

Abstract
Crassostrea gigas originated from the Pacific coast of Asia, but was introduced into several European countries in the early 1970s. Natural populations have now spread across the length of the western seaboard of Europe. To elucidate the demographic and selective processes at play during this rapid expansion, genome-scan analysis was performed on different populations. High diversities and low differentiation were observed overall, but significant genetic differentiation was found among newly established populations and between the newly established northern group and a nearly panmictic group composed of southern European populations and a population from Japan. Loss of genetic diversity was also seen in the north, likely caused by founder events during colonization. The few strongly supported outlier loci revealed a genetic structure uncorrelated with the north/south differentiation, but grouping two samples from the Danish fjords (northern group) and one from the Dutch Scheldt estuary (southern group) with the one from Japan. These findings might reflect the following: (i) parallel adaptation to similar environmental pressures (fjord-like environment) within each of the two groups or (ii) a footprint of a secondary introduction of an alternative genomic background maintained by multifarious isolation factors. Our results call for a closer examination of adaptive genetic structure in the area of origin.
Keywords: AFLPs, Crassostrea gigas, genome scan, invasive species, microsatellites, SNPs
Introduction
During the last century, many species were intentionally translocated beyond their natural geographical ranges for commercial reasons, including cultivation. Some of these species were able to spread autonomously and have caused economic and/or environmental damage (Keller and Lodge 2007), that is, they became invasive. The Pacific oyster, Crassostrea gigas, is endemic to the Pacific coast of Asia, but has been translocated and introduced into many countries worldwide, mainly for aquaculture purposes (CIESM 2000). C. gigas was massively introduced into France at the beginning of the 1970s (Grizel and Héral 1991) and propagated in northern European countries by hatchery-based seed production (Troost 2010). For more than 20 years, C. gigas was only reproductive in France. The species failed to settle in northern Europe until the 1990s, when populations were reported to extend as far north as Norway. This expansion may have been the result of (i) natural dispersion, (ii) an increased ability of transplanted stocks to reproduce locally or (iii) a combination of both these processes. Hence, from the 1990s, the area of reproduction started to expand. Pacific oysters began to reproduce in Brittany (France) and now form large reefs in soft sediments and on rocks in this area (Lejart and Hily 2011). A northward range expansion was then observed in the Netherlands (Nehring 2006), throughout the Wadden Sea (Bruins 1983; Reise 1998; Nehring 1999; Dankers et al. 2004), and as far north as Sweden, where dense populations of recently settled oyster populations have been observed in several shallow-water sites (Strand et al. 2012). In Norway, populations can now be found in the shallow waters of the southern and western coasts (Wrange et al. 2010).
The colonization of these regions could have resulted from transplantation. Hence, in France, as soon as the Pacific oyster began to establish in southern bays, large spat and stock exchanges were started to enhance cultivation in both southern and northern areas of the Atlantic coast and were also made between the Atlantic Ocean and the Mediterranean Sea (Grizel and Héral 1991). In Scandinavia, attempts to cultivate oysters were made in Limfjorden, Denmark (Jensen and Knudsen 2005), in Sweden (oyster spat imported from Wales between 1973 and 1976; Eklund et al. 1977) and in Norway (since 1979; Strand and Vollstad 1997). Trials indicated that the environmental conditions were suitable for cultivation, but reproduction was not reported. In the 1980s, farmers imported Pacific oyster seed from Scotland to Norway. After strict restrictions were put on the importation of molluscs for cultivation purposes, local hatcheries started to produce their own spat in there from 1987 to 1990 (Strand and Vollstad 1997). Consequently, some of these animals may have founded local populations when environmental conditions became favourable, which then formed the basis for regional expansion. Indeed, climate modifications over the last 40 years may have allowed the species to reproduce in more areas. Hence, warm summers and mild winters may have contributed to the settlement of the species in some northern areas (Troost 2010). Northern European fjords could represent a favourable environment for this phenomenon, allowing them to maintain a sufficient adult population size, as the tidal forces are negligible in these areas and the oysters would be less exposed to conditions of extreme cold. Furthermore, the Pacific oyster is known to have a very wide range of tolerance to several factors, such as temperature, salinity and pathogens, with a high competitive ability that contributes to its invasive success. Hence, the temperatures at which the species can survive range from sub-zero (Quayle 1969; Diederich et al. 2005) to approximately +30°C (Le Gall and Raillard 1988; Bougrier et al. 1995). Furthermore, this species was successfully introduced into Europe and appeared tolerant to the iridovirus associated with the decline of the Portuguese oyster, Crassostrea angulata (Grizel and Héral 1991). Lastly, Crassostrea gigas was shown to have a high production yield in the natural environment in France (His 1972; Héral et al. 1986), and double the growth rate observed for the Portuguese oyster (Bougrier et al. 1986).
In this context, one might ask whether the success of this species, especially the northward expansion of its geographical range, could be explained solely by the opportunity to colonize an empty niche, in which case the spread would have depended mainly on demographic processes, possibly facilitated by global warming, or whether local adaptation was also necessary. A genome scan of differentiation (Lewontin and Krakauer 1973; Beaumont and Nichols 1996) between the area of origin, site of introduction and newly colonized sites might be one approach to conjointly identify loci affected by selection and examine demographic effects during introduction and colonization using a subset of markers that do not deviate from neutral expectations. Furthermore, outlier loci might under some circumstances prove useful to characterize repeated introductions that might not be visible with neutral markers if random genetic drift is not strong enough or if high propagule pressure and recurrent introduction from several sources have brought the full diversity of the native stock (Facon et al. 2008; Estoup and Guillemaud 2010; Riquet et al. 2013). Numerous examples exist showing that when population differentiation is found to be higher than the observed genomic average, this can be attributed to locally variable selection (Beaumont 2005; Nosil et al. 2009). However, alternative scenarios might also produce FST outliers (Excoffier and Ray 2008; Bierne et al. 2011, 2013b). Among such scenarios, the gene-surfing effect produced in the wave front of an expanding population (Excoffier et al. 2009b; Hofer et al. 2009) needs to be seriously considered when studying invasive species. Neutral allele surfing, like selection, would also occur at just a few loci and would therefore not affect all loci uniformly, unlike other demographic factors such as demographic expansions, inbreeding or bottlenecks. Furthermore, there have only been ∼40 generations since C. gigas was first introduced into Europe. This seems a short time lag for adaptation to have arisen from new mutations (i.e. the best situation to detect the signature of selection), although high fecundities and the large size of oyster populations would allow an appreciable influx of new mutations in each generation. During invasion, local adaptation is likely to produce so-called soft sweeps (Pennings and Hermisson 2006) because it should proceed from standing genetic variation through small allele frequency changes at many loci and would, thus, not result in a large FST at any locus (Le Corre and Kremer 2003, 2012). Shifts in allele frequency are thus expected only to be visible for strong selection coefficients and with markers very closely linked to selected loci or directly affected by selection.
However, the short time lag of invasion does not necessarily hold back adaptation if multiple introductions occur (Facon et al. 2006). Multiple introductions can introduce genotypes adapted to different environments, or partially isolated genomic backgrounds that have had sufficient time to diverge in the native range via multifarious evolutionary processes, which may replicate the adaptive differentiation in the invaded range. The mixing of differentiated genotypes by multiple introductions is thought to be an important process in adaptation during invasion (Facon et al. 2008; Estoup and Guillemaud 2010).
To date, very few studies of invasive species have used genome scan to investigate the potential role of adaptation during invasion (Prentis and Pavasovic 2013; Riquet et al. 2013). Interestingly, a recent scan of differentiation in an invasive marine invertebrate, the gastropod Crepidula fornicata, identified differentiated backgrounds characterized by a high rate of outliers in the native range of the species in America, but only one background was proved to have invaded Europe, and no outliers have been identified in the invaded range (Riquet et al. 2013). These authors suggested that genome scans with an insufficient marker density are unlikely to reveal adaptation during invasion from the introduction of a single background, even if adaptation has occurred, and advocated that other methods should be used in such cases (e.g. the analysis of phenotypic traits or gene expression, Mayrose et al. 2011; Hodgins et al. 2013).
The present article investigates whether selection has occurred in different European populations of C. gigas. Genetic analyses employed 8 microsatellites, 240 AFLPs and 30 SNP genetic markers. We used several methods to strengthen our detection of outliers and avoid the detection of false positives. These methods vary in stringency and have different potential biases. Lewontin and Krakauer (1973) developed the first test to disentangle the effects of neutral events from natural selection acting on particular loci, based on the distribution of FST across loci and the identification of outlier loci. In the approach of Beaumont and Nichols (1996), the distribution of FST across loci is plotted as a function of heterozygosity between populations, and neutral expectations are simulated under an infinite island model. Both methods rely on the island model of population structure, which can be a problem when the spatial structure generates correlation in co-ancestry (Nei and Maruyama 1975; Robertson 1975; Bonhomme et al. 2010; Bierne et al. 2013b; Fourcade et al. 2013). Excoffier et al. (2009a) modified the method of Beaumont and Nichols (1996) by introducing a hierarchical island model to perform simulations and account for correlation in co-ancestry within groups of a hierarchical structure of populations. Furthermore, Bonhomme et al. (2010) attempted to minimize false positives by accounting for more complex demographic structures using a statistic based on Lewontin and Krakauer's TLK and added phylogenetic estimation of the population's kinship matrix to account for historical branching. Foll and Gaggiotti (2008) proposed a Bayesian approach in which allele frequencies within populations are assumed to follow a multinomial Dirichlet distribution. In their method, FST is broken down into a population-specific component shared by all loci, and a locus-specific component is shared by all populations. Departure from neutrality at a given locus is assumed when the locus-specific component is necessary to explain the observed pattern of diversity. In this model, sampled populations are allowed to receive unequal numbers of migrants from the migrant pool, but can still lead to biases if migrant genes did not originate from the same pool (Excoffier et al. 2009a; Fourcade et al. 2013). Taking into account the different potential biases of those methods, we took a cautious conservative approach by combining the results of several methods, as recommended by Pérez-Figueroa et al. (2010).
In the present article, we report the study of C. gigas populations sampled across Europe and in the native range of the species using a variety of molecular markers and aim to document their genetic variability and population structure and investigate their adaptive response using a genome-scan approach.
Materials and methods
Sampling and marker amplification
For each of the 746 individuals sampled across 16 populations (Table 1, Fig. 1), DNA was extracted from gill tissue using a chloroform extraction followed by purification with the Wizard(R) DNA Clean-Up System (Promega, Madison, WI, USA), according to Wilding et al. (2001). Concentrations were adjusted to 100 ng/μL.
Table 1.
Sample characteristics and analysis effort
| Population | Country | Label | Sampling size | AFLPs | SNPs | Microsatellites | Sampling date |
|---|---|---|---|---|---|---|---|
| Oshima | Japan | OSH | 48 | X | X | X | 2008 |
| Cap d'Agde | France | AGD | 46 | X | X | X | 2008 |
| Marennes | France | MAR | 48 | X | X | X | 2008 |
| Quiberon | France | QUI | 48 | X | X | 2005 | |
| Squiffiec | France | SQU | 48 | X | X | X | 2005 |
| Arcouest | France | ARC | 48 | X | X | 2005 | |
| Normandie | France | NOR | 42 | X | X | 2008 | |
| Oosterschelde | The Netherlands | OOS | 48 | X | X | X | 2006 |
| Grevelingen | The Netherlands | GRE | 46 | X | X | 2006 | |
| Dutch Wadden Sea | The Netherlands | WAD | 48 | X | X | X | 2006 |
| Munkmarsch | Germany | MUN | 48 | X | X | X | 2006 |
| Danish Wadden Sea | Denmark | DWS | 48 | X | X | X | 2008 |
| Limfjord | Denmark | LIM | 48 | X | X | X | 2008 |
| Isefjord | Denmark | ISE | 36 | X | X | X | 2008 |
| Tjarno | Sweden | TJA | 48 | X | X | X | 2007 |
| Kristenberg | Sweden | KRI | 48 | X | X | X | 2007 |
Figure 1.
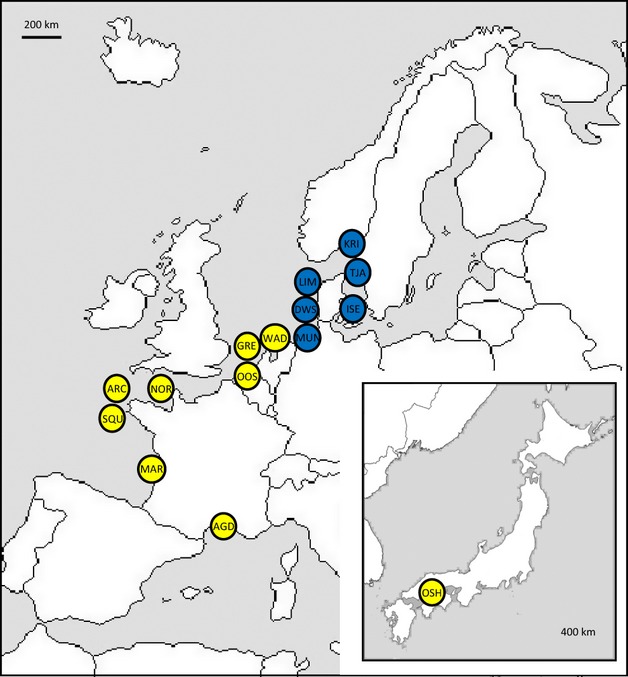
Sampling locations of Crassostrea gigas in Europe and Japan. AGD, Agde; ARC, Arcouest; DWS, Danish Wadden Sea; GRE, Grevelingen; ISE, Isefjord; KRI, Kristinberg; LIM, Limfjord; MAR, Marennes; MUN, Munkmarsch; NOR, Normandie; OOS, Oosterschelde; OSH, Iroshima; QUI, Quiberon; SQU, Squiffiec; TJA, Tjarno; WAD, Dutch Wadden Sea. The two colours represent the two different groups to which the populations belong according to the correspondence analysis and clustering analysis.
The 30 single nucleotide polymorphism markers (Table 2) were developed as described in Sauvage et al. (2007) and genotyped using MALDI-TOF mass spectrometry (matrix-assisted laser desorption/ionization time of flight, Griffin and Smith 2000) at the Max Planck Institute genotyping platform in Berlin (Germany). Briefly, primers were designed using Primer 3 (Rozen and Skaletsky 2000) in ESTs of interest related to summer mortality, identified through a suppression subtractive hybridization approach (Huvet et al. 2004a), or were selected from existing databases such as GigasBase (http://public-contigbrowser.sigenae.org:9090/Crassostrea_gigas/index.html, Fleury et al. 2009) and Genbank.
Table 2.
List of single nucleotide polymorphisms studied
| Number | Usual and full names | Accession Number | Reference |
|---|---|---|---|
| 1 | ATPase H+ C. gigas ATPase H+ transporting lysosomal protein gene, partial cds | EF694094 | Sauvage et al. (2007) |
| 2 | Glutaryl-CoA dehydrogenase–like protein C. gigas glutaryl-CoA dehydrogenase–like protein gene, partial cds | EF694089 | Sauvage et al. (2007) |
| 3 | Amylase gene B C. gigas alpha amylase (amy) gene, amy-B1 allele, partial cds | EF682217 | Sauvage et al. (2007) |
| 4 | Gonadal Transforming Growth factor (ogTGFb) cDN21 Crassostrea gigas cDNA clone cDN21P0003A17 5-, mRNA sequence | AM856765 | Fleury et al. (2009) |
| 5 | NADH dehydrogenase 6 EST016 CgG2RSMorest C. gigas cDNA clone HA26 similar to NADH dehydrogenase 6, mRNA sequence | CK172316 | Huvet et al. (2004a) |
| 6 | Glucose 6 phosphatase C. gigas glucose-6-phosphatase gene, partial cds | EF694098 | Sauvage et al. (2007) |
| 7 | Glycogen synthase C. gigas glycogen synthase gene, partial sequence | EF694079 | Sauvage et al. (2007) |
| 8 | HA114_1 C. gigas clone HA114 genomic sequence | EF694082 | Sauvage et al. (2007) |
| 9 | Ik cytokine-down regulator of HLA_2 C. gigas IK cytokine gene, partial cds | EF999946 | Sauvage et al. (2007) |
| 10 | Laccase_1 C. gigas laccase gene, partial cds | EF999948 | Sauvage et al. (2007) |
| 11 | Notch C. gigas notch3-like protein gene, partial cds | EF999949 | Sauvage et al. (2007) |
| 12 | Superoxide dismutase C. gigas superoxide dismutase gene, partial cds | EF694097 | Sauvage et al. (2007) |
| 13 | RNA helicase C. gigas partial mRNA for RNA helicase | AJ557014 | A. Herpin (unpublished) |
| 14 | Amylase gene A C. gigas alpha amylase (amy) gene, amy-A1 allele, partial cds | EF694074 | Sauvage et al. (2007) |
| 15 | Astacin C. gigas astacin metalloprotease gene, partial cds | EF694085 | Sauvage et al. (2007) |
| 16 | Ferritin GF2 C. gigas ferritin GF2 (GF2) mRNA, complete cds | AY321300 | Gueguen et al. (2003) |
| 17 | Bcl-2C. gigas predicted Bcl-2 protein mRNA | EU678310 | Renault et al. (2011) |
| 18 | BQ426586 C. gigas Hemocytes Lambda Zap Express Library Crassostrea gigas cDNA, mRNA sequence | BQ426586 | Gueguen et al. (2003) |
| 19 | BQ427367 C. gigas putative immune protein gene, partial cds | EF694095 | Sauvage et al. (2007) |
| 20 | Calcium dependant protein kinase C. gigas calcium-dependent protein kinase-like gene, partial sequence | EF694100 | Sauvage et al. (2007) |
| 21 | Glutathione S-transferase C. gigas glutathione S-transferase sigma class gene, partial cds | EF694090 | Sauvage et al. (2007) |
| 22 | Drac 3 C. gigas Drac3-like protein gene, partial cds | EF694083 | Sauvage et al. (2007) |
| 23 | Flavin-containing mono oxygenase 2 C. gigas mRNA for flavin-containing monooxygenase 2 (fmo-2 gene) | AJ585074 | Boutet et al. (2004) |
| 24 | BQ426639 C. gigas Hemocytes Lambda Zap Express Library Crassostrea gigas cDNA, mRNA sequence | BQ426639 | Gueguen et al. (2003) |
| 25 | Glycoprotein hormone receptor C. gigas glycoprotein hormone receptor gene, partial cds | EF694088 | Sauvage et al. (2007) |
| 26 | HA114_2 C. gigas clone HA114 genomic sequence | EF694082 | Sauvage et al. (2007) |
| 27 | Ik cytokine-down regulator of HLA_1 C. gigas IK cytokine gene, partial cds | EF999946 | Sauvage et al. (2007) |
| 28 | Laccase_2 C. gigas laccase gene, partial cds | EF999948 | Sauvage et al. (2007) |
| 29 | Sodium/glucose cotransporter_2 C. gigas sodium/glucose cotransporter mRNA, complete cds | AY551098 | Huvet et al. (2004a) |
| 30 | Tubulin C. gigas tubulin gene, partial cds | EF694087 | Sauvage et al. (2007) |
Outlier loci are indicated in bold.
Microsatellite analysis was performed with 8 markers, according to the initial protocols for L48 (Huvet et al. 2000a), CGE09 (Yu and Li 2007), AMY (Sellos et al. 2003), sili29 and sili44 (Sauvage et al. 2009). A multiplex protocol was used for L10, CG49 and CG108 (Taris et al. 2005).
AFLP analyses for 4 primer pairs (Table S1) were performed using a modified version of Vos et al. (1995). For each sample, 250 ng of genomic DNA was digested with 1.25 units EcoRI (NEB) and MseI (NEB) and ligated with 100 units T4 DNA ligase (NEB), 0.1 μm EcoRI adapter and 1 μm MseI adapter [sequences in Vos et al. (1995)] in 25 μL 1X NEB buffer 2, 1X BSA and 1X manufacturer's ligase buffer for 16 h at 16°C. Preselective PCRs were performed on 2.5 μL diluted ligation (1:9 in 0 X TE) in 25 μL of 0.5 μm preselective primers, 0.2 mm of each dNTP, 1.5 mm MgCl2 and 0.5 units GoTaq (Promega) in manufacturer's buffer. Selective PCRs were performed on 5 μL diluted preselective amplification product (1:9 in 0 X TE) in 20 μL of 5 ng selective primers, 0.2 mm of each dNTP, 1.5 mm MgCl2 and 0.4 units GoTaq (Promega) in manufacturer's buffer. Conditions for preselective and selective PCRs were as given in Vos et al. (1995).
Electrophoresis and data collection were carried out on an ABI3100 sequencer (Applied Biosystems, Foster City, CA, USA) for both AFLP and microsatellite loci.
Diversity and genetic structure
After automatic analysis with GeneMapper® software, AFLP genotypes were filtered using AFLPScore (Whitlock et al. 2008) to minimize error rate (<10%).
A between-class analysis, which is a specific type of correspondence analysis (CA) where each class is a population, was performed for all loci (AFLP, microsatellite and SNP) with ade4 package implemented in R (Chessel et al. 2004). The significance of observed structure was tested using a Monte Carlo test on the between-groups inertia percentage with 1000 randomizations.
Nonbiased heterozygosity (Hnb) (Nei 1978) and pairwise FST (1000 bootstraps) were computed using genetix 4.03 (Belkhir et al. 2001) for SNPs and microsatellites. Allelic richness was computed using FSTAT2.9.3 (Goudet 2001) for microsatellite loci.
For AFLPs, allele frequencies were first estimated using a Bayesian method with nonuniform prior distribution of allele frequencies (Zhivotovsky 1999). Expected heterozygosity under Hardy–Weinberg genotypic proportions (Hj) and pairwise FST were then computed using AFLP-SURV 1.0 (Vekemans 2002). Significance of observed differences in diversities between northern and southern populations was tested with Mann–Whitney–Wilcoxon tests (implemented in R 2.15; R Core Team 2012).
To infer the number of major genetic clusters, the Bayesian MCMC clustering approach was used, implemented in structure 2.3.1 (Pritchard et al. 2000). The admixture model with correlated allele frequencies was used to indicate the most likely pattern of population connectivity. Ten independent trials were run for each predefined number of clusters (K), with K = 1–10 and a burn-in of 50 000 iterations followed by 300 000 MCMC repetitions. We considered both raw probability values of lnP(X|K) and the ΔK estimate (Evanno et al. 2005). Codominant (microsatellites and SNPs) and dominant (AFLPs) markers were analysed separately.
Outlier detection
For technical reasons related to the quality and quantity of DNA, AFLP genotyping was only possible on 12 of the 16 available samples. Detection of AFLP outlier loci was performed using a version of FDIST2 software (Beaumont and Nichols 1996) modified for dominant markers (DFDIST program; http://www.rubic.rdg.ac.uk/∼mab/stuff), Bayescan v2.0 (Foll and Gaggiotti 2008) and the method of Bonhomme et al. (2010). Frequencies of null alleles were computed using the approach of Zhivotovsky (1999). In DFDIST, a mean ‘neutral’ FST value was calculated after trimming 30% of the highest and lowest FST values (see Gagnaire et al. 2009). The number of demes was set at 100, and 50 000 loci were generated by coalescent simulations under the finite island model. The maximum frequency of the commonest allele allowed was set at 0.99. BayeScan was used with default parameters for the chain and model, with 50 000 iterations. Using the approach of Bonhomme et al. (2010), a matrix of Reynold's distances was computed in AFLP-SURV 1.0 (Vekemans 2002); a population sampled in Japan was used as the out-group and 50 000 iterations were made. The chi-square-approximated P-values were corrected for multiple testing according to the Benjamin–Benjamini-Hochberg method (as suggested in Bonhomme et al. 2010).
Detection of microsatellite and SNP outlier loci was performed using a version of DFDIST modified by r. Vitalis to simulate codominant, bi-allelic data (see Ségurel et al. 2010); the method of Excoffier et al. (2009a), implemented in Arlequin 3.5 (Excoffier and Lischer 2010); the method of Foll and Gaggiotti (2008), implemented in Bayescan v2.0; and the method of Bonhomme et al. (2010). For the last two methods, the parameters were the same as for the AFLPs. In R. Vitalis' modified version of DFDIST, 50 000 simulations were performed with 100 demes. The maximum frequency of the commonest allele allowed was set at 0.99. Because the finite island model has recently been shown to lead to a large fraction of false positives if populations are hierarchically subdivided, we used the modified version of FDIST implemented by Excoffier et al. (2009a) for codominant data that use a hierarchical island population model (as defined by Slatkin and Voelm 1991). Two groups of 100 demes were used (following the genetic structure results), that is, northern populations (Danish Wadden Sea, Isefjord, Kristenberg, Limfjord, Munkmarsch and Tjarno) and southern ones (Arcouest, Cap d'Agde, Dutch Wadden Sea, Grevelingen, Marennes, Normandie, Oosterschelde, Quiberon, Squiffiec and Japan). We used 50 000 coalescent simulations for the hierarchical model.
For all methods and types of markers, outlier detection was performed on all populations and on southern populations and northern populations separately. The threshold for outlier detection with BayeScan was set at FDR = 0.05 for both AFLP and SNP.
Results
Genetic diversity within populations
Genotypes at almost 500 AFLP loci were automatically generated for twelve populations using GeneMapper (R) software. Only 240 were used for further analyses (AFLPScore error rate between 2.3 and 9.8%, Table S1).
Diversity estimators were plotted for the three kinds of markers (Fig. 2). Both microsatellite (Fig. 2A) and SNP (Fig. 2B) loci exhibited the same pattern of reduced diversity in populations from areas north of the Netherlands. For these northern samples (from Germany, Denmark and Sweden), nonbiased expected heterozygosities ranged from 0.8563 to 0.9085 and from 0.2996 to 0.3294 for microsatellites and SNPs, respectively. Mean allelic richness at microsatellite loci was between 14.4 and 18.1. For southern samples (from France to the Netherlands) and samples from Japan, heterozygosities were between 0.9284 and 0.9425 (mean allelic richness: 22.7 to 24.0) for microsatellite loci and between 0.3265 and 0.3573 for SNPs. For both microsatellites and SNPs, heterozygosities were significantly different between northern and southern populations (W = 0, P-value = 0.00025 and W = 1 P-value = 0.00050, respectively). For AFLPs (Fig. 2C), heterozygosities ranged from 0.1882 (Oosterschelde) to 0.2119 (Tjarno). No significant difference was observed between the two groups (W = 33, P-value = 0.99567).
Figure 2.
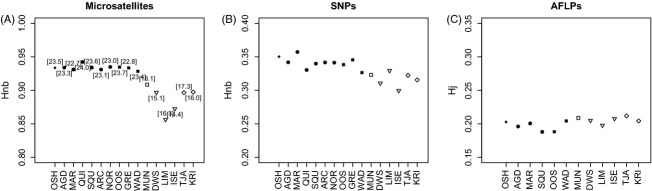
Plot of nonbiased expected heterozygosity (Hnb) and expected heterozygosity under Hardy–Weinberg genotypic proportions (Hj) from the different sample locations. White and black symbols indicate northern and southern populations, respectively. Mean allelic richness is shown in brackets for each population. A: Hnb for the 8 microsatellite loci, B: Hnb for the 30 SNPs, C: Hj for the 240 AFLPs.
Population genetic structure
For all types of loci (i.e. SNPs, microsatellites and AFLPs), global FST values were, respectively, 0.0129, 0.0160 and 0.0294, and all significantly different from zero (P-value > 0.001). Correspondence analysis on populations for all loci (Fig. 3) revealed two groups, which corresponded to the same ones identified on the basis of genetic diversity. The first of these groups included the ‘southern’ locations, with all populations from France, the Netherlands and Japan (endemic location). Global FST values were not significantly different from zero in this group (except for AFLPs when outliers were included). It is interesting to note that there is no genetic differentiation between the sample from Japan and any of those from the southern part of Europe. The second group encompasses the locations the furthest north in Europe, with populations located in Germany, Denmark and Sweden. In this group, global FST values were significantly different from zero (P-value < 0.001); the minimum value was found for microsatellites (0.0107) and the maximum for AFLPs (0.0243). The three populations from Denmark were significantly different from each other (Table S2).
Figure 3.
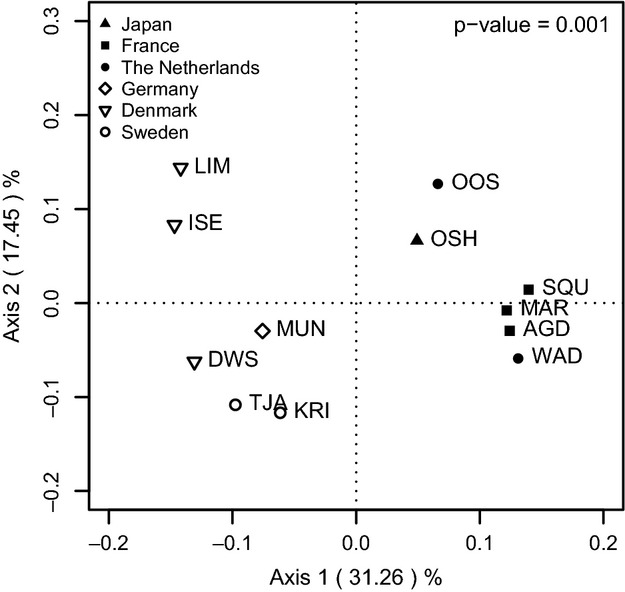
Projection of sample centres of gravity on the first factorial plane of a correspondence analysis (CA) on the matrix of allele frequencies at 278 loci (30 SNPs, 8 microsatellites and 240 AFLPs). White and black symbols indicate northern and southern populations, respectively.
Clustering analysis suggested a partition into two groups (K = 2) as statistically most likely for both codominant and dominant markers (Fig. S1: same colour code as in Fig. 1), thereby confirming the results of the correspondence analysis.
Outlier detection
Results of outlier detection for all methods are presented in Table 3. We did not find any outliers with microsatellite markers. For AFLP loci, detection was performed on all populations using the approach of Bonhomme et al. (2010), and 23 loci were detected as outliers (corrected P value < 0.001). In northern and southern populations, five and eight loci were found to be outliers, respectively, by this method. With Bayescan, eight, three and five outliers were detected in the population groups ‘all’, ‘northern’ and ‘southern’, respectively. With DFDIST, seven, nine and eight outliers were found in all, northern and southern populations, respectively. For all populations, only four loci were identified as outliers with all methods (loci 123, 128, 146 and 186), only one (123) for northern populations and two (146 and 167) for southern ones. No particular geographical gradients in allele frequency were observed (Fig. 4), but four loci (123, 128, 146 and 186) tended to exhibit a similar structure. This is also visible on the second axis of the CA (Fig. 3), which is mainly explained by these four outliers (Fig. 5), and separates four samples with positive coordinates (ISE, LIM, OOS and OSH) from the others. Indeed, the correspondence analysis illustrates beautifully how outliers provide a pattern of genetic structure (axis 2) uncorrelated with the one observed with neutral markers (axis 1). Indeed, the outlier loci that contribute the most to axis 2 group together samples (OOS, OSH, ISE and LIM) that are otherwise differentiated from the other markers: OOS and OSH belonging to the southern group and ISE and LIM belonging to the northern group (which explains the north/south differentiation on axis 1). AFLP markers 123 and 186 in particular show a higher contributions to axis 2 (Fig. 5).
Table 3.
Outlier detection for AFLP and SNP markers
| Methods | AFLP/all | AFLP/North | AFLP/South | SNP/all | SNP/North | SNP/South |
|---|---|---|---|---|---|---|
| Bonhomme et al. (2010)* | 19, 21, 23, 29, 55, 59, 71, 72, 76, 82, 94, 97, 123, 128, 137, 146, 149, 160, 166, 186, 187, 202, 219 | 19, 21, 123, 154, 203 | 72, 82, 97, 146, 149, 160, 166, 167 | ATPaseH+ | None | None |
| BayeScan v2.0 (Foll and Gaggiotti 2008)† | 96, 123, 128, 136, 141, 146, 167, 186 | 123, 128, 186 | 55, 123, 128, 146, 167 | None | None | None |
| DFDIST (Beaumont and Nichols 1996) | 88, 123, 128, 136, 167, 146, 186‡ | 19, 23, 29, 123, 128, 137, 154, 186, 203‡ | 18, 123, 128, 146, 160, 167, 202, 223‡ | None | None | None |
| DFDIST modified by Vitalis | NA | NA | NA | ATPaseH+, glycogen synthase, glutathione S-transferase, HA114_2‡ | ATPaseH+, glutathione S-transferase ‡ | Glutaryl-CoA, BQ427367 ‡ |
| FDIST, hierarchical model (Excoffier et al. 2009a) | NA | NA | NA | ATPaseH+ ‡ | NA | NA |
| Commun to all methods | 123, 128, 146, 186 | 123 | 146, 167 | ATPaseH+ | NA | NA |
Locus with P value after Benjamini–Hochberg correction for multiple tests below 0.001.
Locus detected as outliers with FDR=0.05.
Locus with P value below 0.05.
NA, Not Applicable.
None: No outlier detected.
AFLP outlier loci indicated in bold are shared between all methods.
Figure 4.

Plot of allele frequencies for the 5 AFLP outlier loci and ATPase H+ from the different locations.
Figure 5.
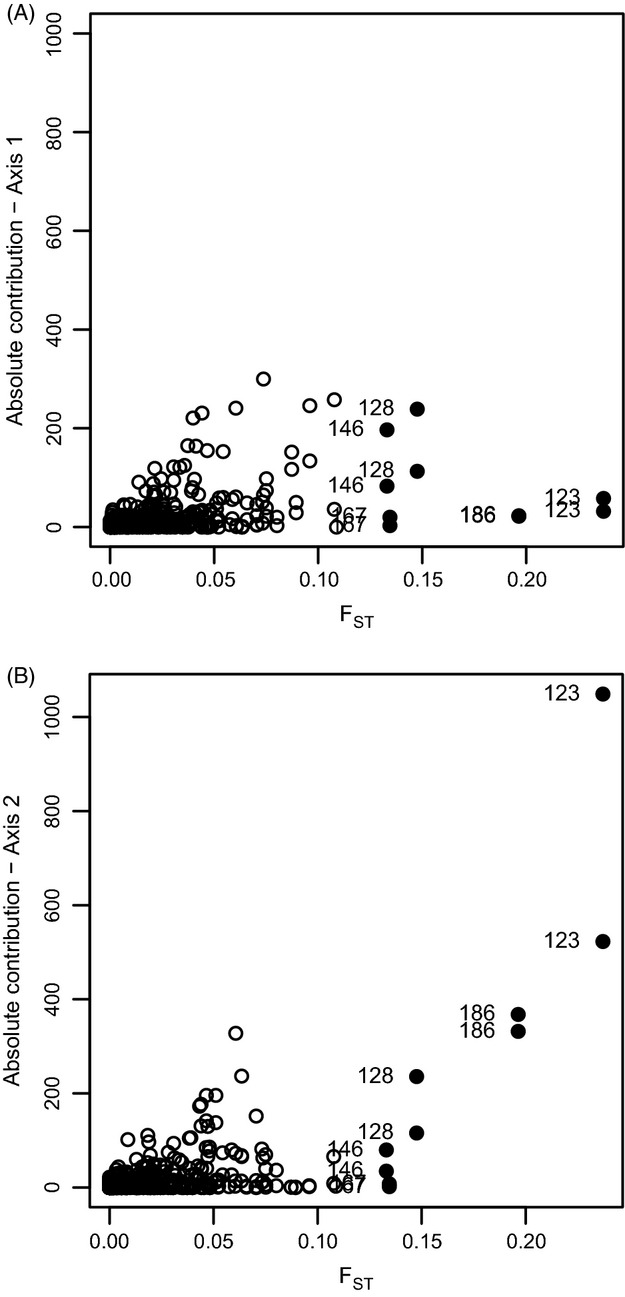
Absolute contributions of the decomposition of contribution for the alleles plotted against FST values for axes 1 (A) and 2 (B) for the 240 AFLP loci. The 5 outlier loci are plotted as black circles.
Such a pattern might have been suspected from the STRUCTURE analysis because OOS and LIM show a more similar pattern with AFLP markers (Fig. S1B), although the clustering analysis did not support a model with more than two clusters as the most likely.
For SNP loci, the method that detected most outliers was DFDIST. With this method, four loci were outside the envelope of neutral expectations for all populations (ATPase H+, glycogen synthase, glutathione S-transferase and HA114_2), two for northern ones (ATPase H+ and glutathione S-transferase) and two for southern ones (glutaryl-CoA dehydrogenase–like protein and BQ427367).
With the other methods, only ATPase H+ appeared to be a significant outlier for all populations. For this locus, allele frequency ranged from 0.6413 (Limfjord) to 0.9894 (Wadden Sea) with a mean of 0.902 (Fig. S1). The differentiation pattern observed at ATPase H+ was partly correlated with the one observed with AFLP outliers, and this locus also contributed substantially to axis 2 of the CA; however, it mainly separated the Danish fjords ISE and LIM from all the other populations. It should be noted that the northern/southern groups were not modified when outlier loci were removed from the structure analysis.
Discussion
Genetic diversity
As previously observed by Huvet et al. (2000b), no loss of diversity was noted when comparing the endemic population from Japan with populations sampled in southern Europe. Following the introduction of non-native species, a reduction in genetic diversity could occur due to founder effects, but this has never been observed in any real cases in bivalves (see Dlugosch and Parker 2008 for review) probably because of their high recruitment rates and recurrent introductions from several sources (Simberloff 2009). In the case of the introduction of C. gigas into France, repeated introductions of hundreds of tons of adults from British Columbia and thousands of tons of spat from Japan were made over the 5 years following the first massive introduction of adults at the beginning of the 1970s (Grizel and Héral 1991). Interestingly, genetic diversities observed in northern populations were lower for both microsatellites and SNPs, although no clear difference was observed with AFLP markers. A recent study performed in the Wadden Sea with a single mitochondrial locus surprisingly showed the opposite trend (Moehler et al. 2011), with a higher haplotype diversity in northern populations, which was attributed to a secondary introduction of hatchery spat from British Columbia. The observed loss of diversity is likely a signature of genetic drift on northern European coasts, where populations are smaller and more isolated than in the south. Previous studies have therefore shown examples of how spatial expansion can induce the structuring of newly colonized areas into distinct sectors of low genetic diversity (Excoffier et al. 2009b).
Genetic structure
Low levels of genetic differentiation were observed in our study, especially within each group (FST = 0.000NS, in the south and FST = 0.0111 in the north). Likewise, marine species with long planktonic larval development (2–4 weeks in Crassostrea gigas according to water temperature and food availability) often exhibit low levels of population genetic structure related to high levels of gene flow (Hellberg et al. 2002). However, some studies have shown small but significant genetic structure in such ‘high gene flow’ marine species (Addison et al. 2008). Our observation is in accordance with previous studies in C. gigas that indicated a lack of genetic structure for mitochondrial and microsatellite DNA (Huvet et al. 2000b) between samples originating from southern Europe and Japan.
Despite this apparently low level of genetic structure, two groups can be distinguished that have higher FST values between them than within each group. The group with southern localities includes areas where C. gigas is currently cultivated: FST values for this group are very low and mainly not significantly different from zero. Farmers' practices with this species commonly involve translocation of stocks. Oysters grown in one region may have come from different origins (hatcheries or natural recruitments), sometimes very distant ones. These practices increase gene flow and therefore reduce genetic structure. The lack of genetic differentiation between Japan and southern Europe and the absence of diversity loss in the latter suggest that the bottleneck related to introduction was not sufficient to affect the genetic diversity, either because the number of founders was sufficiently high or because there have been repeated introductions (large propagule pressure and several sources of introduction; Roman and Darling 2007).
We found the same separation into two groups as Moehler et al. (2011), although this previous study was limited to the Wadden Sea. On the basis of one mitochondrial marker, these authors found virtually no genetic differentiation in the entire southern range of the Wadden Sea or in the putative source from British Columbia they had sampled. Although we do not have such a sample of the putative source in our study, we have a Japanese sample, and this does not show any differentiation from either the southern range of Wadden Sea or the rest of the Southern range sampled here in France. Furthermore, knowing that most of the oysters imported into France at the beginning of the 1970s came from British Columbia and Japan (Grizel and Héral 1991), one may hypothesize that the southern part of Europe up to the southern edge of the Wadden Sea, our sample from Japan and part of the British Columbia stocks may form a group of undifferentiated populations. However, this result also clearly shows the interest of better characterizing the genetic diversity present in the native range, especially the adaptive diversity, and comparing this with the European pattern.
The northern group is characterized by higher estimated FST values and lower diversities than the southern ones. It includes localities where C. gigas is no longer cultivated. These populations are presumed to have a different origin from those of the southern group. In Sylt (at the border between Germany and Denmark), spat was primarily imported from British and Irish hatcheries (Reise 1998; Nehring 1999), and the species began to spread around 1991. In Denmark, a large amount of C. gigas seed oysters were imported from England, the Netherlands and France to different locations for aquaculture experiments (Nehring 2006). In Isefjord, commercial production took place between 1986 and 1999. When oyster farming stopped, some oysters were left at the site, where they survived, but their numbers did not expand (Nehring 2006). The first free-living specimens were observed in 1999, which dispersed from the northern German Wadden Sea by natural means (Reise et al. 2005). The number of oysters that were imported and contributed to present populations in the northern part of Europe is likely to be relatively low compared with those produced and translocated each year in France (about 130 000 tons/year were transported around France, whereas only 100 000–300 000 individual oysters were produced annually in Isefjord). It can therefore be hypothesized that these recently settled northern populations evolved isolated from the southern ones with little or no gene flow caused by human activities. Moreover, among the 16 pairwise FST estimates above 0.05, ten involved populations from Denmark. Large variance in reproductive success, as previously reported in this species (Li and Hedgecock 1998; Boudry et al. 2002), is likely to occur in these relatively isolated populations; this could further enhance genetic drift, decreasing genetic diversity as explained above and increasing the observed genetic differentiation.
Detection of selection
Among the 30 SNPs studied, one was detected as an outlier (3.3%) with all methods used. For AFLPs, four outliers (2%) were detected within the 240 AFLP loci scored, which could appear low compared with that given in the literature for other species. A review by Nosil et al. (2009) on 18 relevant papers dealing with ‘genome scan and selection’ showed substantial, but not extreme, variation in the proportion of outliers, with a range from 0.4% to 24.5% and a mean of 8.5% among the loci analysed. For Littorina saxatilis (an intertidal snail), 5% of 306 AFLP loci were consistently characterized as outliers between ecotypes on three shores (Wilding et al. 2001; Grahame et al. 2006; Wood et al. 2008). For Crassostrea virginica, only 1.4% of outliers were detected with 215 AFLP markers, although only two populations were studied in this work (Murray and Hare 2006). Nosil et al. (2009) concluded that approximately 5–10% of the genome is strongly affected by divergent selection. However, studies on some other marine invertebrates did not detect any outliers, such as in the periwinkle Echinolittorina hawaiiensis (Tice and Carlon 2011) or, interestingly, in the gastropod Crepidula fornicata, which invaded Europe at the same time as with oysters (Riquet et al. 2013). The discrepancy between the different studies on the proportion of outliers might be explained by the variation in the number of populations and individuals analysed, the markers used, the methods used for estimating baseline neutral differentiation or the criteria used for determining outlier status. However, more conceptual explanations have been put forward, such as the spatial structure that some organisms have to cope with, for example, a long tree-like linear habitat (Fourcade et al. 2013), or the timescale at which adaptation could have proceeded (Riquet et al. 2013). Bierne et al. (2013b) emphasized that in any case, outlier tests are simply not designed to account for pervasive selection and that when too high a fraction of the genome is affected by selection, the theory of hybrid zones and genetic barriers should be considered, rather than basic local adaptation. Additionally, in the case of introductions, the time lag can appear too short at first sight to suggest the existence of a genome-wide multifactorial, semipermeable genetic barrier to gene flow, but multiple introductions could well have imported differentiated backgrounds from the area of origin to the invaded zone. In the case of oysters, we already have one such example, as C. angulata was first introduced into Europe several centuries ago and C. gigas was introduced 40 years ago. The two species now hybridize in southern Portugal (Huvet et al. 2004b). Even under a simple model of single introduction and local adaptation, discrepancies among different surveys may also reflect the intensity of divergent selection and/or the time since divergence has been acting between populations (Renaut et al. 2011). Hence, under the hypothesis of a single introduction and spread, selection would have occurred on European populations of C. gigas for about 40 generations. This is a very short period, allowing the detection of very few, if any, outliers, as also recently observed in Crepidula (Riquet et al. 2013).
In the present article, we have presented a stringent detection of AFLP outliers, and our findings are in agreement with the results of a recent comparison between different methods showing that Bayescan usually detects a high percentage of true selective loci as well as <1% false-positive outliers (Pérez-Figueroa et al. 2010). Although the combination of the results of several methods could be considered a cautious approach, the same study highlighted that false positives are common even with a combination of methods and multitest correction, suggesting that the outliers detected should still be considered with extreme caution. Here, we were especially cautious about the north–south comparison for three reasons: (i) it generates a hierarchical structure (Excoffier et al. 2009a), (ii) the sampling was performed along a long linear habitat (Fourcade et al. 2013), and (iii) colonization can produce gene-surfing effects that might bias outlier tests available to date (Klopfstein et al. 2006; Excoffier et al. 2009b; Hofer et al. 2009). As the southern group proved to be panmictic, we repeated outlier tests in comparisons between each of the northern populations and the pooled data from the southern populations. Some outliers were detected in this way, but none were well supported by consistent repetition in multiple comparisons (Fig. S2). However, some detected outliers revealed a genetic structure that was independent of the differentiation between the northern and southern groups, as this latter genetic pattern was the same whether outlier loci were considered or not. These outliers that group populations from the southern and the northern groups are robust candidates for selection because they do not suffer from the problem of an increased variance of neutral FST due to shared co-ancestry (Robertson 1975; Excoffier et al. 2009a; Bonhomme et al. 2010; Bierne et al. 2013b).
Interestingly, among these most differentiated loci, we noticed a tendency for parallel genetic structure, grouping together some samples of the northern group in Denmark (ISE and LIM), a sample of the southern group in the Netherlands (OOS) and the Japanese sample (OSH). An uncorrelated structure between some outliers and the neutral fraction of the genome is clearly visible in the CA, in which the first axis reveals the genome-wide genetic structure between the southern and northern groups, while the second axis reveals the outlier-specific genetic structure that differentiates two samples of the southern group (OOS and OSH) and two samples of the northern group (ISE and LIM). The simplest hypothesis that could explain this pattern would be parallel adaptation to similar environmental pressures in these four populations, which is a hypothesis regularly put forward in the literature (Bradbury et al. 2010; Limborg et al. 2012). By parallel adaptation, we do not necessarily mean that there is independent primary differentiation caused by ecology-driven selection at different locations, but that adaptive polymorphisms associate more with the environment than with the neutral genetic structure (Bierne et al. 2013a). The fact that the Danish samples come from fjords and the Dutch sample from the Scheldt estuary in Zealand (the Netherlands) would point to adaptation to fjord-like environments. In addition to salinity, fjords and estuaries are characterized by multifarious abiotic (e.g. temperature and pH) and biotic (e.g. parasites, predators and competitors) ecological factors. However, we also noticed that each of the four samples individualized by outlier loci came from a site where cultivation has taken place during several periods over the last 40 years. We can therefore speculate that the history of introduction for aquaculture purposes could have interacted with selection to generate the genetic structure observed at outlier loci. Hence, the Danish fjords sampled (Limfjord and Isefjord), as well as the Oosterschelde population in the Netherlands, show higher frequencies at specific markers (Fig. 4), which may reflect the different origins of introduction that occurred in northern Europe and that are partly linked to aquaculture activity, as also concluded by Moehler et al. (2011). This hypothesis of a secondary introduction does not fully explain the uncorrelated effect of selection (outlier-specific) and neutral processes (genome-wide). To understand the genetic structure observed, we need to further assume that the genome of the hatchery stock would have been secondarily swamped by the wave of invasion at neutral markers, erasing the history of introduction at these markers, while the hatchery background would have been partially maintained at selected loci. Our results therefore call for a closer examination of the adaptive genetic structure in the area of the Pacific where this species originated, as well as in other areas where C. gigas is cultivated and that have served as a stepping stone before the introduction into Europe (e.g. British Columbia).
Conclusion and perspectives
This genomic study on the invasiveness of the Pacific cupped oyster in Europe aimed to disentangle the demographic and adaptive factors as well as to consider the impact of aquaculture on the patterns of genetic structure observed. We used numerous markers for the analysis of a specific context of an economically important species during its invasive phase in Europe. We did not observe any loss of diversity during introduction, although some was seen during the northern spread of the species in Europe. There is little, if any, evidence of adaptation following the introduction of Crassostrea gigas into Europe, as there was no genetic differentiation or decrease in diversity between the population from Japan (the origin of European populations) and populations from areas of southern Europe where C. gigas is cultivated. However, northern populations (from Germany, Denmark and Sweden) showed a decrease in diversity, coupled with a stronger genetic structure. This result agrees with the assertions that (i) the number of oysters that participated in the introduction of C. gigas in northern populations was far lower than the large quantities introduced from Japan in the 1970s and (ii) that the invasion wave due to the acclimatization of the species in Europe has yet not reached northern latitudes, but is presently localized somewhere on the North Sea coast of Germany. From a practical point of view, this study demonstrates the genetic impact of aquaculture on a newly introduced species and consequently shows that care must be taken when considering the stocks that are chosen at the beginning of a breeding programme. Indeed, as there are very few means to manage diseases in molluscs (Renault 2011), breeding programmes, together with the modification of farmers' practices (exchanges, densities, etc.), appear to offer a solution to such problems and have begun to develop since 2011. Genetic characterization is clearly of interest for the choice of the initial broodstocks to be used, but also for the genetic diversity to be regularly monitored in such programmes.
Acknowledgments
This work was funded by the Agence National de la Recherche (Hi-Flo project ANR-08-BLAN-0334-01). We thank M. S. Berggren, H. T. Christensen, L. Meistertzheim, P. Norling, J. Petersen, S. Pien and D. B. Toring for providing samples and Helen McCombie for English editing.
Data archiving statement
Data for this study are deposited in the Dryad repository: doi:10.5061/dryad.13jc4.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Bar plot of admixture proportions of individuals, estimated using STRUCTURE with K = 2 groups for microsatellite and SNP markers (A) and AFLP makers (B). Information on sample origin is given below each bar plot.
Table S1. Characteristics of the markers used for AFLP analysis.
Table S2. Estimates of Weir and Cockerham's (1984) θ between pairs of samples.
Literature cited
- Addison JA, Ort BS, Mesa AK, Pogson GH. Range-wide genetic homogeneity in the California sea mussel (Mytilus californianus): a comparison of allozymes, nuclear DNA markers, and mitochondrial DNA sequences. Molecular Ecology. 2008;17:4222–4232. doi: 10.1111/j.1365-294x.2008.03905.x. [DOI] [PubMed] [Google Scholar]
- Beaumont MA. Adaptation and speciation: what can F(st) tell us? Trends in Ecology & Evolution. 2005;20:435–40. doi: 10.1016/j.tree.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Beaumont MA, Nichols RA. Evaluating loci for use in the genetic analysis of population structure. Proceedings of the Royal Society London B. Biological Sciences. 1996;263:1619–1626. [Google Scholar]
- Belkhir K, Borsa P, Goudet J, Chikhi L, Bonhomme F. Genetix v. 4.02. Software for Population Genetics. Montpellier, France: Laboratoire Génome et Populations, University Montpellier 2; 2001. [Google Scholar]
- Bierne N, Welch J, Loire E, Bonhomme F, David P. The coupling hypothesis: why genome scans may fail to map local adaptation genes. Molecular Ecology. 2011;20:2044–2072. doi: 10.1111/j.1365-294X.2011.05080.x. [DOI] [PubMed] [Google Scholar]
- Bierne N, Gagnaire PA, David P. The geography of introgression in a patchy environment and the thorn in the side of ecological speciation. Current Zoology. 2013a;59:72–86. [Google Scholar]
- Bierne N, Roze D, Welch J. Pervasive selection or is it… ? why are Fst outliers sometimes so frequent ? Molecular Ecology. 2013b;22:2061–2064. doi: 10.1111/mec.12241. [DOI] [PubMed] [Google Scholar]
- Bonhomme M, Chevalet C, Servin B, Boitard S, Abdallah JM, Blott S, San Cristobal M. Detecting selection in population trees: The Lewontin and Krakauer test extended. Genetics. 2010;186:241–262. doi: 10.1534/genetics.110.117275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudry P, Collet B, Cornette F, Hervouet V, Bonhomme F. High variance in reproductive success of the Pacific oyster (Crassostrea gigas, Thunberg) revealed by microsatellite-based parentage analysis of multifactorial crosses. Aquaculture. 2002;204:283–296. [Google Scholar]
- Bougrier S, Raguenes G, Bachere E, Tige G, Grizel H. Essai de réimplantation de Crassostrea angulata en France, résistance au chambrage et comportement des hybrides C. Angulata – C. Gigas. Copenhagen, Denmark: ICES; 1986. p. 10. ICES. CM. F/38. [Google Scholar]
- Bougrier S, Geairon P, Desolus-Paoli J, Bacher C, Jonquières G. Allometric relationships and effects of temperature on clearance and oxygen consumption rates of Crassostrea gigas (Thunberg) Aquaculture. 1995;134:143–154. [Google Scholar]
- Boutet I, Tanguy A, Moraga D. Molecular identification and expression of two non-P450 enzymes, monoamine oxidase A and flavin-containing monooxygenase 2, involved in phase I of xenobiotic biotransformation in the Pacific oyster, Crassostrea gigas. Biochimica et Biophysica Acta. 2004;1679:29–36. doi: 10.1016/j.bbaexp.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Bradbury IR, Hubert S, Higgins B, Borza T, Bowman S, Paterson IG, Snelgrove PVR, et al. Parallel adaptive evolution of Atlantic cod on both sides of the Atlantic Ocean in response to temperature. Proceedings of the Royal Society B: Biological Sciences. 2010;277:3725–3734. doi: 10.1098/rspb.2010.0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruins RBW. Crassostrea gigas (Thunberg 1793) op Texel. Correspondentie Blad Nederlands Malacologie Verenigung. 1983;215:1436–1438. [Google Scholar]
- Chessel D, Dufour AB, Thioulouse J. The ade4 package-I- One-table methods. R News. 2004;4:5–10. [Google Scholar]
- CIESM. 2000. Ostreidae oysters - Crassostrea gigas (Thunberg 1793). http://www.ciesm.org/atlas/Crassostrea.html.
- Dankers N, Dijkman EM, Meijboom ML, de Jong G, de Kort A. De verspreiding en uitbreiding van de Japanse Oester in de Nederlandse Waddenzee. Alterra, Wageningen: Alterra-rapport; 2004. [Google Scholar]
- Diederich S, Nehls G, Reise J, van Beusekom K. Introduced Pacific oysters (Crassostrea gigas) in the northern Wadden Sea: invasion accelerated by warm summers? Helgoland Marine Research. 2005;59:97–106. [Google Scholar]
- Dlugosch KM, Parker IM. Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Molecular Ecology. 2008;17:431–449. doi: 10.1111/j.1365-294X.2007.03538.x. [DOI] [PubMed] [Google Scholar]
- Eklund U, Håkansson M, Haamer J. En undersökning om förutsättningarna för ostronodling vid svenska västkusten. Göteborg: Geologiska Institutionen, CTH; 1977. [Google Scholar]
- Estoup A, Guillemaud T. Reconstructing routes of invasion using genetic data: why, how and so what? Molecular Ecology. 2010;19:4113–4130. doi: 10.1111/j.1365-294X.2010.04773.x. [DOI] [PubMed] [Google Scholar]
- Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources. 2010;10:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Ray N. Surfing during population expansions promotes genetic revolutions and structuration. Trends in Ecology & Evolution. 2008;23:347–351. doi: 10.1016/j.tree.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Foll M, Petit RJ. Detecting loci under selection in a hierarchically structured population. Heredity. 2009a;103:285–98. doi: 10.1038/hdy.2009.74. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Foll M, Petit RJ. Genetic Consequences of Range Expansions. Annual Review of Ecology, Evolution, and Systematics. 2009b;40:481–501. [Google Scholar]
- Facon B, Genton BJ, Shykoff J, Jarne P, Estoup A, David P. A general eco-evolutionary framework for understanding bioinvasions. Trends in Ecology and Evolution. 2006;21:130–135. doi: 10.1016/j.tree.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Facon B, Pointier JP, Jarne P, Sarda V, David P. High genetic variance in life-history strategies within invasive populations by way of multiple introductions. Current Biology. 2008;18:363–367. doi: 10.1016/j.cub.2008.01.063. [DOI] [PubMed] [Google Scholar]
- Fleury E, Huvet A, Lelong C, Boulo J, de Lorgeril V, Gueguen Y, Bachère E, et al. Generation and analysis of a 29,745 unique Expressed Sequence Tags from the Pacific oyster (Crassostrea gigas) assembled into a publicly accessible database: the GigasDatabase. BMC Genomics. 2009;10:341. doi: 10.1186/1471-2164-10-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foll M, Gaggiotti OE. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics. 2008;180:977–993. doi: 10.1534/genetics.108.092221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourcade Y, Chaput-Bardy A, Secondi J, Fleurant C, Lemaire C. Is local selection so widespread in river organisms? Fractal geometry of river networks leads to high bias in outlier detection. Molecular Ecology. 2013;22:2065–2073. doi: 10.1111/mec.12158. [DOI] [PubMed] [Google Scholar]
- Gagnaire PA, Albert V, Jónsson B, Bernatchez L. Natural selection influences AFLP intraspecific genetic variability and introgression patterns in Atlantic eels. Molecular Ecology. 2009;18:1678–1691. doi: 10.1111/j.1365-294X.2009.04142.x. [DOI] [PubMed] [Google Scholar]
- Goudet J. 2001. FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3).Available from http://www.unil.ch/izea/softwares/fstat.html. (accessed on 15 October 2011)
- Grahame JW, Wilding CS, Butlin RK. Adaptation to a steep environmental gradient and an associated barrier to gene exchange in Littorina saxatilis. Evolution. 2006;60:268–278. [PubMed] [Google Scholar]
- Griffin TJ, Smith LM. Single-nucleotide polymorphism analysis by MALDI-TOF mass spectrometry. Trends in Biotechnology. 2000;18:77–84. doi: 10.1016/s0167-7799(99)01401-8. [DOI] [PubMed] [Google Scholar]
- Grizel H, Héral M. Introduction into France of the Japanese oyster (Crassostrea gigas. Journal du Conseil International de L'Exploration de la Mer. 1991;47:399–403. [Google Scholar]
- Gueguen Y, Cadoret JP, Flament D, Barreau-Roumiguière C, Girardot AL, Garnier J, Hoareau A, et al. Immune gene discovery by expressed sequence tags generated from hemocytes of the bacteria-challenged oyster, Crassostrea gigas. Gene. 2003;303:139–145. doi: 10.1016/s0378-1119(02)01149-6. [DOI] [PubMed] [Google Scholar]
- Hellberg ME, Burton RS, Neigel JE, Palumbi SR. Genetic assessment of connectivity among marine populations. Bulletin of Marine Science. 2002;70:273–290. [Google Scholar]
- Héral M, Deslous-Paoli JM, Prou J. Population Dynamics and Biomass of Cultivated Cupped Oysters (Crassostrea angulata and Crassostrea gigas) in the Marennes–Oleron Bay on a Century. Conseil International de l'Exploration de la Mer; 1986. p. 23. Copenhagen, Denmark, CM, F/41. [Google Scholar]
- His E. Premiers éléments de comparaison entre l'huître portugaise et l'huître japonaise. Science et Pêche. Bulletin de L'Institut des Pêches Maritimes. 1972;219:1–9. [Google Scholar]
- Hodgins KA, Lai Z, Nurkowski K, Huang J, Rieseberg LH. The molecular basis of invasiveness: differences in gene expression of native and introduced common ragweed (Ambrosia artemisiifolia) in stressful and benign environments. Molecular Ecology. 2013;22:2496–2510. doi: 10.1111/mec.12179. [DOI] [PubMed] [Google Scholar]
- Hofer T, Ray N, Wegmann D, Excoffier L. Large allele frequency differences between human continental groups are more likely to have occurred by drift during range expansions than by selection. Annals of Human Genetics. 2009;73:95–108. doi: 10.1111/j.1469-1809.2008.00489.x. [DOI] [PubMed] [Google Scholar]
- Huvet A, Boudry P, Ohresser M, Delsert C, Bonhomme F. Variable microsatellites in the Pacific oyster Crassostrea gigas and other cupped oyster species. Animal Genetics. 2000a;31:71–72. doi: 10.1111/j.1365-2052.2000.579-5.x. [DOI] [PubMed] [Google Scholar]
- Huvet A, Lapègue S, Magoulas A, Boudry P. Mitochondrial and nuclear DNA phylogeography of Crassostrea angulata, the Portuguese oyster endangered in Europe. Conservation Genetics. 2000b;1:251–262. [Google Scholar]
- Huvet A, Herpin A, Dégremont L, Labreuche Y, Samain J-F, Cunningham C. The identification of genes from the oyster Crassostrea gigas that are differentially expressed in progeny exhibiting opposed susceptibility to summer mortality. Gene. 2004a;343:211–20. doi: 10.1016/j.gene.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Huvet A, Fabioux C, McCombie H, Lapègue S, Boudry P. Natural hybridization between genetically differentiated populations of Crassostrea gigas and C. angulata highlighted by sequence variation in flanking regions of a microsatellite locus. Marine Ecology Progress Series. 2004b;272:141–152. [Google Scholar]
- Jensen K, Knudsen J. A summary of alien marine invertebrates in Danish waters. Oceanological and Hydrobiological Studies. 2005;34(Suppl. 1):137–162. [Google Scholar]
- Keller RP, Lodge DM. Species invasions from commerce in live aquatic organisms: problems and possible solutions. BioScience. 2007;57:428–436. [Google Scholar]
- Klopfstein S, Currat M, Excoffier L. The fate of mutations surfing on the wave of a range expansion. Molecular Biology and Evolution. 2006;23:482–490. doi: 10.1093/molbev/msj057. [DOI] [PubMed] [Google Scholar]
- Le Corre V, Kremer A. Genetic variability at neutral markers, quantitative trait land trait in a subdivided population under selection. Genetics. 2003;164:1205–1219. doi: 10.1093/genetics/164.3.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Corre V, Kremer A. The genetic differentiation at quantitative trait loci under local adaptation. Molecular Ecology. 2012;21:1548–66. doi: 10.1111/j.1365-294X.2012.05479.x. [DOI] [PubMed] [Google Scholar]
- Le Gall J, Raillard O. Influence de la temperature sur la physiologie de l'huitre Crassostrea gigas. Océanis. 1988;14:603–608. [Google Scholar]
- Lejart M, Hily C. Differential response of benthic macrofauna to the formation of novel oyster reefs (Crassostrea gigas, Thunberg) on soft and rocky substrate in the intertidal of the Bay of Brest, France. Journal of Sea Research. 2011;65:84–93. [Google Scholar]
- Lewontin RC, Krakauer J. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics. 1973;74:175–195. doi: 10.1093/genetics/74.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Hedgecock D. Genetic heterogeneity, detected by PCR-SSCP, among samples of larval Pacific oysters (Crassostrea gigas) supports the hypothesis of large variance in reproductive success. Canadian Journal of Fisheries and Aquatic Sciences. 1998;55:1025–1033. [Google Scholar]
- Limborg MT, Helyar SJ, Taylor M, De Bruyn MI, Nielsen EE, Ogden R, Carvalho GR, et al. Environmental selection on transcriptome-derived SNPs in a high gene flow marine fish, the Atlantic herring (Clupea harengus. Molecular Ecology. 2012;21:3686–3703. doi: 10.1111/j.1365-294X.2012.05639.x. [DOI] [PubMed] [Google Scholar]
- Mayrose M, Kane NC, Mayrose I, Dlugosch KM, Rieseberg LH. Increased growth in sunflower correlates with reduced defences and altered gene expression in response to biotic and abiotic stress. Molecular Ecology. 2011;20:4683–4694. doi: 10.1111/j.1365-294X.2011.05301.x. [DOI] [PubMed] [Google Scholar]
- Moehler J, Wegner KM, Reise K, Jacobsen S. Invasion genetics of pacific oyster Crassostrea gigas shaped by aquaculture stocking practices. Journal of Sea Research. 2011;66:256–262. [Google Scholar]
- Murray MC, Hare MP. A genomic scan for divergent selection in a secondary contact zone between Atlantic and Gulf of Mexico oysters, Crassostrea virginica. Molecular Ecology. 2006;15:4229–4242. doi: 10.1111/j.1365-294X.2006.03060.x. [DOI] [PubMed] [Google Scholar]
- Nehring S. Oyster beds and Sabellaria reefs. In: De Jong F, Bakker JF, van Berkel CJM, Dankers N, Dahl K, Gätje C, Marencic H, Potel P, editors. Wadden Sea Quality Status Report. Wadden Sea Ecosystem. Wadden Sea Ecosystem No 9. Wilhelmshaven, Germany: Common Wadden Sea Secretariat, Trilateral Monitoring and Assessment Group, Quality Status Report Group; 1999. pp. 146–147. [Google Scholar]
- Nehring S. 2006. NOBANIS – Invasive Alien Species Fact Sheet – Crassostrea gigas. From: Online database of the North European and Baltic Network on Invasive Alien Species – NOBANIS http://www.nobanis.org. (accessed on 27 May 2010)
- Nei M. Estimation of average heterozygosity and genetic distance from a small number of Individuals. Genetics. 1978;89:583–590. doi: 10.1093/genetics/89.3.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Maruyama T. Letters to the editors: Lewontin–Kraka-er test for neutral genes. Genetics. 1975;80:395. doi: 10.1093/genetics/80.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosil P, Funk DJ, Ortiz-Barrientos D. Divergent selection and heterogeneous genomic divergence. Molecular Ecology. 2009;18:375–402. doi: 10.1111/j.1365-294X.2008.03946.x. [DOI] [PubMed] [Google Scholar]
- Pennings PS, Hermisson J. Soft Sweeps III: the Signature of Positive Selection from Recurrent Mutation. PLoS Genetics. 2006;2:e186. doi: 10.1371/journal.pgen.0020186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Figueroa A, García-Pereira MJ, Saura M, Rolán-Alvarez E, Caballero A. Comparing three different methods to detect selective loci using dominant markers. Journal of Evolutionary Biology. 2010;23:2267–2276. doi: 10.1111/j.1420-9101.2010.02093.x. [DOI] [PubMed] [Google Scholar]
- Prentis PJ, Pavasovic A. Understanding the genetic basis of invasiveness. Molecular Ecology. 2013;22:2366–2368. doi: 10.1111/mec.12277. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of Population Structure Using Multilocus Genotype Data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quayle D. Pacific Oyster Culture in British Columbia. Ottawa: The Queen's printer; 1969. p. 192. Fisheries Research Board of Canada, Bulletin 169. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2012. ISBN 3-900051-07-0, URL http://www.R-project.org/ [Google Scholar]
- Reise K. Pacific oysters invade mussel beds in the European Wadden Sea. Senckenbergiana Maritima. 1998;28:167–175. [Google Scholar]
- Reise K, Dankers N, Essink K. Introduced Species Wadden Sea Quality Status Report. Wilhelmshaven, Germany: Common Wadden Sea secretariat; 2005. [Google Scholar]
- Renault T. Viruses infecting marine molluscs: increased mortality outbreaks and herpes viruses. Bulletin De L'Académie Vétérinaire De France. 2011;164:359–364. [Google Scholar]
- Renault T, Faury N, Barbosa-Solomieu V, Moreau K. Suppression substractive hybridisation (SSH) and real time PCR reveal differential gene expression in the Pacific cupped oyster, Crassostrea gigas, challenged with Ostreid herpesvirus 1. Developmental and Comparative Immunology. 2011;35:725–35. doi: 10.1016/j.dci.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Renaut S, Nolte AW, Rogers SM, Derome N, Bernatchez L. SNP signatures of selection on standing genetic variation and their association with adaptive phenotypes along gradients of ecological speciation in lake whitefish species pairs (Coregonus spp.) Molecular Ecology. 2011;20:545–559. doi: 10.1111/j.1365-294X.2010.04952.x. [DOI] [PubMed] [Google Scholar]
- Riquet F, Daguin-Thiébaut C, Ballenghien M, Bierne N, Viard F. Contrasting patterns of genome-wide polymorphism in the native and invaded distribution of the marine invasive mollusc Crepidula fornicata. Molecular Ecology. 2013;22:1003–1018. doi: 10.1111/mec.12161. [DOI] [PubMed] [Google Scholar]
- Robertson A. Gene frequency distributions as a test of selective neutrality. Genetics. 1975;81:775–785. doi: 10.1093/genetics/81.4.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman J, Darling JA. Paradox lost: genetic diversity and the success of aquatic invasions. Trends in Ecology and Evolution. 2007;22:454–464. doi: 10.1016/j.tree.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods in Molecular Biology. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Sauvage C, Bierne N, Lapègue S, Boudry P. Single Nucleotide polymorphisms and their relationship to codon usage bias in the Pacific oyster Crassostrea gigas. Gene. 2007;406:13–22. doi: 10.1016/j.gene.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Sauvage C, Boudry P, Lapègue S. Identification and characterization of 18 novel polymorphic microsatellite makers derived from expressed sequence tags in the Pacific oyster Crassostrea gigas. Molecular Ecology Resources. 2009;9:853–855. doi: 10.1111/j.1755-0998.2009.02525.x. [DOI] [PubMed] [Google Scholar]
- Ségurel L, Lafosse S, Heyer E, Vitalis R. Frequency of the AGT Pro11Leu polymorphism in humans: does diet matter? Annals of Human Genetics. 2010;74:57–64. doi: 10.1111/j.1469-1809.2009.00549.x. [DOI] [PubMed] [Google Scholar]
- Sellos D, Moal J, Dégremont L, Huvet A, Daniel JY, Nicoulaud S, Boudry P, et al. Structure of amylase genes in populations of Pacific Cupped oyster (Crassostrea gigas): tissue expression and allelic polymorphism. Marine Biotechnology. 2003;5:360–72. doi: 10.1007/s10126-002-0089-7. [DOI] [PubMed] [Google Scholar]
- Simberloff D. The role of propagule pressure in biological invasions. Annual Review of Ecology Evolution and Systematics. 2009;40:81–102. [Google Scholar]
- Slatkin M, Voelm L. FST in a hierarchical island model. Genetics. 1991;127:627–629. doi: 10.1093/genetics/127.3.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand O, Vollstad JH. The Molluscan fisheries and culture of Norway. In: MacKenzie CL Jr, Burrell V, Rosenfield A, Hobart WL, editors. The History, Present condition, and Future of the Molluscan Fisheries of North America and Europe. U.S. Dep. Commerce; 1997. pp. 7–24. Seattle, WA. NOAA Tech. Rep. NMFS 129. [Google Scholar]
- Strand Å, Blanda E, Bodvin T, Davids JK, Jensen LF, Holm-Hansen TH, Jelmert A, et al. Impact of an icy winter on the Pacific oyster (Crassostrea gigas Thunberg, 1793) populations in Scandinavia. Aquatic Invasions. 2012;7:433–440. [Google Scholar]
- Taris N, Baron S, Sharbel TF, Sauvage C, Boudry P. A combined microsatellite multiplexing and boiling DNA extraction method for high throughput parentage analyses in the Pacific Oyster (Crassostrea gigas. Animal Genetics. 2005;36:516–518. [Google Scholar]
- Tice KA, Carlon DB. Can AFLP genome scans detect small islands of differentiation? The case of shell sculpture variation in the periwinkle Echinolittorina hawaiiensis. Journal of Evolutionary Biology. 2011;24:1814–1825. doi: 10.1111/j.1420-9101.2011.02314.x. [DOI] [PubMed] [Google Scholar]
- Troost K. Causes and effects of a highly successful marine invasion: case-study of the introduced Pacific oyster Crassostrea gigas in continental NW European estuaries. Journal of Sea Research. 2010;64:145–165. [Google Scholar]
- Vekemans X. AFLP-SURV version 1.0. Belgium: Laboratoire de Génétique et Ecologie Végétale, Université Libre de Bruxelles; 2002. Distributed by the author. [Google Scholar]
- Vos P, Hogers R, Bleeker M, Reijans M, Hornes T, van de Lee M, Frijters A, et al. AFLP: a new technique for DNA fingerprinting. Nucleic Acids Research. 1995;23:4407–4414. doi: 10.1093/nar/23.21.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock R, Hipperson H, Mannarelli M, Butlin RK, Burke T. An objective, rapid and reproducible method for scoring AFLP peak-height data that minimizes genotyping error. Molecular Ecology Resources. 2008;8:725–735. doi: 10.1111/j.1755-0998.2007.02073.x. [DOI] [PubMed] [Google Scholar]
- Wilding CS, Butlin RK, Grahame J. Differential gene exchange between parapatric morphs of Littorina saxatilis detected using AFLP markers. Journal of Evolutionary Biology. 2001;14:611–619. [Google Scholar]
- Wood HM, Grahame J, Humphray S, Rogers J, Butlin RK. Sequence differentiation in regions identified by a genome scan for local adaptation. Molecular Ecology. 2008;17:3123–3135. doi: 10.1111/j.1365-294X.2008.03755.x. [DOI] [PubMed] [Google Scholar]
- Wrange AL, Valero J, Harkestad LS, Strand O, Lindegarth S, Christensen HT, Dolmer P, et al. Massive settlements of the Pacific oyster, Crassostrea gigas, in Scandinavia. Biological Invasions. 2010;12:1145–1152. [Google Scholar]
- Yu H, Li Q. EST-SSR markers from the Pacific oyster, Crassostrea gigas. Molecular Ecology Notes. 2007;7:860–862. [Google Scholar]
- Zhivotovsky LA. Estimating population structure in diploids with multilocus dominant DNA markers. Molecular Ecology. 1999;8:907–13. doi: 10.1046/j.1365-294x.1999.00620.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.