has higher potency for VAChT (Ki = 1.68 ± 0.14 nM) and has suitable lipophilicity (ALog D = 1.56) for crossing the BBB. Introducing methyl group(s) on the aniline nitrogen by N-methylation is a conventional way to convert compound 6 into carbon-11 radiolabeled tracer using [11C]methyl iodide. Monomethylation and dimethylation afforded 24a (Ki = 0.93 ± 0.09 nM) and 24b (Ki = 3.03 ± 0.48 nM). Their VAChT binding affinities are comparable to that of compound 6 (Ki = 1.68 ± 0.14 nM). The ALog D values of 24a and 24b are 3.24 and 2.61 respectively, which suggests they have suitable lipophilicity for the BBB penetration. We experimentally measured the Log D value of 24b and found to be 2.60.
To test whether an amino group in the 5’-position or 8’-position of the 3’-hydroxy-1’,2’,3’,4’-tetrahydronaphthalen-2’-yl group affects affinity for VAChT, compounds 30a-d and 31a were synthesized and their binding affinities were determined. Compound 30a (Ki = 38.7 ± 7.0 nM) displayed 60-fold higher VAChT binding affinity than that of 31a (Ki = 2310 ± 390 nM). This result is consistent with the reported results in which the 8’-amino analogues displayed much higher potency than that of 5’-amino analogues.4 When aniline compounds 30a, 30b, 30c and 30d are compared with the non-aniline counterparts, 19a, 19c, 19d and 19e, there is no any significant change in binding affinities towards VAChT. This suggests that introducing the 5’-amino group into heteroaromatic carbonyl containing analogues may not be beneficial to VAChT binding affinity. Nevertheless, a large difference in binding affinity between the regioisomeric pairs 30a and 31a was observed.4
These substituted heteroaromatic carbonyl containing VAChT analogues have moderate to high VAChT binding affinities. Consequently, we screened their sigma receptor binding affinity. The in vitro data suggest that these new analogues had very low affinities for the sigma receptors. Except for compound 24a with Ki = 40.9 ± 8.2 nM, all other new analogues exhibit greater than 300 nM for σ1 receptor, as shown in Table 1. Among these new analogues, 6 compounds 19a, 19e, 19g, 19k, 24a and 24b have Ki values less than 20 nM toward VAChT. Compounds 19a and 19g have >70-fold selectivity ratios for VAChT vs either sigma receptor type. Importantly, compounds 19e, 19k and 24b not only display high affinity toward VAChT with Ki values of 8.36 ± 0.68, 12.7 ± 1.1 and 3.03 ± 0.48 nM respectively, but they also display greater than 100-fold selectivity ratios for VAChT versus sigma receptors. Compound 24a has the highest potency for VAChT (0.93 ± 0.09 nM) and is highly selective for VAChT versus σ1 (44-fold) and σ2 (4400-fold) receptors. Thus, compound 24a can be a good blocking agent for validating the selective binding of other VAChT radioligands.
To further test the feasibility of the lead compounds reported in this manuscript to sever as PET tracer for imaging VAChT in vivo in living animal, racemic compound 24b was resolved by HPLC using chiralcel OD column to obtain the (+)-24b and (-)-24b. The in vitro data revealed that (-)-24b (Ki-VAChT = 0.78 nM) is the more potent isomer than the (+)-24b (Ki-VAChT = 19.0 nM). This is consistent to that ligand binding to VAChT is stereoselective.
In Vivo Evaluation in Rats
To check the in vivo distribution of (-)-[11C]24b and its washout kinetics from brain regions of interest and peripheral tissues, ex-vivo biodistribution was conducted in male Sprague-Dawley rats (185 – 205 gram). Rats were euthanized at 5 and 30 min post i.v. injection of (-)-[11C]24b. To test the ability of (-)-[11C]24b to cross the blood brain barrier (BBB), rats were pretreated with Cyclosporine A (CycA) 30 min prior to injection of the radiotracer dose and then euthanized at 30 min post injection of the radiotracer. The distribution data obtained in this study were shown in Figure 3 and 4. For the normal rats, the uptake of radioactivity at 5 min was 0.34, 0.49, 1.77, 0.22, 0.18, 0.89, 1.19, 1.47, 3.45, 0.57 % injected dose per gram (%I.D./g) in blood, heart, lung, muscle, fat, pancreas, spleen, kidney, liver and brain respectively post intravenous (i.v.) injection and the liver has the highest uptake; the radioactivity decreased in all the tissues that were collected in the studies from 5 to 30 min as shown and the liver retained the highest uptake as 2.44 at 30 min. For the rats pretreated with CycA, except the uptake (%I.D./g) in kidney displayed slightly increase (0.77 in control rats, 1.21 in rats pretreated with CycA), the other peripheral tissues didn’t show significant change (Figure 3). For the brain regions of interest, the uptake (%I.D./g) at 30 min in cerebellum, striatum, brain stem, cortex, thalamus, hippocampus, and brain were 0.157, 0.593, 0.245, 0.262, 0.293, 0.265 and 0.272 respectively for the control rats; for the rats pretreated with CycA, the uptakes (%I.D.) were 0.404, 1.020, 0.577, 0.637, 0.600, 0.562 and 0.596 respectively for corresponding brain regions of interest. The uptake in brain was observed to increase 2.2-fold when the rats were pretreated with CycA. P-glycoprotein (P-gp) is a 170-kDa protein that is able to binding with compounds with diversify structures and it has widespread tissue distribution belonging to the adenosine triphosphate (ATP)-binding cassette (ABC) transporters42, 43 and it is expressed in the capillary endothelial cells which comprise the blood-brain barrier.42 CycA, is a modulator/inhibitor of the adenosine triphosphate (ATP)-binding cassette (ABC) transporters including P-gp. It was frequently used to evaluate the ABC transporter-mediated efflux of PET radiotracers and it suggested that ABC transporter-mediated efflux of PET radiotracers has species difference.44 If a radioligand is a substrate of P-gp, rats pretreated with CycA will results in significantly increase of the uptake in the brain of animals, for example, radioligands, [11C]verapramil,45, 46 [11C]GR21823147 and benzamide D3 radioligands.48 For radioligand, (-)-[11C]24b, using CycA pretreated the rats displayed only 2.2-fold increase in the brain uptake of the rats. Considering the brain uptake (%I.D./g) of radioactivity reached 0.57 at 5 min. we concluded that the P-gp modulating the brain uptake of (-)-[11C]24b is very weak and (-)-[11C]24b is able to penetrate the BBB and displayed sufficient uptake in the brain of the rats.
Figure 3.
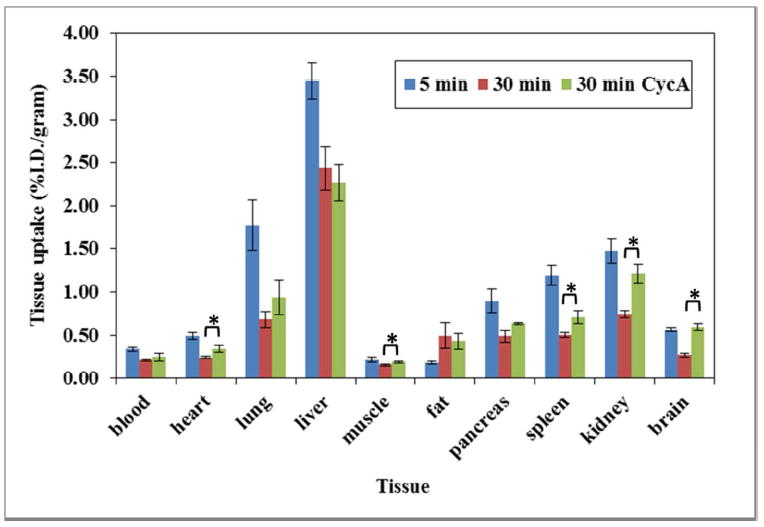
The biodistribution of (-)-[11C]24b in Sprague-Dawley Rats (185 – 205 gram). Control rats were euthanized at 5 and 30 min. Rats pretreated with at 25 mg/Kg of CycA 30 min prior to injection of the radiotracer. The star (*) in the figure are denoted that the %I.D./gram value from heart, muscle, spleen and brain were significantly different between non-CycA treated and CycA treated group (n= 4, student t test, p-value <0.05).
Figure 4.
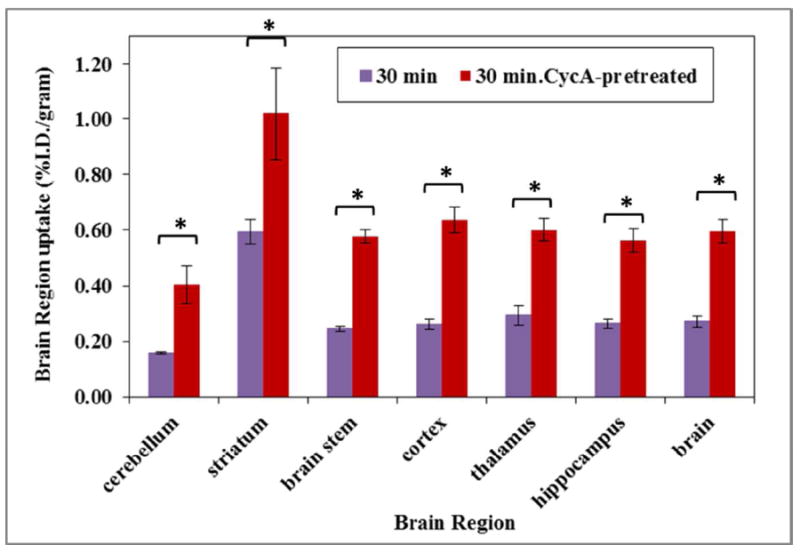
The brain regional uptake of (-)-[11C]24b in Sprague-Dawley rats at 30 min in control rats and rats pretreated with 25 mg/Kg of CycA 30 min prior to injection of the radiotracer. The star (*) in the figure are denoted that the % I.D./g value from all brain regions were significantly different between non-CycA treated and CycA treated group (n= 4, student t test, p-value <0.05).
MicroPET Studies in Monkeys
To further confirm that (-)-[11C]24b is able to bind with VAChT in the brain in vivo, microPET studies of (-)-[11C]24b in the brain of nonhuman primate, male cynomolgus monkey were performed (n = 3). The microPET studies demonstrated that (-)-[11C]24b is able to enter the brain and has highest accumulation in striatum, the VAChT enriched area in the regions of interest in the brain (Figure 5). (-)-[11C]24b is able to give sufficient contrast for the striatum versus other non-target regions. The tissue-time activity curves post injection of (-)-[11C]24b revealed that the radioactivity accumulation was the highest at 10 min post-injection. The ratio of radioactivity accumulation in the target (striatum) and non-target (cerebellum) reaches > 2.5 fold after 70 min post injection (Figure 5). This data suggests that (-)-[11C]24b is a promising PET tracer to quantify VAChT in vivo.
Figure 5.
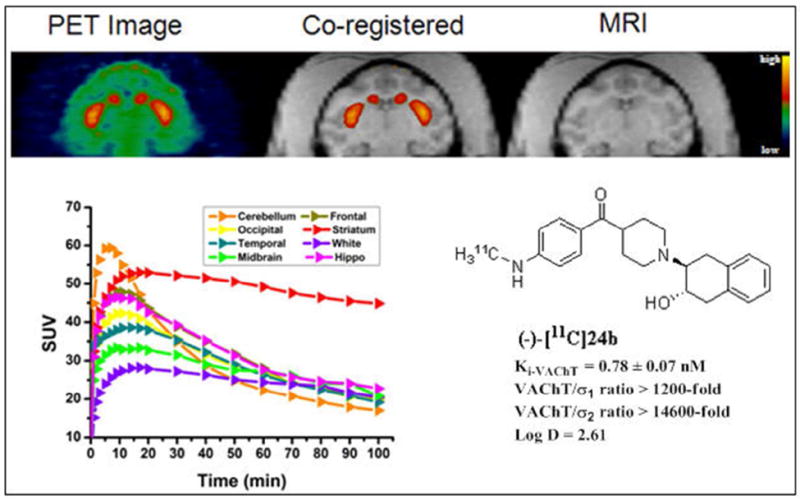
MicroPET imaging studies of (-)-[11C]24b in cynomolgus monkey. MicroPET image (top left), co-registered image (top middle), MRI image (top right), time tissue-activity curve (left bottom).
Conclusion
In the present study, we successfully synthesized a series of new VAChT inhibitors by replacing the benzoyl group in 5 with a heteroaromatic carbonyl group or replacing the 4-aminobenzoyl group with the 4-methylaminobenzoyl group or the 4-N,N-dimethylaminobenzoyl group. The six compounds 19a, 19e, 19g, 19k, 24a and 24b displayed high VAChT binding affinities (Ki values ranging from 0.93 nM to 18.4 nM) and good selectivity for VAChT over sigma receptors. In particular, compounds 19e, 19k and 24b are very potent for VAChT with Ki values of 8.36 ± 0.68, 12.7 ± 1.1 and 3.03 ± 0.48 nM, respectively; and binding selectivity ratios equal to or greater than 100-fold for VAChT over sigma receptors. The racemate 24b was Successfully resolved on HPLC to obtain the minus and plus isomers. In vivo validation of (-)-[11C]24b in rodents and nonhuman primates demonstrated that (-)-[11C]24b is able to penetrate the BBB and has highest accumulation in the striatum, the VAChT enriched area in the brain. The time tissue-activity of (-)-[11C]24b in the brain of cynomolgus monkey revealed that it has favorable washout pharmacokinetics in the brain. Further imaging studies of (-)-[11C]24b in nonhuman primate are warranted to test the feasibility of (-)-[11C]24b to be a promising candidate for assessing the level of VAChT in the brain.
Experimental Section
General
All reagents and chemicals were purchased from commercial suppliers and used without further purification unless otherwise stated. All anhydrous reactions were carried out in oven-dried glassware under an inert nitrogen atmosphere unless otherwise stated. When the reactions involved extraction with methylene chloride (CH2Cl2), chloroform (CHCl3), or ethyl acetate (EtOAc), the organic solutions were dried with anhydrous Na2SO4 and concentrated on a rotary evaporator under reduced pressure. Melting points were determined on the MEL-TEMP 3.0 apparatus and left uncorrected. 1HNMR spectra of majority of the compounds were recorded at 300 MHz on a Varian Mercury-VX spectrometer with CDCl3 as solvents and tetramethylsilane (TMS) was used as the internal standard. Varian 400 MHz NMR was used for some compounds and was reported in the experimental section where appropriate. Elemental analyses (C, H, N) were determined by Atlantic Microlab, Inc. and were found to be within 0.4% of theoretical values. Chiralcel OD column was used for normal phase HPLC to resolve enantiomers. Phenomenex Luna C18 column was used for reverse phase HPLC conditions to purify the radioactive product on HPLC, and Phenomenex Prodigy was used for QC analysis of radiotracer.
Resolution of (4-aminophenyl)(1-(3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)-piperidin-4-yl)metha-none (6) to obtain the minus isomer (-)-6 and the plus isomer (+)-6
Approximately 105.0 mg of racemate 6 was resolved on HPLC using a Chiralcel OD column (250 mm × 10 mm) under isocratic conditions (34% isopropanol in hexane) at a flow rate of 4.0 mL/min and UV wave length at 254 nm to give 41.0 mg of (+)-6 (Rt = 20.8 min) and 48.0 mg of (-)-6 (Rt = 33 min). The specific rotation was determined on an automatic polarimeter (Autopol 111, Rudolph Research, Flanders, NJ). The optical rotation was [α]D = -50° for (-)-6 and [α]D = +63° for (+)-6 at the concentration of 1.0 mg/mL in dichloromethane at 20 °C.
tert-Butyl 4-(methoxy(methyl)carbamoyl)piperidine-1-carboxylate (11)
To a solution of 10 (2.5 g, 10.9 mmol) in CH2Cl2 (30 mL) at room temperature, CDI (1.77 g, 11.0 mmol) was added. The mixture was stirred at room temperature for 1 h and N, O-dimethylhydroxyamine hydrochloride (1.3 g, 13.3 mmol) followed by Et3N were added. The reaction mixture was stirred overnight and was washed with aqueous Na2CO3, water, dried and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give 11 as a white solid (2.70 g, 90%). mp: 54 °C. 1H NMR (CDCl3): δ 1.46 (s, 9H), 1.64-1.72 (m, 4H), 2.78 (s br, 3H), 3.18 (s, 3H), 3.71 (s, 3H), 4.13 (s br, 2H).
Procedure A: General Method for the Synthesis of Compounds 15h-i
4-Bromo-1-methylpyridin-2(1H)-one (15h)
A solution of 4-bromopyridin-2-amine 12h (0.60 g, 3.5 mmol) in a mixture of 2 N H2SO4 (20 mL) and 2 N NaNO2 (10 mL) was stirred at 0-5 °C for 2 h. The reaction mixture was extracted with CH2Cl2. Crude product was used in the next step without further purification. To the above crude product, potassium carbonate (0.50 g, 3.6 mmol) and methyl iodide (0.53 g, 3.7 mmol) were added and heated at 80 °C in acetone (100 mL) in a sealed tube for 4 h. The reaction mixture was cooled, and potassium carbonate was filtered off. Acetone was evaporated and a small amount of water was added to the residue. This solution was extracted with CH2Cl2 and the product was purified with silica gel column chromatography to afford 15h as a yellow solid (355 mg, 57%). 1H NMR (CDCl3): δ 3.49 (s, 3H), 6.31 (d, J = 6.0 Hz, 1H), 6.82 (s, 1H), 7.14 (d, J = 6.0 Hz, 1H).
6-Bromo-1-methylpyridin-2(1H)-one (15i)
Compound 15i as a yellow solid was synthesized starting with compound 12i as described in procedure A. mp: 105 °C. Yield, 54 %. 1H NMR (CDCl3,): δ 3.73 (s, 3H), 6.46-6.52 (m, 2H), 7.10-7.16 (m, 1H).
tert-Butyl 4-(1-methyl-1H-pyrrole-2-carbonyl)piperidine-1-carboxylate (16a)
To a schlenk flask containing N-methylpyrrole (0.49 g, 6.08 mmol), 20 mL of THF was added while stirring at room temperature until a clear solution was formed. The solution was cooled down to -78 °C for 15 min and 12.0 mL (6.08 mmol) of n-BuLi was added. The solution was allowed to warm up to 0 °C for 20 min, resulting in the formation of the yellow lithium intermediate. The resulting mixture was added via cannula into a solution of 11 (0.83 g, 3.04 mmol) in THF (20 mL) at the same temperature and the reaction mixture was stirred at -78 °C for 4 h, then quenched with saturated aqueous NH4Cl and allowed to warm to room temperature. The mixture was washed with brine and dried over Na2SO4, filtered, and evaporated under reduced pressure to give the crude product. The crude compound was purified by silica gel chromatography to obtain the desired compound 16a (0.61 g, 34%). 1H NMR (CDCl3): δ 1.47 (s, 9H), 1.66-1.80 (m, 4H), 2.83 (s, 2H), 3.17 (m, 1H), 3.94 (s, 3H), 4.18 (s br, 2H), 6.15 (dd, J = 4.2, 2.4 Hz, 1H), 6.84 (pseudo t, J = 1.8 Hz, 1H), 6.99 (dd, J = 4.2, 1.5 Hz, 1H).
tert-Butyl 4-(1-methyl-1H-pyrrole-4-carbonyl)piperidine-1-carboxylate (16b)
To a solution of 3-bromo-1-methyl-1H-pyrrole (2.19 g, 13.8 mmol) in anhydrous THF at -78 °C, n-BuLi (1.6 M, 10 mL) was added. The solution was stirred for 45 minutes at -78 °C. The resulting mixture was cannulated into a solution of 11 (2.5 g, 9.2 mmol) in THF (80 mL) at the same temperature and the reaction mixture was stirred at -78 °C for 4 h, then quenched with sat. aqueous NH4Cl and allowed to warm to room temperature. The mixture was washed with brine and dried over Na2SO4, filtered, and evaporated under reduced pressure to give the crude product. The crude compound was purified by silica gel chromatography to obtain the desired compound 16b (1.07 g, 40%). 1H NMR (CDCl3): δ 1.47 (s, 9H), 1.66-1.77 (m, 4H), 2.83 (s, 2H), 3.15-3.18 (m, 1H), 3.70 (s, 3H), 4.13 (s br, 2H), 6.57-6.61 (m, 2H), 7.26-7.29 (m, 1H).
tert-Butyl 4-nicotinoylpiperidine-1-carboxylate (16c)
To a solution of 3-bromopyridine (1.13 g, 7.2 mmol) in anhydrous THF (45 mL) at -40 °C under argon atmosphere, lithium dibutyl(isopropyl)magnesate (5.2 mL, 0.7 M, 3.6 mmol) was added. Stirring was continued at the same temperature for 1 h. The resulting mixture was cannulated into a solution of 11 (1.5 g, 5.5 mmol) in THF (50 mL) at -78 °C. The solution was maintained at -78 °C for 1 h, and quenched with saturated aqueous NH4Cl and allowed to warm to room temperature. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with water, dried over Na2SO4, filtered, and evaporated under reduced pressure to give the crude product. The crude compound was purified by silica gel chromatography to obtain the desired compound 16c (0.49 g, 31%). 1H NMR (CDCl3): δ 1.47 (s, 9H), 1.63-1.89 (m, 4H), 2.92 (m, 2H), 3.38 (m, 1H), 4.20 (m, 2H), 7.44 (ddd, J = 8.1, 5.1, 0.9 Hz, 1H), 8.22 (dt, J = 8.1, 2.1 Hz, 1H), 8.79 (dd, J = 5.1, 1.8 Hz, 1H), 9.16 (d, J = 1.8 Hz, 1H).
tert-Butyl 4-(6-methylnicotinoyl)piperidine-1-carboxylate (16d)
Compound 16d was prepared from 5-bromo-2-methylpyridine following the procedure described above for the preparation of 16c. Yield, 42%. 1H NMR (CDCl3): δ 1.49 (s, 9H), 1.68-1.87 (m, 4H), 2.63 (s, 3H), 2.90 (m, 2H), 3.35 (m, 1H), 4.18 (s br, 2H), 7.28 (d, J = 8.4 Hz, 1H), 8.11 (dd, J = 8.4, 2.1 Hz, 1H), 9.04 (d, J = 2.1 Hz, 1H).
tert-Butyl 4-(6-methoxynicotinoyl)piperidine-1-carboxylate (16e)
Compound 16e was prepared from 5-bromo-2-methoxypyridine following the procedure described above for the preparation of 16b. Yield, 61%. 1H NMR (CDCl3): δ 1.47 (s, 9H), 1.62-1.86 (m, 4H), 2.88 (m, 2H), 3.31 (m, 1H), 4.01 (s, 3H), 4.19 (s br, 2H), 6.81 (d, J = 8.7 Hz, 1H), 8.13 (dd, J = 8.7, 2.4 Hz, 1H), 8.79 (d, J = 2.4 Hz, 1H).
tert-Butyl 4-(3-methoxypicolinoyl)piperidine-1-carboxylate (16f)
Compound 16f was prepared from 2-bromo-3-methoxypyridine following the procedure described above for the preparation of 16b. Yield, 36%. 1H NMR (CDCl3): δ 1.47 (s, 9H), 1.66-1.87 (m, 4H), 2.89 (s, 2H), 3.67-3.81 (m, 1H), 3.90 (s, 3H), 4.07 (s br, 2H), 7.35-7.45 (m, 2H), 8.23-8.25 (m, 1H).
tert-Butyl 4-(3-fluoronicotinoyl)piperidine-1-carboxylate (16g)
Compound 16g was prepared from 3-bromo-5-fluoropyridine following the procedure described above for the preparation of 16b. Yield, 41%. 1H NMR (CDCl3): δ 1.44 (s, 9H), 1.56-1.61 (m, 2H), 1.86-1.90 (m, 2H), 2.87 (s, 2H), 3.17-3.24 (m, 1H), 4.06 (s br, 2H), 7.54-7.58 (m, 1H), 8.53-8.60 (m, 2H).
tert-Butyl 4-(1-methyl-2-oxo-1,2-dihydropyridine-4-carbonyl)piperidine-1-carboxylate (16h)
Compound 16h was prepared from compound 15h following the procedure described above for the preparation of 16b. Yield, 38%. 1H NMR (CDCl3): δ 1.47 (s, 9H), 1.56-1.87 (m, 4H), 2.82 (s, 2H), 3.05-3.07 (m, 1H), 3.50 (s, 3H), 4.17 (s br, 2H), 6.49 (d, J = 6.0 Hz, 1H), 6.73 (d, J = 6.0 Hz, 1H), 7.36 (s, 1H).
tert-Butyl 4-(1-methyl-6-oxo-1,6-dihydropyridine-2-carbonyl)piperidine-1-carboxylate (16i)
Compound 16i was prepared from compound 15i following the procedure described above for the preparation of 16b. Yield, 35%. 1H NMR (CDCl3): δ 1.45 (s, 9H), 1.58-1.87 (m, 4H), 2.81 (s, 2H), 3.05-3.07 (m, 1H), 3.47 (s, 3H), 4.14 (s br, 2H), 6.44-6.47 (m, 1H), 6.69-6.72 (m, 1H), 7.30-7.36 (m, 1H).
Procedure B: General Method for Synthesis of Compounds 17a-i
(1-Methyl-1H-pyrrol-2-yl)(piperidin-4-yl)methanone (17a)
To a solution of 16a (0.67 g, 2.3 mmol) in CH2Cl2 (30 mL), TFA (2 mL) was added. The reaction mixture was stirred at room temperature for 4 h. The solvent was removed under reduced pressure and the residue was neutralized with 1 N NaOH solution and extracted with CH2Cl2 (2 × 15 mL). The organic layer was washed with brine, dried and concentrated to give crude product. The crude product was purified by silica gel column chromatography to afforded 17a (0.32 g, 72%). 1H NMR (CDCl3): δ 1.76-1.87 (m, 4H), 2.81 (m, 2H), 3.17-3.23 (m, 4H), 3.94 (s, 3H), 6.14 (m, 1H), 6.83 (m, 1H), 6.98 (m, 1H).
(1-Methyl-1H-pyrrol-3-yl)(piperidin-4-yl)methanone (17b)
Compound 17b was prepared from compound 16b as described in procedure B. Yield, 80 %. 1H NMR (CDCl3): δ 1.76-1.80 (m, 4H), 2.65-2.73 (m, 2H), 3.01-3.17 (m, 4H), 3.68 (s, 3H), 6.57-6.59 (m, 2H), 7.25 (s, 1H).
Piperidin-4-yl(pyridin-3-yl)methanone (17c)
Compound 17c was prepared from compound 16c as described in procedure B. Yield, 85%. 1H NMR (CDCl3): δ 1.62-1.77 (m, 2H), 1.86-1.89 (m, 3H), 2.79 (td, J = 12.6, 2.7 Hz, 2H), 3.21 (dt, J = 12.9, 3.0 Hz, 2H), 3.37 (tt, J = 11.4, 3.9 Hz, 1H), 7.43 (ddd, J = 8.1, 4.8, 0.9 Hz, 1H), 8.22 (ddd, J = 4.8, 2.1, 1.5 Hz, 1H), 8.78 (dd, J = 4.5, 1.5 Hz, 1H), 9.16 (dd, J = 2.4, 0.9 Hz,1H).
(6-Methylpyridin-3-yl)(piperidin-4-yl) methanone (17d)
Compound 17d was prepared from compound 16d as described in procedure B. Yield, 70%. 1H NMR (CDCl3): δ 1.62-1.87 (m, 5H), 2.63 (s, 3H), 2.77 (m, 2H), 3.16-3.22 (m, 2H), 3.33 (m, 1H), 7.27 (d, J = 8.4 Hz, 1H), 8.12 (dd, J = 8.1, 2.4 Hz, 1H), 9.04 (d, J = 2.4 Hz, 1H).
(6-Methoxypyridin-3-yl)(piperidin-4-yl)methanone (17e)
Compound 17e was prepared from compound 16e as described in procedure B. Yield, 77%. 1H NMR (CDCl3): δ 1.65-1.89 (m, 4H), 2.40 (s, 1H), 2.78 (td, J = 12.0, 2.7 Hz, 2H), 3.21 (dt, J = 9.0, 3.6 Hz, 2H), 3.31 (m, 1H), 4.00 (s, 3H), 6.79 (d, J = 8.4 Hz, 1H), 8.13 (dd, J = 8.4, 2.4 Hz, 1H), 8.78 (d, J = 2.4 Hz, 1H).
(3-Methoxypyridin-2-yl)(piperidin-4-yl)methanone (17f)
Compound 17f was prepared from compound 16f as described in procedure B. Yield, 78%. 1H NMR (CDCl3): δ 1.51-1.81 (m, 4H), 2.69 (s, 2H), 3.08-3.25 (m, 3H), 3.49-3.58 (m, 1H), 3.83 (s, 3H), 7.27-7.36 (m, 2H), 8.16-8.18 (m, 1H).
(5-Fluoropyridin-3-yl)(piperidin-4-yl)methanone (17g)
Compound 17g was prepared from compound 16g as described in procedure B. Yield, 79%. 1H NMR (CDCl3): δ 1.56-1.61 (m, 2H), 1.86-1.90 (m, 2H), 2.87 (m, 2H), 3.17-3.21 (m, 4H), 7.56 (s, 1H), 8.53-8.60 (m, 2H).
1-Methyl-5-(piperidine-4-carbonyl)pyridin-2(1H)-one (17h)
Compound 17h was prepared from compound 16h as described in procedure B. Yield, 79%. 1H NMR (CDCl3): δ 1.57-1.88 (m, 4H), 2.83 (s, 2H), 3.03-3.09 (m, 4H), 3.50 (s, 3H), 6.50 (d, J = 6.0 Hz, 1H), 6.72 (d, J = 6.0 Hz, 1H), 7.38 (s, 1H).
1-Methyl-6-(piperidine-4-carbonyl)pyridin-2(1H)-one (17i)
Compound 17i was prepared from compound 16i as described in procedure B. Yield, 75%. 1H NMR (CDCl3): δ 1.58-1.87 (m, 4H), 2.82 (s, 2H), 3.02-3.10 (m, 4H), 3.47 (s, 3H), 6.42-6.48 (m, 1H), 6.67-6.72 (m, 1H), 7.31-7.36 (m, 1H).
Procedure C: General Method of Preparing (19a-i) and Their Corresponding Oxalates
(1-(3-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(1-methyl-1H-pyrrol-2-yl)methanone (19a)
A mixture of 17a (0.27 g, 1.4 mmol), 18 (0.10 g, 0.68 mmol) and Et3N (0.3 mL, 2.2 mmol) in ethanol (5 mL) was stirred at 75 °C for 36 h. After cooling to room temperature, the reaction mixture was poured into water and extracted with EtOAc (3 × 15 mL). The residue was purified by silica gel column chromatography to give 19a as a white solid (0.11 g, 47%). 1H NMR (CDCl3, free base): δ 1.81-2.03 (m, 4H), 2.37 (m, 1H), 2.76-3.13 (m, 8H), 3.30 (m, 1H), 3.88 (m, 1H), 3.95 (s, 3H), 4.45 (s br, 1H), 6.14 (m, 1H), 6.83 (m, 1H), 6.98 (m, 1H), 7.09-7.16 (m, 4H). Free base was converted to the corresponding oxalate salt by adding oxalic acid in ethyl acetate to 19a in CH2Cl2. mp: 212 °C (decomposed). Anal. (C21H26N2O2 • H2C2O4) C, H, N.
(1-(3-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(1-methyl-1H-pyrrol-3-yl)methanone (19b)
Compound 19b was prepared from compound 17b as described in procedure C. Yield, 50%. 1H NMR (CDCl3, free base): δ 1.83-2.00 (m, 4H), 2.38 (m, 1H), 2.76-3.13 (m, 8H), 3.29-3.35 (m, 1H), 3.71 (s, 3H), 4.32 (s br, 1H), 6.61 (m, 2H), 7.12-7.16 (m, 4H), 7.30-7.31 (m, 1H). The free base was converted to the oxalate salt. mp: 212.6 °C (decomposed). Anal. (C21H26N2O2• H2C2O4•0.25H2O) C, H, N.
(1-(3-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(pyridin-3-yl)methanone (19c)
Compound 19c was prepared from compound 17c as described in procedure C. Yield, 40%. 1H NMR (CDCl3, free base): δ 1.77-1.98 (m, 4H), 2.43 (m, 1H), 2.75-3.02 (m, 7H), 3.20-3.38 (m, 2H), 3.88 (m, 1H), 7.08-7.15 (m, 4H), 7.42-7.46 (m, 1H), 8.21-8.25 (m, 1H), 8.77-8.79 (m, 1H), 9.16 (s, 1H). The free base was converted to the oxalate salt. mp: 214 °C (decomposed). Anal. (C21H24N2O2 • H2C2O4) C, H, N.
(1-(3-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(6-methoxypyridin-3-yl)methanone (19d)
Compound 19d was prepared from compound 17d as described in procedure C. Yield, 44%. 1H NMR (CDCl3, free base): δ 1.80-2.02 (m, 4H), 2.43 (m, 1H), 2.62 (s, 3H), 2.78-3.06 (m, 7H), 3.20-3.38 (m, 2H), 3.89 (m, 1H), 6.83 (d, J = 8.7 Hz, 1H), 7.11-7.18 (m, 4H), 8.17 (dd, J = 8.7, 2.4 Hz, 1H), 8.82 (d, J = 2.4 Hz, 1H). The free base was converted to the oxalate salt. mp: 223 °C (decomposed). Anal. (C22H26N2O2 • 2H2C2O4) C, H, N.
(1-(3-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(6-methylpyridin-3-yl) methanone (19e)
Compound 19e was prepared from compound 17e as described in procedure C. Yield, 42%. 1H NMR (CDCl3, free base): δ 1.74-1.96 (m, 4H), 2.41 (m, 1H), 2.75-3.01 (m, 7H), 3.26-3.33 (m, 2H), 3.86 (m, 1H), 4.04 (s, 3H), 4.17 (s br, 1H), 7.06-7.14 (m, 4H), 7.27 (d, J = 8.1 Hz, 1H), 8.11 (dd, J = 8.1, 2.1 Hz, 1H), 9.03 (d, J = 2.1 Hz, 1H). The free base was converted to the oxalate salt. mp: 240 °C (decomposed). Anal. (C22H26N2O3 • H2C2O4) C, H, N.
(1-(3-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(3-methoxypyridin-2-yl)methanone (19f)
Compound 19f was prepared from compound 17f as described in procedure C. Yield, 36%. 1H NMR (CDCl3, free base): δ 1.72-1.96 (m, 4H), 2.33-2.41 (m, 1H), 2.72-2.96 (m, 7H), 3.26-3.33 (m, 1H), 3.55-3.63 (m, 1H), 3.81-3.90 (m, 3H), 4.30 (s br, 1H), 7.06-7.14 (m, 4H), 7.26-7.39 (m, 2H), 8.23-8.25 (m, 1H). The free base was converted to the oxalate salt. mp: 146.6 °C (decomposed). Anal. (C22H26N2O3• H2C2O4•0.25H2O) C, H, N.
(5-Fluoropyridin-3-yl)(1-(3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)methanone (19g)
Compound 19g was prepared from compound 17g as described in procedure C. Yield, 44%. 1H NMR (CDCl3, free base): δ 1.70-2.04 (m, 4H), 2.34-2.43 (m, 1H), 2.75-3.34 (m, 8H), 3.82-3.90 (m, 1H), 4.13 (s br, 1H), 7.06-7.11 (m, 4H), 7.57-7.61 (m, 1H), 8.56-8.62 (m, 2H). The free base was converted to the oxalate salt. mp: 203.7 °C (decomposed). Anal. (C21H23FN2O2• H2C2O4) C, H, N.
4-(1-(3-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidine-4-carbonyl)-1-methylpyridin-2(1H)-one (19h)
Compound 19h was prepared from compound 17h as described in procedure C. Yield, 37%. 1H NMR (CDCl3, free base): δ 1.72-1.99 (m, 4H), 2.33-2.39 (m, 1H), 2.75-3.01 (m, 6H), 3.27-3.34 (m, 2H), 3.50 (s, 3H), 3.82-3.91 (m, 1H), 4.08 (s br, 1H), 6.45 (d, J = 6.0 Hz, 1H), 6.72 (d, J = 6.0 Hz, 1H), 7.12-7.13 (m, 4H), 7.35 (s, 1H). The free base was converted to the oxalate salt. mp: 93.8 °C (decomposed). Anal. (C22H26N2O3•H2C2O4•1.5H2O) C, H, N.
6-(1-(3-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidine-4-carbonyl)-1-methylpyridin-2(1H)-one (19i)
Compound 19i was prepared from compound 17i as described in procedure C. Yield, 39%. 1H NMR (CDCl3, free base): δ 1.72-2.04 (m, 4H), 2.32-2.39 (m, 1H), 2.81-3.01 (m, 6H), 3.27-3.35 (m, 2H), 3.50 (s, 3H), 3.82-3.89 (m, 1H), 4.06 (s br, 1H), 6.43-6.46 (m, 1H), 6.70-6.73 (m, 1H), 7.06-7.14 (m, 4H), 7.31-7.37 (m, 1H). The free base was converted to the oxalate salt. mp: 122.4 °C (decomposed). Anal. (C22H26N2O3•H2C2O4•1.5H2O) C, H, N.
Procedure D: General Method for the Synthesis of Compounds (19j-k) and Their Corresponding Oxalates
(6-Fluoropyridin-3-yl)(1-(3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)methanone (19j)
To a solution of 17j (2.42 g, 13.8 mmol) in anhydrous THF at -78 °C, n-BuLi (1.6 M, 10 mL) was added. The solution was stirred for 45 minutes at -78 °C. The resulting mixture was cannulated into a solution of 21 (3.97 g, 9.2 mmol) in THF (80 mL) at the same temperature and the reaction mixture was stirred at -78 °C for 4 h, then quenched with sat. aqueous NH4Cl and allowed to warm to room temperature. The mixture was washed with brine and dried over Na2SO4, filtered, and evaporated under reduced pressure to give the crude product. The crude compound was purified by chromatography on silica gel to obtain the TBDMS-protected intermediate (1-(3-(tert-butyldimethylsilyloxy)-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(6-fluoropyridin-3-yl) methanone (1.55 g, 36%). 1H NMR (CDCl3): δ 0.12 (s, 6H), 0.91 (s, 9H), 1.65-1.77 (m, 4H), 2.46-3.05 (m, 9H), 4.13 (s br, 2H), 7.04-7.12 (m, 4H), 8.23-8.37 (m, 2H), 8.78 (s, 1H).
A mixture of the intermediate (1-(3-(tert-butyldimethylsilyloxy)-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(6-fluoropyridin-3-yl) methanone (1.55 g, 3.3 mmol) and concentrated HCl (5 mL) in THF were stirred for 4 h at room temperature until TLC indicated that deprotection was complete, and then it was carefully neutralized with 1 N NaOH and extracted with CH2Cl2. The crude product was purified by column chromatography to afford 19j as white solid (0.92 g, 79%). 1H NMR (CDCl3, free base): δ 1.84-2.03 (m, 4H), 2.38-2.46 (m, 1H), 2.75-3.02 (m, 6H), 3.21-3.34 (m, 2H), 3.82-3.89 (m, 1H), 4.30 (s br, 1H), 7.03-7.11 (m, 4H), 8.33-8.39 (m, 2H), 8.79 (s, 1H). Free base was converted to the corresponding oxalate salt by adding oxalic acid in ethyl acetate to 19j in CH2Cl2. mp: 204.3 °C (decomposed). Anal. (C21H23FN2O2• H2C2O4•0.5H2O) C, H, N.
(2-Fluoropyridin-3-yl)(1-(3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)methanone (19k)
Compound 19k was prepared from compound 17k as describe in procedure D to give TBDMS-protected intermediate (1-(3-(tert-butyldimethylsilyloxy)-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(2-fluoro-pyridin-3-yl) methanone (1.68 g, 39%). 1H NMR (CDCl3): δ 0.11 (s, 6H), 0.90 (s, 9H), 1.63-1.78 (m, 4H), 2.45-3.08 (m, 9H), 4.13 (s br, 2H), 7.04-7.11 (m, 4H), 7.31-7.39 (m, 1H), 8.23-8.30 (m, 1H), 8.38-8.40 (m, 1H). Removal of TBDMS with HCl gave 19k (0.87 g, 75%). 1H NMR (CDCl3, free base): δ 1.84-2.03 (m, 4H), 2.38-2.46 (m, 1H), 2.75-2.99 (m, 6H), 3.25-3.33 (m, 2H), 3.80-3.90 (m, 1H), 4.31 (s br, 1H), 7.03-7.13 (m, 4H), 7.30-7.41 (m, 1H), 8.22-8.31 (m, 1H), 8.38-8.40 (m, 1H). The free base was converted to the oxalate salt. mp: 205.8 °C (decomposed). Anal. (C21H23FN2O2• H2C2O4•H2O) C, H, N.
1-(3-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)-N-methoxy-N-methylpiperidine-4-carboxamide (20)
TFA (2 mL) was added into a solution of 11 (0.38 g, 1.4 mmol) in CH2Cl2 (30 mL). The reaction mixture was stirred at room temperature for 4 h. The solvent was removed under reduced pressure and the residue was neutralized with 1 N NaOH solution and extracted with CH2Cl2 (2 × 15 mL). The organic layer was washed with brine, dried and concentrated to give the crude compound, which was used in the next step without further purification. Crude compound, 18 (0.10 g, 0.68 mmol) and Et3N (0.3 mL, 2.2 mmol) in ethanol (5 mL) were stirred at 75 °C for 36 h. After cooling to room temperature, the reaction mixture was poured into water and extracted with EtOAc (3 × 15 mL). The organic layer was dried and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give 20 as a white solid (86.4 mg, 40%). 1H NMR (CDCl3): δ 1.81-1.93 (m, 4H), 2.28-2.31 (m, 1H), 2.75-2.97 (m, 8H), 3.27-3.20 (s, 3H), 3.27-3.34 (m, 1H), 3.72 (s, 3H), 3.85-3.90 (m, 1H), 4.27 (s br, 1H), 7.09-7.13 (m, 4H).
1-(3-((tert-Butyldimethylsilyl)oxy)-1,2,3,4-tetrahydronaphthalen-2-yl)-N-methoxy-N-methylpiperidine-4-carboxamide (21)
To a solution of 20 (0.4 g, 1.25 mmol) and imidazole (0.4 g, 5.88 mmol) in CH2Cl2 (50 mL), TBDMSCl (0.38 g, 2.5 mmol) was added. The reaction mixture was stirred overnight at room temperature until TLC indicated that the reaction was complete. The reaction mixture was washed with brine and 1 N NaH2PO4 (30 mL × 3). The organic layer was dried and concentrated to a residue. The crude product was purified by silica gel column chromatography to afford 21 as a white solid (0.38 g, 70 %). 1H NMR (CDCl3,): δ 0.12 (s, 6H), 0.91 (s, 9H), 1.65-1.77 (m, 4H), 2.46-3.05 (m, 9H), 3.17 (s, 3H), 3.69 (s, 3H), 4.13 (s br, 2H), 7.04-7.12 (m, 4H).
3-(4-(4-Acetamidobenzoyl)piperidin-1-yl)-1,2,3,4-tetrahydronaphthalen-2-yl acetate (22)
Acetic anhydride (1.89 mL, 18.5 mmol) was added dropwise over 15 min to a magnetically stirred solution of 6 (0.54 g, 1.54 mmol) and Et3N (3.12 g, 30.8 mmol) in dry
CH2Cl2 (10 mL) overnight. The reaction was monitored by TLC. After the reaction was complete, the solvent was evaporated under reduced pressure. Water was added to wash the resulting solid and the residue was purified by silica gel column chromatography (ethyl acetate: hexane = 3:2) to give 22 (0.55 g, 82%). 1H NMR (CDCl3): δ 1.24-1.28 (m, 2H), 1.70-1.84 (m, 2H), 2.12 (s, 3H), 2.22 (s, 3H), 2.44-2.60 (m, 2H), 2.84-3.05 (m, 6H), 3.16-3.23 (m, 2H), 5.27-5.34 (m, 1H), 7.05-7.16 (m, 4H), 7.49 (s br, 1H), 7.62 (d, J = 8.4 Hz, 2H), 7.92 (d, J = 8.4 Hz, 2H).
N-(4-(1-(3-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidine-4-carbonyl)phenyl)-N-methylacetamide (23)
Sodium hydride (96 mg, 2.4 mmol) was added to 3-(4-(4-acetamidobenzoyl)piperidin-1-yl)-1,2,3,4-tetrahydronaphthalen-2-yl acetate 22 (0.87 g, 2 mmol) in anhydrous THF, and iodomethane (0.34 g, 2.4 mmol) was added dropwise to the mixture, which was maintained below 5 °C for 0.5 h and then stirred at room temperature for 1 h. The reaction was monitored by TLC. After the reaction was complete, the mixture was partitioned between saturated aqueous NH4Cl and ethyl acetate. The organic layer was separated and the aqueous layer was extracted with ethyl acetate. The combined organic layers dried and concentrated. The crude product was purified by silica gel column chromatography (ethyl acetate: hexane = 3:2) to give 23 (0.63 g, 78%). 1H NMR (CDCl3): δ 1.77-1.90 (m, 4H), 1.96 (s, 3H), 2.44 (t, J = 10.3 Hz, 1H), 2.76-3.03 (m, 7H), 3.27-3.35 (m, 2H), 3.31 (s, 3H), 3.84-3.89 (m, 1H), 4.21 (s br, 1H), 7.09-7.28 (m, 4H), 7.32 (d, J = 8.1 Hz, 2H), 8.01 (d, J = 7.8 Hz, 2H).
(4-(Dimethylamino)phenyl)(1-(3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)methanone (24a)
Sodium hydride (96 mg, 2.4 mmol) was added to 6 (0.87 g, 2 mmol) in anhydrous THF, and iodomethane (4.8 mmol) was added dropwise to the mixture, which was stirred at room temperature for 0.5 h. The reaction was monitored by TLC. After the reaction was complete, the mixture was partitioned between saturated aqueous NH4Cl and ethyl acetate. The organic layer was separated and the aqueous layer was extracted with ethyl acetate. The combined organic layers were dried and contentrated. The crude product was purified by silica gel column chromatography to give 24a as a white solid (0.63 g, 83%). 1H NMR (CDCl3, free base): δ 1.81-1.98 (m, 4H), 2.36-2.44 (m, 1H), 2.74-3.06 (m, 13H), 3.23-3.33 (m, 2H), 3.83-3.91 (m, 1H), 4.46 (s br, 1H), 6.66 (d, J = 9 Hz, 2H), 7.09-7.15 (m, 4H), 7.88 (d, J = 9 Hz, 2H). Free base was converted to the corresponding oxalate salt by adding oxalic acid in ethyl acetate to 24a in CH2Cl2. mp: 228.9 °C (decomposed). Anal. (C24H30N2O2 • H2C2O4) C, H, N.
(1-(3-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(4-(methylamino)phenyl) methanone (24b)
Concentrated HCl (12 M, 0.25 mL) was added to a stirred solution of 23 (0.4 g, 1.0 mmol) in ethylene glycol (0.75 mL). The reaction mixture was heated to reflux for 3 h and the reaction was monitored by TLC. When the reaction was complete, the mixture was partitioned between water and ethyl acetate. The organic layer was separated and the aqueous layer was extracted. The combined organic layers dried and contentrated. The crude product was purified by silica gel column chromatography (ethyl acetate/hexane, 1/4) to give 24b (0.3 g, 82%). 1H NMR (CDCl3): δ 1.85-1.92 (m, 4H), 2.40-2.44 (m, 1H), 2.77-2.99 (m, 8H), 3.27-3.34 (m, 2H), 3.81-3.89 (m, 2H), 4.19-4.23 (m, 1H), 4.46 (s br, 1H), 6.59 (d, J = 8.1 Hz, 2H), 7.09-7.15 (m, 4H), 7.86 (d, J = 8.1 Hz, 2H). Free base was converted to the corresponding oxalate salt by adding oxalic acid in ethyl acetate to 24b in CH2Cl2. mp: 215.7 °C (decomposed). Anal. (C23H28N2O2• H2C2O4) C, H, N.
Resolution of (1-(3-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(4-(methylamino)phenyl) methanone (24b) to obtain the entantiomerically minus isomer, (-)-24b and plus isomer (+)-24b
Approximately 200 mg of (±)-24b was separated on chiral HPLC using a Chiralcel OD column (250 mm × 10mm). The mobile phase used was 35% isopropanol in hexane at a flow rate of 4.0 mL/min to give 83.3 mg of (+)-24b (Rt = 15 min) and 94.9 mg of (-)-24b (Rt = 30 min). The specific rotation was determined on an automatic polarimeter (Autopol 111, Rudolph Research, Flanders, NJ). The optical rotation of (-)-24b was [α]D = -30.5° at the concentration of 1.8 mg/mL in dichloromethane and that of (+)-24b was [α]D = +21.8 ° at the concentration of 1.1 mg/ mL in dichloromethane at 20 °C. The (-)-24b and (+)-24b were converted to oxalates by treating one equivalent of (-)-24b or (+)-24b with one equivalent of oxalic acid. The salt obtained was used for the in vitro studies. mp of (+)-24b: 199.8 °C (decomposed), mp of (-)-24b: 200.2 °C (decomposed).
(1--3-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(6-hydroxypyridin-3-yl)methanone (25)
Into an oven-dried argon-purged 50 mL round bottom flask was placed 19e (73.3 mg, 0.2 mmol) and 3.0 mL chloroform was added followed by TMSI (57 μL, 0.4 mmol). The reaction mixture was heated to 55 °C for 1 h until TLC (EtOAc/hexane, 1/1) indicated the disappearance of starting material. The reaction mixture was cooled to room temperature and 0.6 mL methanol was added. The reaction mixture was concentrated. Ethyl acetate was added followed by aqueous sodium bicarbonate solution. The ethyl acetate layer was separated and the aqueous layer was extracted with ethyl acetate. Combined organic layers were washed with brine, dried over anhydrous sodium sulfate, and concentrated to give compound 25 as a white solid (69 mg, 97%). 1H NMR (CDCl3): δ 1.40-2.00 (m, 3H), 2.20-2.25 (m, 1H), 2.60-3.10 (m, 8H), 3.30 (dd, J = 6.2, 13.9 Hz, 1H), 3.80-4.00 (m, 1H), 4.20-4.25 (m, 1H), 6.60 (d, J = 9.6 Hz, 1H), 7.00-7.20 (m, 4 H), 8.05 (dd, J = 2.7, 9.6 Hz, 1H), 8.16 (s, 1H).
(6-(2-Fluoroethoxy)pyridin-3-yl)(1-(3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)-piperidin-4-yl)methanone (26a)
To the mixture of 25 (69 mg, 0.195 mmol) and 1-bromo-2-fluoroethane (30 μL, 0.4 mmol) in DMF (2 mL) was added potassium carbonate (60 mg, 0.43 mmol). The resultant reaction mixture was stirred at the room temperature for 72 h. Ethyl acetate was added to the reaction mixture and washed with aqueous sodium bicarbonate solution. The organic layer was dried over anhydrous sodium sulfate and concentrated. The crude product was purified by silica gel chromatography to give colorless sticky solid (40 mg, 51%). 1H NMR (CDCl3): δ 1.75 -1.99 (m, 1H), 2.20 - 2.50 (m, 1H), 2.70-3.10 (m, 11H), 3.30 (dd, J = 16.0, 6.0 Hz, 1H), 3.80-3.95 (m, 1H), 4.27 (t, J = 4.5 Hz, 1H), 4.36 (t, J = 4.5 Hz, 1H), 4.67 (t, J = 4.5 Hz, 1H), 4.83 (t, J = 4.5 Hz, 1H), 6.61 (d, J = 9.6 Hz, 1H), 7.00-7.20 (m, 4H), 7.88 (dd, J = 2.7, 9.6 Hz, 1H), 8.15 (s, 1H). Free base was converted to the corresponding oxalate salt by adding oxalic acid in ethyl acetate to 26a in CH2Cl2. mp: 84 °C (decomposed). Anal. (C24H27FN2O3• H2C2O4•H2O) C, H, N.
(3-(2-Fluoroethoxy)pyridin-2-yl)(1-(3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)methanone (26b)
To a solution of 28 (1.60 g, 7.3 mmol) in THF was added n-BuLi (1.6 M, 7 mL) at -78 °C under N2 atmosphere. The solution was stirred at -78 °C for 1 h and a solution of tert-butyl-4-(methoxy(methyl)carbamoyl)piperidine-1-carboxylate 11 (1.0 g, 3.6 mmol) in THF was added. The solution was maintained at -78 °C for 4 h, and then quenched with saturated aqueous NH4Cl that was allowed to warm to room temperature with stirring. The organic layer was separated and the aqueous layer was extracted with ethyl acetate. The combined extracts were washed with water, dried over Na2SO4, filtered, concentrated, and the residue chromatographed on a silica gel column with ethyl acetate : hexane (1:4, v/v) to give the t-Boc protected compound tert-butyl-4-(3-(2-fluoroethoxy)picolinoyl)piperidine-1-carboxylate (0.51 g, 40%), 1H NMR (CDCl3): δ 1.47 (s, 9H), 1.62-1.70 (m, 4H), 2.83 (s, 2H), 3.64-3.73 (m, 1H), 4.17 (s br, 2H), 4.25-4.36 (m, 2H), 4.68-4.87 (m, 2H), 7.23-7.26 (m, 1H), 7.38-7.40 (m, 1H), 8.25-8.30 (m, 1H).
To a solution of the above compound (0.49 g, 1.4 mmol) in CH2Cl2 (30 mL), TFA (2 mL) was added. The reaction mixture was stirred at room temperature for 4 h. The solvent was removed under reduced pressure, and the residue was neutralized with 1 N NaOH solution and extracted with CH2Cl2 (2 × 15 mL). The organic layer was washed with brine, dried and concentrated to give crude 4-(3-(2-fluoroethoxy)picolinoyl)piperidine. The crude product was used in the next step without further purification.
A mixture of the above compound, 18 (0.10 g, 0.68 mmol) and Et3N (0.3 mL, 2.2 mmol) in ethanol (5 mL) was stirred at 75 °C for 36 h. After cooling to room temperature, the mixture was poured into water, extracted with EtOAc (3 × 15 mL). The organic layer was dried and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give 26b (0.12 g, 43%). 1H NMR (CDCl3, free base): δ 1.73-1.98 (m, 4H), 2.32-2.40 (m, 1H), 2.71-2.96 (m, 8H), 3.31-3.33 (m, 1H), 3.49-3.56 (m, 1H), 3.80-3.89 (m, 1H), 4.24-4.36 (m, 2H), 4.69-4.87 (m, 2H), 7.09-7.25 (m, 4H), 7.34-7.38 (m, 2H), 8.27-8.29 (m, 1H). The corresponding oxalate salt was obtained by adding oxalic acid in ethyl acetate the solution of 26b in CH2Cl2 and then recrystallized. For the oxalate salt, mp: 119.9 °C (decomposed). Anal. (C23H27FN2O3•H2C2O4•H2O) C, H, N.
2-Bromo-3-(2-fluoroethoxy)pyridine (28)
A mixture of 2-bromopyridin-3-ol 27 (0.17 g, 1.0 mmol), 1-bromo-2-fluoroethane (0.25 g, 2.0 mmol) and K2CO3 (0.55 g, 4.0 mmol) in acetonitrile were stirred at reflux for one day until TLC indicated that the reaction was complete. The reaction mixture was filtered, dried and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography to afford 28 (0.16 g, 68%). 1H NMR (CDCl3): δ 4.25-4.34 (m, 2H), 4.73-4.89 (m, 2H), 7.20-7.22 (m, 2H), 8.01 (s, 1H).
Procedure E: General Method of Preparing 30a, 30c-d and 31a
(1-(8-Amino-3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(1-methyl-1Hpyrrol-2-yl)methanone (30a)
A mixture of 29 (0.20 g, 0.78 mmol), 17a (0.44 g, 2.3 mmol) and Et3N (0.5 mL) in ethanol (10 mL) was stirred at 60 °C for 48 h. To the mixture, 1 N NaOH (3 mL) was added and the stirring was continued overnight. The solvent was removed, the residue was extracted with EtOAc (3 × 30 mL) and the organic layer was washed with aqueous Na2CO3 solution, dried and concentrated. The crude product was purified by column chromatography to give 30a (0.11 g, 37%). 1H NMR (CDCl3, free base): δ 1.85-1.96 (m, 4H), 2.43-2.53 (m, 2H), 2.67-2.89 (m 5H), 2.96-3.28 (m, 3H), 3.60 (s br, 2H), 3.85 (m, 1H), 3.95 (s, 3H), 6.14 (m, 1H), 6.57 (m, 2H), 6.83 (s br, 1H), 6.96-7.02 (m, 2H). Free base was converted to the corresponding oxalate salt by adding oxalic acid in ethyl acetate to 30a in CH2Cl2. mp: 131 °C (decomposed). Anal. (C21H27N3O2 • 2H2C2O4 • 2H2O) C, H, N.
(1-(8-Amino-3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(pyridin-3-yl)methanone (30c)
Compound 30c was prepared from compound 17c as described in procedure E. Yield, 61%. 1H NMR (CDCl3, free base): δ 1.80-2.05 (m, 4H), 2.36-2.53 (m, 2H), 2.68-2.92 (m, 5H), 3.01-3.35 (m, 4H), 3.61 (s br, 2H), 3.87 (m, 1H), 6.54-6.60 (m, 2H), 6.99 (td, J = 7.5, 3.3 Hz, 1H), 7.45 (dd, J = 8.1, 4.8 Hz, 1H), 8.24 (dt, J = 8.1, 1.8 Hz, 1H), 8.80 (dd, J = 4.8, 1.8 Hz, 1H), 9.17 (d, J = 2.1 Hz, 1H). The free base was converted to the oxalate salt. mp: 109 °C (decomposed). Anal. (C21H25N3O2 • 2H2C2O4 • 0.5H2O) C, H, N.
(1-(8-Amino-3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(6-methylpyridin-3-yl)methanone (30d)
Compound 30d was prepared from compound 17d as described in procedure E. Yield, 44%. 1H NMR (CDCl3, free base): δ 1.79-1.99 (m, 5H), 2.35-2.53 (m, 2H), 2.64 (s, 3H), 2.67-3.28 (m, 8H), 3.62 (s br, 2H), 3.87 (m, 1H), 6.57 (m, 2H), 6.99 (m, 1H), 7.29 (d, J = 8.1 Hz, 1H), 8.13 (dd, J = 8.1, 2.1 Hz, 1H), 9.04 (d, J = 2.1 Hz, 1H). The free base was converted to the oxalate salt. mp: 98 °C (decomposed). Anal. (C24H29N3O4 • H2C2O4) C, H, N.
(1-(8-Amino-3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(6-methoxypyridin-3-yl)methanone (30e)
Compound 30e was prepared from compound 17e as describe in procedure E. Yield, 43%. 1H NMR (CDCl3, free base): δ 1.65 (s br, 1H), 1.80-2.02 (m, 4H), 2.42-2.53 (m, 2H), 2.67-3.03 (m, 6H), 3.21-3.28 (m, 2H), 3.61 (s br, 2H), 3.86 (m, 1H), 4.02 (s, 3H), 6.57 (m, 2H), 6.82 (d, J = 8.7 Hz, 1H), 6.99 (t, J = 7.8 Hz, 1H), 8.15 (dd, J = 8.7, 2.4 Hz, 1H), 8.80 (d, J = 2.4 Hz, 1H). The free base was converted to the oxalate salt. mp: 81.4 °C (decomposed). Anal. (C22H27N3O3 • 2H2C2O4 • 1.5H2O) C, H, N.
(1-(5-Amino-3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(1-methyl-1H-pyrrol-2-yl)methanone (31a)
Compound 31a was prepared from compound 17a as describe in procedure E. Yield, 20%. 1H NMR (CDCl3, free base): δ 1.85-1.96 (m, 4H), 2.36-2.45 (m, 2H), 2.76-2.89 (m 5H), 2.96-3.16 (m, 3H), 3.62 (s br, 2H), 3.92 (m, 1H), 3.96 (s, 3H), 6.15 (m, 1H), 6.56 (m, 2H), 6.83 (s br, 1H), 6.96- 7.02 (m, 2H). The free base was converted to the oxalate salt. mp: 114 °C (decomposed). Anal. (C21H27N3O2 • 2H2C2O4 • 1.5H2O) C, H, N.
tert-Butyl (4-(3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidine-4-carbonyl)phenyl)-carbamate, (-)-33
To a solution of (-)-6 (153.6 mg, 0.438 mmol) in THF was added Di-tert-butyl dicarbonate (Boc2O, 287.0 mg, 1.314 mmol), Et3N (0.5 mL) and DMAP (5.4 mg, 0.044 mmol). The reaction mixture was stirred at room temperature while monitoring by TLC. After 1.5 hrs the starting material completely disappeared, at which point the reaction mixture was concentrated on Rotary evaporator and partitioned the residue with brine and ethyl acetate. Aqueous phase was washed with ethyl acetate. The organic phase was dried over sodium sulfate, concentrated and purified on silica gel column (8:1 to 6:1 hexanes: ethyl acetate) to give the tri-Boc protected intermediate (-)-32 which was dissolved in methanol and excess K2CO3 was added and refluxed overnight. The product was partitioned between brine and dichloromethane. Aqueous phase was washed twice with dichloromethane. The organic phase was dried over sodium sulfate and concentrated to give (-)-33 as a white solid in 28% yield. 1H NMR (400 MHz, CDCl3): δ 1.55 (s, 9H), 1.78-1.95 (m, 4H), 2.42 (t, J = 12.0 Hz, 1H), 2.78-3.05 (m, 7H), 3.25-3.35 (m, 2H), 3.83-3.90 (m, 1H), 6.73 (s, 1H), 7.08-7.07 (m, 4H), 7.46 (d, J = 11.6 Hz, 2H), 7.92 (d, J = 12.4 Hz, 2H).
Radiochemistry
[11C]CH3I was produced at our institution from [11C]CO2 using a GE PETtrace MeI Microlab. Up to 1.4 Ci of [11C]CO2 was produced from Washington University’s JSW BC-16/8 cyclotron by irradiating a gas target of 0.5% O2 in N2 for 15-30 min with a 40 μA beam of 16 MeV protons. The GE PETtrace MeI microlab coverts the [11C]CO2 to [11C]CH4 using a nickel catalyst (Shimalite-Ni, Shimadzu, Japan P.N.221-27719) in the presence of hydrogen gas at 360 °C; it was further converted to [11C]CH3I by reaction with iodine that was held in a column in the gas phase at 690 °C. Several hundred millicuries of [11C]CH3I were delivered as a gas at approximately 12 min after end of bombardment (EOB), to the hot cell where the radiosynthesis was accomplished.
Radiosynthesis of (-)-[11C]24b
Approximately 1.2 - 1.5 mg of (-) enantiomer of the precursor was placed in a reaction vessel and 0.2 mL of DMF was added followed by 3.0 μL of 5N aqueous sodium hydroxide. The mixture was thoroughly mixed on a vortex. [11C]CH3I was bubbled into the reaction vessel. The reaction mixture was heated at 85 °C for 5 min. The reaction vessel was removed from heating, and 200 μL trifluoroacetic acid was added. The reaction vessel was mixed well and heated again at 85 °C for 5 min, and then quenched by adding 1.4 mL of HPLC mobile phase (29% acetonitrile in 0.1M ammonium formate buffer pH 4.5) and 200 μL 5N aqueous NaOH to neutralize the reaction mixture. The solution was loaded on to reverese phase C-18 column (Phenomenex Luna C18, 250mm × 9.6 mm, 10 μm with UV wave length at 254 nm); using the above HPLC mobile phase, at a flow rate of 4.0 ml/min, the radioactive product was collected between 17.0 and 19.0 min, and diluted with 50 mL of sterile water. The aqueous solution was passed through C18 Sep-Pak Plus (to trap the product) by applying nitrogen pressure. The collection bottle was rinsed by adding 10 mL water and passed through Sep-Pak plus. The product was collected in a vial by eluting C18 Sep-Pak plus with 0.6 mL of EtOH and 5.4 mL of saline to formulate the injection dose. The injection dose sample was authenticated using analytical HPLC system by co-injecting with cold standard compound (-)-24b. The HPLC system for quality control is: Column: Phenomenex Prodigy C18 analytical column, 250 × 4.6 mm; Mobile phase: 37% acetonitrile in 0.1 M ammonium formate buffer pH=4.5; Flow rate: 1.2 mL/min; the retention time for (-)-[11C]-24b was 4.9 min. The entire process was completed in approximately 1 hr. The radiochemical yield was 40-50% (decay corrected to EOB) with the radiochemical purity of > 99% and the specific activity was > 2000 Ci/mmol (decay corrected to end of synthesis).
Log D Measurement
Partition coefficient was measured by mixing the (-)-[11C]24b sample with 3 mL each of 1-octanol and buffer that is 0.1 M phosphate and pH equals 7.4 in a test tube. The mixture in the test tube were vortexed for 20 s followed by centrifugation for 1 min at room temperature. 2 mL of organic layer was transferred to a second test tube, 1 mL 1-octanol and 3 mL PBS buffer was added. The resulting mixture was vortexed for 20 s followed by centrifugation for 1 min at room temperature. 1 mL of organic and aqueous layer were taken separately for measurement. The radioactivity content values (count per minute) of two samples (1 mL each) from the 1-octanol and buffer layers were counted in a gamma-counter. The partition coefficient Log D7.4 was determined by calculating as the decimal logarithm the ratio of cpm/mL of 1-octanol to that of buffer. The measurements were repeated three times. Value of partition coefficient is 2.60.
In Vitro Biological Evaluation
Vesicular acetylcholine transporter binding assays
In vitro binding assays to VAChT were conducted with human VAChT permanently expressed in PC12 cells at about 50 pmol/mg of crude extract. No significant amounts of σ1 or σ2 receptors were present. The radioligand used was 5 nM (-)-[3H]vesamicol, and the assay was conducted at final concentrations of 10-11 M to 10-5 M novel compounds.4, 19 Unlabeled (-)-vesamicol was used as an external standard, for which Ki = 15 nM, and the mixture was allowed to equilibrate at 23 °C for 20 hours. Duplicate data were averaged and fitted by regression of a rectangular hyperbola to estimate the Ki value of the novel compounds.
Sigma receptor binding assays
The compounds were dissolved in DMF, DMSO or ethanol, and diluted in 50 mM Tris-HCl buffer containing 150 mM NaCl and 100 mM EDTA at pH 7.4 prior to performing the σ1 and σ2 receptor binding assays. The detailed procedures for performing the binding assays have been described.19, 49 For the σ1 receptor assays, guinea pig brain membrane homogenates (~300 μg protein) were the receptor resource and ~5 nM (+)- [3H] pentazocine (34.9 Ci/mmol, Perkin-Elmer, Boston, MA) was the radioligand. The incubation was performed in 96-well plates for 90 min at room temperature. Nonspecific binding was determined from samples that contained 10 μM of nonradioactive haloperidol. After 90 min, the reaction was quenched by addition of 150 μL of ice-cold wash buffer (10 mM Tris-HCl, 150 mM NaCl, pH 7.4). The harvested samples were filtered rapidly through a 96-well fiberglass filter plate (Millipore, Billerica, MA) that had been presoaked with 100 μL of 50 mM Tris-HCl buffer at pH 8.0 for 60 min. Each filter was washed 3 × 200 μL of ice-cold wash buffer, and the filter counted in a Wallac 1450 MicroBeta liquid scintillation counter (Perkin-Elmer, Boston, MA). The σ2 receptor binding assays were determined using rat liver membrane homogenates (~300 μg protein) and ~5 nM [3H]DTG (58.1 Ci/mmol, Perkin-Elmer, Boston, MA) in the presence of 1 μM of (+)-pentazocine to block σ1 sites. The incubation time was 120 min at room temperature. Nonspecific binding was determined from samples that contained 10 μM of nonradioactive haloperidol. All other procedures were same as those described for the σ1 receptor binding assay above.
The IC50 value was determined using nonlinear regression analysis. Competitive curves were best fit with a one-site model and gave pseudo-Hill coefficients of 0.6-1.0. Ki values were calculated using the method of Cheng and Prusoff50 and are presented as the mean (± 1 SEM). For these calculations, we used a Kd value of 7.89 nM for [3H](+)-pentazocine binding to σ1 receptor in guinea pig brain and a Kd value of 30.7 nM for [3H]DTG binding to σ2 receptor in rat liver.
Biodistribution in Rats
All animal experiments were conducted in compliance with the Guidelines for the Care and Use of Research Animals established by Washington University’s Animal Studies Committee. For the biodistribution studies, ~350 μCi of (-)-[11C]24b in about 175 μL of 10% ethanol/saline solution (v/v) was injected via the tail vein into mature male Sprague–Dawley rats (185 - 205 g) under anesthesia (2.5% isoflurane in oxygen at a flow rate of 1 mL/min). A group of at least four rats were used for each time point. For the control group, at 5 and 30 min post injection, the rats were anesthetized and euthanized. For the CycA pretreated group, CycA (Sandimmune diluted 1:1 with saline) at a dose of 25 mg/kg were administrated by intravenously (i.v.) 30 min prior to radioligand injection; at 30 min post injection of the radioligand, (-)-[11C]24b, the rats were euthanized. The whole brain was quickly harvested and various organs dissected comprising cerebellum, brain stem, cortex, striatum, thalamus, hippocampus and the brain. The remainder of the brain was also collected in order to determine total brain uptake. Simultaneously, samples of blood, heart, lung, liver, spleen, pancreas, kidney, muscle, fat, and tail were dissected. All the tissue samples were collected in the tared tubes and counted in an automated gamma counter (Beckman Gamma 8000 well counter) along with the standard solution of (-)-[11C]24b prepared by diluting the injectate. The counted tissues were then weighed, and the %ID/g was calculated.
MicroPET Studies in Nonhuman Primate Brain
A microPET Focus 220 scanner (Concorde/CTI/Siemens Microsystems, Knoxville, TN) was used for the imaging studies of (-)-[11C]24b in male cynomolgus monkey (4–6 kg). The animals were fasted for 12 h before the study. The animals were initially anesthetized using an intramuscular injection with ketamine (10 mg/kg) and glycopyrulate (0.13 mg/kg), and intubated with an endotracheal tube under anesthesia (maintained at 0.75–2.0% isoflurane in oxygen) throughout the PET scanning procedure. After intubation, a percutaneous venous catheter was placed for radiotracer injection. A 10 min transmission scan was performed to check the positioning; once confirmed, a 45 min transmission scan was obtained for attenuation correction. After that, the animal was administrated 5–7 mCi of (-)-[11C]-24b via the venous catheter, Subsequently, a 100 min dynamic PET scan (3 × 1 min, 4 × 2 min, 3 × 3 min, and 20 × 5 min) was acquired. During the whole procedure, core temperature was kept constant at 37 °C with a heated water blanket. In each microPET scanning session, the head was positioned supine in the adjustable head holder with the brain in the center of the field of view. For each subject, at least three independent PET studies were performed (n = 3).
MicroPET Image Data Processing and Analysis
PET image reconstructed resolution was < 2.0 mm full width half maximum for all 3 dimensions at the center of the field of view. Emission scans were corrected using individual attenuation and model-based scatter correction and reconstructed using filtered back projection as described previously.51 The first baseline PET image for each animal acted as the target image with the MPRAGE MRI scan and subsequent PETs coregistered to it using automated image registration program AIR.52, 53 All MPRAGE-based volume of interest (VOI) analyses were done by investigators blinded to the clinical status of the monkeys.4 For quantitative analyses, three-dimensional regions of interest (ROI) (cerebellum, frontal, occipital, striatum, temporal, white matter, midbrain and hippocampus) were transformed to the baseline PET space and then overlaid on all reconstructed PET images to obtain time–activity curves. Activity measures were standardized to body weight and dose of radioactivity injected to yield standardized uptake value (SUV).
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Institute Of Neurological Disorders And Stroke of the National Institutes of Health under Award Number R01NS075527, NS061025, The National Institute of Mental Health (NIMH) under Award Number MH092797, and McDonnell Center for Systems Neuroscience of Washington University at St. Louis.
Abbreviations List
- ADME
absorption, distribution, metabolism, and excretion
- ACh
acetylcholine
- AChE
acetylcholinesterase
- AD
Alzheimer’s disease
- CNS
central nervous system
- ABV
aminobenzovesamicol
- Anal
analysis
- BBB
blood-brain-barrier
- BBr3
boron tribromide
- n-BuLi
n-butyllithium
- CDI
1,1’-carbonyldiimidazole
- ChAT
choline acetyltransferase
- CycA
Cyclosporine A
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- (±)FBBV
(±)-trans-2-hydroxy-3-(4-(4-[18F]fluorobenzoyl)piperidino)tetralin
- IC50
half maximum inhibitory constant
- [123I]IBVM
(-)-5-[123I]iodobenzovesamicol
- NBS
N-bromosuccinimide
- PD
Parkinson’s Disease
- PDD
Parkinson’s disease associated with dementia
- PET
positron emission tomography
- P-gp
Permeability glycoprotein
- σ1 receptor
sigma-1 receptor
- σ2 receptor
sigma-2 receptor
- SPECT
single photon emission computed tomography
- TBDMSCl
tert-butyldimethylsilyl chloride
- t-Boc
tert-butyloxycarbonyl
- THF
tetrahydrofuran
- TLC
thin layer chromatography
- TFA
trifluoroacetic acid
- TMSI
trimethylsilyl iodide
- VAChT
vesicular acetylcholine transporter
Footnotes
Supplementary Material
Supporting Information Available: [Elemental analyses of new analogues and X-ray structural data of 19i]. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.http://www.alz.org/downloads/facts_figures_2012.pdf.
- 2.Qiu C, De Ronchi D, Fratiglioni L. The epidemiology of the dementias: an update. Curr Opin Psychiatry. 2007;20:380–385. doi: 10.1097/YCO.0b013e32816ebc7b. [DOI] [PubMed] [Google Scholar]
- 3.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang G, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 4.Efange SM, Khare AB, von Hohenberg K, Mach RH, Parsons SM, Tu Z. Synthesis and in vitro biological evaluation of carbonyl group-containing inhibitors of vesicular acetylcholine transporter. J Med Chem. 2010;53:2825–2835. doi: 10.1021/jm9017916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masliah E, Mallory M, Hansen L, Alford M, DeTeresa R, Terry R, Baudier J, Saitoh T. Localization of amyloid precursor protein in GAP43-immunoreactive aberrant sprouting neurites in Alzheimer’s disease. Brain Res. 1992;574:312–316. doi: 10.1016/0006-8993(92)90831-s. [DOI] [PubMed] [Google Scholar]
- 6.Davies P, Maloney AJF. Selective loss of central cholinergic neurons in alzheimers-disease. Lancet. 1976;2:1403–1403. doi: 10.1016/s0140-6736(76)91936-x. [DOI] [PubMed] [Google Scholar]
- 7.Dekosky ST, Scheff SW. Synapse loss in frontal-cortex biopsies in alzheimers-disease - correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 8.Masliah E, Ellisman M, Carragher B, Mallory M, Young S, Hansen L, Deteresa R, Terry RD. 3-Dimensional analysis of the relationship between synaptic pathology and neuropil threads in alzheimer-disease. J Neuropathol Exp Neurol. 1992;51:404–414. doi: 10.1097/00005072-199207000-00003. [DOI] [PubMed] [Google Scholar]
- 9.Peterson DA, Sejnowski TJ, Poizner H. Convergent evidence for abnormal striatal synaptic plasticity in dystonia. Neurobiol Dis. 2010;37:558–573. doi: 10.1016/j.nbd.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hilker R, Thomas AV, Klein JC, Weisenbach S, Kalbe E, Burghaus L, Jacobs AH, Herholz K, Heiss WD. Dementia in parkinson disease - functional imaging of cholinergic and dopaminergic pathways. Neurology. 2005;65:1716–1722. doi: 10.1212/01.wnl.0000191154.78131.f6. [DOI] [PubMed] [Google Scholar]
- 11.Castell X, Diebler MF, Tomasi M, Bigaria C, De Gois S, Berrard S, Mallet J, Israel M, Dolezal M. More than one way to toy with ChAT and VAChT. J Physiol Paris. 2002;96:61–72. doi: 10.1016/s0928-4257(01)00081-x. [DOI] [PubMed] [Google Scholar]
- 12.Breakefield XO, Blood AJ, Li Y, Hallett M, Hanson PI, Standaert DG. The pathophysiological basis of dystonias. Nat Rev Neurosci. 2008;9:222–234. doi: 10.1038/nrn2337. [DOI] [PubMed] [Google Scholar]
- 13.Kuhl DE, Fessler JA, Minoshima S, Cho K, Frey KA, Wieland DM, Koeppe RA. In-vivo mapping of cholinergic neurons in the human brain using SPECT and IBVM. J Nucl Med. 1994;35:405–410. [PubMed] [Google Scholar]
- 14.Bohnen NI, Frey KA. Imaging of cholinergic and monoaminergic neurochemical changes in neurodegenerative disorders. Mol Imaging Biol. 2007;9:243–257. doi: 10.1007/s11307-007-0083-6. [DOI] [PubMed] [Google Scholar]
- 15.Albin RL, Cross D, Cornblath WT, Wald JA, Wernette K, Frey KA, Minoshima S. Diminished striatal [I-123]iodobenzovesamicol binding in idiopathic cervical dystonia. Ann Neurol. 2003;53:528–532. doi: 10.1002/ana.10527. [DOI] [PubMed] [Google Scholar]
- 16.Barret O, Mazere J, Seibyl J, Allard M. Comparison of noninvasive quantification methods of in vivo vesicular acetylcholine transporter using [I-123]-IBVM SPECT imaging. J Cereb Blood Flow Metab. 2008;28:1624–1634. doi: 10.1038/jcbfm.2008.53. [DOI] [PubMed] [Google Scholar]
- 17.Rahmim A, Zaidi H. PET versus SPECT: strengths, limitations and challenges. Nucl Med Commun. 2008;29:193–207. doi: 10.1097/MNM.0b013e3282f3a515. [DOI] [PubMed] [Google Scholar]
- 18.Jansen FP, Vanderheyden JL. The future of SPECT in a time of PET. Nucl Med Biol. 2007;34:733–735. doi: 10.1016/j.nucmedbio.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 19.Tu Z, Efange SMN, Xu J, Li S, Jones LA, Parsons SM, Mach RH. Synthesis and in vitro and in vivo evaluation of F-18-labeled positron emission tomography (PET) ligands for imaging the vesicular acetylcholine transporter. J Med Chem. 2009;52:1358–1369. doi: 10.1021/jm8012344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sorger D, Scheunemann M, Vercouillie J, Grossmann U, Fischer S, Hiller A, Wenzel B, Roghani A, Schliebs R, Steinbach J, Brust P, Sabri O. Neuroimaging of the vesicular acetylcholine transporter by a novel 4-[F-18]fluoro-benzoyl derivative of 7-hydroxy-6-(4-phenyl-piperidin-1-yl)-octahydro-benzo[1,4]oxazines. Nucl Med Biol. 2009;36:17–27. doi: 10.1016/j.nucmedbio.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 21.Kilbourn MR, Hockley B, Lee L, Sherman P, Quesada C, Frey KA, Koeppe RA. Positron emission tomography imaging of (2R,3R)-5-[F-18] fluoroethoxybenzovesamicol in rat and monkey brain: a radioligand for the vesicular acetylcholine transporter. Nuc Med Biol. 2009;36:489–493. doi: 10.1016/j.nucmedbio.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zea-Ponce Y, Mavel S, Assaad T, Kruse SE, Parsons SM, Emond P, Chalon S, Giboureau N, Kassiou M, Guilloteau D. Synthesis and in vitro evaluation of new benzovesamicol analogues as potential imaging probes for the vesicular acetylcholine transporter. Bioorg Med Chem. 2005;13:745–753. doi: 10.1016/j.bmc.2004.10.043. [DOI] [PubMed] [Google Scholar]
- 23.Scheunemann M, Sorger D, Wenzel B, Heinitz K, Schliebs R, Klingner M, Sabri O, Steinbach J. Synthesis of novel 4-and 5-substituted benzyl ether derivatives of vesamicol and in vitro evaluation of their binding properties to the vesicular acetylcholine transporter site. Bioorg Med Chem. 2004;12:1459–1465. doi: 10.1016/j.bmc.2003.12.035. [DOI] [PubMed] [Google Scholar]
- 24.Wenzel B, Li Y, Kraus W, Sorger D, Sabri O, Brust P, Steinbach J. Pyrrolovesamicols-synthesis, structure and VAChT binding of two 4-fluorobenzoyl regioisomers. Bioorg Med Chem Lett. 2012;22:2163–2166. doi: 10.1016/j.bmcl.2012.01.127. [DOI] [PubMed] [Google Scholar]
- 25.Sluder A, Shah S, Cassayre J, Clover R, Maienfisch P, Molleyres LP, Hirst EA, Flemming AJ, Shi M, Cutler P, Stanger C, Roberts RS, Hughes DJ, Flury T, Robinson MP, Hillesheim E, Pitterna T, Cederbaum F, Worthington PA, Crossthwaite AJ, Windass JD, Currie RA, Earley FGP. Spiroindolines identify the vesicular acetylcholine transporter as a novel target for insecticide action. Plos One. 2012;7:e34712. doi: 10.1371/journal.pone.0034712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kozaka T, Uno I, Kitamura Y, Miwa D, Ogawa K, Shiba K. Syntheses and in vitro evaluation of decalinvesamicol analogues as potential imaging probes for vesicular acetylcholine transporter (VAChT) Bioorg Med Chem. 2012;20:4936–4941. doi: 10.1016/j.bmc.2012.06.040. [DOI] [PubMed] [Google Scholar]
- 27.Giboureau N, Som IM, Boucher-Arnold A, Guilloteau D, Kassiou M. PET radioligands for the vesicular acetylcholine transporter (VAChT) Curr Top Med Chem. 2010;10:1569–1583. doi: 10.2174/156802610793176846. [DOI] [PubMed] [Google Scholar]
- 28.Kilbourn MR, Hockley B, Lee L, Koeppe RA. Successful [F-18]FEOBV imaging of the VAChT in monkey brain. Neuroimage. 2008;41:T93–T93. [Google Scholar]
- 29.Mach RH, Voytko ML, Ehrenkaufer RLE, Nader MA, Tobin JR, Efange SMN, Parsons SM, Gage HD, Smith CR, Morton TE. Imaging of cholinergic terminals using the radiotracer [18F](+)-4-fluorobenzyltrozamicol: In vitro binding studies and positron emission tomography studies in nonhuman primates. Synapse. 1997;25:368–380. doi: 10.1002/(SICI)1098-2396(199704)25:4<368::AID-SYN8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 30.Waterhouse RN. Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents. Mol Imaging Biol. 2003;5:376–389. doi: 10.1016/j.mibio.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 31.Waring MJ. Lipophilicity in drug discovery. Expert Opin Drug Discov. 2010;5:235–248. doi: 10.1517/17460441003605098. [DOI] [PubMed] [Google Scholar]
- 32.Fichert T, Yazdanian M, Proudfoot JR. A structure-permeability study of small drug-like molecules. Bioorg Med Chem Lett. 2003;13:719–722. doi: 10.1016/s0960-894x(02)01035-1. [DOI] [PubMed] [Google Scholar]
- 33.Pajouhesh H, Lenz GR. Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005;2:541–553. doi: 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tu Z, Wang W, Cui J, Zhang X, Lu X, Xu J, Parsons SM. Synthesis and evaluation of in vitro bioactivity for vesicular acetylcholine transporter inhibitors containing two carbonyl groups. Bioorg Med Chem. 2012;20:4422–4429. doi: 10.1016/j.bmc.2012.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Padakanti PK, Wang R, Kil K, Li S, Perlmutter JS, Zeng D, Mach RH, Parsons SM, Tu Z. Radiosynthesis and in vivo evaluation of (-)-[11C]TZ1-27-2 as a PET tracer for imaging VAChT. J Nucl Med. 2011;52(Supplement 1):223. [Google Scholar]
- 36.Pampillon C, Claffey J, Hogan M, Tacke M. Novel achiral titanocene anti-cancer drugs synthesised from bis-N,N-dimethylamino fulvene and lithiated heterocyclic compounds. Biometals. 2008;21:197–204. doi: 10.1007/s10534-007-9108-5. [DOI] [PubMed] [Google Scholar]
- 37.Forlani L, Cristoni G, Boga C, Todesco PE, Del Vecchio E, Selva S, Monari M. Reinvestigation of the tautomerism of some substituted 2-hydroxypyridines. Arkivoc. 2002:198–215. [Google Scholar]
- 38.Frank J, Katritzky AR. Tautomeric pyridines .15. pyridone-hydroxypyridine equilibria in solvents of differing polarity. J Chem Soc Perkin Trans. 1976;2:1428–1431. [Google Scholar]
- 39.Kuzuya M, Noguchi A, Kamiya S, Okuda T. Reactions of 1-unsubstituted tautomeric 2-pyridones with benzyne. Chem Pharm Bull. 1985;33:2313–2322. [Google Scholar]
- 40.Rogers GA, Parsons SM, Anderson DC, Nilsson LM, Bahr BA, Kornreich WD, Kaufman R, Jacobs RS, Kirtman B. Synthesis, in vitro acetylcholine-storage-blocking activities, and biological properties of derivatives and analogs of trans-2-(4-phenylpiperidino) cyclohexanol (vesamicol) J Med Chem. 1989;32:1217–1230. doi: 10.1021/jm00126a013. [DOI] [PubMed] [Google Scholar]
- 41.Peng Y, Liu H, Tang M, Cai L, Pike V. Highly efficient N-monomethylation of primary aryl amines. Chin J Chem. 2009;27:1339–1344. [Google Scholar]
- 42.Loscher W, Potschka H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx. 2005;2:86–98. doi: 10.1602/neurorx.2.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pearce HL, Winter MA, Beck WT. Structural characteristics of compounds that modulate P-glycoprotein-associated multidrug resistance. Adv Enzyme Regul. 1990;30:357–373. doi: 10.1016/0065-2571(90)90026-x. [DOI] [PubMed] [Google Scholar]
- 44.Syvanen S, Lindhe O, Palner M, Kornum BR, Rahman O, Langstrom B, Knudsen GM, Hammarlund-Udenaes M. Species differences in blood-brain barrier transport of three positron emission tomography radioligands with emphasis on P-glycoprotein transport. Drug Metab Dispos. 2009;37:635–643. doi: 10.1124/dmd.108.024745. [DOI] [PubMed] [Google Scholar]
- 45.Ishiwata K, Kawamura K, Yanai K, Hendrikse NH. In vivo evaluation of P-glycoprotein modulation of 8 PET radioligands used clinically. J Nucl Med. 2007;48:81–87. [PubMed] [Google Scholar]
- 46.Luurtsema G, Molthoff CF, Schuit RC, Windhorst AD, Lammertsma AA, Franssen EJ. Evaluation of (R)-[11C]verapamil as PET tracer of P-glycoprotein function in the blood-brain barrier: kinetics and metabolism in the rat. Nucl Med Biol. 2005;32:87–93. doi: 10.1016/j.nucmedbio.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 47.de Vries EF, Kortekaas R, van Waarde A, Dijkstra D, Elsinga PH, Vaalburg W. Synthesis and evaluation of dopamine D3 receptor antagonist 11C-GR218231 as PET tracer for P-glycoprotein. J Nucl Med. 2005;46:1384–1392. [PubMed] [Google Scholar]
- 48.Tu Z, Li S, Xu J, Chu W, Jones LA, Luedtke RR, Mach RH. Effect of cyclosporin A on the uptake of D3-selective PET radiotracers in rat brain. Nucl Med Biol. 2011;38:725–739. doi: 10.1016/j.nucmedbio.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tu Z, Xu J, Jones LA, Li S, Dumstorff C, Vangveravong S, Chen D, Wheeler KT, Welch MJ, Mach RH. Fluorine-18-labeled benzamide analogues for imaging the σ2 receptor status of solid tumors with positron emission tomography. J Med Chem. 2007;50:3194–3204. doi: 10.1021/jm0614883. [DOI] [PubMed] [Google Scholar]
- 50.Cheng Y, Prusoff WH. Relationship between inhibition constant (K1) and concentration of inhibitor which causes 50 per cent inhibition (IC50) of an enzymatic-reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 51.Criswell SR, Perlmutter JS, Videen TO, Moerlein SM, Flores HP, Birke AM, Racette BA. Reduced uptake of [18F]FDOPA PET in asymptomatic welders with occupational manganese exposure. Neurology. 2011;76:1296–1301. doi: 10.1212/WNL.0b013e3182152830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tabbal SD, Mink JW, Antenor JA, Carl JL, Moerlein SM, Perlmutter JS. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced acute transient dystonia in monkeys associated with low striatal dopamine. Neuroscience. 2006;141:1281–1287. doi: 10.1016/j.neuroscience.2006.04.072. [DOI] [PubMed] [Google Scholar]
- 53.Woods RP, Mazziotta JC, Cherry SR. MRI-PET registration with automated algorithm. J Comput Assist Tomogr. 1993;17:536–546. doi: 10.1097/00004728-199307000-00004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.