

Abstract
Porphyromonas gingivalis lipopolysaccharide (Pg-LPS) circulates systemically in over 50% of periodontal disease (PD) patients and is associated with increased matrix metalloproteinase (MMP)-9. We hypothesized that low systemic Pg-LPS would stimulate an inflammatory response in the left ventricle (LV) through MMP-9, leading to a decrease in cardiac function. Wild-type (WT) and MMP-9 null mice (4–7 months old) were exposed for 1 or 28 days to low dose Pg-LPS or saline (n ≥ 6/group). MMP-9 significantly increased in WT mice LV at 1 and 28 days of exposure, compared to control (P < 0.05 for both). Fractional shortening decreased subtly yet significantly in WT mice by day 28 (31 ± 1%) compared to control (35 ± 1%; P < 0.05), and this decrease was attenuated in null (34 ± 1%) mice. Plasma cardiac troponin I levels were elevated in WT mice at day 28. Macrophage-related factors increased over twofold in WT plasma and LV after day 1 (monocyte chemoattractant protein-5, macrophage inflammatory protein (MIP)-1α, MIP-1γ, stem cell factor, Ccl12, Ccl9, Il8rb, Icam1, Itgb2, and Spp1; all P < 0.05), indicating a moderate inflammatory response. Levels returned to baseline by day 28, suggesting tolerance to Pg-LPS. In contrast, macrophage-related factors remained elevated in day 28 null mice, indicating a sustained defense against Pg-LPS stimulation. Consistent with these findings, LV macrophage numbers increased in both groups at day 1 and returned to baseline by day 28 in the WT mice only. Major histocompatibility complex (MCH) II remained elevated in the null group at day 28, confirming Pg-tolerance in the WT. Interestingly Il-1α, a regulator of macrophage immunosuppression, increased in the plasma of WT mice only on day 28, suggesting that Il-1α plays a role in tolerance in a MMP-9-dependent manner. In conclusion, circulating Pg-LPS induced tolerance in WT mice, resulting in significant LV changes and subtle cardiac dysfunction. MMP-9 played a major role in the regulation of chronic systemic inflammation and associated cardiac dysfunction.
Keywords: Cardiac function, inflammation, matrix metalloproteinase-9, periodontal disease, Porphyromonas gingivalis, proteomics
Introduction
An estimated 75% of the adult population in the United States has periodontal disease (PD), of which 30% has the most severe form. Several oral health measurements, including number of decayed, missing, or filled teeth, mean probing depth, oral hygiene status, and percentage of sites with bleeding on probing, each significantly associate with cardiovascular disease (CVD) incidence (Kaisare et al. 2007; Tonetti et al. 2007). Meta-analysis of more than 200,000 individuals revealed that PD increases CVD risk by 35%, indicating PD has a significant impact on public health (Humphrey et al. 2008). While the oral health and CVD link is quite strong, the mechanistic link between oral health and CVD has not been made.
PD commonly results from a bacterial infection in the gingival tissue surrounding the teeth. As the infection progresses deeper into the tissue, an inflammatory response is activated to combat the infection. Porphyromonas gingivalis is an oral pathogen detected in 80% of periodontal patients (Griffen et al. 1998). Porphyromonas gingivalis lipopolysaccharide (Pg-LPS) has been shown to increase proinflammatory cytokines (Gitlin and Loftin 2009; Gu et al. 2011). Increased expression and activity of matrix metalloproteinases (MMPs) have also been associated with inflammation. MMPs, most notably MMP-9, correlate with chronic PD and CVD (Gu et al. 2011). MMP-9 plays an important role in the inflammatory stimulus by mediating neutrophil and macrophage infiltration (Luttun et al. 2004; Bradley et al. 2012). For these reasons, appropriate management of systemic circulating inflammation may reduce CVD risk factors.
We know that PD patients have systemic levels of Pg-LPS that increase circulating inflammatory cytokine levels. In order to test whether Pg-induced activation of inflammatory cytokines can amplify risk of CVD, we hypothesized that low systemic Pg-LPS would increase the inflammatory response in the left ventricle (LV), leading to a decrease in cardiac function that would be attenuated by MMP-9 deletion.
Material and Methods
Mice
C57BL/6J wild-type (WT) and MMP-9 null male and female mice of 4–7 months of age were used in this study (n ≥ 6/group). The MMP-9 null mice are on a C57BL/6J background. The null mice were generated by Zena Werb's laboratory and backcrossed by Lynn Matrisian's laboratory (Kaisare et al. 2007; Humphrey et al. 2008). Mice were kept in a light-controlled environment with a 12:12 h light-dark cycle and given free access to standard mice chow and water. Both WT and MMP-9 null colonies were bred in-house and maintained in the same room, and the groups were examined simultaneously. All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Texas Health Science Center at San Antonio in accordance with the “Guide for the Care and Use of Laboratory Animals.”
Experimental design
WT and MMP-9 null mice were divided into four groups: no treatment (n = 10/genotype), saline treated (n = 5/genotype), acute (1 day) Pg-LPS (n = 6/genotype), and chronic (28 day) Pg-LPS (n = 20/genotype) treated. No treatment and saline-treated mice were analyzed for all echocardiogram variables, and no significant differences were detected. Based on this analysis, the no treatment and saline-treated groups were combined as a single control group and compared to Pg-LPS treated WT or null mice, for a total of six groups.
Pg-LPS infusions
Porphyromonas gingivalis-LPS (0.8 μg/g body weight/day; InvivoGen, San Diego, CA) was continuously infused using Alzet osmotic minipumps (model 2004; Durect, Cupertino, CA) implanted subcutaneously as described previously (Koch et al. 1994). The Pg-LPS dose is low, consistent with circulating levels in PD patients, and this model is not a sepsis model. Mice were anesthetized with 1–2% isoflurane in an oxygen mix. No mortality was observed, and the saline-treated group served as the negative technical control for this procedure.
Echocardiography
Transthoracic echocardiography was performed using the Visual Sonics Vevo 770 system (VisualSonics, Toronto, Canada) with a 30-MHz image transducer. Mice were anesthetized with 1–2% isoflurane in oxygen, and electrocardiogram and heart rate were monitored throughout the imaging procedure. All images were acquired at heart rates >400 bpm to maintain physiological relevance. Measurements were taken from the parasternal long-axis B- and M-mode views. For each parameter, three images from consecutive cardiac cycles were measured and averaged (Lindsey et al. 2006).
Necropsy
For tissue collection, mice were anesthetized with 1–2% isoflurane in an oxygen mix. Five minutes after heparin administration (4 U/g body weight, i.p.), blood was collected from the common carotid artery and immediately centrifuged for collection of plasma. A 1× proteinase inhibitor (Roche, Indianapolis, IN) was added to the plasma, which was stored at −80°C. Plasma samples (100 μL) were sent to Rules Based Medicine (Austin, TX) for multi-analyte proteomic profiling (Chiao et al. 2011). The coronary vasculature was flushed with cardioplegic solution (69 mmol/L NaCl; 12 mmol/L NaHCO3; 11 mmol/L glucose; 30 mmol/L 2,3-butanedione monoxime; 10 mmol/L EGTA; 0.001 mmol/L Nifedipine; 50 mmol/L KCl; and 100 U Heparin in 0.9% saline, pH 7.4). Hearts were resected, and the LV and right ventricle (RV) were separated and weighed individually. The LV was sliced into apex, middle, and base sections. The apex and base sections were placed in individual tubes and stored at −80°C for real-time polymerase chain reaction (RT2-PCR) or immunoblotting analyses. The middle section was fixed in 10% zinc formalin for histological examination.
Real-time RT2-PCR
RNA extraction was performed using TRIzol® Reagent and Purelink® RNA (Invitrogen, Grand Island, NY) according to the manufacturer's instructions. RNA levels were quantified using the NanoDrop ND-1000 Spectrophotometer (Thermo Scientific, Lafayette, CO). Reverse transcription of equal RNA content (1 μg) was performed using the RT² First Strand Kit (330401; Qiagen, Germantown, MD). Real-Time RT2-PCR gene array for Inflammatory Cytokines and Receptors (PAMM-011A; Qiagen) was performed to quantify mRNA levels. All values were normalized to the hypoxanthine guanine phosphoribosyl transferase 1 (Hprt1) housekeeping gene. Only Hprt1 was used for normalization, because this was the only housekeeping gene that does not change following injury. The mRNA levels for all genes measured are shown in Table 3.
Table 3.
Inflammatory mRNA levels evaluated in WT and MMP-9 null after exposure to saline or Pg-LPS for 1 or 28 days
| WT | MMP-9 null | |||||
|---|---|---|---|---|---|---|
| Control (n = 10) | Day 1 (n = 6) | Day 28 (n = 10) | Control (n = 10) | Day 1 (n = 6) | Day 28 (n = 10) | |
| Abcf1 | 780.0 ± 43.0 | 825.1 ± 45.2 | 756.6 ± 27.6 | 624.4 ± 66.5 | 699.2 ± 62.4 | 669.4 ± 50.9 |
| Bcl6 | 125.7 ± 11.2 | 118.7 ± 14.1 | 139.5 ± 13.9 | 130.5 ± 15.2 | 81.5 ± 9.4d | 116.8 ± 5.7e |
| C3 | 410.2 ± 69.7 | 641.1 ± 76.3a | 395.4 ± 30.5b | 411.2 ± 44.3 | 732.4 ± 35.4d | 689.7 ± 74.1cd |
| Casp1 | 49.4 ± 5.3 | 31.7 ± 2.2d | 43.7 ± 3.0 | 65.4 ± 6.4 | 42.7 ± 4.2d | 74.7 ± 6.9e |
| Ccl11 | 9.1 ± 2.1 | 24.1 ± 4.2a | 14.9 ± 3.9 | 15.0 ± 4.2 | 32.9 ± 4.2d | 15.9 ± 4.0e |
| Ccl12 | 29.2 ± 6.8 | 53.3 ± 11.3a | 19.7 ± 2.1b | 25.9 ± 4.8 | 27.3 ± 4.4 | 39.7 ± 9.9 |
| Ccl17 | 1.3 ± 0.3 | 1.3 ± 0.5 | 1.6 ± 0.2 | 1.1 ± 0.2 | 0.5 ± 0.1 | 1.1 ± 0.3 |
| Ccl19 | 109.8 ± 17.2 | 189.1 ± 31.2a | 108.0 ± 10.5b | 97.6 ± 12.1 | 210.9 ± 12.1d | 128.3 ± 18.7e |
| Ccl2 | 52.8 ± 9.2 | 56.8 ± 12.7 | 53.7 ± 7.6 | 68.5 ± 7.0 | 69.2 ± 12.5 | 81.7 ± 8.2 |
| Ccl22 | 1.2 ± 0.3 | 0.3 ± 0.1a | 0.9 ± 0.2b | 0.8 ± 0.2 | 0.4 ± 0.1 | 0.8 ± 0.2 |
| Ccl24 | 2.9 ± 0.7 | 1.9 ± 0.5 | 3.3 ± 0.5 | 2.1 ± 0.4 | 1.2 ± 0.4 | 3.2 ± 0.5 |
| Ccl25 | 8.2 ± 1.4 | 8.9 ± 1.9 | 10.2 ± 0.9 | 8.0 ± 1.0 | 12.6 ± 0.8d | 8.3 ± 1.4e |
| Ccl3 | 1.4 ± 0.3 | 1.5 ± 0.3 | 0.8 ± 0.2 | 1.6 ± 0.2 | 0.9 ± 0.3 | 2.3 ± 0.4c |
| Ccl4 | 2.7 ± 0.6 | 2.4 ± 0.4 | 2.6 ± 0.4 | 5.2 ± 0.8a | 4.8 ± 0.7b | 5.3 ± 0.7c |
| Ccl5 | 16.6 ± 2.7 | 7.7 ± 0.6a | 12.9 ± 1.4 | 28.4 ± 2.7a | 19.6 ± 2.8b | 30.0 ± 3.2c |
| Ccl6 | 13.4 ± 2.2 | 64.8 ± 6.9a | 15.9 ± 1.8b | 16.9 ± 2.1 | 41.4 ± 2.1d | 19.4 ± 3.0e |
| Ccl7 | 34.4 ± 8.7 | 73.5 ± 21.5 | 30.5 ± 4.6 | 47.2 ± 8.8 | 85.4 ± 16.0 | 53.9 ± 10.9 |
| Ccl8 | 11.1 ± 2.7 | 18.3 ± 5.0 | 11.8 ± 1.7 | 16.9 ± 4.9 | 29.1 ± 4.2 | 18.1 ± 2.7 |
| Ccl9 | 27.6 ± 4.5 | 45.9 ± 4.6a | 29.4 ± 2.7b | 24.1 ± 2.5 | 37.1 ± 6.1d | 35.0 ± 2.1d |
| Ccr1 | 12.2 ± 2.9 | 15.6 ± 2.7 | 9.6 ± 1.4 | 10.4 ± 2.0 | 10.7 ± 2.5 | 11.2 ± 1.2 |
| Ccr10 | 32.1 ± 5.2 | 15.8 ± 2.1 | 21.9 ± 4.5 | 22.1 ± 2.8 | 20.8 ± 1.8 | 32.7 ± 2.7de |
| Ccr2 | 9.1 ± 2.6 | 11.0 ± 2.7 | 9.6 ± 1.2 | 16.7 ± 2.5 | 18.6 ± 1.7 | 27.9 ± 4.4c |
| Ccr3 | 16.7 ± 4.9 | 12.6 ± 3.2 | 11.5 ± 2.2 | 18.2 ± 2.1 | 20.1 ± 1.9 | 30.7 ± 4.5cd |
| Ccr5 | 23.5 ± 5.4 | 25.3 ± 4.8 | 15.7 ± 2.9 | 21.9 ± 2.9 | 38.8 ± 9.3 | 29.1 ± 4.2 |
| Ccr7 | 4.8 ± 0.6 | 1.4 ± 0.3a | 2.5 ± 0.5a | 4.6 ± 0.9 | 2.3 ± 0.5 | 4.3 ± 0.7 |
| Ccr8 | 1.1 ± 0.3 | 0.3 ± 0.1a | 0.5 ± 0.1 | 0.6 ± 0.2 | 0.2 ± 0.1d | 0.5 ± 0.1e |
| Ccr9 | 10.1 ± 1.9 | 6.0 ± 0.9 | 8.3 ± 1.7 | 6.8 ± 0.8 | 7.3 ± 0.6 | 11.5 ± 1.7 |
| Cx3 cl1 | 114.0 ± 8.3 | 71.9 ± 4.7a | 89.9 ± 4.1 | 95.2 ± 7.3 | 91.8 ± 8.2 | 121.8 ± 12.7 |
| Cxcl1 | 5.8 ± 1.4 | 12.6 ± 3.3 | 5.8 ± 0.7 | 7.9 ± 1.2 | 21.2 ± 2.7d | 5.6 ± 1.0e |
| Cxcl10 | 5.8 ± 1.4 | 6.2 ± 2.0 | 6.2 ± 1.1 | 16.5 ± 2.5 | 8.1 ± 1.4 | 17.5 ± 4.3 |
| Cxcl11 | 1.2 ± 0.3 | 0.8 ± 0.1 | 1.6 ± 0.4 | 1.9 ± 0.4 | 1.1 ± 0.3 | 1.9 ± 0.3 |
| Cxcl12 | 1188.2 ± 106.2 | 1055.2 ± 105.2 | 1062.3 ± 102.6 | 1277.0 ± 67.8 | 1308.4 ± 41.4 | 1414.9 ± 39.9c |
| Cxcl13 | 1.9 ± 0.5 | 4.6 ± 1.2 | 3.2 ± 0.9 | 0.9 ± 0.2 | 1.2 ± 0.3 | 2.5 ± 0.6d |
| Cxcl5 | 2.6 ± 0.5 | 2.9 ± 0.5 | 1.7 ± 0.5 | 2.3 ± 0.4 | 4.0 ± 1.0 | 3.2 ± 0.6 |
| Cxcl9 | 35.3 ± 5.4 | 13.6 ± 2.3a | 24.5 ± 4.2 | 38.8 ± 11.1 | 21.2 ± 4.4 | 39.4 ± 8.3 |
| Cxcr3 | 2.2 ± 0.5 | 0.8 ± 0.3a | 1.4 ± 0.2 | 1.7 ± 0.4 | 2.7 ± 0.7b | 3.4 ± 0.6 |
| Cxcr5 | 2.4 ± 0.4 | 1.0 ± 0.1 | 1.6 ± 0.4 | 1.7 ± 0.4 | 1.2 ± 0.4 | 2.0 ± 0.5 |
| Icam1 | 68.9 ± 7.0 | 110.3 ± 16.7a | 72.9 ± 5.5b | 73.3 ± 4.5 | 74.4 ± 6.9 | 116.1 ± 6.3cde |
| Il10 | 1.6 ± 0.3 | 3.0 ± 0.3a | 1.5 ± 0.4b | 2.1 ± 0.3 | 1.7 ± 0.6 | 2.6 ± 0.6 |
| Il10ra | 19.7 ± 4.6 | 11.9 ± 2.0 | 13.4 ± 1.6 | 17.0 ± 2.9 | 16.0 ± 3.0 | 21.2 ± 1.6 |
| Il10rb | 1273.0 ± 108.3 | 1079.6 ± 47.7 | 1152.9 ± 63.5 | 944.1 ± 109.5 | 1162.0 ± 159.5 | 1014.8 ± 60.1 |
| Il11 | 1.6 ± 0.2 | 0.7 ± 0.1a | 1.2 ± 0.2 | 1.2 ± 0.4 | 1.0 ± 0.2 | 1.1 ± 0.3 |
| Il13ra1 | 231.2 ± 15.5 | 241.9 ± 15.8 | 206.3 ± 19.1 | 225.8 ± 21.6 | 177.9 ± 29.5 | 202.4 ± 13.4 |
| Il15 | 310.0 ± 27.7 | 329.8 ± 20.1 | 265.4 ± 23.5 | 278.1 ± 31.6 | 309.9 ± 27.9 | 296.2 ± 21.4 |
| Il16 | 25.2 ± 3.8 | 13.5 ± 1.7 | 22.7 ± 3.7 | 21.4 ± 3.0 | 15.2 ± 1.9 | 27.4 ± 3.0 |
| Il18 | 3.6 ± 1.0 | 5.0 ± 1.3 | 5.6 ± 0.6 | 7.9 ± 1.3 | 7.8 ± 1.0 | 7.9 ± 1.0 |
| Il1a | 1.9 ± 0.3 | 1.3 ± 0.4 | 1.3 ± 0.3 | 1.2 ± 0.3 | 0.6 ± 0.2 | 1.0 ± 0.2 |
| Il1b | 9.9 ± 2.1 | 7.4 ± 2.4 | 6.6 ± 1.0 | 8.5 ± 1.4 | 11.5 ± 1.7 | 9.7 ± 2.1 |
| Il1r1 | 97.0 ± 7.3 | 88.9 ± 4.8 | 87.7 ± 6.0 | 79.6 ± 4.0 | 106.5 ± 11.4d | 106.5 ± 6.0d |
| Il1r2 | 3.8 ± 1.3 | 7.9 ± 1.2a | 2.6 ± 0.6b | 2.5 ± 0.6 | 5.7 ± 0.4d | 3.1 ± 0.6e |
| Il2rb | 3.6 ± 0.8 | 1.9 ± 0.4 | 1.7 ± 0.4 | 3.3 ± 0.5 | 2.1 ± 0.4 | 4.0 ± 0.4c |
| Il2rg | 85.2 ± 7.2 | 62.6 ± 3.1a | 62.4 ± 5.1a | 82.4 ± 6.6 | 70.1 ± 3.4 | 103.6 ± 9.7c |
| Il4 | 1.3 ± 0.2 | 0.5 ± 0.1a | 0.7 ± 0.1a | 0.6 ± 0.1 | 0.7 ± 0.2 | 1.2 ± 0.2cde |
| Il6ra | 46.2 ± 6.0 | 51.1 ± 2.5 | 47.2 ± 2.8 | 46.2 ± 5.1 | 65.9 ± 6.4d | 55.3 ± 3.8e |
| Il6st | 857.9 ± 74.5 | 1158.8 ± 49.7a | 795.9 ± 28.9b | 587.9 ± 65.3 | 988.4 ± 84.3d | 710.6 ± 52.6e |
| Il8rb | 1.8 ± 0.3 | 5.8 ± 1.3a | 3.2 ± 0.7 | 1.7 ± 0.7 | 3.9 ± 1.2 | 3.5 ± 0.9 |
| Itgam | 44.6 ± 8.6 | 64.9 ± 5.2 | 33.4 ± 4.2 | 40.7 ± 4.8 | 73.7 ± 10.6d | 57.9 ± 4.9c |
| Mif | 1590.8 ± 171.0 | 1430.5 ± 131.8 | 1561.5 ± 99.6 | 909.2 ± 162.0 | 1141.3 ± 118.7 | 1151.8 ± 153.2 |
| Pf4 | 110.5 ± 14.4 | 145.3 ± 13.0 | 87.0 ± 7.2 | 92.1 ± 15.4 | 114.2 ± 13.3 | 113.2 ± 10.5 |
| Scye1 | 871.3 ± 86.5 | 932.5 ± 60.1 | 881.4 ± 42.0 | 624.9 ± 65.6 | 836.7 ± 47.6 | 726.6 ± 60.1 |
| Spp1 | 0.5 ± 0.3 | 1.3 ± 0.3a | 0.4 ± 0.1 | 0.2 ± 0.2 | 0.9 ± 0.3d | 0.5 ± 0.2 |
| Tgfb1 | 211.9 ± 13.4 | 178.2 ± 13.2 | 200.8 ± 15.9 | 232.5 ± 15.1 | 185.2 ± 15.1 | 260.1 ± 17.4c |
| Tnf | 3.9 ± 1.0 | 1.4 ± 0.4 | 2.7 ± 0.5 | 5.5 ± 0.8 | 2.6 ± 0.4 | 6.3 ± 1.2 |
| Tnfrsf1a | 122.6 ± 15.7 | 156.6 ± 6.8 | 95.5 ± 4.5 | 85.0 ± 14.8 | 135.7 ± 12.1 | 112.8 ± 17.4 |
| Tnfrsf1b | 36.3 ± 2.5 | 42.7 ± 3.2 | 40.0 ± 4.1 | 30.4 ± 3.5 | 40.3 ± 6.1 | 47.1 ± 3.4d |
| Tollip | 296.4 ± 32.8 | 331.1 ± 8.1 | 258.1 ± 12.8 | 218.0 ± 14.0 | 297.8 ± 29.4d | 235.4 ± 10.1 |
| Xcr1 | 1.9 ± 0.6 | 0.6 ± 0.1 | 1.2 ± 0.3 | 2.2 ± 0.6 | 1.2 ± 0.3 | 2.7 ± 0.6 |
aVersus WT control, bversus WT 1d, cversus WT 28d, dversus Null control, and eversus Null 1d; Values are means ± SEM. Not detectable: Ccl1, Ccl20, Ccr4, Ccr6, Cd40lg, CRP, Cxcl15, Ifng, Il13, Il17b, Il1f6, Il1f8, Il20, Il3, Il5ra, Lta, and Ltb.
Protein extraction
Total protein was extracted by homogenizing the samples sequentially in phosphate-buffered saline (PBS) with 1× protease inhibitor cocktail, and in protein extraction reagent type 4 (7 mol/L urea, 2 mol/L thiourea, 40 mmol/L Trizma® base, and the detergent 1% C7BzO; Sigma, St. Louis, MO) with 1× protease inhibitor cocktail. Protein concentrations were determined by the Quick Start™ Bradford Protein Assay (Bio-Rad, Hercules, CA).
Immunoblotting and ELISA
Plasma protein expression of cardiac troponin I (cTnI) was quantified by immunoblotting (ab19615; 1:1000; Abcam, Cambridge, MA). LV protein expression levels were quantified by immunoblotting using antibodies for MMP-9 (ab38898; 1:1000; Abcam) and major histocompatibility complex (MHC) class II (MABF33; 1:1000; Millipore, Billerica, MA). Total protein (10 μg) was separated on 4–12% Criterion™ XT Bis-Tris gels (Bio-Rad), transferred to a nitrocellulose membrane (Bio-Rad), and stained using MemCode™ Reversible Protein Stain Kit (Thermo Scientific) to verify protein concentration and loading accuracy. After blocking with 5% nonfat milk (Bio-Rad), the membrane was incubated with primary antibody, secondary antibody (PI-1000, 1:5000; Vector Laboratories, Burlingame, CA), and detected with ECL Prime Western Blotting Detection Substrate (GE Healthcare Life Sciences, Pittsburgh, PA) or SuperSignal West Femto Chemiluminescent Substrate (Thermo Scientific). Protein levels were quantified by densitometry using the ImageQuant-TL image analysis software (GE Healthcare). The densitometry of the entire lane of the total protein-stained membrane was used for individual lane loading normalization. The relative expression for each immunoblot was calculated as the densitometry of the protein of interest divided by the densitometry of the entire lane of the total protein-stained membrane.
As a secondary confirmation of the cTnI immunoblotting results, an ELISA for cTnI was performed on undiluted plasma collected from each time point using the Life Diagnostics UltraSensitive ELISA Kit for Mouse Plasma (#0.039-2.5). The ELISA was performed according to manufacturer instructions.
Histology
The middle section of the LV (mid-papillary region) was embedded in paraffin and sectioned at 5 μm. Immunohistochemistry was conducted using the Vectastain ABC Kit (Vector Laboratories). An antibody specific for macrophages (Mac-3, Cedarlane CL8943AP; 1:100) was used to stain macrophages. HistoMark Black (54-75-00; KPL, Gaithersburg, MD) was used to visualize positive staining, with eosin as a counterstain. Negative controls were incubated with no primary antibody. For each LV section, five 60× magnification images were captured. The percentage of the macrophage area was measured using Image-Pro Plus version 6.2 (Media Cybernetics, Inc., Warrendale, PA).
Statistical analyses
Data are presented as mean ± SEM. Multiple group comparisons were analyzed by one-way analysis of variance (ANOVA), followed by the Student–Newman–Keuls when the Bartlett's variation test passed, or by the nonparametric Kruskal–Wallis test, followed by Dunn post hoc test when the Bartlett's variation test did not pass. P < 0.05 was considered statistically significant.
Results
MMP-9 increased twofold after Pg-LPS exposure in WT
Matrix metalloproteinase-9 protein levels were determined in the LV of WT animals by immunoblotting. Pro-MMP-9 values doubled after 1 day of Pg-LPS exposure (Fig. 1; P < 0.05) and returned to baseline levels at day 28. Active MMP-9 protein levels increased by 10-fold compared to control at day 28, demonstrating that chronic exposure to Pg-LPS increased MMP-9 activity.
Figure 1.
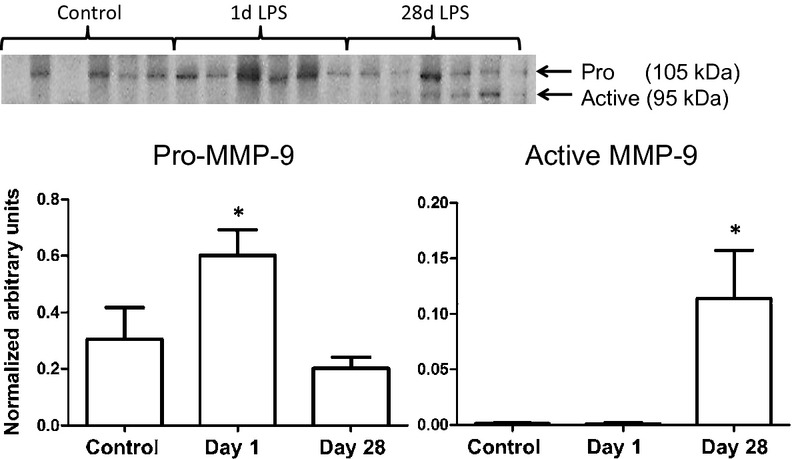
Immunoblotting of MMP-9 in WT and MMP-9 null mice exposed to saline or Pg-LPS for 1 or 28 days indicated circulating Pg-LPS induces a robust increase of MMP-9 levels in the left ventricle. proMMP-9 levels increased in WT mice with acute exposure to Pg-LPS; while active MMP-9 was strongly higher after chronic exposure to Pg-LPS. *P < 0.05 versus control, n = 6/group.
Pg-LPS reduced LV function, and this decrease in function was attenuated in MMP-9 null mice
Cardiac function was measured in WT and MMP-9 null mice using two-dimensional echocardiography (Table 1). MMP-9 null mice showed no overt cardiac dysfunction at baseline, as expected based on previous reports (Ducharme et al. 2000; Lindsey et al. 2006). After 28 days of Pg-LPS exposure, fractional shortening decreased slightly yet significantly in WT, but not in null mice. This decrease in fractional shortening was due to an increase in end systolic dimension (ESD), indicating that systolic dysfunction was the driving force for the decline in cardiac function. Wall thinning was not observed in WT or MMP-9 null mice after Pg-LPS exposure.
Table 1.
LV systolic dysfunction occurs in WT mice, but not MMP-9 Null, exposed to Pg-LPS
| WT | MMP-9 null | |||||
|---|---|---|---|---|---|---|
| Control (n = 15) | Day 1 (n = 6) | Day 28 (n = 20) | Control (n = 15) | Day 1 (n = 6) | Day 28 (n = 20) | |
| Heart rate (bpm) | 469 ± 9 | 448 ± 12 | 445 ± 6 | 446 ± 6 | 461 ± 11 | 454 ± 9 |
| LV wall thickness (mm) | 1.23 ± 0.04 | 1.19 ± 0.03 | 1.14 ± 0.02 | 1.25 ± 0.04 | 1.16 ± 0.04 | 1.19 ± 0.03 |
| End diasolic dimension (mm) | 3.50 ± 0.11 | 3.22 ± 0.16 | 3.69 ± 0.07 | 3.50 ± 0.08 | 3.21 ± 0.08 | 3.68 ± 0.08 |
| End systolic dimension (mm) | 2.28 ± 0.08 | 2.05 ± 0.15 | 2.57 ± 0.071 | 2.27 ± 0.08 | 2.00 ± 0.08 | 2.44 ± 0.07 |
| Fractional shortening% | 35 ± 1 | 37 ± 1 | 31 ± 11 | 35 ± 1 | 38 ± 2 | 34 ± 12 |
Values are means ± SEM.
P < 0.05 versus respective baseline.
P < 0.05 versus WT 28 day LPS. LV, left ventricle; WT, wild type; MMP-9, matrix metalloproteinase-9; Pg-LPS, Porphyromonas gingivalis lipopolysaccharide.
Chronic Pg-LPS exposure resulted in myocardial damage in WT, as shown by an increased release of cardiac troponin I in plasma
In order to further understand the decrease in cardiac function in the WT, plasma levels of cTnI were measured by immunoblotting and ELISA. cTnI is a regulatory protein that controls the calcium-mediated interaction between actin and myosin, and the release of cTnI in the plasma positively correlates with myocardial damage (Adams et al. 1993; Hessel et al. 2008). In the 1 day exposure group, there was no significant change in plasma levels of cTnI in both WT and MMP-9 null mice (Fig. 2). However, by day 28, WT but not null mice showed a slight but significant increase in plasma cTnI indicating chronic exposure of Pg-LPS caused myocardial damage consistent with the decrease in the contractility of the myocardium.
Figure 2.
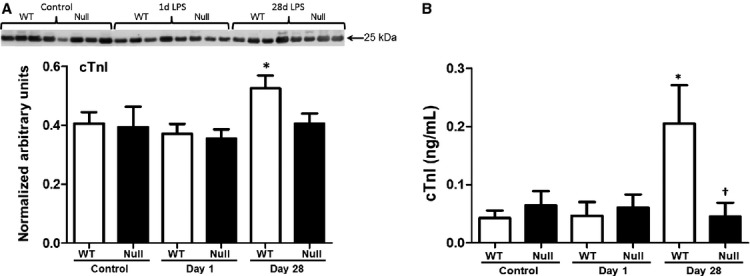
Plasma cardiac troponin I (cTnI) increased in WT at day 28 but not Null as shown by immunoblotting (A) and ELISA (B). *P < 0.05 versus baseline, n = 6/group.
Acute Pg-LPS exposure resulted in significant changes at the gene level in LV in both WT and MMP-9 null mice
Out of 84 inflammatory genes analyzed, only six (Ccl11, Ccl19, Ccl6, Il1r2, Il6st and Spp1) were significantly increased in the LV of both WT and MMP-9 null mice compared to their respective controls (Tables 2 and 3). Of note, these genes were increased only at the 1 day exposure time point. In addition, two genes were decreased at day 1 in both genotypes compared to controls: Casp1 and Ccr8. All eight genes are associated with periodontal health, indicating that the Pg-LPS dose given is representative of changes seen in the setting of PD (McGrory et al. 2004; Sharma and Pradeep 2006; Gemmell et al. 2007; Silva et al. 2007).
Table 2.
Inflammatory Gene Array of WT and MMP-9 null LV. Out of 84 genes evaluated, the 20 shown here were Pg-LPS responsive
| WT | MMP-9 null | |||||
|---|---|---|---|---|---|---|
| Control (n = 10) | Day 1 (n = 6) | Day 28 (n = 10) | Control (n = 10) | Day 1 (n = 6) | Day 28 (n = 10) | |
| C3 | 410.2 ± 69.7 | 641.1 ± 76.3a | 395.4 ± 30.5b | 411.2 ± 44.3 | 732.4 ± 35.4d | 689.7 ± 74.1cd |
| Casp1 | 49.4 ± 5.3 | 31.7 ± 2.2a | 43.7 ± 3.0 | 65.4 ± 6.4 | 42.7 ± 4.2d | 74.7 ± 6.9e |
| Ccl11 | 9.1 ± 2.1 | 24.1 ± 4.2a | 14.9 ± 3.9 | 15.0 ± 4.2 | 32.9 ± 4.2d | 15.9 ± 4.0e |
| Ccl12 | 29.2 ± 6.8 | 53.3 ± 11.3a | 19.7 ± 2.1b | 25.9 ± 4.8 | 27.3 ± 4.4 | 39.7 ± 9.9 |
| Ccl19 | 109.8 ± 17.2 | 189.1 ± 31.2a | 108.0 ± 10.5b | 97.6 ± 12.1 | 210.9 ± 12.1d | 128.3 ± 18.7e |
| Ccl25 | 8.2 ± 1.4 | 8.9 ± 1.9 | 10.2 ± 0.9 | 8.0 ± 1.0 | 12.6 ± 0.8d | 8.3 ± 1.4e |
| Ccl6 | 13.4 ± 2.2 | 64.8 ± 6.9a | 15.9 ± 1.8b | 16.9 ± 2.1 | 41.4 ± 2.1b | 19.4 ± 3.0e |
| Ccl9 | 27.6 ± 4.5 | 45.9 ± 4.6a | 29.4 ± 2.7b | 24.1 ± 2.5 | 37.1 ± 6.1d | 35.0 ± 2.1d |
| Ccr3 | 16.7 ± 4.9 | 12.6 ± 3.2 | 11.5 ± 2.2 | 18.2 ± 2.1 | 20.1 ± 1.9 | 30.7 ± 4.5cd |
| Ccr8 | 1.1 ± 0.3 | 0.3 ± 0.1a | 0.5 ± 0.1 | 0.6 ± 0.2 | 0.2 ± 0.1d | 0.5 ± 0.1e |
| Cxcl1 | 5.8 ± 1.4 | 12.6 ± 3.3a | 5.8 ± 0.7b | 7.9 ± 1.2 | 21.2 ± 2.7d | 5.6 ± 1.0e |
| Cxcl13 | 1.9 ± 0.5 | 4.6 ± 1.2 | 3.2 ± 0.9 | 0.9 ± 0.2 | 1.2 ± 0.3 | 2.5 ± 0.6d |
| Icam1 | 68.9 ± 7.0 | 110.3 ± 16.7a | 72.9 ± 5.5b | 73.3 ± 4.5 | 74.4 ± 6.9 | 116.1 ± 6.3cde |
| Il1r2 | 3.8 ± 1.3 | 7.9 ± 1.2a | 2.6 ± 0.6b | 2.5 ± 0.6 | 5.7 ± 0.4d | 3.1 ± 0.6e |
| Il4 | 1.3 ± 0.2 | 0.5 ± 0.1a | 0.7 ± 0.1a | 0.6 ± 0.1 | 0.7 ± 0.2 | 1.2 ± 0.2cde |
| Il6ra | 46.2 ± 6.0 | 51.1 ± 2.5 | 47.2 ± 2.8 | 46.2 ± 5.1 | 65.9 ± 6.4d | 55.3 ± 3.8d |
| Il6st | 857.9 ± 74.5 | 1158.8 ± 49.7a | 795.9 ± 28.9b | 587.9 ± 65.3 | 988.4 ± 84.3d | 710.6 ± 52.6e |
| Il8rb | 1.8 ± 0.3 | 5.8 ± 1.3a | 3.2 ± 0.7 | 1.7 ± 0.7 | 3.9 ± 1.2 | 3.5 ± 0.9 |
| Spp1 | 0.5 ± 0.3 | 1.3 ± 0.3a | 0.4 ± 0.1 | 0.2 ± 0.2 | 0.9 ± 0.3d | 0.5 ± 0.2 |
| Tnfrsf1b | 36.3 ± 2.5 | 42.7 ± 3.2 | 40.0 ± 4.1 | 30.4 ± 3.5 | 40.3 ± 6.1 | 47.1 ± 3.4d |
aVersus WT control, bversus WT 1d, cversus WT 28d, dversus Null control, eversus Null 1d; Values are means ± SEM. Casp1, caspase 1; Ccl, CC chemokine ligand; Ccr, CC chemokine receptor; Icam1, intracellular adhesion molecule 1; Il1r2, interleukin 1 receptor 2; Il6st, interleukin 6 signal transducer; Spp1, osteopontin; Tnfrsf1b, tumor necrosis factor receptor super family 1b.
Acute systemic inflammation resulted in increased macrophage numbers in the LV
Macrophage numbers increased at day 1 in both WT and MMP-9 null groups. By day 28, macrophage numbers decreased to baseline levels in the WT but remained elevated in the null mice, giving evidence that MMP-9 deletion provided sustained defense against Pg-LPS exposure (Fig. 3). In addition, expression of Itgb2 in the LV of WT and null mice increased at day 1 but decreased back to baseline levels in the WT and not Null (Fig. 3C).
Figure 3.
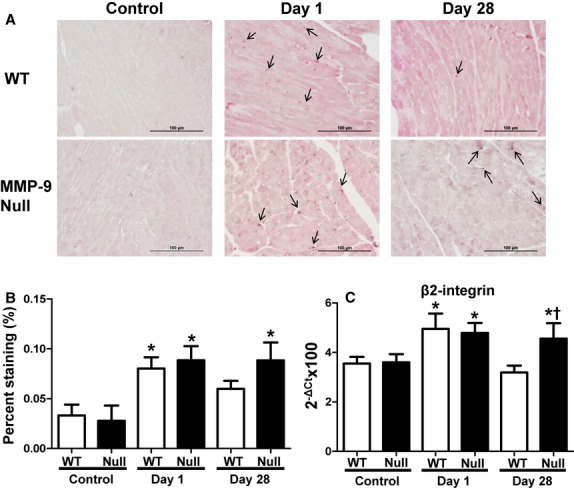
Mac3 staining and Itgb2 expression of WT and MMP-9 null mice exposed to saline or Pg-LPS for 1 or 28 days show macrophages remain elevated in the LV of MMP-9 null but not WT mice, after 28 days exposure to Pg-LPS. (A) representative images; (B) quantification of macrophages in each group n = 6/group; (C) Itgb2 mRNA levels in the LV. *P < 0.05 versus baseline, †P < 0.05 versus WT day 28, n = 6/group.
Chronic Pg-LPS exposure induced macrophage immunosuppression in WT
To determine how MMP-9 regulated the inflammatory response after Pg-LPS exposure, we performed multi-analyte proteomic profiling on plasma and real-time RT2-PCR cytokine gene array on the LV (Fig. 4 and Table 2). After 1 day of Pg-LPS exposure, several factors related to macrophage recruitment and activation increased at least twofold in WT mice compared to controls (all P < 0.05), including plasma analytes: C-reactive protein (CRP), CD40 ligand, growth-regulated α-protein (GROα-protein), macrophage inflammatory protein (MIP)-1α, -1γ, -2, monocyte chemotactic protein (MCP)-3, -5, stem cell factor (SCF), and LV genes: C3, Ccl12/MCP-5, Ccl9-MIP-1γ, Ccl19/MIP-3β, Cxcl1/GROα, Il8rb, Icam1, Itgb1, and Itgb2 (De Nichilo and Burns 1993; Koch et al. 1994; Mantovani et al. 2004; Okada et al. 2009; Cassetta et al. 2011; Nelson et al. 2011). However, after 28 days of Pg-LPS exposure these factors returned to baseline levels in the WT, suggesting immune response suppression or endotoxin tolerance (Roth et al. 1994; West and Koons 2008).
Figure 4.

Plasma proteomic profiling of WT and MMP-9 null mice exposed to saline or Pg-LPS for 1 or 28 days. Out of 49 analytes evaluated, the 9 shown here were different among groups. *P < 0.05 versus baseline, †P < 0.05 versus WT day 28, n ≥ 6/group.
Similar to WT groups, macrophage activation or recruitment plasma analytes (CRP, CD40 ligand, GROα-protein, MIP-1α, -1γ, -2, MCP-3, -5, SCF) and LV genes (C3, Ccl9/MIP-1γ, Ccl11/eotaxin-1, Ccl19/MIP-3β, Ccl25, Cxcl1/GROα, Il6ra, Icam1, and Itgb2, P < 0.05 for all compared to control) increased in null mice after 1-day exposure. Of these, CRP, GROα-protein, MCP-5, MIP-1α, MIP-1γ, SCF, C3, Ccl9, Il6ra, and Itgb2 remained elevated with chronic exposure to Pg-LPS, and Ccr3, Cxcl13, Il4, and Tnfrsf1b had significantly higher expression levels at day 28 compared to baseline control. That Ccl9/MIP-1γ and Cxcl1/GROα had a similar pattern at both the systemic and local levels in both WT and MMP-9 null groups give evidence of sustained inflammatory response in the Null LV after chronic exposure to Pg-LPS.
Interestingly, Il-1α increased in WT but not in null mice as shown in Figure 5. Previous studies have indicated Il-1α to have a role in immunosuppression of macrophages (Tomioka et al. 1996). The increase in plasma Il-1α in WT could explain the reduced macrophage recruitment found in the LV at day 28. Overexpression of these plasma analytes and genes in the null group suggests that MMP-9 deletion has a cardioprotective role against chronic Pg-LPS exposure, by sustaining macrophage infiltration and the inflammatory defense system.
Figure 5.
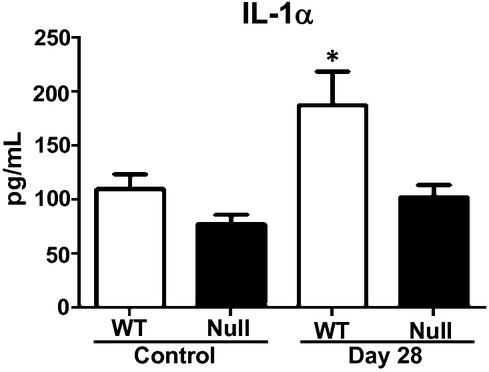
Plasma interleukin-1α (Il-1α) of WT and MMP-9 null mice exposed to saline or Pg-LPS for 1 or 28 days show increased levels in WT at day 28 but not Null. *P < 0.05 versus respective baseline, n ≥ 6/group.
WT mice showed desensitization to the LPS stimulus by day 28
Major histocompatibility complex class II is involved in presentation of extracellular pathogens. Under normal inflammatory conditions, MHC class II expression increase in order to stimulate the immune response. However, during endotoxin tolerance antigen presentation is decreased compared to nontolerant macrophages (Wolk et al. 2000, 2003). We quantified MHC class II protein levels to evaluate if WT mice developed tolerance to Pg-LPS exposure, as suggested by the plasma proteomic profiling and LV gene array results. MHC class II protein values were increased in the null (P < 0.05) but not in the WT mice at day 28 (Fig. 6). Because MMP-9 null animals showed lower protein levels at baseline, we normalized protein values to their respective baseline values. Lower levels of MHC class II are associated with impaired antigen presentation capacity causing a reduction in the reparative immune response (McGrory et al. 2004; Nelson et al. 2011).
Figure 6.
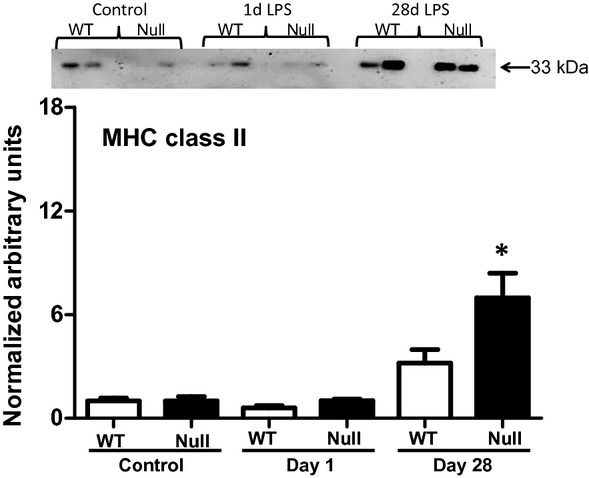
MHC class II immunoblotting of WT and MMP-9 null mice exposed to saline or Pg-LPS for 1 or 28 days reveals MHC class II increased in MMP-9 null but not WT after 28 days Pg-LPS exposure. *P < 0.05 versus baseline, n = 6/group.
Discussion
The goal of this study was to investigate the mechanisms that underlie the connection between PD and CVD incidence. Our data show that circulating Pg-LPS (1) increased MMP-9 protein levels in the WT LV resulting in myocardial damage and a decrease in LV function, (2) stimulated a strong systemic response after 1 day exposure in the WT and MMP-9 null animals, (3) increased macrophage numbers in the LV of both genotypes, and (4) induced desensitization in the WT group after chronic exposure that was MMP-9-dependent. These data suggest that PD plays a key role in CVD by inducing a strong inflammatory response that is regulated by MMP-9 (Fig. 7).
Figure 7.
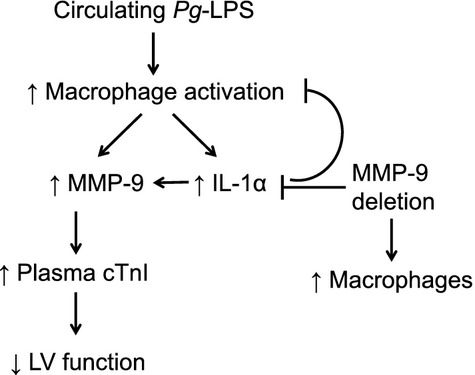
Schematic of MMP-9-dependent Pg-LPS effects in cardiac function.
MMP-9 levels have been correlated with an increase in plasma levels of cTnI (Manginas et al. 2005; Uzuelli et al. 2008), and increased levels of plasma cTnI are associated with myocardial damage (Adams et al. 1993). After 28 days of Pg-LPS exposure, our data show that MMP-9 and plasma cTnI increased in WT only. This increase was accompanied by a decrease in fractional shortening, suggesting that Pg-LPS exposure induces LV dysfunction through MMP-9 effects on systolic function. This finding is in conflict with findings published in Ashigaki et al., however in this study the exposure to P. gingivalis is only 14 days compared to our study which exposed the mice for 28 days to Pg-LPS (Ashigaki et al. 2013). This suggests that longer exposure time may be needed to induce cardiac dysfunction through increased MMP-9.
Circulating Pg-LPS has been shown to increase the inflammatory response and is believed to be the source of CVD risk in PD patients (Scannapieco et al. 2003; Pussinen et al. 2007). This study showed that the Pg-LPS exposure induced a strong inflammatory response in both WT and MMP-9 null. This was evidenced by high levels of inflammatory markers in plasma, and increased expression of proinflammatory cytokines in the LV. Interestingly, plasma and gene levels of inflammatory markers associated with macrophage recruitment and activation returned to baseline values with chronic exposure to Pg-LPS in the WT group, but remained elevated in the Null. Although this increase is modest in the Null group, it is sufficient to help the immune system combat the infection without causing myocardial damage.
Consistent with plasma and gene array results acute exposure increased macrophage numbers in both WT and MMP-9 null mice. Continuous exposure to Pg-LPS caused lower numbers of macrophages in the WT LV at day 28. This was not observed in the null mice, where values remained elevated from day 1 to day 28 compared to control. The increase in plasma Il-1α observed in the WT mice at day 28 most likely played a role in the decrease on macrophage recruitment/activation after chronic exposure, as Il-1α can induce immunosuppression by causing a decrease in antigen presentation on macrophages after chronic microbial exposure (Tomioka et al. 1996).
Based on these results, chronic exposure to Pg-LPS induced endotoxin tolerance or desensitization in the WT group. This was confirmed by an increase in MHC class II protein levels in the LV of MMP-9 null but not WT mice. MMP-9 deletion prevented Pg-LPS tolerance by attenuating the increase in plasma Il-1α and increasing MHC class II in the LV after 28 days of exposure, which resulted in a prolonged inflammatory response but better cardiac function.
In conclusion, PD induces increased risk of CVD by altering the inflammatory response in a MMP-9-dependent manner. MMP-9 plays a major role in the regulation of chronic systemic inflammation and associated cardiac dysfunction. Future studies evaluating the effect of preexisting PD on cardiac injury responses are warranted. Most notably phenotyping of the infiltrated cells would allow for better mechanistic insights on how PD induces cardiac injury. It would be interesting to look at leukocytes obtained from infected mice and determine if antigen-presentation and T-cell stimulatory capability were increased by deletion of MMP-9.
Author Contributions
All authors designed the research, analyzed results, and wrote the manuscript; K. Y. D.-P. performed the experiments.
References
- Adams JE, 3rd, Bodor GS, Davila-Roman VG, Delmez JA, Apple FS, Ladenson JH, et al. Cardiac troponin I. A marker with high specificity for cardiac injury. Circulation. 1993;88:101–106. doi: 10.1161/01.cir.88.1.101. [DOI] [PubMed] [Google Scholar]
- Ashigaki N, Suzuki J, Ogawa M, Watanabe R, Aoyama N, Kobayashi N, et al. Periodontal bacteria aggravate experimental autoimmune myocarditis in mice. Am. J. Physiol. Heart Circ. Physiol. 304:H740–H748. doi: 10.1152/ajpheart.00634.2012. [DOI] [PubMed] [Google Scholar]
- Bradley LM, Douglass MF, Chatterjee D, Akira S, Baaten BJ. Matrix metalloprotease 9 mediates neutrophil migration into the airways in response to influenza virus-induced toll-like receptor signaling. PLoS Pathog. 2012;8:e1002641. doi: 10.1371/journal.ppat.1002641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassetta L, Cassol E, Poli G. Macrophage polarization in health and disease. ScientificWorldJournal. 2011;11:2391–2402. doi: 10.1100/2011/213962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiao YA, Dai Q, Zhang J, Lin J, Lopez EF, Ahuja SS, et al. Multi-analyte profiling reveals matrix metalloproteinase-9 and monocyte chemotactic protein-1 as plasma biomarkers of cardiac aging. Circ. Cardiovasc. Genet. 2011;4:455–462. doi: 10.1161/CIRCGENETICS.111.959981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nichilo MO, Burns GF. Granulocyte-macrophage and macrophage colony-stimulating factors differentially regulate alpha v integrin expression on cultured human macrophages. Proc. Natl Acad. Sci. USA. 1993;90:2517–2521. doi: 10.1073/pnas.90.6.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J. Clin. Invest. 2000;106:55–62. doi: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemmell E, Yamazaki K, Seymour GJ. The role of T cells in periodontal disease: homeostasis and autoimmunity. Periodontol 2000. 2007;43:14–40. doi: 10.1111/j.1600-0757.2006.00173.x. [DOI] [PubMed] [Google Scholar]
- Gitlin JM, Loftin CD. Cyclooxygenase-2 inhibition increases lipopolysaccharide-induced atherosclerosis in mice. Cardiovasc. Res. 2009;81:400–407. doi: 10.1093/cvr/cvn286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffen AL, Becker MR, Lyons SR, Moeschberger ML, Leys EJ. Prevalence of Porphyromonas gingivalis and periodontal health status. J. Clin. Microbiol. 1998;36:3239–3242. doi: 10.1128/jcm.36.11.3239-3242.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Lee HM, Sorsa T, Salminen A, Ryan ME, Slepian MJ, et al. Non-antibacterial tetracyclines modulate mediators of periodontitis and atherosclerotic cardiovascular disease: a mechanistic link between local and systemic inflammation. Pharmacol. Res. 2011;64:573–579. doi: 10.1016/j.phrs.2011.06.023. [DOI] [PubMed] [Google Scholar]
- Hessel MH, Atsma DE, Bax EJ, van der Valk WH, Schalij MJ, van der Laarse A. Release of cardiac troponin I from viable cardiomyocytes is mediated by integrin stimulation. Pflugers Arch. 2008;455:979–986. doi: 10.1007/s00424-007-0354-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey LL, Fu R, Buckley DI, Freeman M, Helfand M. Periodontal disease and coronary heart disease incidence: a systematic review and meta-analysis. J. Gen. Intern. Med. 2008;23:2079–2086. doi: 10.1007/s11606-008-0787-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaisare S, Rao J, Dubashi N. Periodontal disease as a risk factor for acute myocardial infarction. A case-control study in Goans highlighting a review of the literature. Br. Dent. J. 2007;203 doi: 10.1038/bdj.2007.582. E5; discussion 144–145. [DOI] [PubMed] [Google Scholar]
- Koch AE, Kunkel SL, Harlow LA, Mazarakis DD, Haines GK, Burdick MD, et al. Macrophage inflammatory protein-1 alpha. A novel chemotactic cytokine for macrophages in rheumatoid arthritis. J. Clin. Invest. 1994;93:921–928. doi: 10.1172/JCI117097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey ML, Escobar GP, Dobrucki LW, Goshorn DK, Bouges S, Mingoia JT, et al. Matrix metalloproteinase-9 gene deletion facilitates angiogenesis after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2006;290:H232–H239. doi: 10.1152/ajpheart.00457.2005. [DOI] [PubMed] [Google Scholar]
- Luttun A, Lutgens E, Manderveld A, Maris K, Collen D, Carmeliet P, et al. Loss of matrix metalloproteinase-9 or matrix metalloproteinase-12 protects apolipoprotein E-deficient mice against atherosclerotic media destruction but differentially affects plaque growth. Circulation. 2004;109:1408–1414. doi: 10.1161/01.CIR.0000121728.14930.DE. [DOI] [PubMed] [Google Scholar]
- Manginas A, Bei E, Chaidaroglou A, Degiannis D, Koniavitou K, Voudris V, et al. Peripheral levels of matrix metalloproteinase-9, interleukin-6, and C-reactive protein are elevated in patients with acute coronary syndromes: correlations with serum troponin I. Clin. Cardiol. 2005;28:182–186. doi: 10.1002/clc.4960280405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- McGrory K, Flaitz CM, Klein JR. Chemokine changes during oral wound healing. Biochem. Biophys. Res. Commun. 2004;324:317–320. doi: 10.1016/j.bbrc.2004.09.056. [DOI] [PubMed] [Google Scholar]
- Nelson MP, Christmann BS, Werner JL, Metz AE, Trevor JL, Lowell CA, et al. IL-33 and M2a alveolar macrophages promote lung defense against the atypical fungal pathogen Pneumocystis murina. J. Immunol. 2011;186:2372–2381. doi: 10.4049/jimmunol.1002558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M, Saio M, Kito Y, Ohe N, Yano H, Yoshimura S, et al. Tumor-associated macrophage/microglia infiltration in human gliomas is correlated with MCP-3, but not MCP-1. Int. J. Oncol. 2009;34:1621–1627. doi: 10.3892/ijo_00000292. [DOI] [PubMed] [Google Scholar]
- Pussinen PJ, Tuomisto K, Jousilahti P, Havulinna AS, Sundvall J, Salomaa V. Endotoxemia, immune response to periodontal pathogens, and systemic inflammation associate with incident cardiovascular disease events. Arterioscler. Thromb. Vasc. Biol. 2007;27:1433–1439. doi: 10.1161/ATVBAHA.106.138743. [DOI] [PubMed] [Google Scholar]
- Roth J, McClellan JL, Kluger MJ, Zeisberger E. Attenuation of fever and release of cytokines after repeated injections of lipopolysaccharide in guinea-pigs. J. Physiol. 1994;477:177–185. doi: 10.1113/jphysiol.1994.sp020182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannapieco FA, Bush RB, Paju S. Associations between periodontal disease and risk for nosocomial bacterial pneumonia and chronic obstructive pulmonary disease. A systematic review. Ann. Periodontol. 2003;8:54–69. doi: 10.1902/annals.2003.8.1.54. [DOI] [PubMed] [Google Scholar]
- Sharma CG, Pradeep AR. Gingival crevicular fluid osteopontin levels in periodontal health and disease. J. Periodontol. 2006;77:1674–1680. doi: 10.1902/jop.2006.060016. [DOI] [PubMed] [Google Scholar]
- Silva TA, Garlet GP, Fukada SY, Silva JS, Cunha FQ. Chemokines in oral inflammatory diseases: apical periodontitis and periodontal disease. J. Dent. Res. 2007;86:306–319. doi: 10.1177/154405910708600403. [DOI] [PubMed] [Google Scholar]
- Tomioka H, Maw WW, Sato K, Saito H. The role of tumour necrosis factor-alpha in combination with interferon-gamma or interleukin-1 in the induction of immunosuppressive macrophages because of Mycobacterium avium complex infection. Immunology. 1996;88:61–67. doi: 10.1046/j.1365-2567.1996.d01-654.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonetti MS, D'Aiuto F, Nibali L, Donald A, Storry C, Parkar M, et al. Treatment of periodontitis and endothelial function. N. Engl. J. Med. 2007;356:911–920. doi: 10.1056/NEJMoa063186. [DOI] [PubMed] [Google Scholar]
- Uzuelli JA, Dias-Junior CA, Tanus-Santos JE. Severity dependent increases in circulating cardiac troponin I and MMP-9 concentrations after experimental acute pulmonary thromboembolism. Clin. Chim. Acta. 2008;388:184–188. doi: 10.1016/j.cca.2007.11.001. [DOI] [PubMed] [Google Scholar]
- West MA, Koons A. Endotoxin tolerance in sepsis: concentration-dependent augmentation or inhibition of LPS-stimulated macrophage TNF secretion by LPS pretreatment. J. Trauma. 2008;65:893–898. doi: 10.1097/TA.0b013e3181877fde. discussion 898–900. [DOI] [PubMed] [Google Scholar]
- Wolk K, Docke WD, Volk V, von Baehr HD, Sabat R. Impaired antigen presentation by human monocytes during endotoxin tolerance. Blood. 2000;96:218–223. [PubMed] [Google Scholar]
- Wolk K, Kunz S, Crompton NE, Volk HD, Sabat R. Multiple mechanisms of reduced major histocompatibility complex class II expression in endotoxin tolerance. J. Biol. Chem. 2003;278:18030–18036. doi: 10.1074/jbc.M207714200. [DOI] [PubMed] [Google Scholar]