

SUMMARY
Optogenetic techniques provide effective ways of manipulating the functions of selected neurons with light. In the current study, we engineered an optogenetic technique that directly inhibits neurotransmitter release. We used a genetically encoded singlet oxygen generator, miniSOG, to conduct chromophore assisted light inactivation (CALI) of synaptic proteins. Fusions of miniSOG to VAMP2 and synaptophysin enabled disruption of presynaptic vesicular release upon illumination with blue light. In cultured neurons and hippocampal organotypic slices, synaptic release was reduced up to 100%. Such inhibition lasted >1 hr and had minimal effects on membrane electrical properties. When miniSOG-VAMP2 was expressed panneuronally in Caenorhabditis elegans, movement of the worms was reduced after illumination, and paralysis was often observed. The movement of the worms recovered overnight. We name this technique Inhibition of Synapses with CALI (InSynC). InSynC is a powerful way to silence genetically specified synapses with light in a spatially and temporally precise manner.
INTRODUCTION
Optogenetic approaches allow experimenters to control neurophysiological functions of a genetically defined neuronal population through expression of light-responsive activity-modulating proteins. For example, microbial opsin pumps hyperpolarize membrane potentials of expressing neurons during light illumination, reducing the probability of the neurons to achieve suprathreshold depolarization with excitatory inputs (Han and Boyden, 2007). Chemical-biological optogenetic approaches can also be used to hyperpolarize membrane potential (Levitz et al., 2013; Janovjak et al., 2010). The microbial opsin channels, channelrhodopsins, can be used to achieve suprathreshold depolarization with light pulses in the expressing neurons (Boyden et al., 2005; Lin et al., 2009). When channelrhodopsins are expressed at high levels at the membrane of presynaptic terminals, light can induce direct release of neurotransmitters without triggering action potentials, so that focused illumination can be used to map the synaptic inputs to a neuron (Petreanu et al., 2009). Currently there is no technique that allows direct inhibition of synaptic release with light. Optogenetic inhibition of synaptic transmission would be very valuable to dissect the contribution of individual synapses or defined populations to the behavior of defined circuits and whole animals. Synaptic transmission could be blocked by interference with either presynaptic release or postsynaptic receptors. We chose to target presynaptic release because it occurs by a relatively well-conserved mechanism, in contrast to the enormous diversity of postsynaptic receptors.
Vesicular synaptic release is mediated by the SNARE protein complex located at the presynaptic terminal of neurons. Proteins in the SNARE complex have been well characterized and studied, however, there are also multiple associated proteins with unknown functions (Sudhof, 2004). The SNARE proteins, synaptobrevin 2/VAMP2, SNAP-25, and syntaxin, are believed to be the essential proteins for synaptic release in the central nervous system. During synchronized release, calcium influx from voltage-gated calcium channels triggers the binding of VAMP2 and synaptotagmin on the vesicular membrane to the SNAP-25 and syntaxin on the plasma membrane, allowing for the fusion of the vesicular membrane to plasma membrane and the release of vesicular contents. With asynchronized release, the fusion of vesicles is thought to occur without the involvement of voltage-gated calcium channels and synaptotagmin (Smith et al., 2012). Engineering a method to inhibit synaptic release in neurons with light would require the disruption of the endogenous SNARE complex to inhibit their normal function.
Chromophore-assisted light inactivation (CALI) is a powerful technique that can be used to selectively inactivate proteins during excitation of chromophores placed in the proximity of a protein (Jay, 1988; Marek and Davis, 2002; Tour et al., 2003). The reactive oxygen species generated by the chromophore during illumination oxidize nearby susceptible residues (tryptophan, tyrosine, histidine, cysteine and methionine), interfering with protein function. Synthetic chromophores such as malachite green (Jay, 1988), fluorescein (Beck et al., 2002), FlAsH (Marek and Davis, 2002), ReAsH (Tour et al., 2003), and eosin (Takemoto et al., 2011) have been shown to be effective CALI agents. CALI has also been demonstrated with genetically encoded chromophores such as eGFP (Rajfur et al., 2002) and KillerRed (Bulina et al., 2006), although these fluorescent protein-based techniques are much less efficient (Takemoto et al., 2011). A recently engineered flavoprotein, miniSOG, has been shown to be an effective chromophore for the photo-oxidation of diaminobenzidine to introduce contrast in electron microscopy of fixed tissue (Shu et al., 2011). Singlet oxygen is generated when the flavin mononucleotide within miniSOG is illuminated by light with wavelength <500 nm. Flavin mononucleotide is sufficiently ubiquitous within cells to avoid any need to administer exogenous cofactor molecules. Judicious fusion of miniSOG to a mitochondrial transporter enables photoablation of genetically targeted neurons in Caenorhabditis elegans (Qi et al., 2012). Due to the high quantum efficiency for singlet oxygen photogeneration by miniSOG, it should be a more effective genetically encoded CALI chromophore than eGFP or KillerRed.
In the current study, we fuse miniSOG to the SNARE proteins VAMP2 and synaptophysin (SYP1) to inactivate the SNARE complex with light. We were able to achieve the reduction of synaptic release with 480 nm light with both constructs in hippocampal neurons, with the SYP1-based system achieving greater reduction than the VAMP2-based system. When mammalian VAMP2 fused to miniSOG was expressed pan-neuronally in C. elegans, we were able to induce long-lasting paralysis (>1 hr) with 480 nm light. Animals recovered movement when re-tested 24 hr later. We name this technique Inhibition of Synapses with CALI (InSynC). We believe InSynC is a powerful optogenetic technique for inhibiting neurotransmitter release with light and interrogating neurocircuitry in a spatially precise manner.
RESULTS
Design and Assaying of the CALI-Based Synaptic Inhibition System
To design a CALI-based synapse inhibition system, we chose candidate fusion proteins based on two criteria: (1) the protein is essential for vesicular synaptic release in synapses of the central nervous system; and (2) the engineered protein can achieve inhibition in a dominant-negative manner, without the need to eliminate endogenous protein expression.
The SNARE protein synaptobrevin 2/VAMP2 is the core protein in the vesicular SNARE complex, with a cytosolic N-terminal α-helix capable of binding to the α helices of SNAP-25 and syntaxin during vesicle fusion. The C-terminal of VAMP2 consists of a transmembrane domain that is anchored to the vesicular membrane. Both N and C termini of VAMP2 have previously been fused to fluorescent proteins without disrupting function (Deák et al., 2006). The second protein candidate that we chose was synaptophysin (SYP1), which is closely associated with the VAMP2 protein (Arthur and Stowell, 2007), although its role in vesicular release is still unclear. SYP1 has 4 proposed transmembrane domain helices transversing the vesicular membrane, with both N and C termini facing the cytosol. The C terminus of SYP1 has been previously tagged with fluorescent proteins without affecting its function (Dreosti et al., 2009). We genetically fused miniSOG to the N terminus and C terminus of VAMP2 and SYP1, respectively (Figure 1A). To visualize expressing cells, mCherry was placed after the coding sequence of miniSOG-VAMP2 and SYP1-miniSOG, connected by a cotranslationally self-cleaving Thosea asigna virus 2A-like sequence (T2A) (Osborn et al., 2005). The expression of the tagged synaptic proteins and the cytosolic red fluorescent protein were tightly linked genetically, even though the proteins were not fused to each other.
Figure 1. The Designs and Testing of the CALI-Based Synaptic Inactivation System.
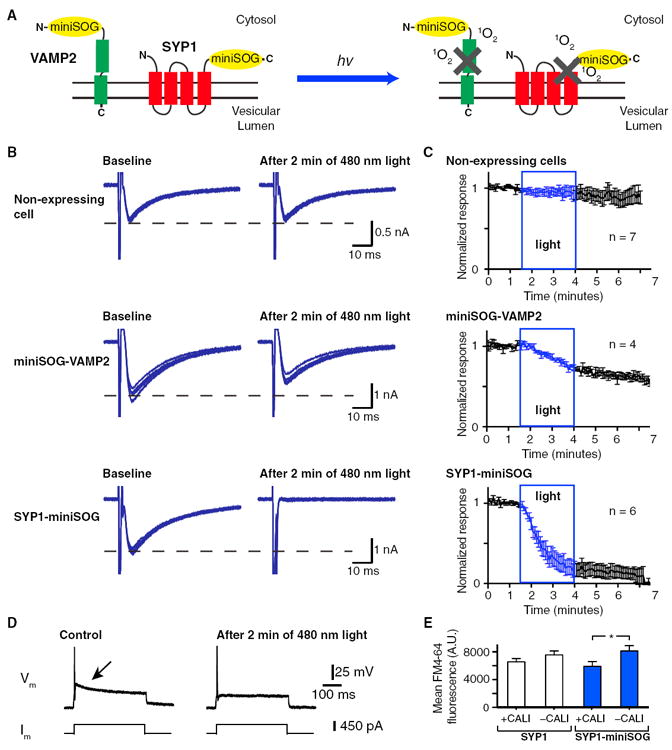
(A) The two designs of CALI-based synaptic inactivation system with miniSOG fused to Vesicular Associated Membrane Protein 2 (VAMP2) or Synaptophysin (SYP1). In the design with VAMP2, miniSOG is fused to the N terminus of VAMP2 facing the cytosolic space. With the SYP1 design, miniSOG is fused to the C terminus of the SYP1 also facing the cytosol. After light illumination, singlet oxygen (1O2) is generated by miniSOG leading to the inactivation of fusion protein.
(B) Representative examples of the effects of light on self-stimulated EPSCs recorded from nonexpressing control neurons (top), miniSOG-VAMP2-expressing neurons (middle), and SYP1-miniSOG-expressing neurons (bottom) before and after 2 min of 480 nm light illumination (9.8 mW/mm2). Each panel shows the overlap of 6 events (30 s). The synaptic release is induced with a 2 ms voltage step (−60 to 0 mV) at 0.2 Hz.
(C) Summaries of the inhibition of the self-stimulated EPSC amplitudes of the non-expressing control (top), miniSOG-VAMP2-expressing (middle), and SYP1-miniSOG-expressing (bottom) with blue light of 2.5 min duration.
(D) Current-clamp recordings from a SYP1-miniSOG expressing neuron in response to current injection. Action potential and self-stimulated excitatory post-synaptic potential (arrowed) can be evoked by the current step (left). After 2 min of 480 nm light illumination, the self-stimulated excitatory postsynaptic potential is abolished but the action potential remains (right).
(E) Mean FM4-64 fluorescence of SYP1-eGFP (SYP1) and SYP1-miniSOG-eGFP/Citrine (SYP1-miniSOG) puncta inside and outside the regions of CALI with 488 nm laser scan (25 mW and cumulative pixel illumination time of 13.86 ms).
See Figure S1 for example of FM4-64 images used for quantification. * indicates difference of p < 0.05. The error bars indicate SEM.
To assay the effects of miniSOG fused to VAMP2 and SYP1 on synaptic release, cultured hippocampal neurons were plated on microislands to induce autaptic synapse formation. The self-stimulated excitatory postsynaptic potential (EPSP) was typically observed as a prolonged depolarization after an action potential in current-clamp recording in response to a depolarizing current injection pulse (Wyart et al., 2005; Figure 1D). In voltage-clamp recording, a depolarizing voltage step can evoke a self-stimulated excitatory postsynaptic current (EPSC; Figure 1B). After establishing a stable baseline with repetitive stimulation, the recorded cell was illuminated for 2.5 min with 9.8 mW/mm2 of 480 nm light. In the nonexpressing cells, a gradual rundown of EPSCs was often observed with repetitive voltage steps independent of light illumination (6.5% ± 7.0% reduction in amplitudes, n = 7). In the miniSOG-VAMP2 expressing cells, a decrease of 29.4% ± 3.3% (n = 4, p = 0.006) in EPSC amplitude was observed after light illumination (Figures 1B and 1C). In the SYP1-miniSOG expressing cells, the reduction of EPSC amplitude was significantly greater at 82.6% ± 8.5% (n = 6, p = 0.0002). In two of the cells that exhibited the reduction of EPSC with SYP1-miniSOG, a stable recording was maintained for 45 min to 1 hr and no recovery in EPSC amplitude was observed. The reduction of EPSC amplitudes was associated with insignificant changes of electrophysiological properties such as membrane resistance (105.6 ± 35.6 MΩ to 73.6 ± 23.2 MΩ, n = 6, p = 0.20) or capacitance (62.5 ± 13.9 pF to 56.9 ± 6.7 pF, n = 6, p = 0.55). The reduction of EPSC amplitude did not alter the ability of the cell to fire action potentials in response to depolarizing current injections (Figure 1D; n = 5). Active and continual synaptic release during illumination was not essential for the inhibition of synaptic release, as the inhibition was still observed in cells where the self-stimulation was discontinued during light illumination (63.7% and 45.7% inhibition in two cells tested with SYP1-miniSOG).
In order to examine whether we can selectively inhibit specific synapses with high spatial resolution, we performed an imaging-based assay with the membrane dye FM4-64 in combination with SYP1-miniSOG. In this assay, SYP1-miniSOG fused to eGFP or Citrine was expressed in cultured cortical neurons. A quadrant of the field of view (159 μm × 159 μm) was scanned with 488 nm laser to conduct CALI of the presynaptic terminals. Vesicular release was induced with 40 mM KCl solution containing 10 μM FM4-64. The high potassium solution and dye solution was subsequently washed out to repolarize the membrane and remove membrane bound FM4-64 (Cousin, 2008). We then measured and averaged the FM4-64 fluorescence of the puncta positive for both FM4-64 and SYP1-miniSOG-eGFP/Citrine inside and outside the CALI region (Figure 1E). The SYP1-min-iSOG-eGFP/Citrine positive puncta within the CALI region had significant less FM4-64 dye uptake compared to the puncta outside the CALI region (5911.2 ± 687.5 and 8118.3 ± 763.2 arbitrary units, n = 78 and n = 95, respectively; p = 0.037). However, this measurement is likely an underestimate of the true level of synaptic inhibition, as we only quantified puncta that are positive for both eGFP/Citrine and FM4-64 and omitted eGFP/Citrine positive puncta that did not have FM4-64 fluorescence. This selection was necessary as not all fluorescent protein-positive puncta were functional presynaptic boutons capable of dye uptake even in the absence of light illumination (see Figure S1 available online). No significant difference was detected between the puncta inside and outside the CALI region in SYP1-eGFP positive puncta (6562.6 ± 466.9 and 7551.1 ± 560.6 arbitrary units, n = 114 and n = 157, respectively; p = 0.20). These results demonstrated that we were able to inhibit presynaptic terminals with high spatial specificity.
Overall, the fusion of miniSOG to the synaptic proteins VAMP2 and SYP1 functionally inhibited synaptic release, with SYP1-miniSOG demonstrating greater effects under the same expression system in the cultured hippocampal neurons. We named this approach Inhibition of Synapses with CALI (InSynC).
Validation of InSynC in Synaptic Connections of Hippocampal Organotypic Slices
To test whether InSynC can depress synaptic connections in a nonautaptic system and whether illumination of presynaptic terminals is sufficient to inhibit vesicular release, we infected the CA3 region of hippocampal organotypic slices with recombinant adenoassociated virus (rAAV) containing SYP1-miniSOG under the human synapsin promoter and assayed the synaptic inputs in the CA1 region with field potential recordings and electrical stimulation. We fused the yellow fluorescent protein variant Citrine (Griesbeck et al., 2001) at the C terminus of SYP1-miniSOG, which enabled us to directly visualize the expression of InSynC at the CA3 presynaptic terminals projecting to CA1 (Figures 2A and 2B). When CA1 neurons were independently infected with Sindbis virus expressing the red fluorescent protein tdTomato, SYP1-miniSOG-Citrine punctate fluorescence signals were detected in the proximity of the tdTomato-expressing dendritic shaft (Figures 2A and 2B). Illumination of the local dendritic recording site in CA1 with 480 nm light led to 86.64% ± 8.55% depression in field excitatory postsynaptic potential (n = 6, p < 0.0001) while the amplitude of the fiber volley remained unchanged after light illumination (100.04% ± 10.38%), indicating minimal effects on the action potentials at presynaptic terminals (Figures 2C, 2D, and 2F). No significant reduction in field excitatory postsynaptic potential was detected in slices infected with rAAV expressing SYP1 directly fused to Citrine (96.30% ± 10.85%, n = 10; Figure 2E). We also expressed SYP1-miniSOG fused with T2A-mCherry sequence, and we observed 82.06% ± 1.99% reduction in electrically evoked EPSC amplitudes in whole-cell recordings of CA1 cells after 5 min illumination of 480 nm light (n = 8; p < 0.0001) (Figure 2G), whereas the slices expressing cytosolic mCherry alone (mCherry; 0.60% ± 6.45% increase, n = 9), cytosolic miniSOG and mCherry (miniSOG-T2A-mCherry; 11.49% ± 10.72%, n = 8) did not have significant decreases in EPSC amplitude (Figures 2G and 2H). Interestingly, in slices expressing miniSOG fused to membrane anchored mCherry (miniSOG-mCherry-CAAX), light caused a nonsignificant increase in electrically evoked EPSC amplitude (32.48% ± 10.61%, n = 12) (Figure 2H and S2). As synaptophysin overexpression had previously been reported to change release probability at presynaptic terminals (Alder et al., 1995), we measured the pair-pulse facilitation from cells in SYP1-miniSOG expressing slices, and compared to the cells from slices expressing mCherry, miniSOG-T2A-mCherry, and miniSOG-mCherry-CAAX (Figure S2 and Table 1). Despite the slight increase in pair-pulse facilitation in SYP1-miniSOG-expressing slices, the values between the five conditions were not significantly different. Light illumination did not change pair-pulse facilitation in any of the conditions. We also observed a slight increase in mEPSC frequency but not the mEPSC amplitude in SYP1-miniSOG-expressing slice (Figures 2J, S2, and Table 1). Both the frequency and the amplitude were nonsignificantly different from values in nonexpressing, mCherry, miniSOG-T2A-mCherry, and miniSOG-mCherry-CAAX-expressing slices prior to light illumination (Figures 2J, S2, and Table 1). Light illumination greatly increased the mEPSC frequencies in slices expressing miniSOG-mCherry-CAAX, (p < 0.0001), SYP1-miniSOG (p = 0.012), and miniSOG-T2A-mCherry (p = 0.047), whereas the mEPSC frequencies were not affected by light in mCherry-expressing slices (Figures 2I, 2J, and S2; Table 1). We were unable to accurately measure the mEPSC amplitudes after light illumination as the increased frequency of mEPSC leads to the superimposition of many events. In the miniSOG-mCherry-CAAX and SYP1-miniSOG recordings, the membrane resistance of the postsynaptic cells were not altered by light illumination (post-light/before light ratio of 0.95 ± 0.06 and 1.01 ± 0.04, respectively). To investigate the effects of membrane targeted miniSOG and its effects on EPSC, we expressed miniSOG-mCherry-CAAX in cultured cortical neurons and conducted whole-cell patch-clamp recordings. In two cells expressing miniSOG-mCherry-CAAX at high level, blue light illumination leads to the appearance of an inward current (129.0 and 57.4 pA) that is associated with a decrease in membrane resistance (14.3% and 55.4% decrease), indicative of increased permeability of the plasma membrane (Figure S2). This effect was not seen in non-expressing cells with light illumination.
Figure 2. Validation of InSynC in Hippocampal Synapses.
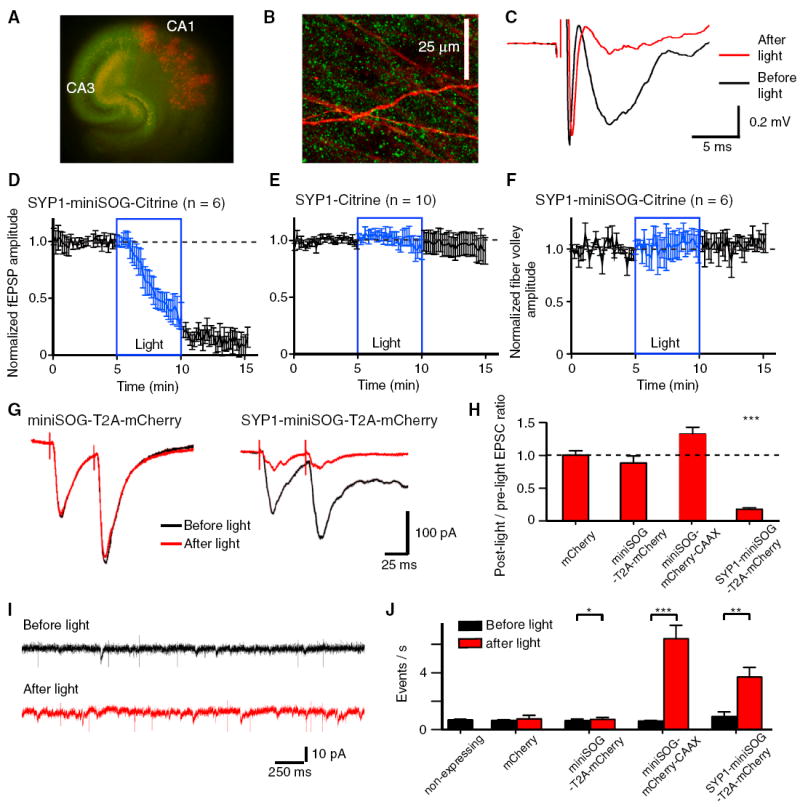
(A) Organotypic hippocampal slice culture coexpressing SYP1-miniSOG-Citrine (green) in presynaptic CA3 neurons and tdTomato (red) in sparsely labeled postsynaptic CA1 neurons. Recombinant AAV expressing SYP1-miniSOG-Citrine was injected into the CA3 region and yellow-green fluorescence can be detected in the CA1 region along the Schaffer collateral pathway. Sindbis virus expressing tdTomato was injected into CA1 to indicate region of field EPSP recording.
(B) High-magnification image of the stratum radiatum of CA1, punctate fluorescence of Citrine signal (green) can be detected throughout the field of view in close proximity of the tdTomato labeled apical dendrites (red).
(C) Example of field excitatory postsynaptic potential (EPSP) recording of the stratum radiatum of CA1 from SYP1-miniSOG-Citrine expressing slice before (black) and after (red) 480 nm light illumination showing the inhibition of field EPSP by light.
(D) Quantification of the light-induced inhibition of field EPSP recorded from SYP1-miniSOG-Citrine-expressing slices.
(E) Quantification of the light-induced inhibition of field EPSP recorded from SYP1-Citrine-expressing slices. Light-induced inhibition of field EPSP was observed in SYP1-miniSOG-Citrine-expressing slices but not in SYP1-Citrine-expressing slices.
(F) Quantification of the fiber volley amplitude in SYP1-miniSOG-Citrine-expressing slices before and after light illumination showing light has no effect on the presynaptic action potential as measured with fiber volley amplitude.
(G) Examples of electrically-evoked EPSCs recorded in CA1 neurons from slices expressing cytosolic miniSOG and mCherry (miniSOG-T2A-mCherry; left) and SYP1-miniSOG-T2A-mCherry (right) before (black) and after (red) light illumination.
(H) Summary graph of the EPSC amplitude ratio (post-light/pre-light) in slices expressing mCherry, cytosolic miniSOG/mCherry (miniSOG-T2A-mCherry), membrane tethered miniSOG and mCherry (miniSOG-mCherry-CAAX) and SYP1-miniSOG/cytosolic mCherry (SYP1-miniSOG-T2A-mCherry).
(I) Example traces of miniature EPSC recordings of CA1 neurons from SYP1-miniSOG-T2A-mCherry-expressing slice before (black) and after (red) light illumination. The fast events that lacked the characteristic profiles of EPSCs were electrical noises.
(J) Summary graph of miniature EPSC frequencies from slices expressing mCherry, miniSOG-T2A-mCherry, miniSOG-mCherry-CAAX, and SYP1-miniSOG-T2A-mCherry.
*, **, and *** indicate differences of p < 0.05, 0.01, and 0.001, respectively. The error bars indicate SEM. See Figure S2 for additional examples and analysis.
Table 1.
Summary of Pair-Pulse Facilitation Ratio and Miniature EPSC (mEPSC) Properties of Nonexpressing Organotypic Slices and Organotypic Slices Expressing mCherry or miniSOG Fusion Proteins
| Pair-Pulse Facilitation Ratio | mEPSC Amplitude before Light (pA) | mEPSC Frequency before Light (Hz) | mEPSC Frequency after Light (Hz) | |
|---|---|---|---|---|
| Nonexpressing | 1.58 ± 0.10 | 12.67 ± 0.46 pA | 0.68 ± 0.06 Hz | Not tested |
| n = 7 | n = 7 | n = 7 | ||
|
| ||||
| mCherry | 1.64 ± 0.13 | 9.78 ± 1.36 pA | 0.65 ± 0.05 Hz | 0.76 ± 0.25 Hz |
| n = 9 | n = 6 | n = 6 | ||
|
| ||||
| miniSOG-T2A-mCherry | 1.55 ± 0.15 | 10.61 ± 0.63 pA | 0.63 ± 0.12 Hz | 0.72 ± 0.14 Hz |
| n = 8 | n = 6 | n = 6 | ||
|
| ||||
| miniSOG-mCherry-CAAX | 1.61 ± 0.09 | 11.25 ± 0.74 pA | 0.61 ± 0.03 Hz | 6.4 ± 0.94 Hz |
| n = 12 | n = 7 | n = 7 | n = 7 | |
|
| ||||
| SYP1-miniSOG-T2A-mCherry | 1.93 ± 0.17 | 12.70 ± 1.50 pA | 0.92 ± 0.33 Hz | 3.7 ± 0.68 Hz |
| n = 8 | n = 8 | n = 8 | n = 8 | |
Validation of InSynC In Vivo in C. elegans
To test whether we can utilize InSynC in behaving animals in vivo, we expressed InSynC in the nematode Caenorhabditis elegans. Mammalian VAMP2 shares 62.9% overall homology with C. elegans synaptobrevin and 86.4% homology in the N-terminal α helices that interact with the SNAP-25 and syntaxin. VAMP2 was chosen over SYP1, because mammalian SYP1 has no homologs in C. elegans. We expressed mammalian VAMP2 fused to miniSOG pan-neuronally in C. elegans. To confirm expression and trafficking of mammalian VAMP2 in C. elegans, Citrine was fused to the luminal C-terminal of miniSOG-VAMP2. Pan-neural-miniSOG-VAMP2-Citrine showed punctate expression in the nerve cords, corresponding to presynaptic terminals (Figure 4A). When miniSOG-VAMP2-Citrine was expressed in the synaptobrevin (snb-1) mutant worm strain md247 (Nonet et al., 1998), the movement phenotype of this strain was rescued (9.31 ± 3.14 bends/min in md247, n = 7 to 26.70 ± 5.19 bends/min in md247 + miniSOG-VAMP2, n = 6, p = 0.013) (Figure 3A). This functional rescue of the movement phenotype of snb-1(md247) mutant strain confirms the incorporation of mammalian VAMP2 into the C. elegans SNARE complex. Illumination of single worms carrying the miniSOG-VAMP2-Citrine transgene with 480 nm light (5.4 mW/mm2) for 3 and 5 min reduced the movements by 68.2% ± 4.2% and 89.9% ± 3.7%, respectively (7.87 ± 1.07 bends/min, p = 0.008 and 3.07 ± 1.19 bends/min, p = 0.003 for 3 and 5 min illumination, respectively) (Figure 3A). Complete paralysis was observed in three of the six worms illuminated with light. The animals were returned to agar plates containing bacteria for recovery, and after 2–3 hr some gradual recovery of movements was noticeable. When the animals were re-tested in identical condition 24 hr later, some recovery of the movements was observed (10.34 ± 3.48 bends/min, n = 4, p = 0.08 compared to before illumination).
Figure 4. Validation of InSynC on Postsynaptic Currents at Neuromuscular Junctions of C. elegans.
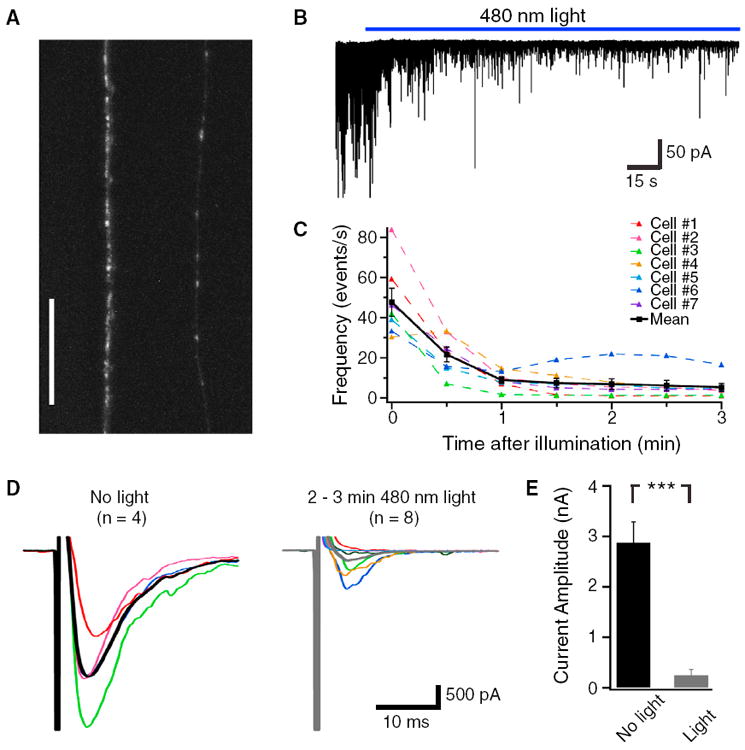
Expression of miniSOG-VAMP2-Citrine fusion protein in C. elegans neurons as visualized by Citrine fluorescence on a confocal microscopy (N2 wild-type background, strain number CZ13748). Punctate fluorescence at synaptic sites of the ventral and dorsal cords of the nematodes can be observed. See Figure S3 for miniSOG-Citrine expression.
(B) An example electrophysiological recording of the muscle of miniSOG-VAMP2 expressing C. elegans (CZ13748) and the effects of 480 nm light (30 mW/mm2) on spontaneous synaptic events.
(C) Quantification of the spontaneous event frequency with the illumination of light.
(D) Example traces of the electrically-evoked postsynaptic current before and after light illumination.
(E) Quantification of the peak amplitudes of electrically-evoked postsynaptic current before and after light illumination.
In (C) and (D), responses in individual cells are shown in different colors, and the mean responses before and after illumination are in black and gray, respectively. The scale in (A) is 25 mm. *** indicates difference at p < 0.001. The error bars indicate SEM.
Figure 3. The Effects of InSynC on Locomotion of C. elegans.
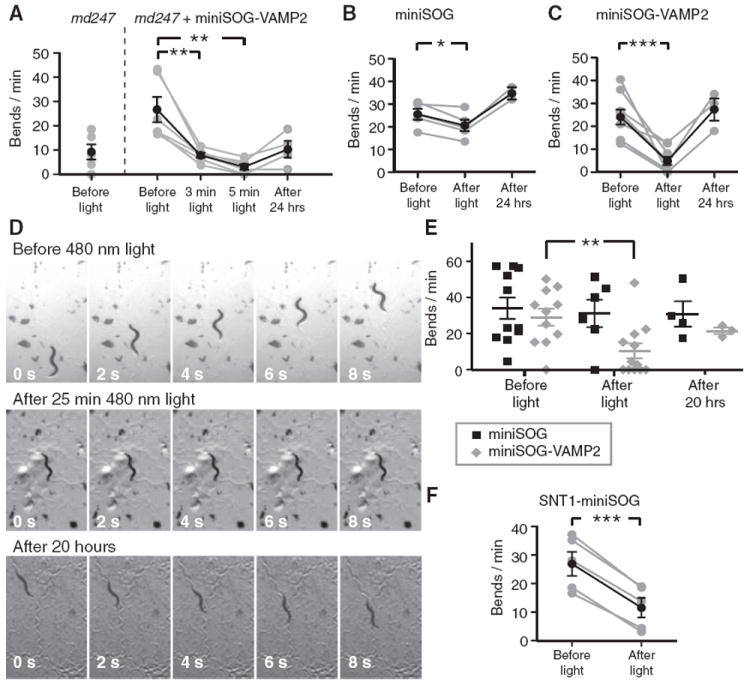
(A) Expression of miniSOG-VAMP2-Citrine (miniSOG-VAMP2) in synaptobrevin mutant worm strain md247 functionally rescued the movement phenotype of md247 as quantified with body bends/min. Illumination of miniSOG-VAMP2-Citrine expressing md247 worms with 480 nm light (5.4 mW/mm2, 3 and 5 min) reduced the movements of the worms. Some recovery was observed when the same worms were re-tested after 24 hr.
(B) The effects of 480 nm light illumination (5.4 mW/mm2, 5 min) on the movements of miniSOG-Citrine (miniSOG)-expressing worms of the wild-type background.
(C) The effects of 480 nm light illumination (5.4 mW/mm2, 5 min) on miniSOG-VAMP2-Citrine (miniSOG-VAMP2; B2)-expressing worms of the wild-type background. Light illumination strongly reduced the movements of the miniSOG-VAMP2-Citrine-expressing worms, with full recovery of movements 24 hr later. See also Movie S1.
(D) Examples of images extracted from movies of miniSOG-VAMP2-Citrine expressing C. elegans before (top) and after (middle) illumination with 480 nm light (0.7 mW/mm2, 25 min), and after 20 hr of recovery in the dark (bottom). Worms successfully transferred after the testing were re-tested for the recovery of movements 20 hr later on agar plate containing bacteria after initial testing.
(E) Quantification of the C. elegans movements on the agar dish before and after light illumination (0.7 mW/mm2, 25 min) and after recovery period.
(F) The effects of 480 nm light illumination (5.4 mW/mm2, 5 min) on the movements of SNT-1-miniSOG-Citrine-expressing worms (SNT1-miniSOG). In (A), (B), and (C), the same worms were tested before and after light illumination, and allowed to recover for 24 hr on agar dishes seeded with bacteria before retesting on bacteria-free agar. The gray lines and symbols represent results from individual worms and the black lines and symbols represent the mean. In (D), multiple worms on the same plate were simultaneously tested. *, **, and *** indicate differences at p < 0.05, p < 0.01, p < 0.001, respectively. The strain numbers for miniSOG-VAMP2-Citrine expressing animal in (A) is CZ13749 and CZ13748 for (C) and (D). The strain number for snt-1-miniSOG-Citrine-expressing animal in (E) is CZ13750 and the miniSOG-Citrine animal in (B) and (D) is CZ14344. The error bars indicate SEM.
A more challenging test of CALI was to determine whether illumination of miniSOG-VAMP2-Citrine could inhibit synaptic release in the presence of normal endogenous VAMP2. When miniSOG-VAMP2-Citrine was expressed in wild-type (N2) worms, the animals showed normal movement under standard culture condition. When single miniSOG-VAMP2-Citrine expressing worms were illuminated for 5 min with 480 nm light (5.4 mW/mm2), we were able to achieve 80.6% ± 7.3% reduction of movements (24.06 ± 3.28 bends/min before light and 4.89 ± 1.76 bends/min after light; n = 9, p = 0.0004) (Figure 3C and Movie S1). Four of the nine worms tested were paralyzed after illumination. Partial recovery of movements was observed in some of the worms re-tested after 6 hr (16.69 ± 5.74 bends/min, 52.7% ± 32.4% of movements before illumination, n = 3). Full recovery of the movement was observed after 24 hr when the same worms were retested (27.38 ± 4.84 bends/min, 140.0% ± 4.0% of movements before illumination; n = 3) (Figure 3C). Control worms expressing miniSOG fused to Citrine without VAMP2 showed a smaller 19.8% ± 4.0% reduction in movements after illumination (25.55 ± 2.40 bends/min and 20.61 ± 2.52 bends/min before and after light illumination, respectively, p = 0.01; n = 5) (Figure 3B). As expected, the fluorescence of miniSOG-Citrine was located at soma and not presynaptic terminals (Figure S3).
We also tested whether we can achieve the same effect with weaker illumination intensity for longer duration (480 nm light for 25 min at 0.7 mW/mm2) in a population of expressing worms. In this experiment, multiple worms were moved to a bacteria-free agar plate and the entire plate was illuminated. The movements of different worms in multiple regions on the plates were imaged and quantified separately. Illumination of the agar plate significantly reduced the movements of the miniSOG-VAMP2-Citrine-expressing worms from 29.04 ± 4.66 body bends/min before light, (n = 11) to 10.49 ± 4.18 bends/min after light, (n = 12), a 63.9% reduction (p = 0.007) (Figures 3D and 3E). In 5 of 12 worms (42%), movement was eliminated by the illumination. We did not observe significant changes in the movement of control worms expressing miniSOG-Citrine after light illumination (34.01 ± 5.91 bends/min before light, n = 11, and 31.15 ± 7.61 bends/min after light, n = 6, 8.4% reduction, p = 0.77) (Figure 3E). The recovery of movement in miniSOG-VAMP2-expressing worms was observed after 20–22 hr in miniSOG-VAMP2 expressing worms (21.76 ± 1.79 bends/min, p = 0.44) on bacteria containing agar plates (Figure 3E). In multiple animals on the recovery dish, the worms aggregated in groups on the bacterial lawn and the movements were not quantified. However, tracks from the animals could be seen on the dish, indicative of movements prior to aggregation. In some animals, the movements were interrupted when they encountered other animal and these were not quantified.
We then performed patch-clamp recording of the C. elegans muscles to confirm the reduction of synaptic inputs onto muscles after illumination with 480 nm light (15 or 30 mW/mm2). The recordings were done on miniSOG-VAMP2-Citrine worms of wild-type background. The spontaneous EPSC frequency was reduced from 47.67 ± 7.00 to 5.22 ± 1.98 events/s after 3 min of light illumination (89.1% reduction, n = 7; p < 0.0001) (Figures 4B and 4C). The inhibition of spontaneous EPSCs occurred largely within 1 min of illumination. There was also a significant reduction in the mean amplitude in electrically evoked EPSCs after 2–3 min of light (0.247 ± 0.12 nA, n = 8) compared to the mean amplitudes without light illumination (2.88 ± 0.41 nA, n = 4; p < 0.0001) (Figures 4D and 4E). In 4 of 8 animals, the electrically evoked EPSCs were abolished by illumination. No effects of light were observed in the nonexpressing progeny from the same parent. To test whether overexpression of miniSOG-VAMP2-Citrine altered vesicular fusion mechanisms, we compared the amplitudes, frequency and the kinetics of spontaneous release in non-expressing and miniSOG-VAMP2-Citrine-expressing worms. None of the parameters measured were significantly different between the two groups without blue light illumination (Figure S4 and Table S1).
Specificity of the CALI Approach
To test the specificity of the InSynC approach, we made additional worms expressing miniSOG-Citrine fused to the C terminus of C. elegans synaptotagmin (SNT-1) (Figure S3). Whereas the synaptobrevin deletion mutation in C. elegans is lethal (Nonet et al., 1998), the snt-1(md290) deletion mutant is viable and retains the ability to move, although at reduced capacity (Nonet et al., 1993). When SNT-1-miniSOG was expressed on wild-type background illumination (5.4 mW/mm2, 5 min) reduced movement by only 60.7% ± 7.4% (27.13 ± 4.2 bends/min and 11.78 ± 3.4 bends/min before and after illumination, respectively, n = 5; p = 0.0001), and complete paralysis was not observed in any of the five worms tested (Figure 3F). The milder effect of CALI on synaptotagmin versus synaptobrevin is consistent with the reduced penetrance of the respective genetic deletions, as if CALI were preferentially inactivating the protein species to which the miniSOG was directly fused, although this could also be explained by other factors such as the expression level of the fusion protein and the susceptibility of the fusion protein to oxidation.
To further test the extent of singlet oxygen mediated CALI in living cells, we expressed singlet-oxygen sensitive fluorescent protein IFP1.4 (Shu et al., 2011) in cultured neurons fused directly to SYP1, SYP1-miniSOG, rat synaptotagmin-1 (SYT1) or expressed as a plasma membrane tethered form (pm-IFP) (Figure 5). In cells expressing SYT1-IFP and pm-IFP, SYP1-Citrine or SYP1-miniSOG-Citrine were coexpressed to test the bleaching of the IFP by differentially-located miniSOG. Exogenously expressed SYT1 with fluorescent protein at the C-terminal has previous been shown to localize to synaptic vesicles but not incorporated in the SNARE complex (Han et al., 2005). IFP fused to SYP1-miniSOG had significant greater bleaching after 93 s of cumulative 495 nm light illumination compared to SYP1-IFP (49.7% ± 1.5% versus 28.0% ± 1.0% bleaching, n = 85 and n = 85, respectively; p < 0.0001). The bleaching of SYT1-IFP in the presence of miniSOG fused to SYP1 (34.6% ± 1.5%, n = 81) was greater than SYP1 control (14.4% ± 1.4%, n = 56; p < 0.0001). The bleaching of pm-IFP in the presence of miniSOG fused to SYP1 (21.5% ± 1.0%, n = 144) was also significant greater than SYP1 control (15.6% ± 1.1%, n = 102; p < 0.0001). However, the difference of pm-IFP bleaching between the SYP1 control and SYP1-miniSOG (5.9%; 95% confidence interval of 3.0% to 8.8%) was smaller than the difference of SYT1-IFP bleaching between the SYP1 control and SYP1-miniSOG (20.2%; 95% confident interval of 16.0% to 24.5%) or the difference of bleaching between SYP1-IFP and SYP1-miniSOG-IFP (21.7%; 95% confidence interval of 18.1% to 25.4%). These results demonstrated singlet oxygen generated by SYP1-miniSOG can oxidize other synaptic proteins on the vesicles, and to a smaller extent, the proteins located on the plasma membrane, although this could potentially due to the plasma membrane localization of exogenously-expressed SYP1 (Li and Tsien, 2012) or the vesicular uptake of some pm-IFP.
Figure 5. The Extent of Singlet Oxygen-Mediated Photo-oxidation as Measured with IFP Photobleaching.
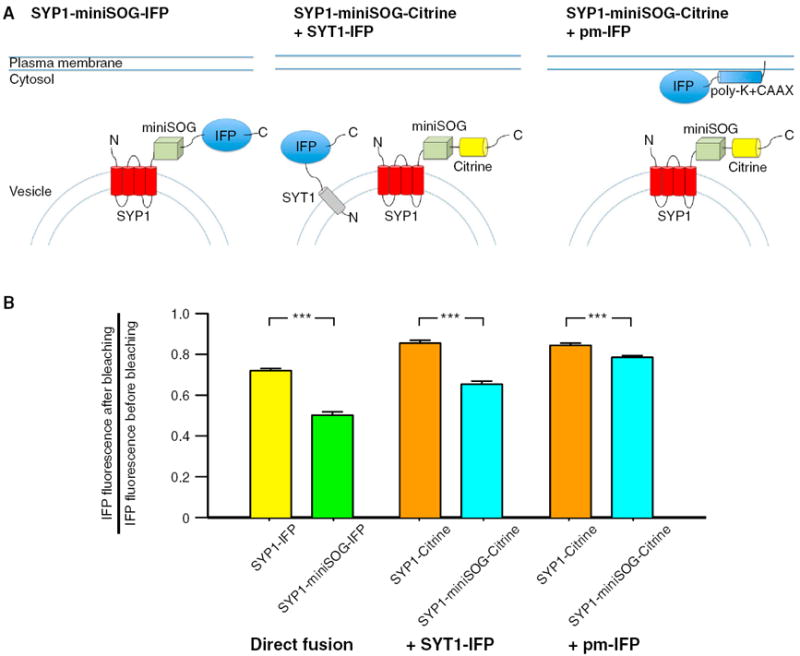
(A) Schematic drawing showing the three conditions tested in the IFP bleaching experiments. IFP was expressed in neurons either fused to the C terminus of the SYP1 or SYP1-miniSOG (left panel). In the other conditions, IFP was coexpressed as synaptotagmin-1 (SYT1) fusion (middle panel) or tethered to the plasma membrane (pm-IFP) in neurons expressing SYP1-miniSOG-Citrine or SYP1-Citrine.
(B) Summary graph showing the IFP bleaching in the six conditions tested. Significant greater IFP bleaching were observed in all three conditions. A smaller difference was observed when IFP was expressed on the plasma membrane and SYP1-miniSOG or SYP1 were expressed on the vesicles. MiniSOG was excited by 93 s of 495 nm light at 20 mW/mm2 and IFP was imaged with 665 nm excitation light.
*** indicates difference at p < 0.001. The error bars indicate SEM.
DISCUSSION
In the current study, we developed an optogenetic technique, InSynC, to inhibit synaptic release with light using chromophore-assisted light inactivation. InSynC with synaptophysin (SYP1) is much more efficient than the corresponding VAMP2 version in the mammalian system. The exact function of synaptophysin in synaptic release is unclear, although it is known to be closely associated with VAMP2 (Washbourne et al., 1995). Both exogenously expressed VAMP2 and synaptophysin tagged with fluorescent proteins are known to incorporate into endogenous v-SNARE (Deák et al., 2006; Dreosti et al., 2009). It has been proposed that synaptophysins are assembled with a connexon-like structure (Arthur and Stowell, 2007), and membrane channels such as connexins or calcium channels have previously been shown to be sensitive to CALI (Tour et al., 2003). Gratifyingly, we were able to inhibit synaptic release efficiently by illuminating miniSOG fusion proteins without replacing the endogenous proteins, either due to the dominant-negative effect of inactivated miniSOG-fusion proteins within the SNARE complex, or the extension of the CALI effect beyond the fusion protein. In the current study, we cannot conclusively distinguish between one mechanism over the other, and it is possible that both mechanisms play a role in inactivating the synaptic release. Our IFP bleaching results demonstrated that the effects of singlet oxygen can extend beyond the fusion protein. However, the concentration of singlet oxygen decreases exponentially from the site of generation, and its effect should be strongest on the fusion protein. The different efficiency with SYP1-miniSOG and miniSOG-VAMP2 in hippocampal culture, and miniSOG-VAMP2 and SNT-1-miniSOG in C. elegans, supported this hypothesis, although this could also be potentially explained by the difference in expression level or the residues susceptible to oxidation on the proteins. The estimated exponential space constant for singlet oxygen diffusion in the cytosol is 70 nm (Hatz et al., 2007), which is greater than the diameter of an average synaptic vesicle (~50 nm) (Kim et al., 2000). The inhibition of synaptic response is only observed when miniSOG is tethered to synaptophysin or VAMP2 and not with membrane-tethered miniSOG, suggesting the inhibition of synaptic release with InSynC requires the specific inhibition of vesicular proteins. It is interesting to note that mEPSC frequency is increased by light in both membrane-tethered miniSOG and SYP1-miniSOG after light illumination, possibly due to the localization of some SYP1-miniSOG onto the plasma membrane (Li and Tsien, 2012) or the oxidation of membrane protein by singlet oxygen diffused to the membrane (see IFP bleaching). The enhanced mEPSC and electrically evoked EPSC by membrane targeted miniSOG after illumination is likely to result from the inward current and the potential depolarization associated with illumination. The mechanism responsible for this inward current is unknown and requires further investigation. The physiological functions of spontaneous release in neuronal signaling are not known, although it has been suggested that spontaneous release stabilizes synapse (McKinney et al., 1999) and tune the sensitivity of the postsynaptic membrane to neurotransmitters (Sutton et al., 2006). The users of the InSynC technology need to be aware of these possible effects when interpreting the results, especially in long-term behavior experiments. Our results also indicated that synaptophysin may have distinct roles in synchronous and asynchronous release at presynaptic terminals as has been suggested with other SNARE proteins (Deitcher et al., 1998; Schulze et al., 1995).
Due to the cuticle, C. elegans resists the introduction of many chemicals. However, genetic modification of C. elegans is relatively easy. C. elegans is an ideal model for the use of InSynC. Mammalian VAMP2 shares a high degree of homology to C. elegans synaptobrevin, and the miniSOG-VAMP2 protein can rescue the behavioral abnormality of the synaptobrevin mutant strain md247, suggesting that mammalian VAMP2 can efficiently incorporate into the C. elegans SNARE complex. The stronger inhibitory effects of mSOG-VAMP2 in C. elegans compared to the mammalian system is likely to be associated with the stronger expression of miniSOG-VAMP2 in C. elegans than in primary hippocampal cultures with human synapsin promoters. We were also able to reduce the movements of worms with synaptotagmin (SNT-1)-miniSOG but its effect was weaker than miniSOG-VAMP2. Therefore, the best InSynC system to utilize will depend on the organism and the phenotype the experimenters wish to achieve.
The replacement of inactivated proteins with newly synthesized proteins is likely the mechanism of recovery. Presynaptic proteins are believed to be synthesized in the soma and transported down the axon, with minimal local protein translation at the presynaptic terminal (Hannah et al., 1999). In our experiments with primary cultured hippocampal neurons and in C. elegans, we illuminated the whole neuron or the whole worm, potentially destroying the newly synthesized protein at the soma and the protein en route to the presynaptic terminal, in addition to the proteins already present in the presynaptic vesicles. It is likely the recovery of the synaptic function can be quicker if illumination is focused on the presynaptic terminal only. In the organotypic slices, only the presynaptic terminals were illuminated, and this is sufficient to inhibit presynaptic vesicular release efficiently. The time required for recovery may also depend on the axon length if the whole neuron is illuminated. The long duration of the effect can be advantageous in experiments where the behavior tested is complex and long lasting.
Compared to current techniques of inhibiting neuronal activities with microbial opsin pumps, InSynC has the following differences: (1) InSynC inhibits synaptic release and not the firing of action potentials and therefore can be used to inhibit a single, spatially distinct axonal innervation without inhibiting other axonal projections made by the same cell. (2) InSynC takes more time to build up but has a long-lasting effect (>1 hr) that persists after the termination of the light pulse. The slower kinetics of InSynC will prevent some biophysical applications requiring precision timing but should facilitate experiments in which synapses are sequentially inactivated to titrate effects on circuit dynamics. (3) Effective light illumination for InSynC is on the presynaptic site and not the soma, potentially reducing light-mediated toxicity to the cell. (4) The effects of InSynC can be graded and not all-or-none. As with all other optogenetic techniques, the efficiency of such techniques depends heavily on the expression level and the properties of the cells targeted. Given the widespread distribution of VAMP2 and synaptophysin in the mammalian nervous system (Marquèze-Pouey et al., 1991; Trimble et al., 1990), it is likely that InSynC will be applicable to the majority of neurons targeted.
In conclusion, we have demonstrated that it is possible to use a genetically-encoded singlet oxygen generator to conduct CALI experiments in vitro and in vivo, and that CALI can be used to engineer new optogenetic techniques by inhibiting the function of specific proteins. Our optogenetic technique, InSynC, is a powerful method for inhibiting synaptic release with light, and is currently the only optogenetic approach that can efficiently inhibit a specific axonal projection in vivo and in vitro. This approach complements the existing optogenetic tools and can be used to study the function of specific projections.
EXPERIMENTAL PROCEDURES
Constructs and Molecular Cloning
Complementary DNA (cDNA) encoding Vesicle-associated membrane protein (VAMP2), C. elegans synaptotagmin 1 (SNT-1) and synaptophysin (SYP1) were fused to miniSOG by polymerase chain reaction with Phusion (New England Biolabs). VAMP2 and SYP1 fused with miniSOG were inserted into a lentiviral vector (gift from Ed Boyden, MIT) with the hSynapsin promoter and Woodchuck Postranscription Regulatory Element (WPRE). A Thosea asigna virus 2A (T2A) sequence was fused in frame with mCherry in the lentiviral vector at 3′ end of the transgene. The AAV2 vector (gift from Dr. Lin Tian, University of California, Davis) contained the hSynapsin promoter and WPRE flanking the SYP1-miniSOG or SYP1. The sequence coding for Citrine was inserted in frame at the 3′ end. VAMP2 cDNA was provided by Dr. S. Andrew Hires (Janelia Farm Research Campus) and SYP1 cDNA was amplified by RT-PCR from rat brain RNA (Clontech). The C. elegans synaptotagmin 1 (snt-1) cDNA was provided by Dr. Erik Jorgensen (University of Utah).
For the worm constructs, miniSOG-VAMP2, miniSOG, and snt-1-miniSOG were fused to the Citrine cDNA at the 3′ end and inserted into the Gateway entry vector (Life Technologies). LR reaction (Life Technologies) was used to introduce this insertion into the Prgef-1 destination vector PCZGY66 vector for injection into C. elegans.
The annotated DNA and protein sequences of InSynC are provided in Supplemental Information.
Recombinant Adenoassociated Virus Production
Recombinant adenoassociated virus with serotype 8 containing the SYP1-miniSOG-Citrine, SYP1-Citrine, SYP1-miniSOG-T2A-mCherry, mCherry, miniSOG-T2A-mCherry, and miniSOG-mCherry-CAAX were produced according to the protocols at http://vectorcore.salk.edu/protocols.php with minor modifications. In brief, AAV2 plasmid and helper plasmids XX6-80 and XR8 (National Vector Biorepository) were transfected into 293A cells (Life Technology) with calcium phosphate precipitation (Clontech). Recombinant AAV2/8 were released from the cells by freeze-thawing and purified with iodix-anol gradient purification. The virus was further concentrated using 50 kDa Amicon Ultra centrifugal filters (Millipore). The rAAV-SYP1-miniSOG-Citrine titer was measured by quantitative PCR to be 6.6 × 1013 genome copy (GC)/ml (Salk Vector Core). The rAAV-SYP1-miniSOG-T2A-mCherry titer was estimated to be 2.3 × 1013 GC/ml with Quant-IT picogreen dsDNA dye (Life Technologies).
Sindbis Virus Production
Sindbis virus containing the tdTomato transgene is produced as described previously (Malinow et al., 2010). In brief, BHK cells were electroporated with RNAs transcribed from pSinRep5-tdTomato and DH(26S) plasmids. The media was collected 40 hr later and centrifuged to obtain the concentrated virus.
Hippocampal and Cortical Neuronal Culture and Electrophysiological Recording
Hippocampal microisland cultures were made by a protocol modified from Bekkers (2005). In brief, a collagen (0.5mg/ml, Affymetrix)/poly-d-lysine (0.1mg/ml) mixture was sprayed onto the glass surface of glass bottom dishes (MatTek) with an atomizer. Hippocampal and cortical neurons were extracted from P2 Sprague-Dawley rat pups with papain digestion and mechanical trituration. Hippocampal neurons were transfected by electroporation (Lonza) and plated at 1.5–3 × 104 cells per dish. Cortical neurons were plated on poly-d-lysine coated dish and infected with rAAV three days after plating. The procedures of extracting cultured neurons and organotypic slices (below) from rat pups were approved by the UCSD Institutional Animal Care and Use Committee.
Cultured hippocampal neurons were placed on an Olympus IX71 microscope with 20× air phase contrast objective for the recording (Olympus). Illumination (9.8 mW/mm2) from a xenon arc lamp (Opti-quip) was filtered through a 480/40 nm filter and reflected to the specimen with a full-reflective mirror (Chroma). Illumination was controlled with a mechanical shutter (Sutter Instrument). Recordings were performed with an Axopatch 200B patch amplifier, Digidata 1332A digitizer, and pCLAMP 9.2 software (Molecular Devices). EPSCs were evoked with a 2 ms voltage step from −60 mV to 0 mV at 0.2 Hz. Illumination was initiated after 1.5 min of stable baseline (changes <10%) of EPSC amplitude. One hundred percent response for each cell was the mean EPSC amplitude of the 1 min prior to light illumination and the amplitudes of each EPSC were normalized to this 100% response. Reduction of EPSC amplitudes was measured as the mean amplitudes of 6 EPSCs (25 s) after light illumination. Only cells with series resistance <10 MΩ and changes of series resistance <20% after light illumination were analyzed. The external solution contained 118 mM NaCl, 3 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 20 mM glucose (pH 7.35, 315 mOsm). The intracellular pipette solution contained 110 mM K-gluconate, 30 mM KCl, 5 mM NaCl, 2 mM MgCl2, 0.1 mM CaCl2, 2 mM MgATP, 0.3 mM TrisGTP, and 10 mM HEPES (pH 7.25, 285 mOsm). Cortical neurons were recorded with intracellular solution containing 110 mM Cs methanesulfonate, 30 mM tetraethylam-monium chloride, 10 mM EGTA, 10 mM HEPES, 1 mM CaCl2, 1 mM MgCl2, 2 mM Mg-ATP, 0.15 mM Na3-GTP (pH 7.25, 285 mOsm). With cortical neuron recording, the extracellular solution contained 1 μM TTX. Light intensity was measured with a calibrated photometer with an integrating sphere detector (International Light Technologies) placed on the objective. A glass slide with a semispherical lens was used to direct the light into the integrating sphere. The area of illumination was measured with a stage micrometer for illumination intensity.
Hippocampal Organotypic Slice Culture and Electrophysiological Recording
Organotypic hippocampal slices were prepared from 2 days old rat pups as described previously (Shi et al., 1999). The various constructs were expressed using rAAV virus in CA3 neurons in 2 DIV slice cultures. Cells were allowed to express for 14–17 days before being used for recordings. CA3 region was surgically removed to prevent stimulus induced bursting. Recordings were done in ACSF containing 119 mM NaCl, 2.5 mM KCl, 26 mM NaHCO3, 1 mM NaH2PO4, 11 mM glucose, 4 mM MgCl2, and 4 mM CaCl2, 100 μM picrotoxin and 4 μM 2-chloroadenosine (pH 7.4) at 24°C–28°C.
Organotypic hippocampal slices were placed in a recording chamber on an Olympus BX50WI microscope with 60× water immersion objective (Olympus). Extracellular field potential was recorded in stratum radiatum with glass electrodes (1–2 MΩ) filled with the perfusion solution. Synaptic responses were evoked by stimulating two independent pathways using bipolar stimulating electrodes (Frederick Haer) placed 150–250 μm down the apical dendrites, 100 μm apart, and 150–200 μm laterally in opposite directions. Field EPSP was measured by averaging the response over a 5 ms fixed window covering the peak amplitude. Results from two pathways were averaged and analyzed as n = 1. Whole-cell patch-clamp recordings were done with intracellular solution containing 115 mM Cs-Methanesulfonate, 20 mM CsCl, 10 mM HEPES, 2.5 mM MgCl2, 4 mM Na2ATP, 0.4 mM Na3GTP, 10 mM Na-phosphocreatine, 0.6 mM EGTA(pH 7.25). Electrically evoked EPSCs and miniature EPSCs were recorded under voltage clamping (Vhold = −60 mV; junctional potential not corrected). Recording and analysis were done with IGOR Pro (WaveMetrics). For the analysis of mEPSC frequency, EPSCs that had the characteristic excitatory EPSC profile (Bekkers et al., 1990) was manually identified over 1 min period of recording. Light illumination was provided from a 100 W mercury arc lamp filtered through a eGFP filter set with 480/40 nm excitation filter (Olympus). The illumination area was 360 μm in diameter.
Confocal and Fluorescence Imaging
Hippocampal organotypic slices infected with rAAV and Sindbis virus were imaged under low magnification with a MVX10 Macroview microscope (Olympus). Citrine fluorescence was imaged with the eGFP filter set and tdTomato was imaged with a Texas Red filter set. For high magnification, the slices were fixed with 4% paraformaldehyde and imaged with a Zeiss LSM 780 confocal microscope (Zeiss).
C. elegans Expression and Behavior
Germline transmission of Prgef-1-miniSOG-VAMP2-Citrine, Prgef-1-snt-1-miniSOG-Citrine, and Prgef-1-miniSOG-Citrine was achieved following standard procedures (Mello and Fire, 1995). The plasmids were injected at 5-15 ng/μl into N2 or NM467 SNB-1 (md247) (Nonet et al., 1998) using Pmyo2-mCherry or Pttx3mCherry as coinjection markers. For behavioral experiments, a single worm (L4 stage) was transferred onto glass bottom culture dishes (MatTek) coated with a thin layer of Nematode Growth Medium (NGM) agar. The worms were illuminated (480/40 filter) with a 20× air objective on an Olympus IX71 inverted microscope (Olympus). The illumination is reflected onto the specimen plane with a 518 nm dichroic to allow visualization of the worms for tracking during the illumination period (5 min). The light intensity was measured to be 5.4 mW/mm2 with the procedure described under the Hippocampal Neuronal Culture and Electrophysiological Recording section. The worms were manually tracked during illumination for continuous illumination. After testing, the worms were transferred back to the NGM agar dish seeded with bacteria for recovery. The animals successfully transferred between the dishes were used for testing recovery the next day. For experiment with weaker light intensity and longer illumination duration, multiple L4 worms were placed on a 35 mm dish coated with agar were illuminated under a Zeiss Discovery V12 dissecting microscope with an eGFP filter set (480/40 nm excitation) at 9.3× magnification for 25 min. The light intensity was measured to be 0.7 mW/mm2 with a planar light detector connected to a power meter (Newport). Movement was recorded using a Nikon DS-QiMc camera and NIS elements F3.0 software (Tokyo, Japan). In some of the regions, multiple worms aggregate in regions with bacteria carried over with the animal transfer, the movements of these worms were not used for quantification. The quantification of movement was done according to Sawin et al. (2000). The strain numbers for N2 worms with pan-neuronal expression of miniSOG-VAMP2-Citrine, snt-1-miniSOG-Citrine, and miniSOG-Citrine are CZ13748, CZ13750, and CZ14344, respectively. The strain number for miniSOG-VAMP2-Citrine expression in md247 background is CZ13749.
C. elegans Electrophysiology
Dissection methods were described as previously (Stawicki et al., 2011). Adult worms were immobilized on Sylgard-coated coverslips with cyanoacrylate glue. A dorsolateral incision was made with a sharp glass pipette and the cuticle flap was glued down to expose the ventral medial body wall muscles. The preparation was then treated by collagenase type IV (0.4 mg/ml; Sigma-Aldrich) for ~30 s.
The bath solution containing (in mM): 127 NaCl, 5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 4 MgCl2, 10 glucose, and sucrose to 340 mOsm, bubbled with 5% CO2, 95% O2 at 20°C. The pipette solution containing (in mM): 120 Cs-methanesulfonate, 4 CsCl, 15 CsF, 4 MgCl2, 5 EGTA, 0.25 CaCl2, 10 HEPES, and 4 Na2ATP, adjusted to pH 7.2 with CsOH. Whole-cell recordings (Vhold = −60 mV) from muscle cells were performed at 20°C with 2–3 MΩ pipettes. An EPC-10 patch-clamp amplifier was used with the Patchmaster software package (HEKA Electronics). An electrode positioned close to the ventral nerve cord was used to stimulate evoked release by applying a 0.5 ms 85 μA square pulse with a stimulus current generator (WPI). Further analysis was done with IGOR Pro (WaveMetrics). Illumination was provided with a Sutter Instrument Lambda LS with a filter wheel for shuttering. The excitation light was filtered with 480 nm excitation filter (N41012; Chroma) and focused on the specimen with a 63× water immersion objective (Olympus). Light intensity measurement was done similarly to that described for cell culture recordings.
FM4-64 Dye Imaging and Analysis
Cultured cortical neurons (15 DIV) plated on a glass-bottom culture dish were imaged on a Zeiss Live 5 Confocal Microscope with a 20× air objective. The neurons were electroporated with the indicated plasmids prior to plating. Prior to imaging, the cells were treated briefly with balanced saline solution containing 40 mM KCl before placed in the same external solution used in cell culture recordings. Five to seven regions (318 μm × 318 μm) on each dish were tested per experiment. After acquiring the initial image of eGFP/Citrine fluorescence of each region, a quadrant of the field of view (159 μm × 159 μm) was scanned with 488 nm laser (25% of 100 mW) with pixel dwelling time of 154 μs for 90 frames, given each pixel the cumulative illumination time of 13.86 ms. The sample was then perfused for 2.5 min (~3.5 ml) of 40 mM KCl containing-external solution containing 10 μM FM4-64FX (Life Technologies) before washout with standard external solution with no FM4-64 for 7.5 min (~10 ml). The same regions were then re-imaged for FM4-64 fluorescence. The eGFP/citrine fluorescence was excited with 488 nm laser (5% intensity of 100 mW) and imaged with a 505 LP emission filter and pixel dwell time of 154 μs. The FM4-64 fluorescence was excited with 532 nm laser (10% of intensity of 75 mW) and imaged with a 650 LP emission filter (pixel dwell time of 131 μs). For both imaging and CALI, 5 optical slices with 0.84 μm spacing were acquired in the z series.
For the analysis of the results, the CALI regions were identified on the images and the FM4-64 fluorescence of the puncta inside and outside the CALI region were quantified separately. As not all eGFP or Citrine positive puncta are presynaptic boutons capable of vesicular release (Figure S1), only puncta positive for both eGFP/Citrine and FM4-64 were used for quantification to avoid false negative. However, this criterion also underestimates the inhibition of vesicular release with InSynC, as presynaptic boutons that were strongly inhibited and failed to take up FM4-64 were not quantified. The mean fluorescence value from a region with no fluorescent structures in the FM4-64 image was chosen to provide the background value to be substracted. The fluorescence values were measured on ImageJ. For statistical comparison, unpaired Student’s t tests were performed on the mean FM4-64 fluorescence values inside and outside the CALI region.
IFP Bleaching
Cultured cortical or hippocampal neurons plated on glass-bottom culture dish (7 DIV) were imaged on an inverted microscope with a 40× oil objective (Zeiss). IFP (Shu et al., 2009) was imaged with a 665/45 nm excitation filter, 725/50 nm emission filter, and 695 nm dichroic mirror. Citrine and miniSOG were excited with 495/10 nm excitation filter, 515 nm dichroic mirror, and imaged with 535/25 nm emission filter. The light intensity of 495 nm excitation was 20 mW/mm2. The field of view and focus were adjusted with IFP fluorescence. The images of IFP and citrine were acquired at 512 × 512 pixel resolution (150 ms exposure time) with a Cascade 1024 EMCCD camera (Photometric). IFP bleaching and imaging were mediated by alternating 495 nm (3 s) and 665 nm excitation (150 ms) for 31 frames given accumulative 495 nm excitation of 93 s and 665 nm excitation of 4.65 s. For the analysis of the bleaching, puncta positive for both IFP and citrine were selected and the mean fluorescence was measured. The IFP fluorescence was normalized to the initial fluorescence intensity. Image acquisition and analysis were on performed on the Slidebook 5.0 software (Intelligent Imaging Innovations, Inc.). The fluorescence intensities of the corresponding treatment pairs were compared with unpaired Student’s t tests.
Plasma membrane targeting of IFP (pm-IFP) was achieved with a C terminus CaaX motif preceded by a lysine-rich sequence (KKKKKKSKTK). For C-terminal fusion of IFP to SYP1 and SYT1, flexible linkers (>25 amino acids) were used to ensure expression and trafficking. Both SYT1-IFP and pm-IFP were expressed under the control of the truncated hSynapsin promoter and a WPRE sequence was inserted after the stop codon. Expression in the neurons was achieved with electroporation prior to plating. For IFP imaging, the cells were incubated with 5 μM billiverdin in neurobasal media for 15 min prior to imaging.
Statistical Analysis
All values are expressed as mean ± SEM. Two-tailed paired student’s t tests were used for the comparison of the same sample before and after light illumination. Two-tailed unpaired Student’s t tests were used to compare two unmatched samples. For multiple comparisons, one-way ANOVA was used followed by Tukey’s multiple comparison tests between all pairs. The exact p values reported were not adjusted for multiple comparisons. Statistical tests were done with Graphpad Prism 5.
Supplementary Material
Acknowledgments
J.Y.L. was funded by Foundation of Research, Science and Technology New Zealand. S.B.S. was funded by Ruth L. Kirschstein National Research Service Awards (NIH NINDS NS067891). C.D.P. was funded by Instituts de Recherche en Santé du Canada. The project was supported by National Institutes of Health grants to R.M. (MH091119), Y.J. (NS035546), and R.Y.T. (NS027177). R.Y.T. and Y.J. are Investigators of the Howard Hughes Medical Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Neurological Disorders and Stroke or the National Institutes of Health.
Footnotes
J.Y.L. and S.B.S. designed the InSynC constructs. J.Y.L. conducted and analyzed the hippocampal microisland recordings. J.Y.L. and S.B.S. conducted and analyzed the worm movement and imaging experiments. K.Z. conducted and analyzed the electrophysiological recordings from C. elegans muscle cells. S.N. conducted and analyzed the organotypic slice experiments. R.Y.T., C.D.P., R.M., and Y.J. contributed to the design and analysis of the experiments. All authors contributed to the writing and discussion of the manuscript.
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures, one table, one movie, and Supplemental Text and can be found with this article online at http://dx.doi.org/10.1016/j.neuron.2013.05.022.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alder J, Kanki H, Valtorta F, Greengard P, Poo MM. Overexpression of synaptophysin enhances neurotransmitter secretion at Xenopus neuromuscular synapses. J Neurosci. 1995;15:511–519. doi: 10.1523/JNEUROSCI.15-01-00511.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur CP, Stowell MH. Structure of synaptophysin: a hexameric MARVEL-domain channel protein. Structure. 2007;15:707–714. doi: 10.1016/j.str.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck S, Sakurai T, Eustace BK, Beste G, Schier R, Rudert F, Jay DG. Fluorophore-assisted light inactivation: a high-throughput tool for direct target validation of proteins. Proteomics. 2002;2:247–255. doi: 10.1002/1615-9861(200203)2:3<247::aid-prot247>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Bekkers JM. Presynaptically silent GABA synapses in hippocampus. J Neurosci. 2005;25:4031–4039. doi: 10.1523/JNEUROSCI.4969-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Richerson GB, Stevens CF. Origin of variability in quantal size in cultured hippocampal neurons and hippocampal slices. Proc Natl Acad Sci USA. 1990;87:5359–5362. doi: 10.1073/pnas.87.14.5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- Bulina ME, Chudakov DM, Britanova OV, Yanushevich YG, Staroverov DB, Chepurnykh TV, Merzlyak EM, Shkrob MA, Lukyanov S, Lukyanov KA. A genetically encoded photosensitizer. Nat Biotechnol. 2006;24:95–99. doi: 10.1038/nbt1175. [DOI] [PubMed] [Google Scholar]
- Cousin MA. Use of FM1-43 and other derivatives to investigate neuronal function. Curr Protoc Neurosci. 2008;Chapter 2(Unit 2.6) doi: 10.1002/0471142301.ns0206s43. http://dx.doi.org/10.1002/0471142301.ns0206s43. [DOI] [PubMed] [Google Scholar]
- Deák F, Shin OH, Kavalali ET, Südhof TC. Structural determinants of synaptobrevin 2 function in synaptic vesicle fusion. J Neurosci. 2006;26:6668–6676. doi: 10.1523/JNEUROSCI.5272-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitcher DL, Ueda A, Stewart BA, Burgess RW, Kidokoro Y, Schwarz TL. Distinct requirements for evoked and spontaneous release of neurotransmitter are revealed by mutations in the Drosophila gene neuronal-synaptobrevin. J Neurosci. 1998;18:2028–2039. doi: 10.1523/JNEUROSCI.18-06-02028.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreosti E, Odermatt B, Dorostkar MM, Lagnado L. A genetically encoded reporter of synaptic activity in vivo. Nat Methods. 2009;6:883–889. doi: 10.1038/nmeth.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbeck O, Baird GS, Campbell RE, Zacharias DA, Tsien RY. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J Biol Chem. 2001;276:29188–29194. doi: 10.1074/jbc.M102815200. [DOI] [PubMed] [Google Scholar]
- Han X, Boyden ES. Multiple-color optical activation, silencing, and desynchronization of neural activity, with single-spike temporal resolution. PLoS ONE. 2007;2:e299. doi: 10.1371/journal.pone.0000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Rhee JS, Maximov A, Lin W, Hammer RE, Rosenmund C, Südhof TC. C-terminal ECFP fusion impairs synaptotagmin 1 function: crowding out synaptotagmin 1. J Biol Chem. 2005;280:5089–5100. doi: 10.1074/jbc.M408757200. [DOI] [PubMed] [Google Scholar]
- Hannah MJ, Schmidt AA, Huttner WB. Synaptic vesicle biogenesis. Annu Rev Cell Dev Biol. 1999;15:733–798. doi: 10.1146/annurev.cellbio.15.1.733. [DOI] [PubMed] [Google Scholar]
- Hatz S, Lambert JD, Ogilby PR. Measuring the lifetime of singlet oxygen in a single cell: addressing the issue of cell viability. Photochem Photobiol Sci. 2007;6:1106–1116. doi: 10.1039/b707313e. [DOI] [PubMed] [Google Scholar]
- Janovjak H, Szobota S, Wyart C, Trauner D, Isacoff EY. A light-gated, potassium-selective glutamate receptor for the optical inhibition of neuronal firing. Nat Neurosci. 2010;13:1027–1032. doi: 10.1038/nn.2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay DG. Selective destruction of protein function by chromophore-assisted laser inactivation. Proc Natl Acad Sci USA. 1988;85:5454–5458. doi: 10.1073/pnas.85.15.5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Atwood HL, Cooper RL. Assessing accurate sizes of synaptic vesicles in nerve terminals. Brain Res. 2000;877:209–217. doi: 10.1016/s0006-8993(00)02641-x. [DOI] [PubMed] [Google Scholar]
- Levitz J, Pantoja C, Gaub B, Janovjak H, Reiner A, Hoagland A, Schoppik D, Kane B, Stawski P, Schier AF, et al. Optical control of metabotropic glutamate receptors. Nat Neurosci. 2013;16:507–516. doi: 10.1038/nn.3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Tsien RW. pHTomato, a red, genetically encoded indicator that enables multiplex interrogation of synaptic activity. Nat Neurosci. 2012;15:1047–1053. doi: 10.1038/nn.3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JY, Lin MZ, Steinbach P, Tsien RY. Characterization of engineered channelrhodopsin variants with improved properties and kinetics. Biophys J. 2009;96:1803–1814. doi: 10.1016/j.bpj.2008.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Hayashi Y, Maletic-Savatic M, Zaman SH, Poncer JC, Shi SH, Esteban JA, Osten P, Seidenman K. Introduction of green fluorescent protein (GFP) into hippocampal neurons through viral infection. Cold Spring Harb Protoc. 2010 doi: 10.1101/pdb.prot5406. http://dx.doi.org/10.1101/pdb.prot5406. [DOI] [PMC free article] [PubMed]
- Marek KW, Davis GW. Transgenically encoded protein photoinactivation (FlAsH-FALI): acute inactivation of synaptotagmin I. Neuron. 2002;36:805–813. doi: 10.1016/s0896-6273(02)01068-1. [DOI] [PubMed] [Google Scholar]
- Marquèze-Pouey B, Wisden W, Malosio ML, Betz H. Differential expression of synaptophysin and synaptoporin mRNAs in the post-natal rat central nervous system. J Neurosci. 1991;11:3388–3397. doi: 10.1523/JNEUROSCI.11-11-03388.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney RA, Capogna M, Dürr R, Gähwiler BH, Thompson SM. Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nat Neurosci. 1999;2:44–49. doi: 10.1038/4548. [DOI] [PubMed] [Google Scholar]
- Mello C, Fire A. DNA transformation. Methods Cell Biol. 1995;48:451–482. [PubMed] [Google Scholar]
- Nonet ML, Grundahl K, Meyer BJ, Rand JB. Synaptic function is impaired but not eliminated in C. elegans mutants lacking synaptotagmin. Cell. 1993;73:1291–1305. doi: 10.1016/0092-8674(93)90357-v. [DOI] [PubMed] [Google Scholar]
- Nonet ML, Saifee O, Zhao H, Rand JB, Wei L. Synaptic transmission deficits in Caenorhabditis elegans synaptobrevin mutants. J Neurosci. 1998;18:70–80. doi: 10.1523/JNEUROSCI.18-01-00070.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn MJ, Panoskaltsis-Mortari A, McElmurry RT, Bell SK, Vignali DA, Ryan MD, Wilber AC, McIvor RS, Tolar J, Blazar BR. A picornaviral 2A-like sequence-based tricistronic vector allowing for high-level therapeutic gene expression coupled to a dual-reporter system. Mol Ther. 2005;12:569–574. doi: 10.1016/j.ymthe.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Petreanu L, Mao T, Sternson SM, Svoboda K. The subcellular organization of neocortical excitatory connections. Nature. 2009;457:1142–1145. doi: 10.1038/nature07709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi YB, Garren EJ, Shu X, Tsien RY, Jin Y. Photo-inducible cell ablation in Caenorhabditis elegans using the genetically encoded singlet oxygen generating protein miniSOG. Proc Natl Acad Sci USA. 2012;109:7499–7504. doi: 10.1073/pnas.1204096109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajfur Z, Roy P, Otey C, Romer L, Jacobson K. Dissecting the link between stress fibres and focal adhesions by CALI with EGFP fusion proteins. Nat Cell Biol. 2002;4:286–293. doi: 10.1038/ncb772. [DOI] [PubMed] [Google Scholar]
- Sawin ER, Ranganathan R, Horvitz HR. C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron. 2000;26:619–631. doi: 10.1016/s0896-6273(00)81199-x. [DOI] [PubMed] [Google Scholar]
- Schulze KL, Broadie K, Perin MS, Bellen HJ. Genetic and electrophysiological studies of Drosophila syntaxin-1A demonstrate its role in nonneuronal secretion and neurotransmission. Cell. 1995;80:311–320. doi: 10.1016/0092-8674(95)90414-x. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Shu X, Royant A, Lin MZ, Aguilera TA, Lev-Ram V, Steinbach PA, Tsien RY. Mammalian expression of infrared fluorescent proteins engineered from a bacterial phytochrome. Science. 2009;324:804–807. doi: 10.1126/science.1168683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu X, Lev-Ram V, Deerinck TJ, Qi Y, Ramko EB, Davidson MW, Jin Y, Ellisman MH, Tsien RY. A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol. 2011;9:e1001041. doi: 10.1371/journal.pbio.1001041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Chen W, Vyleta NP, Williams C, Lee CH, Phillips C, Andresen MC. Calcium regulation of spontaneous and asynchronous neurotransmitter release. Cell Calcium. 2012;52:226–233. doi: 10.1016/j.ceca.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stawicki TM, Zhou K, Yochem J, Chen L, Jin Y. TRPM channels modulate epileptic-like convulsions via systemic ion homeostasis. Curr Biol. 2011;21:883–888. doi: 10.1016/j.cub.2011.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Takemoto K, Matsuda T, McDougall M, Klaubert DH, Hasegawa A, Los GV, Wood KV, Miyawaki A, Nagai T. Chromophore-assisted light inactivation of HaloTag fusion proteins labeled with eosin in living cells. ACS Chem Biol. 2011;6:401–406. doi: 10.1021/cb100431e. [DOI] [PubMed] [Google Scholar]
- Tour O, Meijer RM, Zacharias DA, Adams SR, Tsien RY. Genetically targeted chromophore-assisted light inactivation. Nat Biotechnol. 2003;21:1505–1508. doi: 10.1038/nbt914. [DOI] [PubMed] [Google Scholar]
- Trimble WS, Gray TS, Elferink LA, Wilson MC, Scheller RH. Distinct patterns of expression of two VAMP genes within the rat brain. J Neurosci. 1990;10:1380–1387. doi: 10.1523/JNEUROSCI.10-04-01380.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washbourne P, Schiavo G, Montecucco C. Vesicle-associated membrane protein-2 (synaptobrevin-2) forms a complex with synaptophysin. Biochem J. 1995;305:721–724. doi: 10.1042/bj3050721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyart C, Cocco S, Bourdieu L, Léger JF, Herr C, Chatenay D. Dynamics of excitatory synaptic components in sustained firing at low rates. J Neurophysiol. 2005;93:3370–3380. doi: 10.1152/jn.00530.2004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.