

Abstract
Enzyme promoted assembly offers a simple and straightforward means to construct monodisperse molecular objects too large for classical organic synthesis and too small for top-down techniques. This communication outlines the design and construction of a heterobifunctional protein building block, HaloTag-cutinase, that reacts rapidly and selectively with an appropriately functionalized small molecule linker; and describes the step-wise combination of these building blocks to generate a 300 kDa “megamolecule” that is precisely-defined with respect to domain orientation, connectivity, and composition.
Keywords: megamolecules, protein-based materials, nanostructured materials, modular synthesis, bioconjugation
The ability to synthesize molecules with precise control over structure gives chemists a powerful tool for studies in a broad range of disciplines. The synthesis of structures with molecular weights of up to a few thousand Daltons is now routine, though not always efficient. The preparation of larger structures, in contrast, remains challenging and the synthesis of molecules with molecular weights greater than 100,000 Da with absolute precision is essentially out of reach. The development of methods that can reliably access molecules in this latter size range and allow for precise control over structure could have substantial implications for new areas of study in the molecular sciences. In this paper, we describe a method based on the selective reaction of bivalent linkers with recombinant proteins to prepare molecules having molecular weights near 300,000 Da and that, like small molecule targets, have precisely-defined structures.
Polymers represent the most common examples of synthetic molecules having extremely large molecular weights. Methods of polymerization yield structures that have well-defined chemical composition but exhibit a distribution of sizes. Dendrimer synthesis, by extension, has enabled the preparation of extremely large (~108 Da) discrete molecules with persistant dimensions approaching those of biological entities such as viral capsids.[1[ With respect to precision however, a dendrimer, wherein every covalent bond in the final structure is known with certainty, can be prepared with molecular weights up to ~105 Da.[2[ In contrast, biological macromolecules synthesized in vivo such as proteins and DNA, are in this respect, essentially perfectly defined. It is this precise control over structure that permits the critical fidelity of biomacromolecular function in living systems. This understanding has led to the routine use of oligonucleotides to create synthetic nanoscaled objects with a high degree of structural precision.[3[ Proteins have similarly been employed as the building blocks of nanostructured objects, yet such approaches have relied largely on physical self-assembly in analogy with DNA.[4[
In this communication, we introduce a modular approach that uses recombinant proteins and synthetic linkers to assemble very large molecules that have precisely-defined structures. The approach is based on the selective reaction of an electrophilic molecule with an active-site residue in an enzyme. For example, the serine esterase cutinase reacts specifically with phosphonate ligands to give phosphonate esters of an active-site serine residue.[5[ Similarly, the haloalkane dehalogenase, HaloTag, reacts with α-chloroalkanes to esterify an active-site aspartate residue,[6[ and treatment of a mutant O6-alkylguanine DNA alkyltransferase, SnapTag, with O6- benzylguanine derivatives results in benzylation of an active site cysteine residue.[7[ Hence, fusion proteins comprising two enzymes can react with bivalent cross-linkers that present the appropriate ligands to generate large molecules having defined structures.[8[ In this paper we demonstrate this strategy with the synthesis of a 300 kDa molecule from five HaloTag-cutinase (HC) fusion proteins.
We first constructed a plasmid vector that coded for an N- terminal HaloTag domain that was joined to a C-terminal cutinase domain by an (EAAAK)4 sequence. This linker adopts a partially helical conformation and was used to reduce the possibility of stericcongestion of the enzyme active sites.[9[ This construct was transformed into Origami B(DE3) e. coli and expressed by induction with IPTG. The fusion protein, HC, was isolated by Ni-NTA affinity chromatography and purified by size exclusion chromatography (SEC). Separately, we synthesized a linker molecule having two hexa(ethylene glycol) segments with a p-nitrophenyl phosphonate ligand on one end and a hexylchloride group on the other end (Figure 1). The bifunctional linker was synthesized convergently in 14 steps starting from monobenzyl hexaethylene glycol and is outlined in the Supporting Information.
Figure 1.
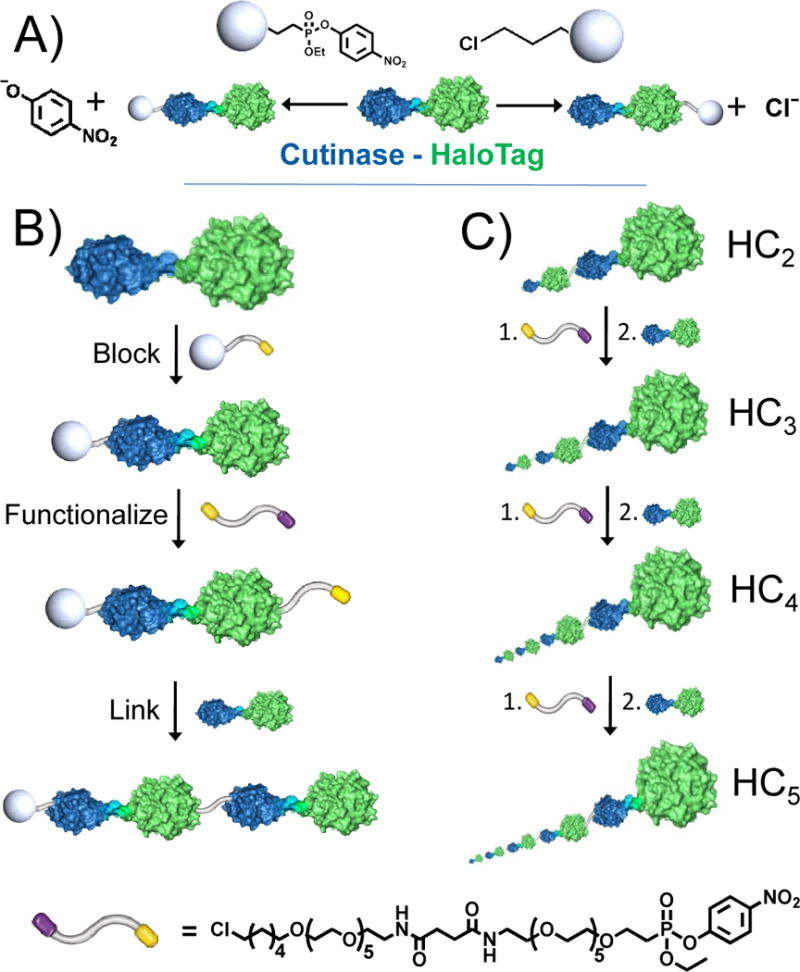
This paper describes an approach for synthesizing megamolecules through a step-wise joining of a fusion protein prepared from cutinase (blue) and HaloTag (green) and of a bifunctional linker terminated in irreversible inhibitors for the proteins. A) The fusion protein (HC) can be blocked at the cutinase or HaloTag domain by reaction with a phosphonate or hexylchloride group, respectively. B) A dimeric structure can be assembled by allowing the fusion protein to react with the linker followed by another equivalent of HC protein. C) Repetition of this cycle gives oligomeric forms of the HC protein. The bifunctional linker is terminated in p-nitrophenylphosphonate (yellow) and an alkylchloride (purple) group (bottom). The proteins in panels a and b are shown to scale and those in panel c are shown in perspective with respect to the plane of the page.
We measured the rate constants for reaction of the HaloTag and cutinase domains of the fusion protein with the bifunctional linker. In one example, we first blocked the cutinase domain of the HC fusion protein with 4-nitrophenyl-(6-carboxyhexyl)-1-phosphonate (4-NPCP) and then allowed the HaloTag domain (at 1 μM) to react with the linker (20 μM) in separate reactions for times ranging from 0 to 600 seconds. We stopped the reactions by adding a large excess of a chloroalkyl-AlexaFluor 488 conjugate to label the unreacted HaloTag with a fluorophore. The reaction mixtures were then resolved with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The amount of fluorescently-labeled protein was measured on a gel scanner and used to determine the yield for each reaction (Figure 2). Reaction of the protein (E) with the linker (L) proceeds by way of an enzyme-substrate complex (eq 1). Because the rate constant for the chemical step (k2) is slow relative to that for dissociation of the E-L complex (k−1), the rate law for the reaction is first order with respect to both the enzyme and the linker (eq 2). Further, because the concentration of linker was 20-fold greater than that of the protein, we fit the kinetic data to a pseudo-first order model to obtain a second order rate constant, keff, of 715 ± 79 M−1s−1 for the reaction of the HaloTag domain with the linker. We repeated this experiment with HC that was first treated with an alkylchloride inhibitor to block the HaloTag domain and found that cutinase reacts with the bifunctional linker with a second order rate constant of 30 ± 5 M−1s−1.
Figure 2.
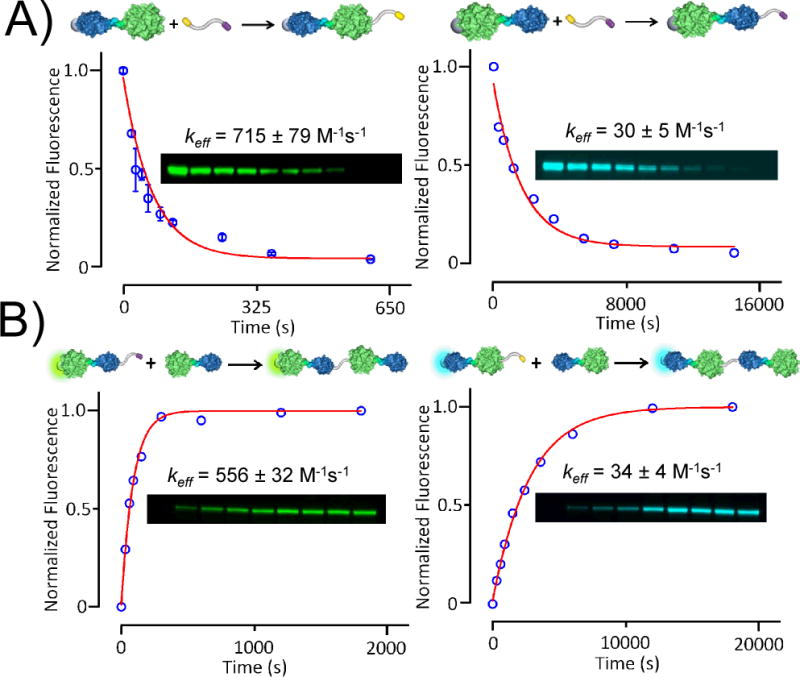
Kinetic analysis of the reactions of HaloTag-Cutinase (HC) fusions and linker molecules. A) The HC fusion protein was treated with a ligand to block either the cutinase (left) or HaloTag (right) domain and then treated with the linker for specified times before treating with an excess of an inhibitor-fluorophore conjugate to label the remaining unreacted protein. The yields for each reaction were determined by monitoring the loss of the starting protein by gel electrophoresis and the data were fit to determine the first order rate constant for the reaction. B) This experiment was repeated to measure the rate constant for the coupling of two fusion proteins, as described in the text.
(1).
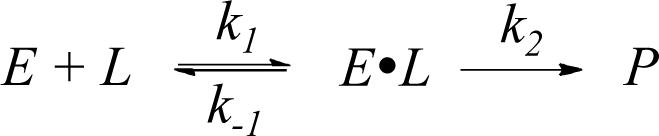
(2).
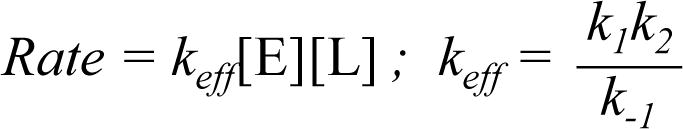
We performed a similar set of experiments to determine the rate constants for the reaction of one HC protein with a second HC protein that was blocked on one domain with a ligand-fluorophore conjugate (*HC or HC*) and reacted with the linker on the other domain. We did so because the rate constant of a reaction can show a complex dependence on the molecular weight of the reactants. We obtained rate constants of 556 ± 32 M−1s−1 and 34 ± 4 M−1s−1 for the reaction of HC with *HC-alkylchloride and phosphonate-HC*, respectively. Hence, the rate constants are of the same order as those observed for reaction of HC with the linker. The effective rate constants are also significantly larger than those for common organic reactions and are important for enabling the assembly of large molecules. Our strategy is based on a reaction wherein the two reactants associate to give a non-covalent intermediate prior to their reaction. In this way, we do not require concentrations of reactants in the mM range, which is typical for reactions of small molecules, but in practice difficult to achieve with macromolecules. This non-covalent complex also has the benefit of making the reaction with the desired nucleophile—as opposed to other potentially reactive sites on the protein surface—more selective, as evidenced by the lack of oligomeric products in the reactions (See Supporting Info Figure S1).
We next illustrate the synthesis of a molecule from five HC fusion proteins and four linkers. We first treated the HC protein with 4-NPCP to block the cutinase domain and then the bifunctional linker to functionalize the HaloTag domain, and finally with one equivalent of HC to prepare the dimer (HC2). We isolated this product in 77% yield after purification by SEC. The dimer was then treated with one equivalent of the linker followed by one equivalent of HC to give the HC trimer (HC3) in 55% isolated yield. The tetramer (HC4) and pentamer (HC5) were synthesized by repeating this cycle and were isolated in 30% and 9% yields, respectively. We characterized the oligomeric products by SDS-PAGE and found that the molecular weights of the products were consistent with the expected values (Figure 3A). We also analyzed the products with MALDI-MS and found that the experimental values obtained were in good agreement with the expected values (Figure 3B and Table 1), though molecular weights of the larger oligomers exceed the range that MALDI can reliably address due to poor ionization efficiency of molecules > 200 kDa and the lack of commercially available mass calibrants above 160 kDa.[10[
Figure 3.
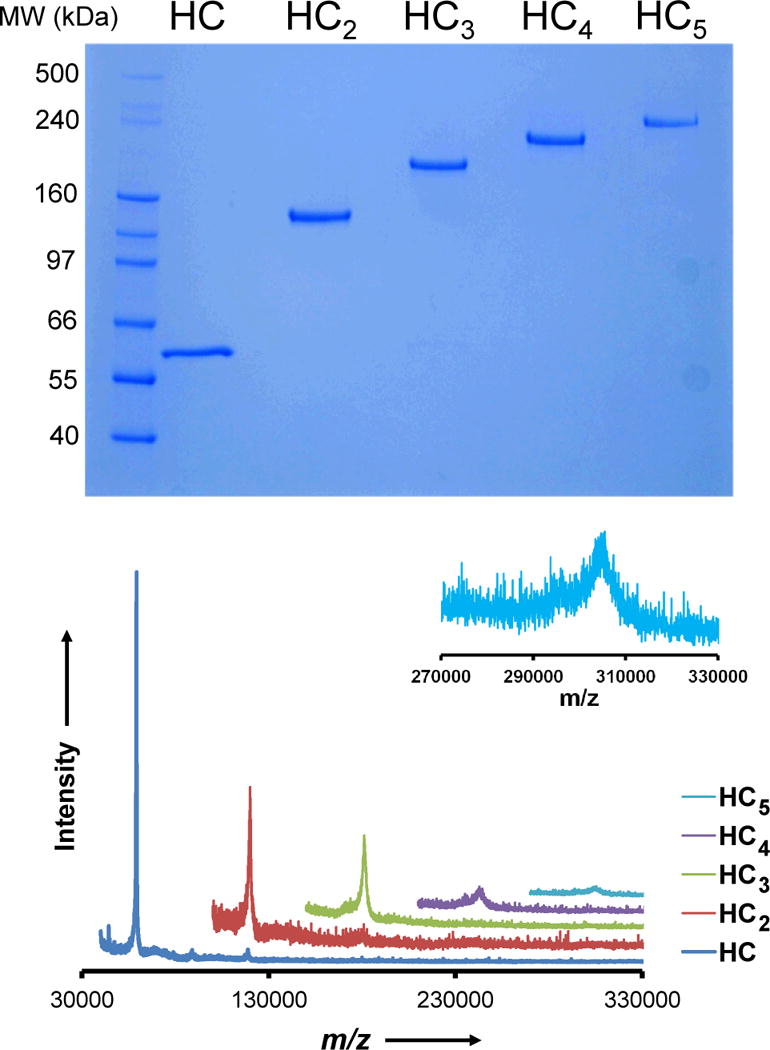
The molecular weights of the oligomers that were prepared as outlined in Figure 1C were determined using (A) SDS-PAGE analysis and (B) MALDI mass spectrometry. The inset is a zoom of the MALDI spectrum for the pentameric product around the expected m/z value.
Table 1.
Calculated and experimentally determined oligomer molecular weights and their corresponding Hydrodynamic Radii.
| Species | Calculated MW (Da) | Experimental MW (m/z)[a] | Hydrodynamic Radius (Å)[b] |
|---|---|---|---|
| HC | 59211 | 59354 | 38 |
| HC2 | 119432 | 119203 | 52 |
| HC3 | 179445 | 180926 | 62 |
| HC4 | 239458 | 242773 | 69 |
| HC5 | 299471 | 305455 | 74 |
From MALDI. M/z values were assigned by fitting the centroid to each peak.
Determined from size exclusion chromatography.
We characterized the hydrodynamic properties of the HC oligomers using size exclusion chromatography (SEC) (Figure 4). The hydrodynamic radii (Rh) for the complexes are given in Table 1. A log-log plot of the dependence of the hydrodynamic radius on molecular weight is linear, which is consistent with polymeric systems (Supporting Info Figure S2). The slope of the best-fit line gives a scaling coefficient of ν = 0.41. Polymer theory predicts limiting values of this coefficient to be 0.33 for spherical molecules and 1.0 for rigid rods, with intermediate values intermediate between these geometries.[11[ For example, studies of globular proteins and dendrimers have yielded coefficients of ν = 0.29[12[ and and 0.3,[13[ respectively, while values of ν = 0.6 have been found for long segments of double-stranded DNA.[14[ Hence, our scaling coefficient suggests that the HC chains deviate significantly from globularity while maintaining a somewhat compact structure in solution. Small-angle X-ray scattering studies of tetraubiquitin, a signaling polyprotein, have revealed a similar compact geometry even when the dimensions of the polymer are highly anisotropic.[15[
Figure 4.
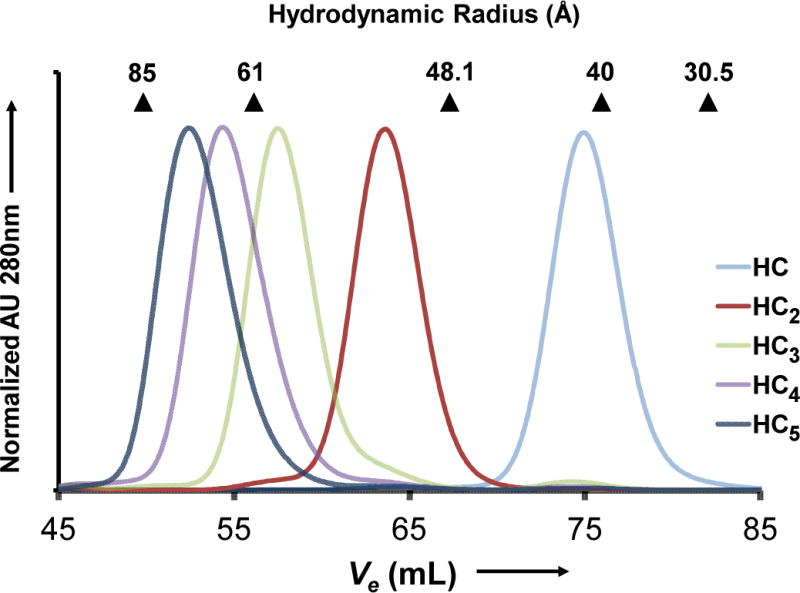
Hydrodynamic characterization of oligomeric reaction products. Size exclusion chromatography traces are shown for each species. Hydrodynamic radii were determined using globular protein calibrants.
This work introduces a strategy to prepare megamolecules having molecular weights greater than 100 kDa and having precisely defined structures in the sense that each covalent bond in the final molecule is defined. One consequnce of using recombinant building blocks is that the e. coli ribosome exhibits an error rate in amino acid incorporation of about 0.03 %.[16[ It is also significant that the pentameric HC molecule described here has a mass greater than 250 kDa which generally exceeds that which can be prepared using standard e. coli expression systems. The use of enzyme-substrate reaction pairs to join the protein building blocks is significant because the binding of these partners overcomes the slow rate constants that would be expected for biomolecular reactions of macromolecules. To address our modest isolated yields of products, we are actively investigating solid-phase synthetic approaches that may decrease the amount of needed reagent in each assembly reaction and aid in the purification of such structures.
This modular approach to preparing megamolecules allows the use of additional enzyme-substrate pairs that expands the synthetic flexibility of this technique, the use of domains that undergo conformational changes in response to specific analytes that can dynamically manipulate such architectures,[17[ and the preparation of scaffolds that incorporate nanoparticles, polymers and other biomolecules. Additionally, the use of multi-functional linkers and the incorporation of self-assembling protein domains provides the possibility of preparing 2- and 3-dimensional structures having a variety of supramolecular geometries. Finally, chemistries developed for the site-specific functionalization of proteins should enable the further elaboration of these structures.[18[
The method described here offers a route to generate precisely- defined structures that are too large to be prepared by the strategies found in chemistry and too small for the top-down strategies that underlie microfabrication. We expect that this route will enable studies in a number of areas, including the preparation of synthetic extracellular matrices for biology and of defined nanoparticle aggregates for sensing and catalysis.
Supplementary Material
Acknowledgments
This research was supported by DARPA. The authors thank Dr. Merav Tsubery for assistance with developing the protein constructs, Prof. Richard Jordan for helpful discussions, and Meghan Carrel for assistance with graphics.
Footnotes
Experimental Section: Experimental details including synthesis of ligands, protein expression and purification; and experimental protocols can be found in the supporting information section.
References
- 1.Tolic LP, Anderson GA, Smith RD, Brothers HM, Spindler R, Tomalia DA. Int J Mass Spectrom. 1997;165:405–418. [Google Scholar]
- 2.Schluter AD, Zhang BZ, Wepf R, Fischer K, Schmidt M, Besse S, Lindner P, King BT, Sigel R, Schurtenberger P, Talmon Y, Ding Y, Kroger M, Halperin A. Angew Chem. 2011;123:763–766. doi: 10.1002/anie.201005164. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2011;50:737–740. doi: 10.1002/anie.201005164. [DOI] [PubMed] [Google Scholar]
- 3.a) Rothemund PW. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]; b) Zhang YW, Seeman NC. J Am Chem Soc. 1994;116:1661–1669. [Google Scholar]
- 4.Ringler P, Schulz GE. Science. 2003;302:106–109. doi: 10.1126/science.1088074. [DOI] [PubMed] [Google Scholar]; b Padilla JE, Colovos C, Yeates TO. Proc Natl Acad Sci USA. 2001;98:2217–2221. doi: 10.1073/pnas.041614998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kwon Y, Han ZZ, Karatan E, Mrksich M, Kay BK. Anal Chem. 2004;76:5713–5720. doi: 10.1021/ac049731y. [DOI] [PubMed] [Google Scholar]
- 6.Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV. ACS Chem Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 7.Juillerat A, Gronemeyer T, Keppler A, Gendreizig S, Pick H, Vogel H, Johnsson K. Chem Biol. 2003;10:313–317. doi: 10.1016/s1074-5521(03)00068-1. [DOI] [PubMed] [Google Scholar]
- 8.Johnsson K, Chidley C, Mosiewicz K. Bioconjugate Chem. 2008;19:1753–1756. doi: 10.1021/bc800268j. [DOI] [PubMed] [Google Scholar]
- 9.Arai R, Ueda H, Kitayama A, Kamiya N, Nagamune T. Protein Eng Des Sel. 2001;14:529–532. doi: 10.1093/protein/14.8.529. [DOI] [PubMed] [Google Scholar]
- 10.Breuker K. Principles of Mass Spectrometry Applied to Biomolecules. John Wiley & Sons, Inc.; Weinheim: 2006. pp. 177–212. [Google Scholar]
- 11.Waigh TA. Applied Biophysics. John Wiley & Sons, Ltd.; West Sussex: 2007. pp. 171–203. [Google Scholar]
- 12.Wilkins DK, Grimshaw SB, Receveur V, Dobson CM, Jones JA, Smith LJ. Biochemistry. 1999;38:16424–16431. doi: 10.1021/bi991765q. [DOI] [PubMed] [Google Scholar]
- 13.a) Scherrenberg R, Coussens B, van Vliet P, Edouard G, Brackman J, de Brabander E, Mortensen K. Macromolecules. 1998;31:456–461. [Google Scholar]; b) Wong S, Appelhans D, Voit B, Scheler U. Macromolecules. 2001;34:678–680. [Google Scholar]
- 14.Rawat N, Biswas P. J Chem Phys. 2009;131:165104. doi: 10.1063/1.3251769. [DOI] [PubMed] [Google Scholar]
- 15.Datta AB, Hura GL, Wolberger C. J Mol Biol. 2009;392:1117–1124. doi: 10.1016/j.jmb.2009.07.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kramer EB, Farabaugh PJ. RNA. 2007;13:87–96. doi: 10.1261/rna.294907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall WP, Anker JN, Lin Y, Modica J, Mrksich M, Van Duyne RP. J Am Chem Soc. 2008;130:5836–5837. doi: 10.1021/ja7109037. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Murphy WL, Dillmore WS, Modica J, Mrksich M. Angew Chem. 2007;119:3126–3129. doi: 10.1002/anie.200604808. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:3066–3069. doi: 10.1002/anie.200604808. [DOI] [PubMed] [Google Scholar]
- 18.Witus LS, Moore T, Thuronyi BW, Esser-Kahn AP, Scheck RA, Iayarone AT, Francis MB. J Am Chem Soc. 2010;132:16812–16817. doi: 10.1021/ja105429n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.