

Abstract
G-protein-coupled receptors initiate a wide range of signalling pathways in cells. It seems that both a G protein and an agonist molecule must bind to the receptors to persistently activate them.
G-protein-coupled receptors (GPCRs) constitute a large family of proteins — consisting of more than 800 members in humans — that are embedded in the plasma membranes of most cells. They bind specifically to a diverse range of ‘agonist’ molecules, which convey sensory information or communicate changes in physiological states1. Once stimulated by agonists, GPCRs bind to and activate G proteins, which are attached to the inner surface of cell membranes. The G proteins then initiate signal cascades that control a wide variety of important biological processes. In this issue, two papers from Kobilka and colleagues2,3 and another from the groups of Tate and Schertler4 report structural, biophysical and computational studies that provide insight into GPCR activation by agonists.
The membrane-embedded domains of GPCRs consist of a bundle of seven trans-membrane (TM) α-helices5 (Fig. 1). Agonists bind to a cavity girded by four of these helices near the extracellular surface of the receptor. Agonist-induced structural changes are propagated through the transmembrane region of the receptor by rotation of certain TM helical segments, to open a G-protein binding site on the intracellular surface. One class of GPCRs are the β-adrenergic receptors, which activate a G protein known as Gs. Activation of Gs initiates the ‘fight-or-flight’ response by increasing the force of cardiac contraction (causing a pounding heart), relaxing smooth muscle and mobilizing glucose in skeletal muscle.
Figure 1. Activation of G-protein-coupled receptors.
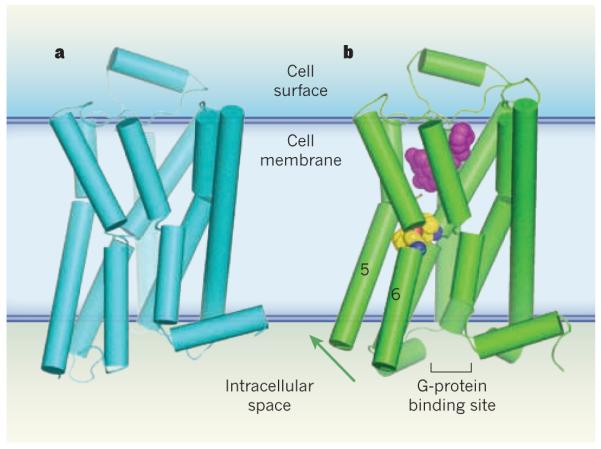
G-protein-coupled receptors (GPCRs) are a family of membrane-bound proteins. They activate G proteins when bound by agonist molecules, but are inactive in the presence of inverse agonists. a, The structure of the β-adrenergic receptor β2AR — a GPCR — is shown here in its inactive (R) state8. The receptor is bound to the inverse agonist carazolol (not shown). Cylinders indicate α-helices. b, Rasmussen et al.2 report the crystal structure of β2AR in complex with an agonist (purple) and with an antibody fragment (not shown), both of which stabilize the receptor’s active (R*) state. The antibody binds at the G-protein binding site, and acts as a substitute for β2AR’s G protein (Gs). The transition from the R to the R* state, induced by agonist binding, probably results from a contraction of the agonist binding site. The contraction promotes changes in the packing of amino-acid residues (yellow/blue/red spheres) between transmembrane helices 5 and 6. These changes coincide with rotation of parts of these helices (green arrow). Together, the conformational shifts create a binding site for the carboxy terminus of the G protein (or, in this case, the antibody fragment). This crystal structure2, along with structures and molecular simulations in two other papers3,4, suggest that both a G protein and an agonist must bind to GPCRs to stabilize the R* state.
To a first approximation, GPCRs are dynamic proteins that switch between an inactive (R) state and an active (R*) conformation that can engage G proteins. Agonists drive the conformational equilibrium towards R*; partial agonists do the same, but less efficiently, whereas inverse agonists drive the receptor towards the R state. In the absence of any such molecules, the equilibrium is biased towards R, although a small fraction of receptors remain active.
A major challenge for receptor biochemists has been to understand how agonists and inverse agonists control the conformational equilibrium. An important advance was made with the discovery that opsin, a form of the GPCR involved in vision but lacking its activating cofactor, adopts a presumed R* conformation in which the TM5 and TM6 helices are splayed apart, forming a cavity that accepts the carboxy terminus of a G protein known as transducin6,7. But attempts to crystallize agonist-bound R* states for other, more flexible GPCRs have met with frustration: crystal structures of the β-adrenergic receptors β1AR and β2AR have been obtained only for complexes of the receptors bound to inverse agonists8,9.
Until now. In the first of the three papers published in this issue, Rasmussen et al.2 (page 175) report that when llamas are inoculated with phospho lipid vesicles that contain agonist-bound β2ARs, they produce antibodies that selectively bind to the R* state of the receptors. The authors identified a fragment of these antibodies — the ‘nanobody’ Nb80 — that binds to the R* state, and found that it has remarkably similar properties to those of Gs. Specifically, the affinity of β2AR for an agonist (isoprenaline) increases nearly a hundredfold in the presence of Nb80, indicating that the nano-body is an activator of agonist binding, like Gs. The authors did not see this effect with inverse agonists.
Using techniques honed in earlier studies8, Rasmussen et al.2 obtained crystals of β2AR (modified in such a way as to promote crystallization) in a lipid-like environment, bound to both Nb80 and a potent β2AR agonist. The medium-resolution (3.5-Å) structure revealed a conformation similar to that observed for opsin6,7, but with a poly-peptide loop of Nb80 inserted into the cavity that, in opsin, binds the carboxy-terminal fragment of transducin. By comparing their crystal structure2 with that in which β2AR is bound to an inverse agonist9, the authors observed that the transition between the R* and R states involves repacking of hydrophobic amino-acid residues in TM5 and TM6 at the ‘waist’ of the receptor (Fig. 1). The repacking is coupled with small rotations of TM5 and TM6 at hinge points within both helices, which cause the G-protein binding site to open. The rotations are facilitated by an evolutionarily conserved proline amino-acid residue in each helix: proline residues cannot participate in the hydrogen bonds that rigidify α-helices, and thus provide flexible hinge points for rotation about bonds that link adjacent peptides.
But the structural changes induced at the agonist binding site itself are small. So which agonist–receptor interactions are essential to receptor activation? Warne et al.4 (page 241) have addressed this question by solving four crystal structures in which a β1AR is bound to either an agonist or a partial agonist. Remarkably, they found that all of the complexes are in the inactive R state. Even so, both agonists and partial agonists pull TM7 and TM5 together, slightly constricting the agonist binding site — an effect also seen in Rasmussen and colleagues’ β2AR–agonist–Nb80 structure2, but not in complexes of GPCRs with inverse agonists8–10. Furthermore, Warne et al.4 observed that full agonists were able to form a hydrogen bond to a conserved serine amino-acid residue in TM5. The formation of this interaction seems to be the tipping point that precipitates receptor activation, and may reduce the energy barrier to the residue repacking and helix rotation that is observed in the β2AR–agonist–Nb80 complex2.
Rosenbaum et al.3 (page 236) describe their attempt to trap an agonist-activated R* conformation by covalently anchoring a potent agonist to β2AR. When tethered to its binding site, the agonist cannot dissociate from the receptor and efficiently activates Gs — even in the presence of an inverse agonist that can displace the analogous non-covalently tethered agonist from its complex with β2AR. Nevertheless, the authors found that the resulting agonist–β2AR adduct crystallizes in the R state.
The caveat here3, as in Warne and colleagues’ β1AR structure4, is that the lipid components used as surrogates for membranes in the crystals may not be as effective as native membrane bilayers in stabilizing the R* state when only agonist is present. Rosenbaum et al.3 therefore performed very long molecular-dynamics simulations — extending to 30 microseconds, a record for simulations of transmembrane proteins — of the agonist-bound receptor from Rasmussen and colleagues’ study2, but with the nano-body removed. These simulations suggest that agonist-bound R* states of the receptor are intrinsically unstable in the absence of a G protein.
The three papers2–4 in this issue reveal that persistent activation of a GPCR is achieved through the binding of both an agonist and a G protein at opposite ends of the receptors relative to the lipid bilayer, where the combined binding interactions reduce the energy barriers to the formation of the R* state. Nevertheless, the conformational range and dynamics of GPCRs differ from one receptor to another. Opsin, for example, is locked completely in the R state until it is fully converted to the R* state by the light-induced isomerization of its cofactor; this accounts in part for the very high signal-to-noise ratio of visual signalling. On the other hand, β2ARs have constitutive activity (caused by the small fraction of the receptors that exist in the R* state at equilibrium) that may confer resistance to coronary artery disease11. The instability of R* in the absence of G proteins might also slow down agonist-dependent desensitization, a process in which prolonged exposure to agonists inactivates receptor–G-protein signalling. It will therefore be crucial to discover the molecular determinants of receptor dynamics and conformational equilibria, and how these are so finely tuned by receptor ligands.
References
- 1.Pierce KL, Premont RT, Lefkowitz RJ. Nature Rev. Mol. Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 2.Rasmussen SGF, et al. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenbaum DM, et al. Nature. 2011;469:236–240. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Warne T, et al. Nature. 2011;469:241–244. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenbaum DM, Rasmussen SGF, Kobilka BK. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 7.Scheerer P, et al. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 8.Cherezov V, et al. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenbaum DM, et al. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 10.Warne T, et al. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piscione F, et al. J. Am. Coll. Cardiol. 2008;52:1381–1388. doi: 10.1016/j.jacc.2008.07.034. [DOI] [PubMed] [Google Scholar]