

Abstract
From the 19th century to the present, the complex indole alkaloid strychnine has engaged the chemical community. In this review, we examine why strychnine has been and remains today an important target for directed synthesis efforts. A selection of the diverse syntheses of strychnine is discussed with the aim of identifying their influence on the evolution of the strategy and tactics of organic synthesis.
Keywords: natural products, strychnine, synthesis strategy, total synthesis
1. Introduction
Strychnine is one of the most famous chemical compounds, being known to chemists and the public alike. It is the major alkaloid isolated from the seeds of the trees Strychnos nux-vomica and Strychnos ignatii Bergius (Saint-Ignatiu’s bean), which are broadly distributed in the Asian tropics.[ 1 ] The powerful and dangerous physiological properties of powdered nuts of Strychnos nux-vomica have been known since the middle ages.[ 2 ] This preparation (nux vomica or vomiting nuts) was investigated haphazardly by doctors in Germany in the 16th and 17th centuries and more systematically by physicians in France and England during the 19th century.[3] Strychnine was first isolated in pure form from Strychnos ignatii Bergius by Pelletier and Caventou in 1818,[4] and subsequently purported to have various benefits including to increase appetite, tone skeletal musculature, increase memory, and cure snakebites.[3,5] At one time, strychnine was even taken as a ‘performance-enhancing drug.’[6] However, its only actual utility is as a poison. The ease of its isolation led to strychnine being a widely used pest control agent in the 18th century, with its use as a poison for rodents continuing today.[5,7] Thankfully, its modern use is dwindling as a dose as small as 50 mg can be fatal to an adult human and more effective rodenticides with lower human toxicities are now available. Regardless, the throes and seizures symptomatic of strychnine poisoning have made it irresistible to artists looking to portray dramatic deaths, including masters such as Alfred Hitchcock in the movie Psycho and Agatha Christie in her novel The Mysterious Affair at Styles.
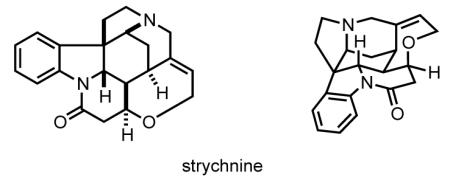
Strychnine is a powerful neurotoxin whose mode of action is well characterized. The alkaloid competitively binds to the glycine receptor chloride channel (also named the strychnine-sensitive glycine receptor), eliminating glycinergic inhibition in the spinal cord.[8,9] The resulting over-excitation of nerves causes muscular convulsions, the inability to control respiration, and eventually asphyxiation. There is no direct antidote for strychnine poisoning, which is treated by administering anticonvulsants and muscle relaxants to stem the convulsions.[10] Strychnine has played an important role in elucidating the chemistry of the nervous system. As a result of its high affinity binding to strychnine (Kd in the nanomolar range), the glycine receptor was the first nicotinoid receptor isolated from mammalian tissue.[11] Strychnine’s covalent attachment to the transmembrane α1 subunit upon UV irradiation helped identify the ligand binding site of the glycine receptor. In addition, autoradiographical mapping of [3H]strychnine binding sites allowed mapping of the location of glycine receptors in the central nervous system.[8,9]
1.1. Importance of strychnine in organic chemistry
Strychnine stands out among classical natural products as one of the first to be isolated and purified, yet one of the last to have its structure established.[12] As such, it has played a central role in the development of several major movements in organic chemistry.
Extensive investigations towards characterizing the structure of strychnine were pursued throughout the early 20th century, led by Leuchs and Robinson who published nearly 400 papers addressing strychnine’s structure using degradative methods.[ 13 ] Finally, Woodward and Brehm proved the correct structure in 1948 from a combination of degradative evidence and a key use of ultraviolet spectroscopy.[14,15] The connectivity and absolute configuration of strychnine were soon confirmed by X-ray crystallography.[ 16 ] Strychnine was one of the last natural product structures to be solved using largely classical methods, although Woodward’s use of ultraviolet spectroscopy foreshadowed the preeminent role that spectroscopic techniques would play in structure elucidation.
With the structure of strychnine finally resolved, organic chemists hoped to produce strychnine in the flask. Remarkably, this daunting feat was accomplished merely six years later when R. B. Woodward announced the first total synthesis of strychnine in 1954.[17] At the time, strychnine was by far the most complex molecule to be produced by chemical synthesis. Woodward’s monumental achievement is often considered the beginning of a golden age of total synthesis. It is certain that this synthesis inspired many of the organic chemists who contributed to the remarkable surge of natural product total syntheses in the 1970’s and 80’s.
1.2. Strychnine biosynthesis
Nearly 30 naturally occurring Strychnos alkaloids are now recognized.[1] As with all monoterpene indole alkaloids, their biosynthesis originates from the union of tryptamine and secologanin (Scheme 1).[18,19] Pictet–Spengler type condensation of these precursors generates strictosidine, which further converts to geissoschizine after deglycosylation.
Scheme 2.

Conversion of isostrychnine and the Wieland–Gumlich aldehyde to strychnine.
Geissoschizine is a major branching point in indole alkaloid biosynthesis. Elaboration of geissoschizine without rearrangement of the fused tetracyclic ring system generates the yohimbine, heteroyohimbine and corynantheine subgroups of indole alkaloids. However, alkaloids of the strychnos group derive from geissoschizine by a cascade sequence believed to originate by oxidative rearrangement of the indole ring to generate spiroxindole (1), which subsequently condenses with the pendant 1,3-dicarbonyl side chain to arrive at dehydropreakuammicine. Decarboxylation of dehydropreakuammicine generates norfluorocurarine. Further oxidation of the terminal allylic position and reduction of the vinylogous formamide give rise to the Wieland–Gumlich aldehyde, a natural product also known as caracurine VII. Confirming earlier postulates by Woodward[20] and Robinson,[21] the intermediacy of the Wieland–Gumlich aldehyde in the biosynthesis of strychnine was established by Heimberger and Scott in 1973.[22] Strychnine’s final piperidone ring is installed by condensation with acetic acid.[23]
1.3. Natural Products and Total Synthesis: Bound Together in the History of Organic Chemistry and a Pertinent Union Today
Nature’s molecules and organic chemical synthesis have been intimately linked since Wöhler’s rational synthesis of urea in 1828. [ 24 – 27 ] For the next 150 years, chemical synthesis played an indispensible role in structure elucidation and verification. This function has been supplanted largely by analytical methods. Nonetheless, the consequential role that total synthesis stills plays in structure elucidation is seen in the large number of natural products whose structures have been revised recently as a result of total synthesis endeavors.[28]
Natural products continue to play a major role in human health, as both therapeutic agents and the inspiration for marketed drugs.[29] Although natural product drugs are rarely produced entirely by chemical total synthesis,[29c] for rare natural products isolated from sources other than plants or microorganisms, total synthesis can provide initial supplies for preclinical studies (Figure 1).[29b] For example, Novartis’s evaluation of discodermolide, an antitumor agent that binds to microtubules with higher affinity than paclitaxel, was enabled by Paterson’s total synthesis.[30] Another example was the total synthesis of crambescidin 800 developed in our laboratories, which was employed by Pharma Mar to obtain preclinical supplies of this structurally novel antineoplastic marine natural product.[31]
Figure 1.
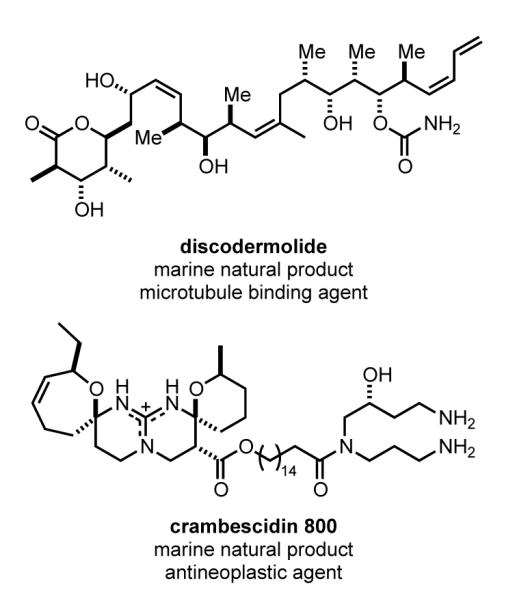
Representative natural products whose preclinical evaluation was enabled by total synthesis.
Natural product total synthesis endeavors often provide intermediates and chemistry that allows the natural product’s structure to be modified. Such changes can improve biological and physical properties, and allow the natural product’s molecular target to be identified. One salient example is Danishefsky’s “diverted total synthesis” of migrastatin, a macrolide antibiotic that inhibits tumor metastasis by blocking human tumor cell migration. These efforts identified a number of migrastatin analogues having enhanced drug characteristics (Figure 2).[32] Another fine example is Wender’s synthesis and biological evaluation of simplified analogs (bryologs) of the powerful antitumor agent bryostatin.[33 ] The crowning recent achievement in this arena was the launch of eribulin (HalavenTM) for the treatment of metastatic breast cancer in 2010. Kishi’s remarkable total synthesis of halichondrin B[34] led to the identification of the macrocyclic fragment of this dauntingly complex natural product as the essential anti-cancer pharmacophore, and provided much of the chemistry that has allowed eribulin to be produced commercially by chemical synthesis.[35]
Figure 2.

Examples of recent drugs and potential therapeutics developed as the result of total synthesis efforts.
Thus far our discussion of the relationship of natural products and target-directed total synthesis has not touched on the vital role played by natural products in advancing the logic and tools of synthetic organic chemistry. Natural products having unique structures for years have inspired the development of new chemical synthesis strategies and chemical transformations.[25] Illustrative examples of the latter include Barton’s invention of the deoxygenation of secondary alcohols in connection with the synthesis of aminoglycoside antibiotics,[36] our invention of the aza-Cope–Mannich reaction during the synthesis of gephyrotoxin,[37] and Baran’s recent invention of silver-catalyzed arylation of electron-deficient aromatics with boronic acids as an outgrowth of the synthesis of palau’amine.[38] Polyfunctional natural products moreover serve as an ideal platform for evaluating the selectivity, scope and utility of synthetic methods, knowledge that is essential for effective synthesis planning and execution.
Our aim in this review is to illustrate the fundamental role that natural products total synthesis plays in the continuing evolution of the science of chemical synthesis by considering a selection of past and very recent total syntheses of strychnine. Strychnine is an ideal focus for such an analysis. Its structure presents a serious challenge for total synthesis, while its seven distinctly different rings provide much latitude in synthesis strategy. Moreover strychnine’s popularity as a synthesis target—stimulated in part by its historical significance to the field of organic chemistry—makes this molecule ideal for benchmarking the utility of new chemical transformations and new chemical synthesis strategies.
2. Overview of Strychnine as a Synthetic Target
At the time of its first synthesis, Robinson remarked about strychnine: “For its molecular size, it is the most complex substance known.”[39] Even by modern standards, strychnine poses significant challenges for total synthesis. Foremost is construction of the umbrella-like CDE-tricyclic unit (Figure 3). This fragment is highly congested, containing five of the six stereogenic carbons of strychnine and its sole quaternary carbon stereocenter. If the F ring is to be constructed late in a synthesis, a secondary challenge is stereocontrolled assembly of the (E)-C19–C20 double bond.
Figure 3.
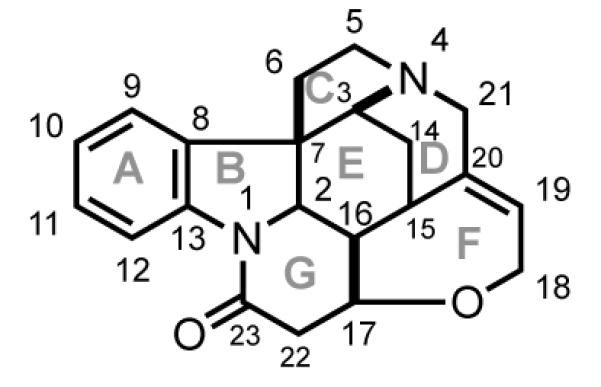
Numbering scheme for strychnine.
To date, 17 total and formal syntheses of strychnine have been reported.[40] All syntheses of strychnine have intercepted one of two common intermediates: isostrychnine (2) or the Wieland–Gumlich aldehyde (3, Scheme 2). Both intermediates can be converted in one step to strychnine. As first defined by Prelog, strychnine and isostrychnine (2) are equilibrated under basic conditions by fragmentation and reformation of the oxepene ring.[41] However, this equilibrium favors the ring-opened isomer in approximately a 3:1 ratio. As a result, the highest reported yield of strychnine from isostrychnine is 28%.[42] The Wieland–Gumlich aldehyde (3) can be converted to strychnine by condensation with malonic acid and decarboxylation, a transformation that takes place in high yield, typically on the order of 80%.[43] Thus, the Wieland-Gumlich aldehyde is the most advantageous strychnine precursor in terms of synthetic planning; however, isostrychnine can be a viable alternative, particularly if it is accessed in high efficiency.
Scheme 2.
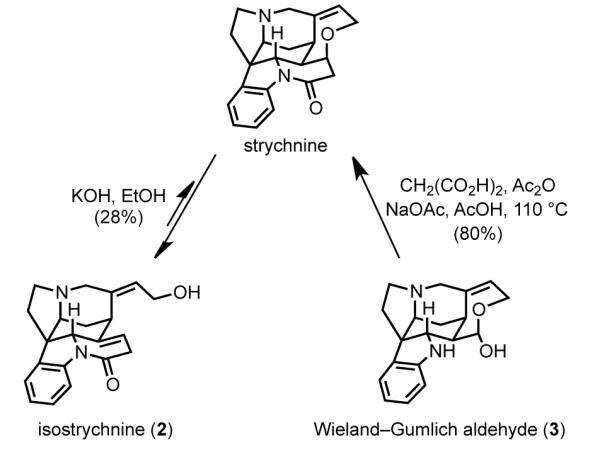
Conversion of isostrychnine and the Wieland–Gumlich aldehyde to strychnine.
In light of the large number of syntheses of strychnine reported, we have chosen only a subset for discussion. We consider syntheses that typically introduce new ways to construct the complex strychnine skeleton and teach general lessons about chemical synthesis strategy and/or tactics. When a general strategy has been utilized in several syntheses, we have chosen the ones that we feel best illustrate the advantages of the approach. We will consider the syntheses in chronological order, which will provide some insight into the progress of chemical synthesis over this period. Our selection is admittedly highly personal. The syntheses we do not discuss have all contributed substantially to synthetic art in this area and are discussed in previous comprehensive reviews.[40,44]
3. A Selection of Total Syntheses of Strychnine and Lessons Learned
3.1. Woodward’s Landmark Synthesis (1954)
The total synthesis of strychnine by Woodward in 1954 was a milestone in chemical synthesis.[17] Woodward’s approach was based on his proposal for strychnine’s biosynthesis, a suggestion that was shown later to be incorrect.[45] Nonetheless, this hypothesis provided a roadmap for a successful total synthesis. Woodward targeted isostrychnine (2), which was seen arising from α-keto lactam 4 by partial reduction of the pyridone and elaboration at the ketone carbonyl group (Scheme 3). The piperidine D ring would derive from γ-amino acid 5, with the E ring of this intermediate being formed by Dieckmann cyclization of diester 6. Woodward envisioned a 3,4-dimethoxyphenyl (veratryl) ring serving as a latent precursor of the G ring during Pictet–Spengler-type cyclization of an iminium ion derived from glyoxal and 2-veratryltryptamine 8. Early introduction of the veratryl group was a strategic decision to direct this cyclization to the β carbon of the indole to produce spiroindoline 7.
Scheme 3.

Retrosynthetic analysis of Woodward’s route to strychnine.
Woodward’s strychnine synthesis began with the preparation of 2-veratrylindole (9) from 3,4-dimethoxyphenyl methyl ketone and phenylhydrazine by Fischer indole synthesis (Scheme 4). In four subsequent steps, the aminoethyl side chain of tryptamine intermediate 8 was incorporated. Closure of the C ring and formation of the quaternary carbon was achieved by activation under neutral conditions of the imine formed by condensation of 8 with ethyl glyoxal by reaction with p-toluenesulfonyl chloride and pyridine. The resulting N-sulfonyliminium ion (10) cyclized to form spiroindole 7 with high stereoselectivity.[46] Stereoselection in this step undoubtedly arose from destabilizing steric interactions between the ethyl ester and the veratryl group. After reduction of the imine of 7 and acetylation of the indoline nitrogen, the veratryl group was cleaved selectively with ozone in aqueous acetic acid to yield diester 11. Heating this product in methanolic HCl cleaved the acetate group and promoted lactonization and double bond isomerization to give pyridone 12. At this stage, four of the seven rings of strychnine were assembled and the stage was set for constructing the congested D and E rings.
Scheme 4.

Synthesis of a tetracyclic intermediate containing the ABCG rings.
From the outset, Woodward foresaw construction of the E ring by Dieckmann cyclization, which would necessitate prior epimerization at C3 of precursor 12 (Scheme 5). Exposure of 12 to NaOMe did result in enolization of the ethyl ester, but subsequent elimination of p-toluenesulfinic acid and eventually glycine ethyl ester led to the unexpected formation of carbazole 13. Woodward attributed this reactivity to the leaving-group ability of the arylsulfonyl substituent on nitrogen of the α-amino ester. Removing this leaving group by converting sulfonamide 12 to acetamide 6 allowed, after epimerization of the aminoester, smooth Dieckmann condensation to form the E ring. The unneeded C14 oxidation of pentacyclic product 14 was removed in four steps to provide carboxylic ester 15. Epimerization of the β ester 15 during its hydrolysis with KOH relieved the destabilizing quasi 1,3-diaxial interactions present in 15 to give equatorial acid 5. This intermediate was resolved with quinidine and served as the first relay point of the synthesis.
Scheme 5.
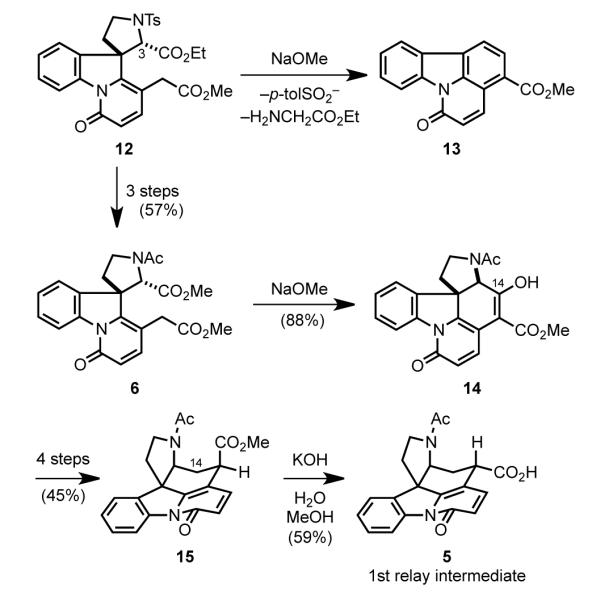
Woodward’s construction of the E ring.
Woodward then approached the installation of the two-carbon bridge that would form the D ring (Scheme 6). Initial attempts to build this ring focused on the possibility of appending a two-carbon dielectrophile between the potentially nucleophilic atoms 4 and 15 of acid 5. However, this strategy was never fruitful and an alternate approach was devised.[47] Carboxylic acid 5 was converted to the methyl ketone by reaction with acetic anhydride, a classical transformation involving intramolecular attack in this case of a C15 enolate on the acetyl carbon of mixed anhydride intermediate 16 with loss of carbon dioxide from the resulting β-lactone (17).[48] Subsequent hydrolysis of the acetamide under acidic conditions provided aminoketone 18. Upon exposure to selenium dioxide, methyl ketone 18 was converted to hexacyclic α-keto lactam 4. The equatorially disposed α-ketoaldehyde 19 produced initially in this oxidation was in equilibrium with its less-favored axial isomer 20. Intramolecular capture of this transient intermediate by the pyrrolidine nitrogen generated a hemiaminal intermediate, which upon further oxidation with selenium dioxide gave lactam 4. Addition of sodium acetylide to the ketone of intermediate 4, followed by partial reduction of the triple bond using Lindlar’s catalyst installed the final carbon atoms of the strychnine skeleton. Although achieved in only low yield (2%), the 5→21 conversion assembled the critical, sterically congested CDE fragment of strychnine.
Scheme 6.

Cyclization to form the D ring and installation of the final two carbon atoms.
Having installed all the required carbon atoms, isostrychnine (2) was accessed from hexacyclic intermediate 21 in three additional steps (Scheme 7). Reduction of the pyridone ring of 21 by lithium aluminum hydride provided dihydro product 23. The regio- and diastereoselectivity of this reduction was justified by initial reaction with the tertiary alcohol to generate aluminum alkoxide 22. This intermediate would allow delivery of hydride selectively to C2 from the more sterically hindered concave face, providing the required stereoisomer 23. Transformation of the tertiary allylic alcohol to a mixture of allylic bromides and subsequent hydrolysis of these intermediates in boiling aqueous sulfuric acid provided isostrychnine (2), with the yield of this sequence being compromised by the formation of the corresponding (Z)-allylic alcohol.
Scheme 7.

Completion of the first strychnine total synthesis.
In this way, Woodward was able to synthesize what was the most complex natural product known in 1954, an event signaling that no natural structure was too complex to be synthesized. Woodward’s success in forging this demanding target is particularly impressive in light of the few selective C–C bond-forming reactions available at the time. Two themes still important in modern organic synthesis strategy are apparent in the Woodward synthesis: use of a masked precursor of a reactive unit—in Woodward’s case an aromatic veratryl antecedent of a dienyl diester—and intramolecular delivery of a reagent. Perhaps most impressive is the fact that nearly forty years transpired before subsequent total syntheses of strychnine were recorded.[44a–c, 49]
3.2. Overman’s Cationic Rearrangement Cascade Approach (1993)
In 1993, nearly 40 years after Woodward’s accomplishment, our group reported the first enantioselective total synthesis of strychnine.[49] Our retrosynthetic analysis focused on the Wieland– Gumlich aldehyde (3) as the most efficient precursor of strychnine (Scheme 8). The Wieland–Gumlich aldehyde, upon disconnection of the B ring, simplifies to tetracyclic keto aniline 24 containing the critical CDE tricyclic unit. The aza-Cope rearrangement/Mannich cyclization cascade reaction developed in our laboratory was seen as delivering 24 from precursor 25, an intermediate containing only two of the final rings of strychnine (A and D).[37] Disconnecting the two heteroatom bonds to the cyclopentane ring of 25 and simplifying the styrene fragment to an aryl ketone yields 26. In the synthetic direction, formation of the piperidine ring and the proper relative configuration of the tertiary allylic alcohol would arise by intramolecular opening of a cyclopentyl epoxide derived from dienone 26, a tactic that had been developed during our earlier synthesis of the simpler Strychnos alkaloid akuammicine.[ 50 ] Intermediate 26 would in turn come from carbonylative Stille coupling of cyclopentene 27 and an aniline precursor. Stannane intermediate 27 was originally envisioned to arise from the 1,4-addition of a fully-fashioned (Z)-butenyl organometallic to an enantioenriched cyclopentenyl electrophile. However, with this coupling never realized in a satisfactory manner, meso-diacetate 28 became the starting material.
Scheme 8.

Retrosynthetic analysis of Overman’s route to strychnine.
The Overman synthesis of (–)-strychnine began with desymmetrization of cis-1,4-diacetoxycyclopent-2-ene 28 using electric eel acetylcholinersterase, following a procedure documented in Organic Syntheses that delivered alcohol 29 in high yield and 99% ee (Scheme 9).[51] Stereospecific palladium-catalyzed coupling of this enantioenriched intermediate with carbon nucleophiles had been described at the time.[52] To arrive at the natural enantiomer of strychnine, the carbonate derivative 30 was prepared and coupled with β-ketoester 31. As expected, this palladium-catalyzed coupling took place with complete site and stereoselectivity to give 32, a 1:1 mixture of ester epimers, with complete retention of enantiomeric purity.[53] In order to introduce an E-double bond, the keto group of β-ketoester 32 needed to be reduced in a stereoselective manner. Although conventional reduction of these β-ketoester epimers to give syn-β-hydroxyester products could not be accomplished,[54,55] addition of sodium cyanoborohydride to a solution of 32 premixed with titanium tetrachloride afforded the anti-β-hydroxyester product of each ester epimer in high stereoselectivity. This unconventional outcome was proposed to result from chelation of titanium to both the ester and the tert-butyl ether to form intermediate 33, with hydride addition from the less hindered axial face then providing the observed products. Stereospecific syn-elimination of intermediates 34 yielded exclusively the E-trisubstituted double bond.[56] Five standard transformations then converted diester 35 to the vinyl stannane 27.
Scheme 9.

Synthesis of vinyl stannane 27.
The aza-Cope–Mannich cyclization precursor was synthesized in eight steps from vinyl stannane 27 (Scheme 10). Carbonylative Stille coupling of cyclopentyl stannane 27 with triazone-protected oiodoaniline 36 provided ketone 26 in high yield.[57] Regioselective epoxidation of enone 26 with t-BuO2H and base took place with high stereoselectivity from the face opposite the butenyl side chain to give epoxide 37. Wittig methylenation of the ketone and standard conversion of the silyl ether to a trifluoroacetamide then delivered epoxy amide 38. Deprotonation of the trifluoroacetamide induced intramolecular epoxide opening to form the piperidine D ring and provide intermediate 25 after cleavage of the trifluoroacetyl group with base.
Scheme 10.

Synthesis of aza-Cope rearrangement/Mannich cyclization precursor.
Assembly of the pivotal CDE tricyclic core of strychnine was accomplished in a single step and nearly quantitative yield by exposure of amino alcohol 25 to paraformaldehyde at 80 °C (Scheme 11). This aza-Cope rearrangement/Mannich cyclization cascade proceeds by initial generation of iminium 39, which undergoes [3,3]-sigmatropic rearrangement to construct the C5–C6 bond and generate iminium ion–enol 40. Formation of the C3–C7 bond by a transannular Mannich cyclization then terminates the cascade process. This process generated two new carbon-carbon bonds as well as a carbon-nitrogen bond while constructing the congested CDE-tricyclic core of strychnine.
Scheme 11.
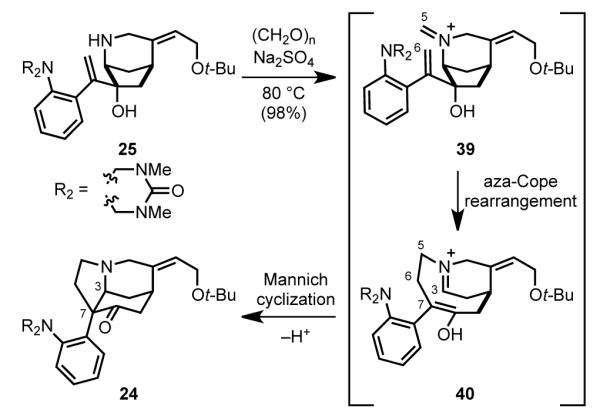
Aza-Cope–Mannich reaction to form the C and E rings.
Tetracyclic intermediate 24 was advanced to the Wieland– Gumlich aldehyde in five steps (Scheme 12). Carbomethoxylation of the enolate derived from 24 and removal of the triazone protecting group with acid yielded hydroxyakuammicine (41).[58] Whereas attempted reduction of the enamine of 41 under neutral or weakly acidic conditions resulted in decomposition, zinc in sulfuric acid successfully saturated the 2,16-double bond. Equilibration of the initially produced axial ester then delivered 42 in good yield (68%). The configurational outcome of these steps had some precedent from related transformations of akuammicine.[59] Partial reduction of the ester of 42 with diisobutylaluminum hydride at − 90 °C afforded the Wieland-Gumlich aldehyde (3), which was transformed to (–)-strychnine in high yield.
Scheme 12.

Synthesis of the Wieland–Gumlich aldehyde.
The Overman 1993 synthesis of (–)-strychnine was both the first enantioselective and the first non-relay total synthesis of strychnine. Although it was of similar length as the Woodward synthesis, it was several orders of magnitude more efficient. As we suggested in our full account,[49b] this substantial increase in efficiency resulted in large part from the progress in synthetic organic chemistry registered in the intervening 40 years, as no more than half of the reactions utilized in the Overman synthesis would have been available to Woodward in 1954. In particular, this synthesis highlighted the utility of palladium-catalyzed reactions (η3- allylpalladium alkylation and carbonylative cross-coupling) and the aza-Cope–Mannich reaction cascade for constructing critical C–C bonds. The use of enzymatic desymmetrization and exploitation of symmetry were illustrated also, the latter underscored by a minor modification of the sequence—employing hydroxy ester 29 in the early Tsuji–Trost coupling—that allowed ent-strychnine to be obtained for the first time.[60,61]
3.3. Rawal’s Diels–Alder and Heck Cyclization Approach (1994)
Rawal envisioned a streamlined route for the synthesis of strychnine by way of isostrychnine (Scheme 13).[62] The plan was to employ an intramolecular Heck reaction to construct the piperidine D ring from pyrrolo[2,3-d]carbazole vinyl iodide 43,[63] itself arising from intramolecular Diels-Alder reaction of enecarbamate 44. The pyrroline ring of 44 would stem from ring-expansion of cyclopropyl imine 45. This sequence was based on Rawal’s earlier synthesis of structurally less-elaborate Strychnos alkaloids.[64]
Scheme 13.
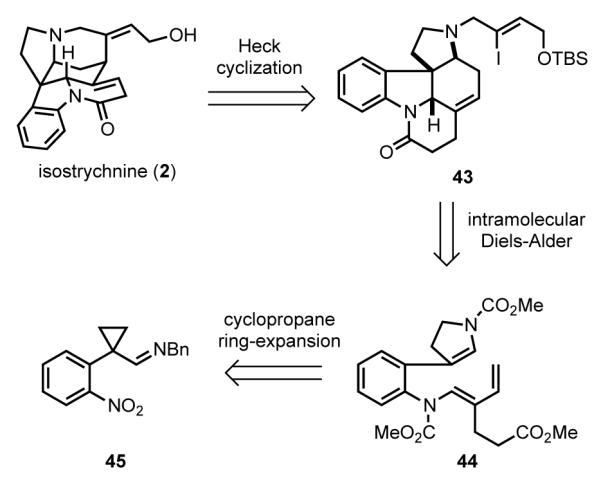
Rawal’s retrosynthetic analysis.
The Diels-Alder substrate 44 was assembled in seven steps from commercially available o-nitrobenzylnitrile 46 (Scheme 14). Initially cyclopropyl imine 45 was synthesized in three standard steps from nitrile 46, 1,2-dibromoethane, and benzylamine. Nucleophilic opening of the cyclopropane with TMSI generated intermediate 47, which upon intramolecular alkylation provided dihydropyrrole 48.[65] Reduction of the nitro group, followed by condensation of the product with methyl 4-formylhex-4-enoate and N-alkoxycarbonylation delivered diene carbamate 44.
Scheme 14.
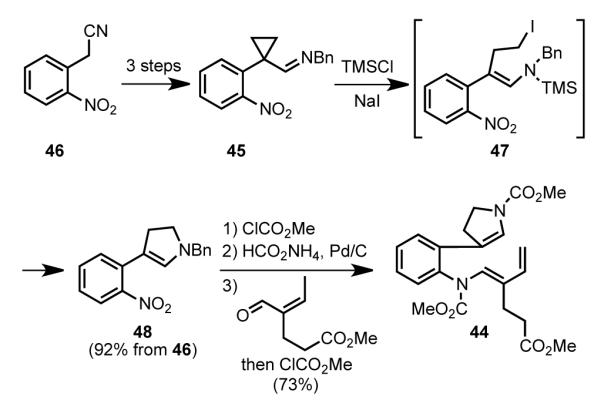
Synthesis of the Diels–Alder precursor.
In five steps, Diels-Alder substrate 44 was transformed into isostrychnine (Scheme 15). Heating 44 at 185 °C induced intramolecular cycloaddition by way of the less sterically hindered exo transition state to provide pyrrolocarbazole 49 in nearly quantitative yield. Dispatch of the carbamate groups of 49 upon reaction with TMSI was accompanied by formation of the G ring to provide lactam 50, which upon alkylation with allyl bromide 51 gave rise to the Heck cyclization substrate 43. In the second pivotal step of the sequence, cyclization of 43 under Jeffery–Heck reaction conditions formed the final piperidine D ring.[66] Deprotection of the silyl ether then delivered isostrychnine (2).
Scheme 15.

Intramolecular Diels–Alder reaction and synthesis of isostrychnine (2).
Rawal’s synthesis of strychnine was remarkable for both its low step count and high efficiency. Isostrychnine (2) was formed in 34% overall yield from 46, corresponding to ~10% overall yield of racemic strychnine over 13 steps. This is the most efficient construction of racemic strychnine reported to date. Like the Overman synthesis, the Rawal synthesis illustrated the utility of transition metal-catalyzed C–C bond-forming transformations, in this case the intramolecular Heck reaction to assemble the challenging piperidine D ring. The high strategic value of this tactic is seen by its use in five subsequent strychnine syntheses. A crucial feature contributing to the efficiency of the Rawal synthesis was the incisive design whereby the intramolecular Diels–Alder reaction produced the six-membered carbocyclic E ring with the double bond in exactly the position required for the subsequent intramolecular Heck closure of the D ring.
3.4. Kuehne’s Cationic Rearrangement Cascade Approach (1998)
Kuehne first described the total synthesis of racemic strychnine in 1993,[42] and five years later the synthesis of (–)-strychnine by a related strategy.[67] Like Rawal, a pyrrolo[2,3-d]carbazole was a central intermediate, which in Kuehne’s synthesis was seen as the product of a cascade cationic rearrangement (Scheme 16). Targeting the Wieland–Gumlich aldehyde in the 1998 synthesis, the retrosynthetic analysis envisioned late stage formation of the F ring and C19–C20 double bond from ketoester precursor 52. Conventional disconnection of the D ring revealed hydroxyketone 53, which would arise from precursor 54. The C and E rings of pyrrolocarbazole 54 were disconnected by a cyclization/rearrangement cascade of iminium 55, an approach that had been developed[68] as an alternative to the biomimetic secodine-like cyclizations Kuehne had previously pioneered to prepare Aspidosperma[69] and some Strychnos alkaloids.[70] Iminium ion intermediate 55 would be generated from an appropriate aldehyde and a tryptophan derivative (56), which would ultimately derive from (S)-tryptophan methyl ester.
Scheme 16.

Kuehne’s retrosynthetic analysis.
Starting with (S)-tryptophan methyl ester, tryptophan derivative 56 was synthesized in five conventional steps (Scheme 17). Acid-promoted condensation of amino ester 56 and 2,4-hexadienal in refluxing benzene proceeded with high diastereoselectivity to generate pyrrolo[2,3-d]carbazole 54 in 84% yield. This impressive transformation is believed to take place by intramolecular Mannich reaction of the initially generated iminium ion via conformer 55-Re in which destabilizing steric interactions between the methyl ester and the indole are minimized. After tautomerization, 1,5-diene 57 undergoes Cope rearrangement by a favored chair geometry that places the propene and ester substituents in pseudo-equatorial positions (58). Intramolecular Mannich cyclization of iminium ion 59 then forms the C3–C7 bond, completing the construction of the C and D rings of strychnine with complete diastereoselectivity.[71]
Scheme 17.

Diastereoselective Mannich cyclization/Cope rearrangement/Mannich cyclization cascade (R = CH=CHCH3).
The final three rings of strychnine were constructed from pyrrolocarbazole 54 as outlined in Scheme 18. Removal of the now unneeded methyl ester substituent—accomplished by way of the corresponding nitrile[72]—and oxidative elaboration of the propenyl side chain delivered hydroxyketone intermediate 53 in seven steps. After conversion of the primary alcohol to a tosylate, hydrogenolytic removal of the benzyl-protecting group took place with concomitant intramolecular alkylation to generate piperidinone 52. Horner– Wadsworth–Emmons olefination of this intermediate provided enoate 60 as nearly exclusively the E alkene stereoisomer. In Kuehne’s earlier synthesis of racemic strychnine,[42] this olefination was carried out on an intermediate having the piperidone G ring already in place and occurred in a nonstereoselective fashion. Kuehne notes that stereoselection in the formation of 60 was critically dependent upon the experimental conditions, although no rational for the high selectivity obtained using the potassium salt in THF was advanced. A four-step sequence involving stepwise reduction of the ester and the vinylogous carbamate then yielded the Wieland–Gumlich aldehyde (3).
Scheme 18.

Closure of the D ring and completion of the synthesis.
The power of iminium ion-initiated cascade reaction sequences is nicely illustrated in the Kuehne synthesis, as the C and E rings of strychnine were fashioned in short order from (S)-tryptophan methyl ester. In this sequence the ultimately unneeded methyl ester played an important role in directing the enantioselective evolution of pyrrolo[2,3-d]carbazole intermediate 54. Although several steps were required to elaborate this intermediate to (–)-strychnine, Kuehne’s contribution remains one of the shorter enantioselective syntheses of strychnine achieved to date.
3.5. Vollhardt’s [2+2+2] Cycloaddition Approach (2000)
Vollhardt envisaged a cobalt-mediated [2+2+2] cycloaddition to quickly assemble the ABEG tetracyclic core of (±)-strychnine (Scheme 19).[73] In this analysis, the D ring of isostrychnine (2) was seen arising from dienone vinyl iodide 61 by an intramolecular Heck or radical cyclization reaction. Disconnection of the pyrrolidine ring of 61 led to cobalt-bound triene 62, which in the central step of the synthesis was seen resulting from [2+2+2]-cycloaddition of acetylene and indolyl enyne 63.
Scheme 19.
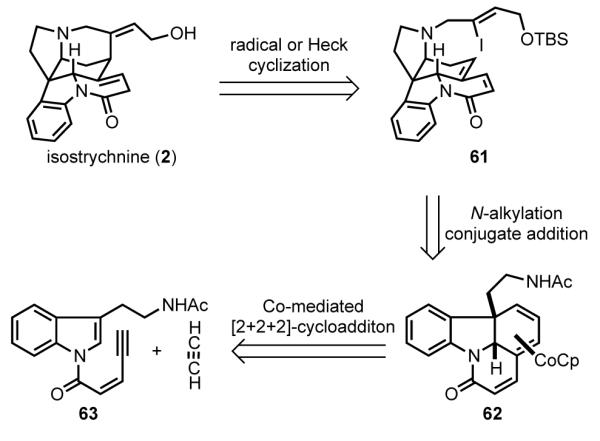
Vollhardt’s retrosynthetic analysis.
The tetracyclic core of (±)-strychnine was rapidly assembled from Nb-acetyltryptamine (64) (Scheme 20). Amide formation between indole 64 and acid chloride 65 under Schotten-Baumann conditions afforded indolyl amide 63. Exposure of this intermediate to CpCo(C2H4)2 under an acetylene atmosphere generated a single stereoisomer of cobalt-bound triene 62 in 46% yield.[ 74 ] This transformation presumably occurs by initial formation of cyclometalated intermediate 66, which undergoes intramolecular [4+2]-cycloaddition and bond reorganization to deliver 62.[ 75 ] Related [2+2+2]-cycloaddition reactions were performed on a number of alkynyl indole substrates.[73,74a] Oxidative removal of the cobalt after cleavage of the acetyl group of 62 resulted in conjugate addition of the β-aminoethyl side chain to form the strychnine C ring (67). Alkylation of the resulting secondary amine with allyl bromide 51 provided intermediate 61.
Scheme 20.
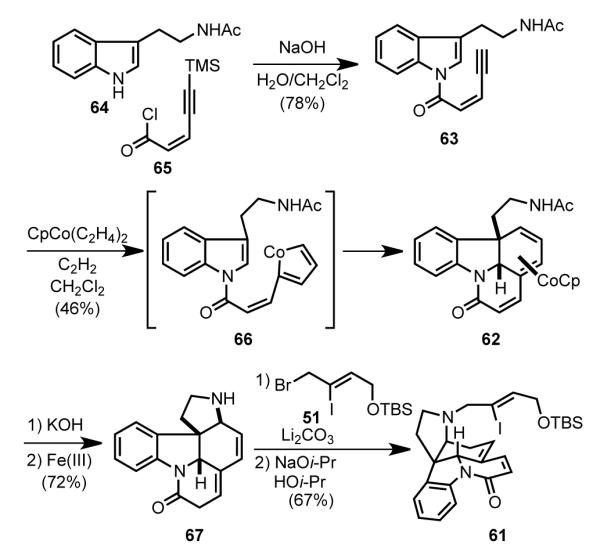
[2+2+2] cycloaddition to form an ABEG tetracyclic intermediate (62).
All that remained to access isostrychnine from intermediate 61 was closure of the piperidine D ring (Scheme 21). Reductive radical-chain cyclization of 61 proceeded in good yield to give a 1:1 mixture of TBS-protected isostrychnine 69 and double bond isomer 68, with the loss of configuration of the C19–C20 double bond arising from rapid isomerization of the vinyl radical intermediate.[70c,76] Alternatively, Heck cyclization of 61 provided pyridone 70, which could be reduced to isostrychnine derivative 69 using lithium aluminum hydride, but the overall yield of the two-step stereoselective process was lower than the yield of pure 69 obtained from radical cyclization. It is noteworthy that the LiAlH4 reduction yielded the correct configuration at C2 without the presence of a directing group, remarkable selectivity the authors attributed to hydride attack from the less-hindered Si face of the pyridone.[73] Acidic cleavage of the TBS group of 69 provided isostrychnine (2) in high yield.[62]
Scheme 21.
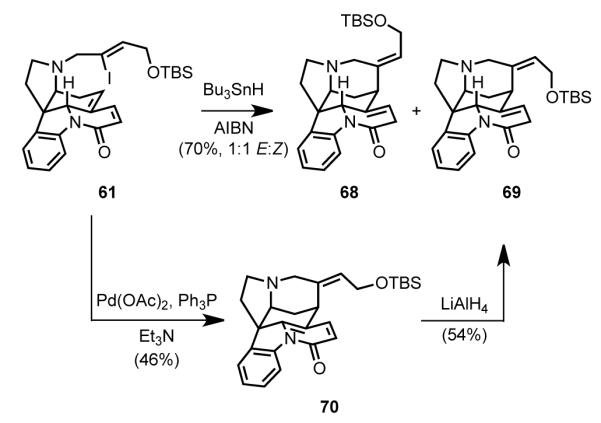
Cyclization of the vinyl iodide side chain to form the D ring.
Vollhardt’s concise total synthesis of (±)-strychnine illustrates the participation of the 2,3-double bond of indole in cobalt-mediated [2+2+2]-cycloadditions.[74] This powerful transformation allows a high degree of complexity to be introduced into an indole starting material in one synthetic step. The success of this step in Vollhardt’s synthesis of (±)-isostrychnine depended critically upon the nature of the oxidant employed in the demetallation step, as Vollhardt had reported earlier that oxidation of closely related complexes can lead to non-demetallative formation of propellane products.[ 77] This synthesis also evaluated the potential to generate the C15–C20 double bond using a radical cyclization; however, as likely expected, generation of the C20 vinyl radical resulted in loss of alkene configuration.
3.6. Martin’s Biomimetic Approach (2001)
In 2001 Martin reported a biogenetically inspired synthesis of strychnine.[78] Hydroxyakuammicine (41) was selected as a late-stage intermediate, because its 3-step elaboration to strychnine was known from the Overman synthesis (Scheme 22).[49] Martin planned to mimic the biogenesis of Strychnos alkaloids (Scheme 1) by forming hydroxyakuammicine from deformylgeissoschizine derivative 71 by an oxidative rearrangement cascade. Intermediate 71 was seen deriving from an intramolecular hetero Diels–Alder reaction of aldehyde 72, which in turn would arise by tandem acylation/vinylogous Mannich reaction of dihydro azacarbazole 73.
Scheme 22.

Martin’s retrosynthetic analysis.
The tetracyclic deformylgeissoschizine analogue 71 was assembled by a general strategy developed earlier by Martin for the synthesis of heteroyohimboid and corynantheoid alkaloids (Scheme 23).[79] Azacarbazole 73, available in two steps from tryptamine, was allowed to react with acryloyl chloride 74 to generate N-acyliminium intermediate 75, which underwent subsequent vinylogous Mannich addition with 1-trimethylsilyloxy-1,3-butadiene. This one-pot sequence rapidly generated intermediate 72, which contains all the carbon atoms of hydroxyakuammicine (41). Intramolecular hetero Diels–Alder cyclization of enal 72 cleanly gave intermediate 76 having a heteroyohimboid ring system, which was elaborated in six additional steps to oxidative rearrangement precursor 71.
Scheme 23.

Synthesis of deformylgeissoschizine-type precursor 71.
Initial attempts to realize the pivotal biomimetic oxidative rearrangement were carried out with several structurally related precursors.[ 80] For example, oxidation of lactam 77 with tertbutylhypochlorite, followed by reaction with AgNO3 and perchloric acid in aqueous methanol promoted the desired carbon-carbon bond migration of chloroindolenine intermediate 79 (Scheme 24).[78b] However, spiroxindole product 80, possessing the opposite configuration at C7 to that found in Strychnos alkaloids, was formed. A related sequence starting with deformylgeissoschizine (81) provided two oxindole products 84 and 85, both of which possessed the unwanted configuration at C3. In this case retro-Mannich fragmentation of the initially formed product(s) (82→83) leads to equilibration at both C3 and C7 to generate 84 and 85. Of the four potential C3/C7 stereoisomers, these two are favored thermodynamically. This preference arises from the piperidine ring preferentially adopting a conformation having the acetic ester side chain axial in order to avoid unfavorable A1,3-interactions with the (E)-ethylidine group, thus favoring an axial orientation of the angular C3-hydrogen.[81]
Scheme 24.

Initial attempts at oxidative rearrangement.
To access hydroxyakuammicine (41), the biomimetic strategy was modified by elaborating the chloroindolenine intermediates under basic conditions (Scheme 25). Chlorination of indole 71 by sequential reaction with SnCl4 and tert-butylhypochlorite is presumed to provide a mixture of epimeric chloroindolenines 86 and 87. Treatment of this mixture with LHMDS generates the ester enolate, which in the β-epimer series (86) cyclizes to form 88. The anti relationship of the chlorine and C2–C3 σ-bond in 88 results in facile 1,2-migration to give 89, which upon treatment with acidic methanol afforded hydroxyakuammicine (41) in 22% yield. It is hypothesized that the related adduct derived from the α-chloroindolenine 87 does not undergo 1,2-rearrangement because of the syn relationship of the chlorine and C2–C3 σ-bond. Early investigation of this sequence with the analogue of 71 lacking the OTBS group had shown that pretreatment with SnCl4 favored formation of the β-chloroindolenine intermediate, which resulted in akuammicine being formed in 52% yield.
Scheme 25.

Biomimetic rearrangement of deformylgeissoschizine derivative 71 to hydroxyakuammicine (41).
The Martin formal total synthesis of (±)-strychnine and total synthesis of (±)-akuammicine represent the first successful biomimetic constructions of Strychnos alkaloids. These syntheses demonstrate both the advantages and challenges of a biomimetic synthesis strategy. Although biomimetic rearrangement cascades can develop a great deal of complexity in a single step, laboratory methods for stereoselective introduction of functionality to initiate such cascades can lack the selectivity and efficiency of the biological counterpart. In this case, the efficiency of the pivotal cascade rearrangement was likely undermined by low stereoselectivity of the oxidation of precursor 71 to generate chloroindolenine intermediate 86. It merits note that intramolecular capture of a chloroindolenine intermediate by a malonate nucleophile to initiate skeletal rearrangement was used by Stork to access a pyrrolo[2,3-d]carbazole intermediate in his unpublished synthesis of (±)-strychnine.[44c]
3.7. Fukuyama’s Transannular Pictet–Spengler Cyclization Approach (2004)
Intramolecular additions to iminium ion intermediates played a central role in four of the syntheses that we have considered thus far. Nonetheless, Fukuyama foresaw orchestrating this tactic in a unique manner.[82] In the retrosynthesis, the CDE fragment of the Wieland– Gumlich aldehyde was simultaneously disconnected at the C3–C7, and C3–N4 bonds to yield tricyclic amino aldehyde 91 (Scheme 26). Further simplification by doubly disconnecting the 9-membered azacyclic ring of 91 to give diol 92 anticipates forming this challenging medium ring by dual Mitsunobu coupling with onitrophenyl sulfonamide (NsNH2), an effective tactic introduced earlier by Fukuyama.[ 83 ] Further simplification of indole intermediate 92 leads to indole malonate 93 and allylic epoxide 94, which were anticipated to be joined stereoselectively by palladium-catalyzed Tsuji–Trost coupling.[84]
Scheme 26.

Fukuyama’s retrosynthetic analysis.
The Fukuyama synthesis begins by preparation of the partners for the palladium-catalyzed coupling reaction from inexpensive starting materials (Scheme 27). In three conventional steps,[85] benzoic acid was converted to bromohydrin 95, which was resolved using lipase AYS and vinyl acetate to give enantiopure acetate 96.[86] In three additional steps, this intermediate was elaborated to enantioenriched allylic epoxide 94. The indole malonate nucleophile 93 was prepared in five steps from quinoline. Addition of water to the quinolinium salt generated by thioacylation of quinoline with thiophosgene (97) resulted in opening of the pyridine ring to provide (Z)-enal isothiocyanate 98, which was directly reduced to (Z)-allylic alcohol 99 with sodium borohydride. The allylic alcohol was protected as the silyl ether, and dimethylmalonate was added to the isothiocyanate to provide thioamide 100. In an indole construction developed earlier by Fukuyama, [87] the α-amino radical, generated from the reaction of the thioamide with borane and tributyltin hydride, cyclized onto the pendant double bond to generate the indole malonate 93.
Scheme 27.

Synthesis of coupling partners.
The union of fragments 93 and 94 via the π-allylpalladium complex generated from 49 was accomplished with a palladium(0)trifurylphosphine catalyst (Scheme 28). As expected, this alkylation took place by double-inversion to produce cyclohexenyl indole malonate 101 in good yield.[84b] Protecting group manipulations and decarboxylation then provided macrocyclization substrate 102 in four additional steps.
Scheme 28.
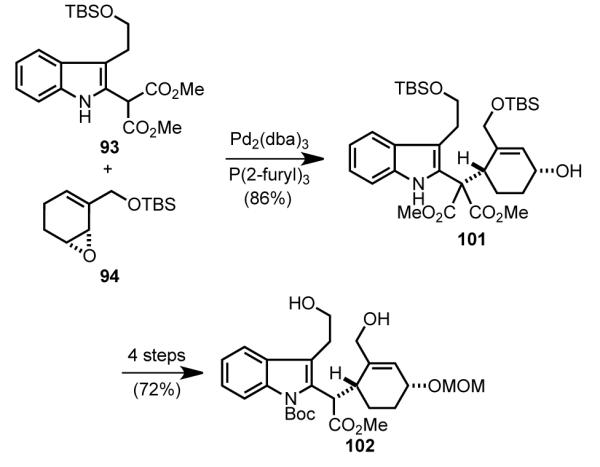
Tsuji–Trost coupling of 93 and 94 and elaboration to diol 102.
Diol 102, containing all of the carbon atoms of the Wieland– Gumlich aldehyde was advanced to tricyclic intermediate 91 in seven steps (Scheme 29). Mitsunobu reaction of diol 102 with onitrobenzene sulfonamide gave nine-membered ring intermediate 103 in 95% yield.[83,88] Epimerization at C16, followed by cleavage of the MOM ether and oxidation of the alcohol yielded enone 104. Rubottom oxidation of the silyl enol ether derived from ketone 104 provided α-hydroxyketone 105.[89] Subsequent reaction with lead tetraacetate in methanol resulted in cleavage of the cyclohexenone ring to give tricyclic aldehyde 91.
Scheme 29.

Double-Mitsunobu reaction and aldehyde synthesis.
Intermediate 91 was properly functionalized to investigate the proposed transannular Mannich cyclization (Scheme 30). Simultaneous removal of the nosyl and Boc protecting groups upon reaction of 91 with thiophenol and TFA revealed the secondary amine, which condensed with the pendant aldehyde to generate iminium ion 90. Intramolecular nucleophilic attack by the indole fashioned the C and E rings as well as the quaternary carbon stereocenter of strychnine, delivering 60 in 84% yield from precursor 105. This transannular Mannich cyclization had precedent from the early Magnus synthesis of strychnine in which the iminium ion intermediate was generated by oxidation of a piperidine ring.[44a,b] Reduction of the vinylogous carbamate and ester functional groups of 60 provided the Wieland-Gumlich aldehyde (3) in four additional steps.[67]
Scheme 30.

Transannular Mannich cyclization.
Fukuyama’s use of Mitsunobu macrocyclization allowed for a strategically unique approach for the synthesis of strychnine. The congested CDE core ring system was constructed in a single step from a macrocyclic precursor (91). The drawback of this approach was the rather lengthy sequence required to assemble this macrocyclic precursor. The utility of enzymatic resolution and palladium-catalyzed C–C bond formation were further illustrated in this route to (–)-strychnine, as was the Fukuyama indole synthesis.
3.8. Reissig’s Ketyl Radical Cyclization Approach (2010)
By intercepting Rawal’s pentacyclic intermediate 50,[62] Reissig completed a concise formal total synthesis of (±)-strychnine. In their strategy, the pyrrolidine ring of 50 was disconnected to arrive at tetracyclic precursor 106 (Scheme 31). Retrosynthetic cleavage of the C2–C16 and C3–C7 bonds of this intermediate led to N-acylindole 107, which was seen evolving to intermediate 106 in one step by a samarium-mediated ketyl-radical cyclization cascade developed earlier in the Reissig laboratories.[90,91]
Scheme 31.
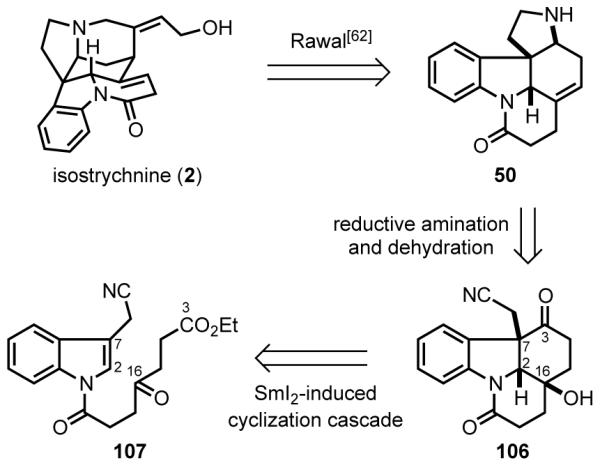
Reissig’s retrosynthetic analysis.
Tetracyclic intermediate 106 was assembled in a three-step sequence from commercially available diethyl 4-oxopimelate 108 (Scheme 32). The synthesis began with lipase-catalyzed selective hydrolysis of diester 108 to provide the monoester of 4-oxoheptanedioic acid,[ 92 ] which was coupled with 3-indolylacetonitrile to provide amide 107. Reaction of this intermediate at room temperature with excess samarium iodide and HMPA initiated a cascade sequence that gave tetracyclic intermediate 106 in 70–75% yield.[93] In this effective sequence, 6-exo-trig cyclization of ketyl radical 109 generates benzyl radical 110, which is further reduced to alkylsamarium intermediate 111. Intramolecular acylation of this organosamarium species then delivers tetracyclic product 106.
Scheme 32.
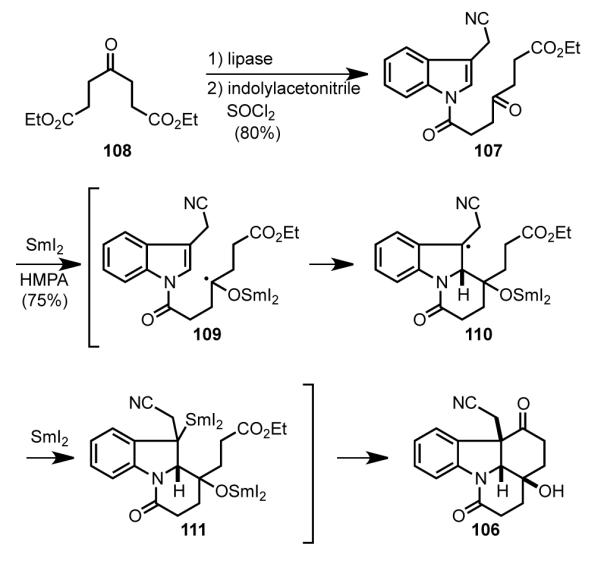
SmI2-induced cyclization cascade to form tetracyclic intermediate 106.
In three to five additional steps, tetracyclic intermediate 106 was transformed to Rawal’s intermediate 43 (Scheme 33). In a crucial step in this sequence, the nitrile of 106 was reduced to the primary amine and intramolecular reductive amination was realized upon reaction of 106 for an extended period with Raney-nickel under a hydrogen atmosphere. The high stereoselectivity observed in the reduction of the imine intermediate, which had some precedent,[94] was suggested to result from shielding of the β-face by the angular hydroxyl group. The final step required to arrive at 115 was regioselective dehydration of the tertiary alcohol. Attempted dehydration of the methyl carbamate derivative (or methyl sulfonamide congener) with the Burgess reagent produced a 2:1:1 mixture of the three possible alkene products. Selectivity was enhanced when this dehydration was accomplished with Martin’s sulfurane, giving isomerically pure 115 in 73% yield over two steps. In two additional steps, this product was converted to Heck precursor 43, completing a formal total synthesis of (±)-strychnine. Alternatively, the product of nitrogen alkylation with vinyl iodide 113 could be similarly dehydrated to directly afford 43, albeit in lower yield as a result of reduced selectivity in the sulfurane-mediated elimination.[95]
Scheme 33.

Transformation to Rawal’s intermediate.
In this approach to strychnine, construction of intermediate 106 containing the ABEG rings of strychnine in only three steps from commercial starting materials showcased the power of the reductive cascade processes developed in the Reissig laboratory. The opening step of the synthesis exemplified another use of enzyme catalysis, in Reissig’s case to selectively desymmetrize a symmetrical achiral precursor. Reissig’s elaboration of tetracyclic intermediate 106 in five additional steps to intercept Rawal’s intermediates 43 and 50 completed a concise formal total synthesis of (±)-strychnine.
3.9. Vanderwal’s Zincke Aldehyde Approach (2011)
The Vanderwal approach to strychnine envisaged a succinct series of disconnections.[ 96 ] Formation of the D ring by intramolecular addition of a butenyl organometallic to an electron-deficient double bond would form the C15–C20 bond (Scheme 34). In contrast to the earlier uses of this bond construction, in this plan the conjugate addition would lead directly to the more efficient precursor of strychnine, the Wieland–Gumlich aldehyde (3). Pyrrolo[2,3-d]carbazole intermediate 116 was seen arising by intramolecular Diels–Alder reaction of indole dienal 117. In a key simplification, this tryptamine dienal (117) would arise from tryptamine and pyridine via the venerable Zincke ring opening of pyridinium salts.
Scheme 34.
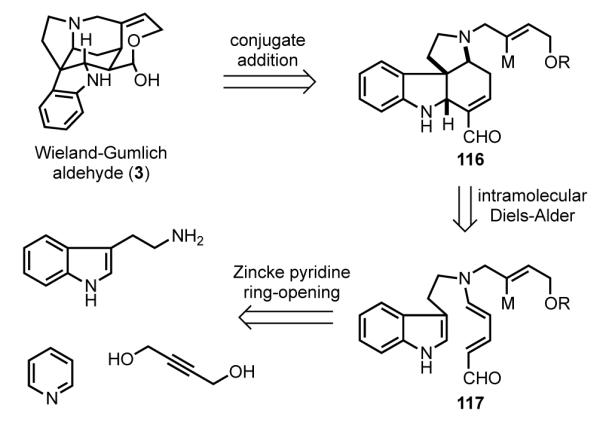
Vanderwal’s retrosynthetic analysis.
The Vanderwal synthesis began with the construction of pyrrolocarbazole 122 in three steps from commercial reagents (Scheme 35). Reaction of Nb-allyltryptamine, available in one step from tryptophyl bromide, with 2,4-dinitrobenzenepyridinium chloride initiates the Zincke pyridine ring-opening to generate imine 120 by electrocyclic ring opening of dihydropyridine aminal 119. Hydrolytic workup then yields the tryptamine-derived Zincke aldehyde 121.[97,98] Because of their stabilized push-pull nature, Zincke aldehydes were known to be poor 4-π components in intermolecular Diels–Alder reactions.[99] Thus, earlier attempts to realize intramolecular cycloaddition of intermediates similar to 121 under thermal or Lewis acid- or protic acid-catalyzed conditions met with failure.[100] The enabling finding of Vanderwal’s investigations was the discovery that the desired bond construction could be achieved under basic conditions by what is likely a step-wise anionic bicyclization. Thus, heating intermediate 121 at 80 °C in the presence of a slight excess of potassium tert-butoxide gave rise to pyrrolocarbazole 122 in 64% yield. An intramolecular Diels–Alder reaction of an indole, in this case with a tethered furan, played a central role in Padwa’s synthesis of (±)-strychnine.[44h–i] The strategic advantage of the Vanderwal approach is that cycloaddition product 122 is generated appropriately functionalized for rapid conversion to the natural product.
Scheme 35.
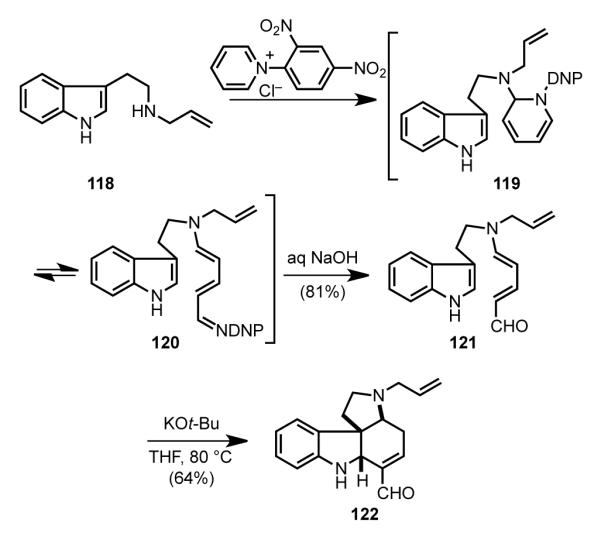
Synthesis of a pyrrolo[2,3-d]carbazole intermediate from a Zincke aldehyde.
In three additional steps, tetracyclic intermediate 122 was elaborated to racemic Wieland–Gumlich aldehyde (3) (Scheme 36). Removal of the allyl-protecting group from 122 proved to be challenging, as reductive, oxidative, and acidic conditions were not compatible with the reactive enal functionality. Ultimately it was found that palladium-catalyzed alkylative deallylation was successful when methyl Meldrum’s acid was employed to trap the allyl fragment. This bulky nucleophile undergoes 1,4-addition or Knoevenagel condensation with the enal only slowly, thus minimizing these side reactions that plagued more conventional deallylation procedures. Exploiting ruthenium-catalyzed transhydrosilylation,[101] the (Z)-2-butenyl fragment 125 was prepared succinctly in three steps from 2-butyne-1,4-diol by way of oxasilacycle 123. Although intermediate 124 was relatively unstable—it contains both nucleophilic and electrophilic functionality—the crude amino aldehyde generated by deallylation could be directly alkylated with allyl bromide 125 to yield the stable tertiary amine 126. After extensive experimentation, it was found that generation of the alkoxide of 126 in the presence of cuprous bromide initiated a Brook rearrangement, with the resulting vinyl copper species (116) undergoing intramolecular conjugate addition to the enal. Unfortunately, the yield of this penultimate step in the Vanderwal total synthesis of (±)-strychnine was low (5–10%) under a variety of surveyed reaction conditions.
Scheme 36.

Alkylation and conjugate addition to give the Wieland– Gumlich aldehyde.
The Vanderwal total synthesis of (±)-strychnine is the most direct reported to date, with the longest linear sequence being only six steps. Even though the final conjugate addition step was low yielding, the overall efficiency of this construction of racemic strychnine was a respectable 2–3%. This synthesis beautifully illustrates the power of incisive synthetic strategy. Its conciseness is a result of each key step delivering a product with exactly the functionality required for the next transformation, with no adjustment of oxidation state being required throughout the sequence.
3.10. MacMillan’s Organocatalytic Cascade Approach (2011)
The Macmillan group envisioned a short enantioselective synthesis of (–)-strychnine as part of an indole alkaloid total synthesis endeavor in which a common enantiomerically enriched intermediate would serve as a precursor of several structurally distinct indole alkaloids.[ 102 ] In the context of (–)-strychnine, MacMillan’s retrosynthetic analysis disconnects the C15–C20 bond to give pyrrolo[2,3-d]carbazole precursor 127, which upon intramolecular Heck reaction would yield directly a protected form of the Wieland–Gumlich aldehyde (Scheme 37). Intermediate 127 was further simplified to pyrrolo[2,3-d]carbazole dienal 128, which in turn was seen arising from 2-alkenyltryptamine 129 by an enantioselective organocatalytic Diels–Alder/amination cascade reaction.
Scheme 37.
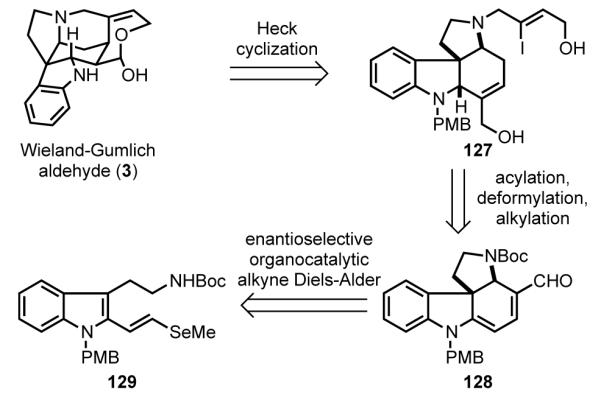
MacMillan’s retrosynthetic analysis.
In the first stage of MacMillan’s synthesis of (–)-strychnine, an enantioselective organocatalytic cascade reaction was utilized to construct pyrrolo[2,3-d]carbazole intermediate 128 rapidly (Scheme 38). Starting with readily available tetrahydro-β-carboline (130), indole vinylselenide 129 was assembled in three conventional steps. Reaction of 2-alkenylindole 129 with propynal in the presence of enantiopure imidazolidinone catalyst 131 gave rise to pyrrolocarbazole 128 in 82% yield and 97% ee. This impressive transformation takes place by initial catalytic enantioselective formation of Diels–Alder adduct 132, which rapidly loses methylselenide to give diiminium intermediate 133. After intramolecular 1,4-addition of the carbamate nitrogen, hydrolysis of iminium ion 134 yields product 128 and regenerates the imidazolidinone catalyst. The regioselectivity of intramolecular addition of the nitrogen was controlled by the leaving group ability of the selenide. In earlier studies, MacMillan had observed that under similar conditions, the methylthio analog of 129 underwent C–N bond formation at C2 to give a pyrrolidinoindoline product that retained the thiomethyl substituent.[103,104]
Scheme 38.

Enantioselective organocatalytic cascade to form a pyrrolo[2,3-d]carbazole intermediate.
In seven additional steps, pyrrolocarbazole dienal 128 was advanced to the Wieland–Gumlich aldehyde (Scheme 39). Refunctionalization of the cyclohexadiene ring was accomplished by deformylation of 128 in the presence of 1 equiv of Wilkinson’s catalyst, and subsequent methoxycarbonylation of the dienamine product by sequential reaction with phosgene and methanol to form 135.[105] The uncommon reactivity of the dienamine intermediate with phosgene at the internal, rather than terminal, position likely reflects steric shielding of the terminal vinylic carbon by the bulky NBoc substituent. Three additional routine steps gave pyrrolo[2,3-d]carbazole intermediate 127. Intramolecular Heck reaction of 127 under Jeffery conditions[66] was found to be optimal for constructing the C15–C20 bond with concomitant formation of the F ring hemiacetal (138). The authors note that the presence of the pmethoxybenzyl substituent was critical in facilitating β-hydride elimination from carbopalladate 136 away from the angular methine hydrogen to form the critical enol precursor 137 of product 138. Removal of the PMB group with trifluoroacetic acid and thiophenol provided the Wieland-Gumlich aldehyde (3).
Scheme 39.

Heck cyclization to form the Wieland–Gumlich aldehyde.
In the MacMillan synthesis, enantioselective cascade organocatalysis was exploited to construct the pyrrolo[2,3-d]carbazole tetracyclic core of (–)-strychnine quickly. The significance of recent advances of enantioselective organocatalysis,[106] in which the MacMillan laboratory has played a major role, are nicely highlighted in this synthesis which achieves the shortest and most efficient route (12 steps and 6% overall yield) to (–)-strychnine to date.
4. Summary and Outlook
A number of trends become apparent when comparing the ten syntheses of strychnine discussed in this review (Table 1). In the nearly sixty years since Woodward’s inaugural total synthesis in 1954, the efficiency with which chemists can construct a molecule of strychnine’s complexity has advanced remarkably. Whereas Woodward’s pioneering synthesis was realized in 0.0002% overall yield and required two relay intermediates, six of the syntheses we have considered proceed in 2% overall yield or greater, with Rawal’s synthesis of (±)-strychnine proceeding in a remarkable 10% overall yield. Equally strikingly, the number of steps required to prepare (±)- and (–)-strychnine has dropped significantly, with five of the syntheses we considered requiring 15 or less linear steps from a commercially available starting material, and the shortest, from Vanderwal, only 6.
Table 1.
Tabulated summary of highlighted syntheses of strychnine.

| Main Author | Year | Target Strychnine Precursor | Carbon-Carbon Bonds Formed[a] | Step Count[b] |
Overall Yield[c] |
|---|---|---|---|---|---|
| Woodward[17] | 1954 | (−)-isostrychnine[d] | C7–C8, C7–C6, C7–C3, C15–C14, C20–C19 | 29 | 0.0002% |
| Overman[49] | 1993 | (−)-Wieland–Gumlich aldehyde | C15–C20, C2–C7/C7–C8, C7–C6, C5–C6/C7–C3, C16–C17 |
25 | 3% |
| Rawal[62] | 1994 | (±)-isostrychnine | C7–C6, C2–C7/C14–C3, C15–C20 | 12 | 10% |
| Kuehne[67] | 1998 | (−)-Wieland–Gumlich aldehyde | C2–C16, C7–C3/C16–C15, C20–C21, C20–C19 | 21 | 4% |
| Vollhardt[73] | 2000 | (±)-isostrychnine | C16–C17, C2–C16/C7–C3/C15–C14, C15–C20 | 14 | 0.7% |
| Martin[78] | 2001 | (±)-Wieland–Gumlich aldehyde | C3–C14, C15–C20, C2–C16/C7–C3 | 16 | 1% |
| Fukuyama[82] | 2004 | (−)-Wieland–Gumlich aldehyde | C2–C7, C16–C15, C7–C3 | 25 | 1% |
| Reissig[90] | 2010 | (±)-isostrychnine | C2–C16/C7–C3, C15–C20 | 9 | 4% |
| Vanderwal[96] | 2011 | (±)-Wieland–Gumlich aldehyde | C2–C16/C7–C3, C15–C20 | 6 | 2–3% |
| MacMillan[102] | 2011 | (−)-Wieland–Gumlich aldehyde | C16–C15, C7–C3/C15–C14, C16–C17, C15–C20 | 12 | 7% |
Bonds are listed in the order that they are formed in the synthesis. Bonds formed in a single transformation are joined by a slash and highlighted in bold.
The longest linear sequence from a readily available commercial precursor to strychnine. A single step is counted when a product is isolated, though not necessarily purified
Overall yield of strychnine was calculated as the combined reported yields of reactions in the longest linear sequence starting from a readily available commercial precursor.
Resolution of an intermediate was used to access the natural enantiomer.
Remarkable also is the diversity of the chemistry and synthesis strategies that have been implemented to address the challenges strychnine poses for chemical synthesis. This variety is apparent in the syntheses discussed in this review, and even more evident in the structures of key intermediates employed in all the strychnine syntheses reported to date (Figure 4).
Figure 4.

Structural diversity of key intermediates employed in strychnine syntheses.
Even with this diversity in approach, some disconnections have been recognized as particularly strategic. Most notably, late-stage construction of the C15–C20 bond, first demonstrated by Stork,[44c] is the most common method for construction of the D-ring; ten of the 17 published syntheses rely on this bond formation. Another common feature is creating the C7 quaternary carbon by formation of the C3–C7 bond, a strategy employed in all but the Rawal synthesis. The Overman and Rawal syntheses were unique in not employing an indole precursor in the transformation that constructs the C7 quaternary carbon center.[107]
It is instructive to consider some of the important factors that have contributed to the recent remarkable increase in our ability to prepare a molecule of strychnine’s complexity (Table 1). Prominent among these is the large number of selective C–C bond-forming transformations that have been introduced since the time of Woodward’s groundbreaking synthesis. The well appreciated central role of palladium-catalyzed C–C bond-forming processes in contemporary organic synthesis is apparent in the use of intramolecular Heck reactions, Tsuji–Trost cross-couplings, and carbonylative cross-coupling in six of the ten syntheses that we considered.[108] This feature is notably highlighted in the Mori synthesis in which four bonds (three C–C and one C–N) were fashioned with palladium-catalyzed reactions.[44g] In addition, the important role that other catalytic reactions play in contemporary organic synthesis is evident in three of the enantioselective syntheses, where enzymatic desymmetrization,[49] enzymatic resolution,[82] or enantioselective organocatalysis[102] were utilized to access a chiral intermediate in high enantiomeric purity. Particularly apparent is the impact of transformations that form more than one C–C bond.[106c,109] Although no reactions of this type were used in the Woodward synthesis, all but one of the subsequent strychnine syntheses that we have considered employed at least one, with three C–C σ-bonds being forged in the central cobalt-promoted [2+2+2]-cycloadditon step of the Vollhardt synthesis (Table 1).
An effective synthesis of a complex organic molecule requires both powerful bond-forming reactions and insightful strategy. Although many of the factors that contribute to synthetic efficiency have been discussed and evaluated,[ 110 ] suffice it to say that minimizing steps that do not form bonds of the target structure is a hallmark of powerful synthesis strategy. The importance of minimizing refunctionalization steps—oxidation, reduction, protection, deprotection—is well illustrated in the most concise strychnine syntheses; redox and protecting group refunctionalization were minimized in the Rawal, Vollhardt and Reissig syntheses of (±)-strychnine and absent in Vanderwal’s short construction.
An additional trend is apparent in the syntheses that we have considered. Until the most recent synthesis by MacMillan, enantioselective constructions of (–)-strychnine were substantially longer than total syntheses of the racemate. We speculate that this is not an isolated example, but a reflection of the fact that for many chiral structural motifs powerful enantioselective multi-bond forming methods are not yet available though a topic of much current research.
Although it is difficult even in one’s own research to recognize all the factors that contributed to consequential developments, we feel confident that a number of the advances in organic synthesis strategy and tactics that underpinned the strychnine total syntheses we have discussed were influenced early on by the structure and historical importance of strychnine. Does strychnine still represent a meaningful target for total synthesis? Yes indeed, with the proviso that consequential future contributions will be more effective than those recorded to date and most likely introduce new directions in organic synthesis strategy and tactics.
Biography
Larry Overman was born in Chicago, Illinois in 1943 and raised in Hammond, Indiana. He obtained a B.A. degree from Earlham College in 1965 and completed his doctoral dissertation in 1969 with Professor Howard W. Whitlock, Jr. at the University of Wisconsin. After a NIH postdoctoral fellowship with Professor Ronald Breslow at Columbia University, he joined the faculty at the University of California, Irvine in 1971 where he is now Distinguished Professor of Chemistry. Professor Overman’s research interests center on the invention of new transformations and strategies in organic synthesis and the total synthesis of natural products and their congeners. Using synthesis strategies developed largely in his laboratory, Professor Overman’s group has completed total syntheses of more than 90 structurally complex natural products.

Jeff Cannon was born in Escondido, CA in 1985. He studied at Occidental College in Los Angeles, CA, where he received his A.B. in chemistry in 2007. He is currently studying for his PhD in the laboratory of Professor Larry Overman at the University of California, Irvine. His research interests are currently focused on the asymmetric total synthesis of complex alkaloid natural products.

Footnotes
L.E.O. thanks NIGMS (30859 and 98601) for funding and J. S. C. BMS for a graduate fellowship.
4.1.1. Abbreviations
Ac – acetyl
AChE – acetylcholinesterase
AIBN – azobisisobutyronitrile
Ar – aromatic
Bn – benzyl
Boc – tert-butoxycarbonyl
Cp – cyclopentadienyl
Cp* – pentamethylcyclopentadienyl
dba – dibenzylidene acetone
DBU – 1,8-diazabicyclo[5.4.0]undec-7-ene
DCC – Dicyclohexylcarbodiimide
DEAD – diethylazodicarboxylate
DIBAL-H – di-iso-butylaluminum hydride
DMAP – N,N-dimethylaminopyridine
DNP – 2,4-dinitrophenyl
Glc – glycoside
HMDS – hexamethyldisilylamide
HMPA – hexamethylphosphoramide
LDA – Lithium di-iso-propylamide
mCPBA – 3-chloroperoxybenzoic acid
MOM – methoxymethyl
Ms – methanesulfonyl
1-Nap – 1-naphthyl
Ns – 4-nitrobenzenesulfonyl
PMB – 4-methoxybenzyl
PPA – polyphosphoric acid
p-tol – p-tolyl
pyr – pyridine
SEM – 2-(trimethylsilyl)ethoxymethyl
TBA – tribromoacetic acid
TBS – tert-butyldimetylsilyl
TFA – trifluoroacetic acid
THF – tetrahydrofuran
TIPS – tri-iso-propylsilyl
TMS – trimethylsilyl
Trp – tryptophan
Ts – p-toluenesulfonyl
References
- [1].Creasey WA. In: Monoterpenoid Indole Alkaloids. Part 4. Saxton JE, editor. Supplement to Vol. 25. Wiley; New York: 1994. pp. 715–754. [Google Scholar]
- [2 ].The first recorded mention of nuts having powerful medical properties is believed to be a treatise by Haly Abbas, the Bagdad physician to Calif Adud al-Dawlah, that became known in the West as Lieber regius or the Pantegni.[3]
- [3 ].For a fascinating scholarly history of strychnine, see: Buckingham J. Bitter Nemensis. The Intimate history of Strychnine. CRC Press; Boca Raton, Florida: 2008.
- [4] a).Pelletier PJ, Caventou JB. Ann. Chim. Phys. 1818;8:323. [Google Scholar]; b) Pelletier PJ, Caventou JB. Ann. Chim. Phys. 1819;10:142. [Google Scholar]
- [5] a).Franz DN. In: The Pharmacological Basis of Therapeutics. 5th ed Goodman LS, Gilman H, editors. MacMillan; New York: 1975. pp. 359–361. [Google Scholar]; b) Barron SE, Guth PS. Trends Pharm. Sci. 1987;8:204–206. [Google Scholar]
- [6] a).Martin D, Gynn R. The Olympic Marathon, Human Kinetics. Champaigne, USA: 2000. [Google Scholar]; b) Pain S. New Scientist. 2004 Aug 7;:46. [Google Scholar]
- [7].Van Heerden PV, Edibam C, Augustson B, Thompson WR, Power BM. Anaesth. Intens. Care. 1993;21:876–877. doi: 10.1177/0310057X9302100624. [DOI] [PubMed] [Google Scholar]
- [8].Aprison MH. In: Glycine Neurotransmission. Otterson OP, Mathisen J, editors. Wiley; New York: 1990. pp. 1–23. [Google Scholar]
- [9] a).For recent reviews, see: Cascio M. J. Biol. Chem. 2004;279:19383–19386. doi: 10.1074/jbc.R300035200. Betz H, Laube B. J. Neurochem. 2006;97:1600–1610. doi: 10.1111/j.1471-4159.2006.03908.x. Lynch JW. Neuropharmacology. 2009;56:303–309. doi: 10.1016/j.neuropharm.2008.07.034.
- [10] a).Jackson G, Ng SH, Diggle GE, Bourke IG. British Med. J. 1971;3:519–520. doi: 10.1136/bmj.3.5773.519. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lambert JR, Byrick RJ, Hammeke MD. Can. Med. Assoc. J. 1981;124:1268–1270. [PMC free article] [PubMed] [Google Scholar]
- [11].Pfeiffer F, Graham D, Betz H. J. Biol. Chem. 1982;257:9389–9393. [PubMed] [Google Scholar]
- [12] a).For historical reviews see: Huisgen R. Angew. Chem. 1950;62:527–534. Robinson R. In: Progess in Organic Chemistry. Cook JW, editor. Vol. 1. Butterworths; London: 1952. p. 12. Smith GF. Alkaloids. 1965;8:591.
- [13] a).Briggs LH, Openshaw HT, Robinson R. J. Chem. Soc. 1946:903. [PubMed] [Google Scholar]; b) Robinson R. Experientia. 1946;2:28–29. doi: 10.1007/BF02154708. [DOI] [PubMed] [Google Scholar]
- [14] a).Woodward RB, Brehm WJ, Nelson AL. J. Am. Chem. Soc. 1947;69:2250. doi: 10.1021/ja01201a526. [DOI] [PubMed] [Google Scholar]; b) Woodward RB, Brehm WJ. J. Am. Chem. Soc. 1948;70:2107–2115. doi: 10.1021/ja01186a034. [DOI] [PubMed] [Google Scholar]
- [15].The correct structure of strychnine first appeared in publications from Robinson’s laboratory. However, his group favoured a different structure.[13]
- [16] a).Robertson JH, Beevers CA. Acta Crystallogr. 1951;4:270–275. [Google Scholar]; b) Bokhoven C, Schoone JC, Bijvoet JM. Acta Crystallogr. 1951;4:275–280. [Google Scholar]; c) Peerdeman AF. Acta Crystallogr. 1956;9:824. [Google Scholar]
- [17] a).Woodward RB, Cava MP, Ollis WD, Hunger A, Daeniker HU, Schenker K. J. Am. Chem. Soc. 1954;76:4749–4751. [Google Scholar]; b) Woodward RB, Cava MP, Ollis WD, Hunger A, Daeniker HU, Schenker K. Tetrahedron. 1963;19:247–288. [Google Scholar]
- [18] a).Herbert RB. In: Monoterpenoid Indole Alkaloids. Part 4. Saxton JE, editor. Supplement to Vol. 25. Wiley; New York: 1994. pp. 1–13. [Google Scholar]; b) Sapi J, Massiot G. In: Monoterpenoid Indole Alkaloids. Part 4. Saxton JE, editor. Supplement to Vol. 25. Wiley; New York: 1994. pp. 279–356. [Google Scholar]
- [19] a).Bisset NG. In: Indole and Biogenetically Related Alkaloids. Phillipson JD, Zenk MH, editors. Academic Press; London: 1980. pp. 27–61. [Google Scholar]; b) Kisakürek MV, Leeuwenberg AJM, Hesse M. In: Alkaloids: Chemical and Biological Perspecitves. Pelletier SW, editor. Vol. 1. Wiley; New York: 1983. pp. 211–376. [Google Scholar]; c) Atta-ur-Rahman, Basha A. Biosynthesis of Indole Alkaloids. Clarendon Press; Oxford: 1983. pp. 45–93. [Google Scholar]; d) Dewick PM. Medicinal Natural Products. A Biosynthetic Approach. Wiley; Chichester: 1998. pp. 324–334. [Google Scholar]
- [20].Woodward RB. Nature. 1948;162:155–156. doi: 10.1038/162155a0. [DOI] [PubMed] [Google Scholar]
- [21].Robinson R. The Structural Relations of Natural Products. Oxford University Press; 1955. p. 112. [Google Scholar]
- [22].Heimberger SI, Scott AI. J. Chem. Soc., Chem. Commun. 1973:217–218. [Google Scholar]
- [23].Schlatter Ch., Waldner EE, Schmid H, Maier W, Gröger D. Helv. Chim. Acta. 1969;52:776–789. doi: 10.1002/hlca.19690520103. [DOI] [PubMed] [Google Scholar]
- [24].Nicolaou KC, Vourloumis D, Winssinger N, Baran PS. Angew. Chem. Int. Ed. 2000;39:44–122. [PubMed] [Google Scholar]; b) Nicolaou KC, Snyder SA. Proc. Nat. Acad. Sci. USA. 2004;101:11929–11936. doi: 10.1073/pnas.0403799101. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Nicolaou KC. J. Org. Chem. 2009;74:951–972. doi: 10.1021/jo802351b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25] a).Corey EJ, Cheng X-M. The Logic of Chemical Synthesis. Wiley; New York: 1989. [Google Scholar]; b) Nicolaou KC, Sorensen EJ. Classics in Total Synthesis. VCH; Weinheim: 1996. [Google Scholar]; c) Nicolaou KC, Sorensen EJ. Classics in Total Synthesis II. Wiley–VCH; Weinheim: 2003. [Google Scholar]
- [26] a).Breslow R. Chemistry: Today and Tomorrow. American Chemical Society; Washington, D.C.: 1996. [Google Scholar]; b) Hanessian S. Why, What, and How of Synthesis. Wiley–VCH; in press. [Google Scholar]
- [27].Wöhler F. Ann. Phys. Chem. 1828;12:253. [Google Scholar]
- [28] a).Weinreb SM. Acc. Chem. Res. 2003;36:59–65. doi: 10.1021/ar0200403. [DOI] [PubMed] [Google Scholar]; b) Nicolaou KC, Snyder SA. Angew. Chem. Int. Ed. 2005;44:1012–1044. doi: 10.1002/anie.200460864. [DOI] [PubMed] [Google Scholar]; c) Usami Y. Mar. Drugs. 2009;7:314–330. doi: 10.3390/md7030314. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Maier ME. Nat. Prod. Rep. 2009;26:1105–1124. doi: 10.1039/b809658a. [DOI] [PubMed] [Google Scholar]
- [29] a).Koehn FE, Carter GT. Nat. Rev. Drug Discov. 2005;4:206–220. doi: 10.1038/nrd1657. [DOI] [PubMed] [Google Scholar]; b) Paterson I, Anderson EA. Science. 2005;310:451–453. doi: 10.1126/science.1116364. [DOI] [PubMed] [Google Scholar]; c) Newman DJ, Cragg GM. J. Nat. Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]; d) Cragg GM, Grothaus PG, Newman DJ. Chem. Rev. 2009;109:3012–3043. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]; e) Kumar K, Waldmann H. Angew. Chem. Int. Ed. 2009;48:3224–3242. doi: 10.1002/anie.200803437. [DOI] [PubMed] [Google Scholar]; f) Kinghorn AD, Pan L, Fletcher JN, Chai H. J. Nat. Prod. 2011;74:1539–1555. doi: 10.1021/np200391c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30] a).Paterson I, Florence GJ, Gerlach K, Scott JP. Angew. Chem. Int. Ed. 2000;39:377–380. doi: 10.1002/(sici)1521-3773(20000117)39:2<377::aid-anie377>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]; b) Paterson I, Florence GJ. Eur. J. Org. Chem. 2003:2193–2208. [Google Scholar]; c) Mickel SJ, Niederer D, Daeffler R, Osmani A, Kuesters E, Schmid E, Schaer K, Gamboni R. Org. Proc. Res. Dev. 2004;8:122–130. [Google Scholar]
- [31] a).Coffey DS, McDonald AI, Overman LE, Rabinowitz MH, Renhowe PA. J. Am. Chem. Soc. 2000;122:4893–4903. [Google Scholar]; b) Aron ZD, Pietraszkiewicz H, Overman LE, Valeriote F, Cuevas C. Bioorg. Med. Chem. Lett. 2004;14:3445–3449. doi: 10.1016/j.bmcl.2004.04.071. [DOI] [PubMed] [Google Scholar]
- [32] a).Njardarson JT, Gaul C, Shan D, Huang X-Y, Danishefsky SJ. J. Am. Chem. Soc. 2004;126:1038–1040. doi: 10.1021/ja039714a. [DOI] [PubMed] [Google Scholar]; b) Gaul C, Njardarson JT, Shan D, Dorn DC, Wu K-D, Tong WP, Huang X-Y, Moore MAS, Danishefsky SJ. J. Am. Chem. Soc. 2004;126:11326–11337. doi: 10.1021/ja048779q. [DOI] [PubMed] [Google Scholar]; c) Shan D, Chen L, Njardarson JT, Gaul C, Ma X, Danishefsky SJ, Huang X-Y. Proc. Nat. Acad. Sci. U.S.A. 2005;102:3772–3776. doi: 10.1073/pnas.0500658102. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Oskarsson T, Nagorny P, Krauss IJ, Perez L, Mandal M, Yang G, Ouerfelli O, Xiao D, Moore MAS, Massagué J, Danishefsky SJ. J. Am. Chem. Soc. 2010;132:3224–3228. doi: 10.1021/ja9101503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wender PA, Verma VA, Paxon TJ, Pillow TH. Acc. Chem. Res. 2008;41:40–49. doi: 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]
- [34] a).Aicher TD, Kishi Y. Tetrahedron Lett. 1987;28:3463–3466. [Google Scholar]; b) Aicher TD, Buszek KR, Forsyth CJ, Fang FG, Jung SH, Kishi Y, Scola PM. Tetrahedron Lett. 1992;33:1549–1552. [Google Scholar]; c) Buszek KR, Fang FG, Forsyth CJ, Jung SH, Kishi Y, Scola PM, Yoon SK. Tetrahedron Lett. 1992;33:1553–1556. [Google Scholar]; d) Fang FG, Kishi Y, Matelich MC, Scola PM. Tetrahedron Lett. 1992;33:1557–1560. [Google Scholar]; e) Aicher TD, Buszek KR, Fang FG, Forsyth CJ, Jung SH, Kishi Y, Matelich MC, Scola PM, Spero DM, Yoon SK. J. Am. Chem. Soc. 1992;114:3162–3164. [Google Scholar]
- [35] a).Towle MJ, Salvato KA, Budrow J, Wels BF, Kuznetsov G, Aalfs KK, Welsh S, Zheng W, Seletsky BM, Palme MH, Habgood GJ, Singer LA, DiPietro LV, Wang Y, Chen JJ, Quincy DA, Davis A, Yoshimatsu K, Kishi Y, Yu MJ, Littlefield BA. Cancer Res. 2001;61:1013–1021. [PubMed] [Google Scholar]; b) Yu MJ, Kishi Y, Littlefield BA. In: Anticancer Agents from Natural Products. Newman DJ, Kingston DGI, editors. Taylor & Francis; Washington D. C.: 2005. [Google Scholar]
- [36] a).Barton DHR, McCombie SW. J. Chem. Soc., Perkin Trans. 1975;1:1574–1585. [Google Scholar]; b) Barton DHR, Parekh SI. Half Century of Free Radical Chemistry. Cambridge University Press; Cambridge, England: 1993. Ch. 4. [Google Scholar]
- [37] a).Kakimoto M, Overman LE. J. Am. Chem. Soc. 1979;101:1310–1312. [Google Scholar]; b) Overman LE. Tetrahedron. 2009;65:6432–6446. doi: 10.1016/j.tet.2009.05.067. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Overman LE, Humphreys PG, Wellmaker GS. Organic Reactions. Vol. 75. Wiley; Hoboken, New Jersey: 2011. pp. 747–820. [Google Scholar]
- [38 ] a).Seiple IB, Su S, Rodriguez RA, Glantassio R, Fujiwara Y, Sobel AL, Baran PS. J. Am. Chem. Soc. 2010;132:13194–13196. doi: 10.1021/ja1066459. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fujiwara Y, Domingo V, Selple IA, Glanatassio R, Del Bel M, Baran PS. J. Am. Chem. Soc. 2011;133:3292–3295. doi: 10.1021/ja111152z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Robinson R. In: Progress in Organic Chemistry. Cook JW, editor. Vol. 1. Butterworths; London: 1952. p. 2. [Google Scholar]
- [40] a).For leading reviews see: Beifuss U. Angew. Chem. Int. Ed. 1994;33:1144–1149. Bonjoch J, Solé D. Chem. Rev. 2000;100:3455–3482. doi: 10.1021/cr9902547. Mori M. Heterocycles. 2010;81:259–292. Roth K. Chem. Unserer Zeit. 2011;45:202–218.
- [41].Prelog V, Battegay J, Taylor WI. Helv. Chim. Acta. 1948;31:2244–2246. doi: 10.1002/hlca.19480310750. [DOI] [PubMed] [Google Scholar]
- [42].Kuehne ME, Xu F. J. Org. Chem. 1993;58:7490–7497. [Google Scholar]
- [43].Anet FAL, Robinson R. Chem. Ind. 1953:245. [Google Scholar]
- [44] a).For the syntheses not discussed, see: Magnus P, Giles M, Bonnert R, Kim CS, McQuire L, Merritt A, Vicker N. J. Am. Chem. Soc. 1992;114:4403–4405. Magnus P, Giles M, Bonnert R, Johnson G, McQuire L, Deluca M, Merritt A, Kim CS, Vicker N. J. Am. Chem. Soc. 1993;115:8116–8129. Stork G. Disclosed at the Ischia Advanced School of Organic Chemistry. Ischia Porto; Italy: Sep 21, 1992. Solé D, Bonjoch J, García-Rubio S, Peidró E. J. Bosch, Angew. Chem. Int. Ed. 1999;38:395–397. doi: 10.1002/(SICI)1521-3773(19990201)38:3<395::AID-ANIE395>3.0.CO;2-5. Solé D, Bonjoch J, García-Rubio S, Peidró E. J. Bosch, Chem. Eur. J. 2000;6:655–665. doi: 10.1002/(sici)1521-3765(20000218)6:4<655::aid-chem655>3.0.co;2-6. Ohshima T, Xu Y, Takita R, Shimizu S, Zhong D, Shibasaki M. J. Am. Chem. Soc. 2002;124:14546–14547. doi: 10.1021/ja028457r. Nakanishi M, Mori M. Angew. Chem. Int. Ed. 2002;41:1934–1936. doi: 10.1002/1521-3773(20020603)41:11<1934::aid-anie1934>3.0.co;2-f. Zhang H, Boonsombat J, Padwa A. Org. Lett. 2007;9:279–282. doi: 10.1021/ol062728b. Boonsombat J, Zhang H, Chughtai MJ, Hartung J, Padwa A. J. Org. Chem. 2008;73:3539–3550. doi: 10.1021/jo8003716. Sirasani G, Paul T, Dougherty W, Jr., Kassel S, Andrade RB. J. Org. Chem. 2010;75:3529–3532. doi: 10.1021/jo100516g.
- [45].For a detailed discussion of the “Woodward Fission”, see: Berson JA. Chemical Discovery and the Logicians’ Program. Wiley-VCH; Weinheim: 2003. Ch. 8. and references therein.
- [46].For a review of the chemistry of N-sulfonyliminium ions, see: Weinreb SM. Top. Curr. Chem. 1997;190:131–184.
- [47].Woodward RB. In: Experientia Supplementum II. Verlag B, editor. Basel and Stuttgart; 1955. p. 213. [Google Scholar]
- [48] a).For precedent of this transformation in 1956 see: King JA, McMillan FH. J. Am. Chem. Soc. 1951;73:4911–4915. King JA, McMillan FH. J. Am. Chem. Soc. 1955;77:2814–2816.
- [49] a).Knight SD, Overman LE, Pairaudeau G. J. Am. Chem. Soc. 1993;115:9293–9294. [Google Scholar]; b) Knight SD, Overman LE, Pairaudeau G. J. Am. Chem. Soc. 1995;117:5776–5788. [Google Scholar]
- [50] a).Fevig JM, Marquis RW, Jr., Overman LE. J. Am. Chem. Soc. 1991;113:5085–5086. [Google Scholar]; b) Angle SR, Fevig JM, Knight SD, Marquis RW, Jr., Overman LE. J. Am. Chem. Soc. 1993;115:3966–3976. [Google Scholar]
- [51] a).Deardorff DR, Matthews AJ, McMeekin DS, Craney CL. Tetrahedron Lett. 1986;27:1255–1256. [Google Scholar]; b) Deardorff DR, Windham CQ, Craney CL. Org. Synth. 1996;73:25. [Google Scholar]
- [52] a).Oppolzer W, Gaudin J-M, Birkinshaw TN. Tetrahedron Lett. 1988;29:4705–4708. [Google Scholar]; (b) Deardorff DR, Linde RG, III, Martin AM, Shulman MJ. J. Org. Chem. 1989;54:2759–2762. [Google Scholar]; (c) Montforts FP, Gesing–Zibulak I, Grammenos W, Schneider M, Lauman K. Helv. Chim. Acta. 1989;72:1852–1859. [Google Scholar]
- [53] a).Tsuji J, Takahashi H, Morikawa M, Y. Tetrahedron Lett. 1965;6:4387–4388. [Google Scholar]; (b) Trost BM. Angew. Chem. Int. Ed. Engl. 1989;28:1173–1192. [Google Scholar]
- [54] a).Reetz MT. Angew. Chem. Int. Ed. Engl. 1984;23:556–569. [Google Scholar]; b) Reetz MT. Acc. Chem. Res. 1993;26:462–468. [Google Scholar]
- [55].Nakata T, Oishi T. Tetrahedron Lett. 1980;21:1641–1644. [Google Scholar]
- [56].Alexandr C, Rouessac F. Bull. Soc. Chim. Fr. 1971:1837. [Google Scholar]
- [57] a).Stille JK. Angew. Chem. Int. Ed. Engl. 1986;25:508–523. [Google Scholar]; b) Farina V, Krishnamurthy V, Scott WJ. Organic Reactions. Vol. 50. Wiley; Hoboken, New Jersey: 1997. pp. 1–652. [Google Scholar]
- [58].Mander LN, Sethi SP. Tetrahedron Lett. 1983;24:5425–5428. [Google Scholar]
- [59].Edwards PN, Smith GF. J. Chem. Soc. 1961:152. [Google Scholar]
- [60].Ent-strychnine is a weak inhibitor of the inhibitory glycine receptor, see: Knight SD, Miledi R, Nguyen Q-T, Overman LE, Pairaudeau G. Bioorg. Med. Chem. Lett. 1965;5:749–752.
- [61].The use of enantioselective hydrolysis of meso-2-alken-1,4-diol derivatives coupled with η3-allylpalladium chemistry to prepare either enantiomer of a substitution product was originally demonstrated by Bäckvall: Schink HE, Bäckvall J-E. J. Org. Chem. 1992;57:1588–1591. Bäckvall J-E, Gatti R, Schink HE. Synthesis. 1993:343–348.
- [62].Rawal VH, Iwasa S. J. Org. Chem. 1994;59:2685–2686. [Google Scholar]
- [63].For selected reviews of the intramolecular Heck reaction see: Link JT. Organic Reactions. Vol. 60. Wiley; Hoboken, NJ: 2002. Chapter 2. Dounay AB, Overman LE. Chem. Rev. 2003;103:2945–2963. doi: 10.1021/cr020039h. Oestreich M, editor. The Mizoroki–Heck reaction. Wiley; Chichester, UK: 2009.
- [64].Rawal VH, Michoud C, Monestel RF. J. Am. Chem. Soc. 1993;115:3030–3031. [Google Scholar]
- [65] a).Stevens RV. Acc. Chem. Res. 1977;10:193–198. [Google Scholar]; b) Boeckman RK, Walters MA. In: Advances in Heterocyclic Natural Product Synthesis. Pearson WH, editor. Vol. 1. JAI Press; New York: 1990. p. 1. [Google Scholar]
- [66].Jeffery T. Tetrahedron Lett. 1985;26:2667–2670. [Google Scholar]
- [67].Kuehne ME, Xu F. J. Org. Chem. 1998;63:9427–9433. [Google Scholar]
- [68].Parsons RL, Beck JD, Kuehne ME. J. Org. Chem. 1993;58:7482–7489. [Google Scholar]
- [69] a).Kuehne ME, Frasier DA, Spitzer TD. J. Org. Chem. 1991;56:2696–2700. [Google Scholar]; b) Kuehne ME, Brook CS, Frasier DA, Xu F. J. Org. Chem. 1994;59:5977–5982. [Google Scholar]; c) Kuehne ME, Brook CS, Frasier DA, Xu F. J. Org. Chem. 1995;60:1864–1867. [Google Scholar]
- [70] a).Kuehne ME, Wang T, Seraphin D. J. Org. Chem. 1996;61:7873–7881. doi: 10.1021/jo961023s. [DOI] [PubMed] [Google Scholar]; d) Kuehne ME, Bandarage UK, Hammach A, Li Y-L, Wang T. J. Org. Chem. 1998;63:2172–2183. [Google Scholar]
- [71].Kuehne ME, Xu F. J. Org. Chem. 1997;62:7950–7960. doi: 10.1021/jo970207j. [DOI] [PubMed] [Google Scholar]
- [72].Yamada S, Akimoto H. Tetrahedron Lett. 1969;10:3105–3108. doi: 10.1016/s0040-4039(01)88360-x. [DOI] [PubMed] [Google Scholar]
- [73].Eichberg MJ, Dorta RL, Lamottke K, Vollhardt KPC. Org. Lett. 2000;2:2479–2481. doi: 10.1021/ol006131m. [DOI] [PubMed] [Google Scholar]
- [74] a).Grotjahn DB, Vollhardt KPC. J. Am. Chem. Soc. 1986;108:2091–2093. [Google Scholar]; b) Boese R, Van Sickle AP, Vollhardt KPC. Synthesis. 1994:1374–1382. [Google Scholar]
- [75].Vollhardt KPC. Angew. Chem. Int. Ed. Engl. 1984;23:539–556. [Google Scholar]
- [76].Stork G, Baine NH. J. Am. Chem. Soc. 1982;104:2321–2323. [Google Scholar]
- [77].Grotjahn DB, Vollhardt KPC. J. Am. Chem. Soc. 1990;112:5653–5654. [Google Scholar]
- [78] a).Martin SF, Clark CW, Ito M, Mortimore M. J. Am. Chem. Soc. 1996;118:9804–9805. doi: 10.1021/ja010935v. [DOI] [PubMed] [Google Scholar]; b) Ito M, Clark CW, Mortimore M, Goh JB, Martin SF. J. Am. Chem. Soc. 2001;123:8003–8010. doi: 10.1021/ja010935v. [DOI] [PubMed] [Google Scholar]
- [79].Martin SF, Benage B, Geraci LS, Hunter JE, Mortimore MP. J. Am. Chem. Soc. 1991;113:6161–6171. and references therein. [Google Scholar]
- [80].Martin SF, Mortimore M. Tetrahedron Lett. 1990;31:4557–4560. and references therein. [Google Scholar]
- [81] a).For earlier studies of stereochemical equilibration of similar structures, see: Finch N, Taylor WI. J. Am. Chem. Soc. 1962;84:1318–1320. Shavel J, Zinnes H. J. Am. Chem. Soc. 1962;84:1320–1321. Awang DVC, Vincent A, Kindack D. Can. J. Chem. 1984;62:2667–2675. Wenkert E, Shi Y-J. Synth. Commun. 1989;19:1071–1079. Stahl R, Borschberg H-J, Acklin P. Helv. Chim. Acta. 1996;79:1361–1378.
- [82].Kaburagi Y, Tokuyama H, Fukuyama T. J. Am. Chem. Soc. 2004;126:10246–10247. doi: 10.1021/ja046407b. [DOI] [PubMed] [Google Scholar]
- [83] a).Kan T, Fukuyama T. J. Synth. Org. Chem. Jpn. 2001;59:779–789. [Google Scholar]; b) Kan T, Fukuyama T. Chem. Commun. 2004:353–359. doi: 10.1039/b311203a. [DOI] [PubMed] [Google Scholar]
- [84] a).Tsuji T, Kataoka H, Kobayashi Y. Tetrahedron Lett. 1981;22:2575–2578. [Google Scholar]; b) Trost BM, Molander GA. J. Am. Chem. Soc. 1981;103:5969–5972. [Google Scholar]
- [85].Boger DL, Patel M, Takusagawa F. J. Org. Chem. 1981;46:1904–1911. [Google Scholar]
- [86].For selected recent reviews of enantioselective enzymatic kinetic resolution using lipases, see: Ghanem A, Aboul-Enein HY. Tetrahedron: Asymm. 2004;15:3331–3351. Chênevert R, Pelchat N, Jacques F. Curr. Org. Chem. 2006;10:1067–1094. Ghanem A. Tetrahedron. 2007;63:1721–1754.
- [87].Tokuyama H, Yamashita T, Reding MT, Kaburagi Y, Fukuyama T. J. Am. Chem. Soc. 1999;121:3791–3792. [Google Scholar]
- [88].Fo reviews of the Mitsunobu reaction, see: Mitsunobu O. Synthesis. 1981:1–28. Swamy KCK, Kumar NNB, Balaraman E, Kumar KVPP. Chem. Rev. 2009;109:2551–2651. doi: 10.1021/cr800278z.
- [89].Rubottom GM, Gruber JM, Juve HD, Jr., Charleson DA. Organic Syntheses. Coll. Vol. VII. Wiley & Sons; New York: 1990. p. 282. [Google Scholar]
- [90].Beemelmanns C, Reissig H-U. Angew. Chem. Int. Ed. 2010;49:8021–8025. doi: 10.1002/anie.201003320. [DOI] [PubMed] [Google Scholar]
- [91] a).Gross S, Reissig H-U. Org. Lett. 2003;5:4305–4307. doi: 10.1021/ol0355581. [DOI] [PubMed] [Google Scholar]; b) Beemelmanns C, Reissig H-U. Org. Biomol. Chem. 2009;7:4475–4480. doi: 10.1039/b913015b. [DOI] [PubMed] [Google Scholar]; c) Beemelmanns C, Blot V, Gross S, Lentz D, Reissig H-U. Eur. J. Org. Chem. 2010:2716–2732. [Google Scholar]
- [92] a).Moriuchi F, Muroi H, Aibe H. Chem. Lett. 1987:1141–1144. [Google Scholar]; b) Moriuchi F, Muroi H, Yasushi Y. No. JP 01132393 Patent. 1989
- [93] a).For reviews on the use of SmI2 promoted reactions see: Molander GA, Harris CR. Chem. Rev. 1996;96:307–338. doi: 10.1021/cr950019y. Berndt M, Gross S, Hölemann A, Reissig H-U. Synlett. 2004:422–438. Edmonds DJ, Johnston D, Procter DJ. Chem. Rev. 2004;104:3371–3403. doi: 10.1021/cr030017a. Gopalaiah K, Kagan H. New J. Chem. 2008;32:607–637. Nicolaou KC, Ellery SP, Chen JS. Angew. Chem. Int. Ed. 2009;48:7140–7165. doi: 10.1002/anie.200902151. Procter DJ, Flowers RA, II, Skrydstrup T. Organic Synthesis by using Samarium Diiodide. Royal Society of Chemistry; Cambridge: 2009.
- [94] a).Azzouzi A, Perrin B, Sinibaldi M-E, Gramain J-C, Lavaud C. Tetrahedron Lett. 1993;34:5451–5454. [Google Scholar]; b) Kraus GA, Bougie D. Tetrahedron. 1994;50:2681–2690. [Google Scholar]; c) Callaghan O, Lampard C, Kennedy AR, Murphy JA. J. Chem. Soc. Perkin Trans. 1999;1:995–1001. [Google Scholar]
- [95].Beemelmanns C, Reissig H-U. 2011. unpublished results.
- [96].Martin DBC, Vanderwal CD. Chem. Sci. 2011;2:649–651. [Google Scholar]
- [97] a).Zincke T. Justus Liebigs Ann. Chem. 1904;330:361–374. [Google Scholar]; b) Zincke T. Justus Liebigs Ann. Chem. 1904;333:296–345. [Google Scholar]; c) König W. J. Prakt. Chem. 1904;69:105–137. [Google Scholar]; d) Zincke T, Wurker W. Justus Liebigs Ann. Chem. 1905;338:107–141. [Google Scholar]
- [98] a).For Vanderwal’s previous studies with Zincke aldehydes, see: Kearney AM, Vanderwal CD. Angew. Chem. Int. Ed. 2006;45:7803–7806. doi: 10.1002/anie.200602996. Steinhardt SE, Silverston JS, Vanderwal CD. J. Am. Chem. Soc. 2008;130:7560–7561. doi: 10.1021/ja8028125. Miches TD, Rhee JU, Vanderwal CD. Org. Lett. 2008;10:4787–4790. doi: 10.1021/ol8020435. Steinhardt SE, Vanderwal CD. J. Am. Chem. Soc. 2009;131:7546–7547. doi: 10.1021/ja902439f. Michels TD, Kier MJ, Kearney AM, Vanderwal CD. Org. Lett. 2010;12:3093–3095. doi: 10.1021/ol101035p. Paton RS, Steinhardt SE, Vanderwal CD, Houk KN. J. Am. Chem. Soc. 2011;133:3895–3905. doi: 10.1021/ja107988b.
- [99].Baldwin JE, Claridge TDW, Culshaw AJ, Heupel FA, Lee V, Spring DR, Whitehead RC. Chem. Eur. J. 1999;5:3154–3161. and references therein. [Google Scholar]
- [100].Martin DBC, Vanderwal CD. J. Am. Chem. Soc. 2009;131:3472–3473. doi: 10.1021/ja900640v. [DOI] [PubMed] [Google Scholar]
- [101].Trost BM, Ball ZT. J. Am. Chem. Soc. 2005;127:17644–17655. doi: 10.1021/ja0528580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Jones SB, Simmons B, Mastracchio A, MacMillan DWC. Nature. 2011;475:183–188. doi: 10.1038/nature10232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Jones SB, Simmons B, Macmillan DWC. J. Am. Chem. Soc. 2009;131:13606–13607. doi: 10.1021/ja906472m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Lévy J, Sapi J, Laronze J-Y, Royer D, Toupet L. Synlett. 1992:601–602. [Google Scholar]
- [105].Prashad M, Lavecchia L, Prasad K, Repic O. Synth. Commun. 1995;25:95–100. [Google Scholar]
- [106] a).For selected recent reviews of organocatalysis, see: Enders D, Grondal C, Hüttl MRM. Angew. Chem. Int. Ed. 2007;46:1570–1581. doi: 10.1002/anie.200603129. Dondoni A, Massi A. Angew. Chem. Int. Ed. 2008;47:4638–4660. doi: 10.1002/anie.200704684. Grondal C, Jeanty M, Enders D. Nature Chem. 2010;2:167–178. doi: 10.1038/nchem.539. Merino P, Marqués-López E, Tejero T, Herrera RP. Synthesis. 2010:1–26.
- [107].The special challenges in forming quaternary carbon centers are discussed in many reviews, see, inter alia: Fuji K. Chem. Rev. 1993;93:2037–2066. Douglas CJ, Overman LE. 2004;101:5363–5367. doi: 10.1073/pnas.0307113101. Christoffers J, Baro A, editors. Quaternary Stereocenters. Wiley-VCH; Weinheim: 2005.
- [108] a).For selected reviews of palladium catalysis in organic synthesis, see: Poli G, Giambastiani G, Heumann A. Tetrahedron. 2000;56:5959–5989. de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. Vols 1 and 2. Wiley-VCH; Weinheim: 2004. Nicolaou KC, Bulger PG, Sarlah D. Angew. Chem. Int. Ed. 2005;44:4442–4489. doi: 10.1002/anie.200500368.
- [109] a).For selected reviews of cascade reactions, see: Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis G. Angew. Chem. Int. Ed. 2002;41:1668–1698. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z. Tietze LF, Brasche G, Gericke KM. Domino Reactions in Organic Synthesis. Wiley-VCH; Weinheim: 2006. Nicolaou KC, Edmonds DJ, Bulger PG. Angew. Chem. Int. Ed. 2006;45:7134–7186. doi: 10.1002/anie.200601872. Nicolaou KC, Chen JS. Chem. Soc. Rev. 2009;38:2993–3009. doi: 10.1039/b903290h.
- [110] a).See, inter alia: Hendrickson JB. J. Am. Chem. Soc. 1975;97:5784–5800. Trost BM. Science. 1991;254:1471–1477. doi: 10.1126/science.1962206. Trost BM. Angew. Chem. Int. Ed. Engl. 1995;34:259–281. Wender PA, Croatt MP, Witulski B. Tetrahedron. 2006;62:7505–7511. Hoffmann RW. Synthesis. 2006:3531–3541. Baran PS, Maimone TJ, Richter JM. Nature. 2007;446:404–408. doi: 10.1038/nature05569. Richter JM, Ishihara Y, Masuda T, Whitefield BW, Llamas T, Pohjakallio A, Baran PS. J. Am. Chem. Soc. 2008;130:17938–17954. doi: 10.1021/ja806981k. Burns NZ, Baran PS, Hoffman RW. Angew. Chem. Int. Ed. 2009;48:2854–2867. doi: 10.1002/anie.200806086. Young IS, Baran PS. Nat. Chem. 2009;1:193–205. doi: 10.1038/nchem.216. Newhouse T, Baran PS, Hoffmann RW. Chem. Soc. Rev. 2009;38:3010–3021. doi: 10.1039/b821200g. Gaich T, Baran PS. J. Org. Chem. 2010;75:4657–4673. doi: 10.1021/jo1006812.