

Abstract
Objective
Lynch syndrome is the most common cause of inherited colorectal cancer (CRC) and is due to germline mutations in mismatch repair (MMR) genes. Early Lynch syndrome diagnosis and appropriate CRC surveillance improves mortality. Traditional qualitative clinical criteria including Amsterdam and Bethesda guidelines may miss mutation carriers. Recently, quantitative predictive models including MMRPredict, PREMM(1,2,6), and MMRPro were developed to facilitate diagnosis. However, these models remain to be externally validated in the US. Therefore, we evaluated the test characteristics of Lynch syndrome predictive models in a multi-center, tertiary referral group at two US academic centers.
Methods
We retrospectively collected data on 230 consecutive individuals who underwent genetic testing for MMR gene mutations at the University of Chicago and University of California at San Francisco's Cancer Risk Clinics. Each individual's risk of mutation was examined using MMRPredict, PREMM(1,2,6), and MMRPro. Amsterdam and Bethesda criteria were also determined. Testing characteristics were calculated for each of the models.
Results
We included 230 individuals in the combined cohort. 113 (49%) probands were MMR mutation carriers. Areas under the receiver operator curves were 0.76, 0.78, and 0.82 for MMRPredict, PREMM(1,2,6), and MMRPro respectively. While similar in overall performance, our study highlights unique test characteristics of these three quantitative models including comparisons of sensitivity and specificity. Moreover, we identify characteristics of mutation carriers who were missed by each model.
Conclusion
Overall, all three Lynch syndrome predictive models performed comparably in our multi-center US referral population. These results suggest that Lynch syndrome predictive models can be used to screen for MMR mutation carriers and can provide improved test characteristics compared to traditional clinical criteria. Identification of MMR mutation carriers is paramount as appropriate screening can prevent CRC mortality in this high-risk group.
Introduction
Lynch syndrome (also called hereditary nonpolyposis colorectal cancer or HNPCC) is the most common cause of familial colorectal cancer (CRC), accounting for 3–5% of all CRC cases.(1) It is an autosomal dominant syndrome that imparts a 60–80% lifetime risk of CRC, 60–70% risk of endometrial cancer and approximately 15% risk for other extracolonic neoplasms including ovarian, small bowel, gastric, biliary, central nervous system, sebaceous gland, renal and renal collecting system.(2, 3) Lynch syndrome is caused by germline mutations in DNA mismatch repair genes including hMLH1 and hMSH2 (80% of cases), hMSH6 (5–10% of cases) and rarely hPMS2. In approximately 10–15% of patients meeting clinical criteria for Lynch syndrome, no mutations in mismatch repair genes are identified.(4) Loss of mismatch repair function results in errors in genomic replication known as microsatellite instability (MSI-H) which can be detected in 90% of Lynch-associated CRC. While suggestive, this finding is not diagnostic, as up to 10–20% of sporadic CRCs also display MSI-H features due to epigenetic silencing by promoter hypermethylation.(5)
Given the diversity in family and personal histories of cancer, lack of exclusive pathologic features, and known mortality benefit of an early diagnosis of Lynch syndrome (6), two qualitative clinical criteria, the Amsterdam and Bethesda guidelines, were developed to help facilitate diagnosis. However, there have been concerns about sensitivity, specificity and predictive values of these original guidelines.(6) Mutation carriers who do not fulfill these criteria, 22% in one large cohort,(7) remain at significantly increased risk for colorectal cancer.(8) Factors such as small families, adoption, unknown family history, newly arisen or undiscovered mutations, and patients without available tumor data limit the value of these clinical and pathologic methods.(9)
With increasing knowledge and understanding of the biologic basis of Lynch syndrome, three new quantitative predictive models, MMRPredict(9), PREMM(1,2,6)(15), and MMRPro(16) have emerged to help identify potential mutation carriers. Similar to predictive models in hereditary breast-ovarian syndrome such as BRCApro, the goal is a simple, accurate, clinically useful tool for predicting the likelihood of Lynch syndrome. Given that CRC mortality can be cost effectively averted in Lynch syndrome patients by early and intensive surveillance(6, 10), these three models represent potential advancement in screening and diagnosis. Overall, previous validation studies outside of the United States in both modest-sized referral populations (11, 12) as well as larger, population-based studies (13, 14) have found similar test characteristics for all three models. However, they have not been systematically validated in larger, US-based referral populations, nor have they examined the updated PREMM(1,2,6) algorithm which now includes hMSH6 (15). Our aim was to evaluate testing characteristics of these models in a multi-center, tertiary referral study group in the US screened through the University of California at San Francisco and the University of Chicago's Cancer Risk Clinics.
Methods
We obtained the pedigrees of 230 consecutive patients who underwent germline mutation testing at the University of California at San Francisco's Colorectal Cancer Prevention Program and the University of Chicago's Cancer Risk Clinic. All patients were referred based on a clinical history or tumor information suggestive of a possible diagnosis of Lynch syndrome. Individuals were evaluated by a genetic counselor who obtained detailed family histories and, using Progeny (Progeny Software, South Bend, IN), recorded pedigree information on the proband and both affected and unaffected relatives. Germline mutation analysis was performed by Myriad Labs (Salt Lake City, UT) or the Mayo Clinic Molecular Genetics Laboratory (Rochester, MN). Mutation risk scores were calculated for each of the three predictive models. MMRPredict and PREMM(1,2,6) are both freely available via web-based, online calculators. (MMRPredict: http://hnpccpredict.hgu.mrc.ac.uk/ & PREMM(1,2,6): http://www.dfci.org/premm/).
MMRPredict was limited to 145 of our 230 probands as it only calculates risk in CRC affected patients. Variables in this model include proband age, sex, tumor location, presence of synchronous or metachronous CRC, presence and age of 1st degree relative's with CRC (diagnosed at age >50 or <50) or endometrial cancer. PREMM(1,2,6) scores were calculated in all 230 probands. Proband-specific variables include presence and youngest age at diagnosis of CRC and endometrial cancer as well as presence of other Lynch-associated tumors. In addition, first- or second-degree relatives with colorectal, endometrial, and other Lynch-associated cancers as well as the youngest age at diagnosis are included. MMRPro requires a complete pedigree to assess risk and was performed on all 230 patients. Proband, first, and 2nd degree relatives data including age at diagnosis of colorectal or endometrial cancer and current age of unaffected relatives was entered into CaGENE 5.2 (CancerGene, Dallas, TX) for MMRPro score calculation.
When data was missing or unknown, a best faith assumption was made. These assumptions were based on routine practice in a genetics clinic, as the model builders do not give guidance on inputting missing data. In the case of MMRPro, when age was unknown, it was estimated based on a 30 year separation between generations. With MMRPredict, when tumor location was unknown, scores were calculated using both proximal and distal locations, then the average between the two was used. When MMR mutation analysis revealed a variant result, these were considered as positive findings (2 probands).
Receiver Operator Characteristic (ROC) curves and area under ROC curves (AUC) were generated for the three models. We calculated specificity, positive likelihood ratios and resultant model scores across a range of sensitivities (90%, 95%, and 98%). We compared characteristics of individuals missed by all three models to those who were captured by the models. Moreover, we compared characteristics of individuals missed exclusively by each of the predictive models. P-values were calculated using two-tailed Fisher's exact or chi2 tests and unpaired T-tests for categorical and continuous data, respectively. All statistical analysis was performed using STATA 10.1 (College Station, TX).
Results
Clinical characteristics of patients included in this study are shown in Table 1. Of the 230 patients evaluated, 113 were found to have germline mutations associated with Lynch Syndrome with an overall prevalence of 49%. Our population included 80 males and 150 females of which 50% and 49%, respectively, were found to be mutation positive. While the majority of patients were Caucasian, 18% were Asian (10 probands), Hispanic (16 probands), or African Americans (15 probands). The mismatch repair mutation spectrum included 42% hMLH1, 45% hMSH2, and 13% hMSH6.
Table 1.
Clinical Characteristics by Mutation Status
| Mutation Positive | Mutation Negative | P-value | |
|---|---|---|---|
| Probands | 113 (49%) | 117 (51%) | |
| UCSF (%) | 71 (56%) | 55 (44%) | |
| UC (%) | 42 (41%) | 62 (59%) | |
| Women (%) | 73 (64%) | 77 (65%) | 0.89 |
| Ethnicity | |||
| Caucasian (%) | 88 (46%) | 102 (54%) | 0.08 |
| African American (%) | 9 (60%) | 6 (40%) | 0.43 |
| Other (%)* | 17 (65%) | 9 (35%) | 0.09 |
| CRC (% probands affected) | 78 (69%) | 67 (57%) | 0.07 |
| Mean age CRC (range in years) | 43 (21–72) | 50 (19–76) | 0.0001 |
| EC (% probands affected) | 27 (23%) | 15 (12%) | 0.03 |
| Mean age EC (range in years) | 45 (29–60) | 51 (32–88) | 0.16 |
| Other Lynch tumor (% probands affected) | 17 (15%) | 12 (10%) | 0.32 |
| hMLH1 | 47 (42%) | ||
| hMSH2 | 51 (45%) | ||
| hMSH6 | 15 (13%) | ||
| 1st degree relatives | |||
| CRC (% FDR affected**) | 32% | 17% | 0.0001 |
| Mean age CRC (years) | 47 | 53 | 0.001 |
| EC (% FDR affected) | 13% | 4% | 0.0002 |
| Other Lynch tumor (% FDR affected) | 9% | 6% | 0.07 |
| 2nd degree relative | |||
| CRC (% SDR affected**) | 18% | 8% | 0.0001 |
| Mean age CRC (years) | 46 | 61 | 0.0001 |
| EC (% SDR affected) | 8% | 4% | 0.06 |
| Other Lynch tumor (% SDR affected) | 5% | 3% | 0.16 |
UCSF, University of California San Francisco; UC, University of Chicago; CRC, colorectal cancer; EC, endometrial cancer
Other includes 10 Hispanic and 16 Asian individuals
%FDR and %SDR is a measure of number of FDR or SDR affected family members divided by total FDR or SDR family members at risk to account for family size
Probands harboring MMR mutations differed from those who did not in several important ways. Probands with mutations trended towards higher rates of CRC (69% vs 57%, p 0.07) and their CRC occurred at a younger age (43 vs 50, p 0.0001). Mutation carriers also had higher rates of endometrial cancer (23% vs 12%, p 0.03) and trended towards a younger age of disease onset (45 vs 51, p 0.16). Rates of other Lynch associated tumors were similar in both groups (15% vs 10%, p 0.32). Family histories were also significantly different between mutation carriers and non-carriers. Families with mutations had higher rates of 1st degree relatives with CRC (32% vs 17%, p 0.0001) at younger mean ages (47 vs 53, p 0.001). Rates of endometrial cancer in 1st degree relatives were also higher in those with mutations (13% vs 4%, p 0.0002). Higher rates of CRC (18% vs 8%, p 0.0001) and younger age of onset (46 vs 61, p=0.0001) were also noted in second degree relatives of mutation carriers.
ROC analysis for all models is shown in Figure 1. For MMRPredict, we found an AUC of 0.76 (95% CI 0.68–0.84). PREMM(1,2,6) demonstrated an AUC of 0.78 (95% CI 0.72–0.84), while MMRPro had an AUC of 0.82 (95% CI 0.74–0.86). In contrast, AUCs for Amsterdam and Bethesda were 0.68 and 0.52, respectively.
Figure 1.
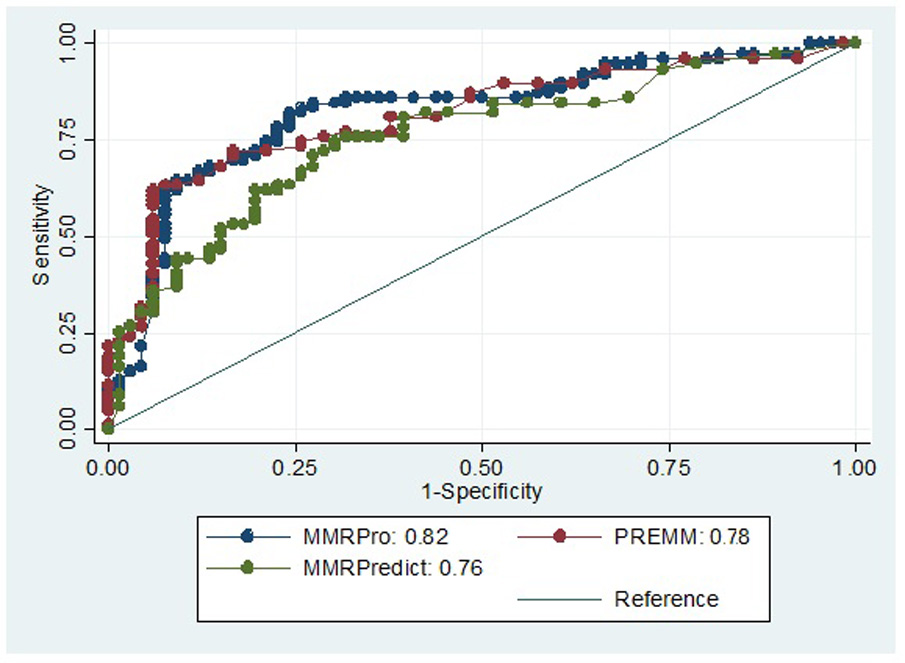
MMRPredict, PREMM(1,2,6) and MMRPro provide a quantitative estimate of MMR mutation risk thus, for a given population, physicians and patients can determine a predicted risk threshold above which consideration for germline genetic testing would be appropriate. We calculated testing characteristics for each model across a range of sensitivities in our referral population. Shown in Table 2 are specificity, positive likelihood ratio, and predicted risk score (%) when sensitivity is set at 90%, 95%, and 98%. For MMRPredict to obtain a sensitivity of 90%, a threshold for mutation testing of >4% would provide a specificity of 29% and a positive likelihood ratio of 1.3. At the highest sensitivity (98%), the specificity drops to 10% with a risk score cutoff of >1%. For PREMM(1,2,6), a cutoff of >6% would be 90% sensitive and 38% specific with a positive likelihood ratio of 1.4. At sensitivities of 95% and 98%, the specificity is 0% with a cutoff risk score of >0%. For MMRPro, a cutoff score of >7% would be 90% sensitive and 36% specific. For a 98% sensitive test using MMRPro, as with PREMM (1,2,6), all patients would require genetic testing as the risk score cutoff is >0%.
Table 2.
Quantitative Prediction Model Testing Characteristics at Sensitivities of 90%, 95% and 98%
| MMRPredict | PREMM(1,2,6) | MMRPro | |
|---|---|---|---|
| >90% Sensitivity | |||
| Specificity | 29% | 38% | 36% |
| LR+ | 1.3 | 1.4 | 1.4 |
| Risk score cutoff* | >4% | >6% | >7% |
| >95% Sensitivity | |||
| Specificity | 21% | 0% | 9% |
| LR + | 1.2 | 1.0 | 1.0 |
| Risk score cutoff | >2% | >0% | >1% |
| >98% Sensitivity | |||
| Specificity | 10% | 0% | 0% |
| LR + | 1.1 | 1.0 | 1.0 |
| Risk score cutoff | >1% | >0% | >0% |
LR+, positive likelihood ratio
MMRPredict & PREMM(1,2,6) output is expressed as predicted percentage risk of MMR mutation, while MMRPro as numerical probability of MMR mutation; for purposes of this table, all risk scores are shown as percentages
The authors of MMRPredict and PREMM (1,2,6) also provide guidance for appropriate cutoffs leading to further evaluation. MMRPredict describes >5% to be the “optimal combination of clinical utility and efficiency.”(9) In our cohort, using >5% as a diagnostic threshold for MMRPredict resulted in a sensitivity of 85%, specificity of 35%, and positive likelihood ratio of 1.3. The authors of PREMM(1,2,6) suggest a cutoff of >5% “may be a reasonable threshold” for further genetic testing and, in our cohort, this resulted in a sensitivity of 90%, specificity of 37%, and positive likelihood ratio of 1.4. (15) The authors of MMRPro report “thresholds should be chosen based on individual circumstances,” but do mention their model performed with higher sensitivity and specificity than Bethesda with MSI in the range of >35% to >62% cutoff. (16) In our cohort, these thresholds would result in a sensitivity of 65–78%, specificity of 68–84%, and positive likelihood ratio of 2.4–4.2 for MMRPro.
Testing characteristics were calculated for the qualitative Amsterdam and Bethesda guidelines. Amsterdam criteria were fulfilled in 148 of 230 probands mandating less genetic testing (147 of 230, 64%) with an improved specificity (52%). These gains, however, were at the expense of sensitivity which proved to be only 81%. Bethesda guidelines presented a different set of limitations requiring almost universal germline testing (216 of 230, 94%) with subsequent high sensitivity (99%) but low specificity (10%). In comparison, the quantitative predictive models would require testing between 76%-85% of the probands in our highly-selected population to obtain a sensitivity of >90%.
In order to determine characteristics of individuals missed by the models, we compared missed mutation carriers to those identified by the models at cut-off scores corresponding to >90% sensitivity. Twenty-two mutation carriers were missed by at least one model, seven by at least two models, and two by all three models. Of the seven mutation carriers missed by at least two models, two had hMLH1 mutations, four had hMSH2 mutations and one had an hMSH6 mutation. Mutation carriers missed by at least two models compared to subjects identified by the models had increased rates of CRC [6/7 (86%) vs 78/113 (69%), respectively] at older ages of onset (52 years vs 43 years, respectively). There were no cases of endometrial cancer or other Lynch Syndrome-associated malignancy in missed mutation carriers. Their family histories revealed fewer 1st and 2nd degree relatives with CRC (14% vs 32% and 15% vs 18%, respectively) at older ages of onset (55 vs 47 and 80 vs 46, respectively). There was no family history of endometrial cancer and 1st and 2nd degree relatives had similar rates of other Lynch Syndrome-associated tumors.
In order to determine if different models fit a pattern of who they missed, we considered characteristics of subjects missed exclusively by each model (Table 3). Four mutation carriers were missed exclusively by MMRPredict at a cut-off score of >4%. Two of these missed subjects had a personal history of endometrial cancer which is not included in the MMRPredict model. PREMM(1,2,6), when output threshold was >6%, missed five patients that were detected by MMRPro and MMRPredict. PREMM(1,2,6) did not miss any patients with personal history of endometrial cancer nor hMSH6 mutation carriers. Only one of five subjects missed by PREMM(1,2,6) had other Lynch-associated tumors. MMRPro missed six mutation carriers at a cut-off score of >7% predicted risk. These patients have increased rates of other Lynch-associated tumors and their family histories revealed fewer individuals affected by CRC. Like PREMM(1,2,6), MMRPro did not miss any hMSH6 mutation carriers.
Table 3.
Characteristics of mutation carriers missed exclusively by each predictive models
| MMRPredict | PREMM(1,2,6) | MMRPRO | ||
|---|---|---|---|---|
| N= | 4 pts | 5 pts | 6 pts | |
| Proband | ||||
| Women | 4/4 | 4/5 | 3/6 | |
| CRC | 4/4 | 3/5 | 4/6 | |
| Mean age CRC (years) | 49 | 44 | 34 | |
| EC | 2/4 | 0/5 | 0/6 | |
| Mean age EC (years) | 48 | NA | NA | |
| Other Lynch tumor | 1/4 | 1/5 | 2/6 | |
| hMLH1 | 2/4 | 1/5 | 3/6 | |
| hMSH2 | 1/4 | 4/5 | 3/6 | |
| hMSH6 | 1/4 | 0/5 | 0/6 | |
| 1st degree relatives | ||||
| CRC (% FDR affected) | 21% | 25% | 18% | |
| Mean age CRC (years) | 50 | 55 | 69 | |
| EC (% FDR affected) | 0% | 0% | 0% | |
| Other Lynch tumor (% FDR affected) | 7% | 4% | 15% | |
| 2nd degree relative | ||||
| CRC (% SDR affected) | 8% | 29% | 1% | |
| Mean age CRC (years) | 48 | 73 | 45 | |
| EC (% SDR affected) | 0% | 20% | 0% | |
| Other Lynch tumor (% SDR affected) | 8% | 10% | 5% | |
CRC, colorectal cancer; EC, endometrial cancer; FDR, first degree relative;
We also considered test characteristics in African Americans given limited information about racial differences in Lynch syndrome. A total of 15 African American probands were tested of which nine (60%) were found to be mutation positive. MMRPredict, PREMM(1,2,6), and MMRPro were all 100% sensitive with AUC's of 0.89, 0.89, and 0.93, respectively in our African American patients.
Discussion
In a large group of consecutive patients who presented for hereditary cancer evaluation and subsequent germline mutation testing at the University of California at San Francisco and University of Chicago's Cancer Risk Clinics, our data suggests that MMRPredict, PREMM(1,2,6) and MMRPro performed similarly in predicting mismatch repair mutation carriers with AUCs between 0.76 – 0.82. One benefit of these newer quantitative models over previous clinical criteria is the clinician's ability to determine a threshold for further genetic evaluation that is appropriate in a given population. In our referral cohort, a sensitivity of >90% resulted in a similar specificity and positive likelihood ratio in all three predictive models. When compared to qualitative Bethesda and Amsterdam criteria, we observed improved testing characteristics. Bethesda, while 99% sensitive, required almost universal testing and had limited specificity. While Amsterdam tested significantly fewer patients, its sensitivity was only 81%, which is too low for screening purposes. These results are similar to those reported in two smaller-scale referral populations from Canada (11, 12) and, while representing a highly selected referral population, remain relevant to gastroenterologists and other health care providers who must risk-stratify patients for Lynch Syndrome in clinical practice.
Seven mutation carriers were missed by two or more models and represent an important cohort of patients that may be difficult to screen using clinical criteria. While these carriers had higher rates of CRC, they occur at an older age of onset, which is less suspicious from the models’ standpoint. Additionally, they exhibited no personal history of endometrial cancer or other Lynch-associated tumors. Their family histories were also less suggestive with lower rates of CRC and older ages of onset, no cases of endometrial cancer, and similar rates of other Lynch-associated tumors. Mutation and gender distribution appeared similar to our overall population, although these findings are limited by the small sample size. While five of seven met Bethesda Criteria, only one met Amsterdam.
Although overall performance was similar among the three models, important differences should be noted as they affect application of these models in clinical practice. These features are summarized in Table 4. MMRPredict only accounts for probands with CRC when calculating risk scores and, subsequently, misses carriers with a personal history of endometrial cancer or any history of other Lynch-associated tumors. MMRPredict's strengths are its fast, simple, web-based platform and ability to incorporate tumor information such as location and MSI / IHC testing when available. It is less suited for probands with strong, but remote, family histories or in those with numerous Lynch-associated tumors beyond CRC.
Table 4.
Summary of characteristics and clinical application of Lynch syndrome predictive models
| Model | Characteristics | Clinical application |
|---|---|---|
| MMRPredict (9) |
|
|
| PREMM 1,2,6 (15) |
|
|
| MMRPro (16) |
|
|
CRC, colorectal cancer; EC, endometrial cancer; MSI, microsatellite instability; IHC, immunohistochemistry; MMR, mismatch repair; LS, Lynch syndrome
includes MSI/IHC and tumor location
PREMM (1,2,6), like MMRPredict, is available as a web-based module and can rapidly assess mutation risk for hMLH1, hMSH2, and, more recently, has been updated to include hMSH6 (15). In our population, it functioned particularly well in hMSH6, missing no carriers uniquely. It is also the only model that accounts for Lynch-associated cancers beyond EC and CRC which is illustrated by its improved performance in patients affected by these tumors. PREMM(1,2,6)’s strengths are its simple, web-based platform and ability to account for other Lynch-associated tumors. PREMM(1,2,6) may be less informative in patients with MSI or IHC testing as, in contrast to MMRPredict and MMRPro, it does not incorporate this tumor data.
For genetic counselors, MMRPro's benefits include its ability to quantify risk for patients even when tumor or germline testing is negative. The algorithm also takes into account age and cancer status of all first and second-degree relatives individually, as is illustrated by the reduced rates of CRC affected family members in its missed carriers. However, it is significantly more time consuming, requiring a complete family pedigree to provide thorough risk estimates. Additionally, MMRPro is unable to incorporate other Lynch-associated malignancies, explaining their increased prevalence in mutation carriers missed by MMRPro. MMRPro is most valuable as an attempt to quantify risk in tumor or germline negative probands or when extensive pedigree information is available. It is less suited for families with significant Lynch-associated tumors beyond endometrial cancer or CRC and when time for assessment is limited.
There are strengths and limitations to our study. We included consecutive individuals from two large, geographically distinct centers in the United States. Subjects’ personal and family histories were well-characterized and included several racial and ethnic backgrounds. Moreover, we included the most recent PREMM(1,2,6) model which has not previously been validated. One limitation is the highly select referral population which, while relevant to many centers evaluating suspected Lynch Syndrome, may not apply to a more generalized patient population. Moreover, we had only a small number of subjects that were missed by the models. In addition, while all patients underwent testing for hMLH1, hMSH2, and hMSH6, these results may not apply to patients with mutations in hPMS2 and EpCAM(TACSTD), as none of our patients had mutations in these genes.
In summary, this is the first study to evaluate Lynch syndrome predictive models in a multi-center and multi-ethnic US referral population. We found that the three models tested, MMRPredict, PREMM(1,2,6), and MMRPro, performed comparably and showed overall improved test characteristics compared to Amsterdam and Bethesda guidelines. Although each model has varying strengths and weaknesses, familiarity with and use of these models can help increase detection of Lynch Syndrome and prevent cancer-related morbidity and mortality.
What is Current Knowledge
Early identification and appropriate surveillance saves lives in Lynch Syndrome.
Traditional qualitative criteria like Amsterdam and revised Bethesda guidelines may miss patients.
What is New Here
Newer quantitative predictive models including PREMM(1,2,6), MMRPro and MMRPRedict performed similarly in a large, high-risk US referral population.
Each model has unique characteristics and clinical application.
These models should be implemented in routine clinical practice to identify individuals and families at risk for Lynch Syndrome.
Acknowledgments
We thank Kisha Hope who helped in creating the University of Chicago database.
Financial support: S.S.K. is supported by the NIH/NCI (K08CA142892-01A1).
Abbreviations
- MMR
mismatch repair
- CRC
colorectal cancer
- AUC
area under curve
- ROC
receiver operator curve
- HNPCC
hereditary non-polyposis colorectal cancer
Footnotes
Guarantor of the article: Sonia S. Kupfer, MD
- Omar Khan: study concept and design; acquisition of data; analysis and interpretation of data; drafting of the manuscript; statistical analysis; approval of final submitted draft
- Amie Blanco: acquisition of data; approval of final submitted draft
- Peggy Conrad: acquisition of data; approval of final submitted draft
- Cassandra Gulden: acquisition of data; approval of final submitted draft
- Tovah Moss: acquisition of data; approval of final submitted draft
- Olufunmilayo I. Olopade: critical revision of the manuscript for important intellectual content; approval of final submitted draft
- Sonia S. Kupfer: study concept and design; acquisition of data; analysis and interpretation of data; critical revision of the manuscript for important intellectual content; approval of final submitted draft
- Jonathan Terdiman: study concept and design; critical revision of the manuscript for important intellectual content; approval of final submitted draft
Conflict of Interest:
Potential competing interests: None.
References
- 1.Lynch HT, Riley BD, Weissman SM, et al. Hereditary nonpolyposis colorectal carcinoma (HNPCC) and HNPCC-like families: Problems in diagnosis, surveillance, and management. Cancer. 2004;100:53–64. doi: 10.1002/cncr.11912. [DOI] [PubMed] [Google Scholar]
- 2.Vasen HF, Watson P, Mecklin JP, et al. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453–1456. doi: 10.1016/s0016-5085(99)70510-x. [DOI] [PubMed] [Google Scholar]
- 3.Desai TK, Barkel D. Syndromic colon cancer: lynch syndrome and familial adenomatous polyposis. Gastroenterol Clin North Am. 2008;37:47–72. vi. doi: 10.1016/j.gtc.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 4.de la Chapelle A. Genetic predisposition to colorectal cancer. Nat Rev Cancer. 2004;4:769–780. doi: 10.1038/nrc1453. [DOI] [PubMed] [Google Scholar]
- 5.Aaltonen LA, Salovaara R, Kristo P, et al. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med. 1998;338:1481–1487. doi: 10.1056/NEJM199805213382101. [DOI] [PubMed] [Google Scholar]
- 6.Jarvinen HJ, Aarnio M, Mustonen H, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118:829–834. doi: 10.1016/s0016-5085(00)70168-5. [DOI] [PubMed] [Google Scholar]
- 7.Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N Engl J Med. 2005;352:1851–1860. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- 8.Aarnio M, Sankila R, Pukkala E, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer. 1999;81:214–218. doi: 10.1002/(sici)1097-0215(19990412)81:2<214::aid-ijc8>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 9.Barnetson RA, Tenesa A, Farrington SM, et al. Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med. 2006;354:2751–2763. doi: 10.1056/NEJMoa053493. [DOI] [PubMed] [Google Scholar]
- 10.Vasen HF, van Ballegooijen M, Buskens E, et al. A cost-effectiveness analysis of colorectal screening of hereditary nonpolyposis colorectal carcinoma gene carriers. Cancer. 1998;82:1632–1637. [PubMed] [Google Scholar]
- 11.Monzon JG, Cremin C, Armstrong L, et al. Validation of predictive models for germline mutations in DNA mismatch repair genes in colorectal cancer. Int J Cancer. 2010;126:930–939. doi: 10.1002/ijc.24808. [DOI] [PubMed] [Google Scholar]
- 12.Pouchet CJ, Wong N, Chong G, et al. A comparison of models used to predict MLH1, MSH2 and MSH6 mutation carriers. Ann Oncol. 2009;20:681–688. doi: 10.1093/annonc/mdn686. [DOI] [PubMed] [Google Scholar]
- 13.Green RC, Parfrey PS, Woods MO, et al. Prediction of Lynch syndrome in consecutive patients with colorectal cancer. J Natl Cancer Inst. 2009;101:331–340. doi: 10.1093/jnci/djn499. [DOI] [PubMed] [Google Scholar]
- 14.Balmana J, Balaguer F, Castellvi-Bel S, et al. Comparison of predictive models, clinical criteria and molecular tumour screening for the identification of patients with Lynch syndrome in a population-based cohort of colorectal cancer patients. J Med Genet. 2008;45:557–563. doi: 10.1136/jmg.2008.059311. [DOI] [PubMed] [Google Scholar]
- 15.Kastrinos F, Steyerberg EW, Syngal S, et al. The PREMM (1,2,6) model predicts risk of MLH1, MSH2 and MSH6 germline mutations based on cancer history. Gastroenterology. 2011 Jan;140(1):73–81. doi: 10.1053/j.gastro.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen S, Wang W, Lee S, et al. Prediction of germline mutations and cancer risk in the Lynch syndrome. JAMA. 2006;296:1479–1487. doi: 10.1001/jama.296.12.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]