

SUMMARY
A “switch” from oxidative phosphorylation (OXPHOS) to aerobic glycolysis is a hallmark of T cell activation and is thought to be required to meet the metabolic demands of proliferation. However, why proliferating cells adopt this less efficient metabolism, especially in an oxygen-replete environment, remains incompletely understood. We show here that aerobic glycolysis is specifically required for effector function in T cells but that this pathway is not necessary for proliferation or survival. When activated T cells are provided with costimulation and growth factors but are blocked from engaging glycolysis, their ability to produce IFN-γ is markedly compromised. This defect is translational and is regulated by the binding of the glycolysis enzyme GAPDH to AU-rich elements within the 3′ UTR of IFN-γ mRNA. GAPDH, by engaging/disengaging glycolysis and through fluctuations in its expression, controls effector cytokine production. Thus, aerobic glycolysis is a metabolically regulated signaling mechanism needed to control cellular function.
INTRODUCTION
Nonproliferating cells metabolize glucose to pyruvate, which enters the mitochondrial tricarboxylic acid (TCA) cycle and generates reducing equivalents for fueling ATP production via oxidative phosphorylation (OXPHOS). However, proliferating cells such as activated T cells and cancer cells engage glycolysis, where pyruvate is fermented to lactate in the cytoplasm even when sufficient oxygen is present to utilize OXPHOS, a process termed the Warburg effect (Fox et al., 2005; Frauwirth et al., 2002; Gerriets and Rathmell, 2012; Jones and Thompson, 2007). Although both processes generate ATP, glycolysis is less efficient, indicating that it might provide other advantages during proliferation. It is thought that the metabolism of proliferating cells is adapted to facilitate uptake and incorporation of nutrients into the biomass needed to produce a daughter cell; i.e., aerobic glycolysis is necessary, both in terms of energy and biosynthesis, for cellular proliferation (Lunt and Vander Heiden, 2011; Vander Heiden et al., 2009). However, cells such as dendritic cells switch from OXPHOS to aerobic glycolysis upon TLR-induced activation but do not proliferate (Krawczyk et al., 2010). This observation suggests that aerobic glycolysis may be necessary for pathways other than, or in addition to, those underlying proliferation. Therefore, we sought to unravel the requirements for OXPHOS and aerobic glycolysis in T cell activation, proliferation, and effector function.
RESULTS
OXPHOS, but Not Aerobic Glycolysis, Is Required for the Activation of Naive T Cells
We measured the extracellular acidification rate (ECAR), an indicator of aerobic glycolysis, and the oxygen consumption rate (OCR), an indicator of OXPHOS, of in-vitro- and in-vivo-activated T cells and found that both had high ECAR and OCR in comparison to naive cells (Figure 1A), indicating that activated T cells use both aerobic glycolysis and OXPHOS (Gatza et al., 2011; Michalek et al., 2011; van der Windt and Pearce, 2012; Wang et al., 2011). To assess whether mitochondrial ATP derived from OXPHOS was necessary for T cell activation, we activated CFSE-labeled naive T cells in the presence of the ATP synthase inhibitor oligomycin and measured subsequent proliferation. Even low concentrations of oligomycin (4.1 nM) inhibited proliferation (Figure 1B) and activation marker expression (Figure 1C). We verified that the concentrations of oligomycin inhibiting activation and proliferation also reduced OCR (Figure 1D) and ATP (Figure S1A available online). These data indicate that T cell activation requires mitochondrial ATP. It is also worth considering that, in addition to inhibiting ATP production directly, it is possible that blocking electron transport chain (ETC) flux in naive T cells with oligomycin will lead to radical oxygen species (ROS) accumulation, thereby inducing cellular stress and contributing to the subsequent drop in ATP production.
Figure 1. OXPHOS, but Not Aerobic Glycolysis, Is Required for the Activation of T Cells.

(A) OCR and ECAR of naive and activated T cells were assessed. Activated CD4+ T cells were obtained from L. monocytogenes-infected mice or after in vitro stimulation with anti-CD3/28 for 72 hr. OCR/ECAR ratio of the cells was also determined; *p = 0.014 and **p < 0.0001.
(B) CFSE-labeled naive T cells were activated with anti-CD3/28 in the presence or absence of oligomycin (Oligo) for indicated times, and then proliferation was measured by CFSE dilution.
(C) Naive T cells were activated with anti-CD3/28 for 72 hr in the absence (no drug) or in the presence of oligomycin. The mean fluorescence intensity (MFI) of the activation markers CD44 and CD25 were determined and normalized to the level expressed in naive cells; *p = 0.001 for CD44, *p = 0.002 for CD25.
(D) OCR of T cells that were activated with anti-CD3/28 and cultured in the presence of low concentrations of oligomycin for 3 days, *p = 0.0001.
(E) CFSE-labeled naive T cells were activated with anti-CD3/28 and cultured in medium supplemented either with (+Glc) or without (−Glc) glucose or with galactose (+Gal),or galactose plus oligomycin (+Gal/Oligo) for 6 days. CFSE dilution was measured 1, 3, or 6 days after activation.
(F) Live-cell (7AAD−) numbers of T cells were determined and normalized to the number of live cells cultured in glucose medium at the indicated times.
(G) OCR and ECAR of T cells that were activated for 3 days with anti-CD3/28 and cultured in medium supplemented either with glucose (+Glc) or galactose (+Gal), *p = 0.0001.
Data are representative of at least two independent experiments and depict mean ± SEM (error bars) of quadruplicates (A), (C), (D), (F), and (G) or of three independent experiments (B) and (E). See also Figure S1.
Glucose is the normal sugar in T cell media and, when activated in glucose, T cells adopt aerobic glycolysis (Frauwirth et al., 2002; Gerriets and Rathmell, 2012). Previous studies demonstrated that T cells cultured without sugar have severe defects (Cham et al., 2008; Cham and Gajewski, 2005; Tripmacher et al., 2008). To specifically address the role of aerobic glycolysis in T cell activation, we activated naive T cells in either glucose or galactose. Cells grown in galactose are forced to respire and do not use aerobic glycolysis (Bustamente et al., 1977; Le Goffe et al., 1999; Rossignol et al., 2004; Weinberg et al., 2010). We found that T cells cultured in galactose activated (Figure S1B), generated ATP (Figure S1C), proliferated—albeit at a slower initial rate than cells grown in glucose (Figure 1E)—and survived (Figure 1F). Cells grown without sugars did not proliferate (Figure 1E), and their survival decreased precipitously (Figure 1F). Importantly, cells grown in galactose with oligomycin neither proliferated (Figure 1E) nor survived (Figure 1F), confirming that T cells activated in galactose fuel proliferation and survival with mitochondrial ATP from OXPHOS (Figure 1G; OCR) and not by aerobic glycolysis (Figure 1G; ECAR).
Activated T Cells Can Use Either Aerobic Glycolysis or OXPHOS to Fuel Proliferation
To determine whether OXPHOS was required for continued proliferation of blasting T cells, we activated naive T cells and added oligomycin 1, 2, or 3 days later. We then assessed continued proliferation 24 and 72 hr following the addition of drug (Figure 2A). By 2 days postactivation, cells were able to proliferate in high concentrations of oligomycin (100 nm—1 μm) (Figure 2A). This was not the case 1 day postactivation, indicating that T cells do not relinquish the need to generate energy from their mitochondria until between 1 and 2 days postactivation (Figure 2A). To confirm this result and to test whether ETC activity was required for proliferation, we cultured activated T cells in the presence of rotenone and antimycin A, complex I and III inhibitors, respectively, and found that neither abrogated proliferation of activated T cells (Figure 2B, left). We did find, however, that myxothiazol, a complex III inhibitor that blocks ROS production from this site, inhibited T cell division (data not shown), agreeing with published observations that ROS can act as a positive signal for cell proliferation (Weinberg et al., 2010). Importantly, addition of oligomycin or antimycin A and rotenone decreased OCR while augmenting ECAR (Figure 2B, right), showing that cells offset ETC inhibition by enhancing aerobic glycolysis and that, over time, cells compensate for the initial drop in ATP caused by oligomycin (Figure S1D). Our metabolite analysis showed increased α-ketoglutarate in T cells cultured with mitochondrial inhibitors (Figure S2A). Although these data do not prove that glutamine is the anaplerotic substrate that replenishes the TCA cycle in this setting, they might be consistent with increased glutamine-dependent metabolism (Wang et al., 2011). Recently published papers show that cells unable to engage in oxidative metabolism due to mutations in the ETC or TCA cycle, hypoxia, or culture with mitochondrial inhibitors use reductive carboxylation of α-ketoglutarate to produce citrate and other precursors during growth and proliferation (Metallo et al., 2012; Mullen et al., 2012). We might speculate that glutamine-dependent reductive carboxylation, rather than oxidative metabolism, also fuels the TCA cycle and supports precursor biosynthesis for the proliferation of T cells cultured with ETC inhibitors in our system.
Figure 2. Activated T Cells Can Use Aerobic Glycolysis or OXPHOS Interchangeably to Fuel Proliferation and Survival.

(A) Naive T cells were activated with anti-CD3/28 for 1, 2, or 3 days and were CFSE labeled and cultured with or without oligomycin. Proliferation was measured by CFSE dilution at 24 and 72 hr post-CFSE labeling.
(B) Naive T cells were activated with anti-CD3/28 for 3 days. Cells were then CFSE labeled and cultured with mitochondrial inhibitors. Proliferation was measured by CFSE dilution at 24, 48, and 72 hr post-CFSE labeling (left), and OCR and ECAR were measured at 72 hr after culturing with or without mitochondrial inhibitors (O, oligomycin; R/A, rotenone and antimycin-A) (right).
(C) Naive T cells were activated with anti-CD3/28 in glucose medium for 3 days, followed by CFSE labeling. The CFSE-labeled activated cells were then cultured in medium supplemented either with glucose (+Glc), glucose plus 1 μM oligomycin (Glc+Oligo), without glucose −Glc), or with galactose (+Gal), or galactose plus 4 nM oligomycin (Gal+Oligo). CFSE dilution was measured at 24 and 72 hr post-CFSE labeling.
(D) Live-cell (7-AAD−) numbers of activated CD4+ T cells were determined at 72 hr and normalized to the number of live cells cultured in glucose medium at 0 hr.
(E and F) T cells were activated with anti-CD3/28 in glucose medium for 3 days. OCR and ECAR were measured 1 hr after these cells were switched into media with glucose (+Glc) or without glucose (−Glc) or with galactose (+Gal).
Data are representative of at least three independent experiments (A), (B, left), and (C) or of two independent experiments and depict mean ± SEM (error bars) of quadruplicates (B, right), (D), (E), and (F); *p = 0.0001. See also Figures S1 and S2.
To determine whether aerobic glycolysis is required for continued proliferation of activated T cells, we activated T cells for 3 days in glucose, and then we either continued culture in glucose or switched cells to galactose. We found that activated T cells proliferated (Figure 2C), survived (Figure 2D), retained activation markers (Figure S1E), and generated ATP (Figure S1F), whether utilizing aerobic glycolysis or OXPHOS. In contrast to T cells growing on glucose, when oligomycin was added to activated T cells forced to grow on galactose, the cells ceased proliferating (Figure 2C), and survival decreased precipitously (Figure 2D), indicating that these cells use OXPHOS (Figure 2E) and not aerobic glycolysis (Figure 2F) to fuel metabolic demands. Unlike T cells cultured in 13C-glucose, cells cultured in 13C-galactose produce low levels of lactate and pyruvate when compared to glucose-cultured cells (Figure S2B), and these cells incorporate low levels of galactose carbon into what are already reduced levels of pyruvate (Figure S2C), lactate, or into TCA cycle intermediates (Figure S2D). Consistent with previously published observations in other cell types (Bustamente et al., 1977; Gohil et al., 2010; Le Goffe et al., 1999; Rossignol et al., 2004; Weinberg et al., 2010), these results indicate that galactose is not as efficiently metabolized as glucose in the glycolysis pathway—or into the TCA cycle—in T cells. We also found that galactose-cultured T cells had higher levels of glutamine and glutamate (Figure S2B). Although these results do not prove whether these cells are using glutamine as an alternate substrate for OXPHOS in this setting, they might suggest this possibility and, if so, would be consistent with previous observations showing that glutamine is the major energy source for cells cultured in galactose (Reitzer et al., 1979). Together, these data show that activated T cells can use OXPHOS or aerobic glycolysis to support proliferation and survival.
Aerobic Glycolysis Is Required for Optimal IFN-γ Cytokine Production in T Cells
We have shown here that T cell activation and proliferation can proceed in the absence of aerobic glycolysis and can be fully supported by OXPHOS. Nevertheless, the observation that activated T cells engage aerobic glycolysis remains (Figure 1A). Therefore, we ask the question: what function does aerobic glycolysis serve in activated T cells? Because activated T cells assume effector functions, such as the production of adaptive cytokines, in addition to enhanced proliferation, we measured cytokine production in activated T cells cultured in either glucose or galactose. We found that T cells cultured in galactose had a severe defect in IFN-γ and IL-2 production (Figures 3A, 3B, S3A, and S3B). Neither mitochondrial ATP nor ETC function was required for cytokine production, as levels were unaffected in cells cultured with mitochondrial inhibitors (Figures 3C and S3A), indicating that aerobic glycolysis—and not OXPHOS—is a critical metabolic pathway for effector function. Neither the expression of IFN-γ or IL-2 messenger RNA (mRNA), nor protein expression of the transcription factor T-bet, differed between glucose- and galactose-cultured cells, suggesting that defective cytokine production was due to a block in translation rather than transcription (Figure 3D and data not shown). To rule out any possibility that the lower amount of IL-2 produced by galactose-cultured T cells was responsible for their lower IFN-γ cytokine expression, all T cells were cultured in the presence of 10 U/ml of exogenous IL-2.
Figure 3. Aerobic Glycolysis Is Required for Optimal IFN-γ Cytokine Production in T Cells.

(A) Intracellular IFN-γ was measured in T cells that were activated with anti-CD3/28 in medium containing either glucose (Glc) or galactose (Gal) for 4 days (top), or for 3 to 4 days in Glc followed by culture in Glc or Gal for 1 day (bottom, (B)–(H); or with oligomycin or rotenone/antimycin A for additional one day (C).
(B) IFN-γ in the culture supernatant was measured by ELISA after restimulation. Frequencies of IFN-γ-producing cells are shown (A) and (C).
(D) IFN-γ mRNA and T-bet expression.
(E) Western blot analysis of p-4EBP1 and p-S6K and p-AMPK and p-eIF2-α were examined.
(F) Hspa5 and Ppp1r15a expression after treatment with 0.1 mM thapsigargin (Thaps) for 4 hr.
(G) Mean cell diameter and volume as measured by laser light-scattering method.
(H) Total protein density (left) and total protein concentration (right) measured by Sypro Ruby staining and BCA assay, respectively.
Plots in (A) and (C) are representative of >3 independent experiments; (B) graph shows the mean ± SEM (*p = 0.001) from two independent experiments; (D) qPCR data are generated from five independent experiments and are shown as mean ± SEM, n.s., not significant, and the FACS plot is representative of two independent experiments; (E) blots are representative of two to four independent experiments; (F) results are presented as mean ± SEM from six independent experiments for cells without Thaps treatment and from one experiment for cells with Thaps treatment; (G) data are representative of two independent experiments; and (H) data are presented as mean ± SEM from three independent experiments. See also Figure S3.
We reasoned that altered cytokine translation could reflect a cellular stress response (Scheu et al., 2006) caused by the substrate transition from glucose to galactose. To explore this possibility, we assessed mTOR target activation and AMPK-α and eIF2-α phosphorylation in cells cultured in glucose versus galactose and found no differences in these pathways (Figure 3E). We also measured the mRNA expression of the integrated stress response genes Hspa5 and Ppp1r15a in glucose- and galactose-cultured cells with or without thapsigargin, which induces ER stress (Scheu et al., 2006). We found that glucose- and galactose-cultured cells expressed BiP or GADD34 to similar levels and not to the high levels expressed when ER stress is induced (Figure 3F). Together, these data indicate that the induction of a classical stress response is not responsible for the block in translation of effector cytokine mRNAs in galactose-cultured T cells. We also found that cell size (Figure 3G), total protein levels (Figure 3H), IFN-γ protein stability (Figure S3C), and ROS production (Figure S3D) were similar between T cells cultured in glucose and galactose, suggesting that the decrease in cytokine production was not due to broad differences in cellular health. Together, these data demonstrate that the engagement of aerobic glycolysis constitutes a physiological mechanism unrelated to ER stress that controls cytokine mRNA translation.
Aerobic Glycolysis Controls the Preferential Translation of IFN-γ mRNA
To establish whether the engagement of aerobic glycolysis preferentially regulates the translation of cytokine mRNAs, we fractionated ribosomes from glucose- and galactose-cultured cells and measured IFN-γ and β-actin transcripts. We found that IFN-γ transcripts were mainly associated with polysomes in activated T cells using aerobic glycolysis, indicating ongoing translation of IFN-γ mRNA, whereas more IFN-γ transcripts were associated with monosomes in galactose-cultured activated T cells, indicating less translation of IFN-γ mRNA in cells using OXPHOS (Figure 4A). Importantly, β-actin mRNAs, which are highly translated and associated with a greater number of polysomes than are IFN-γ transcripts (peaks shift to the right), were equally associated with polysomes in both conditions (Figure 4A). This is consistent with normal expression of β-actin protein regardless of the sugar substrate utilized. These results show that the engagement of aerobic glycolysis specifically permits the translation of IFN-γ mRNA in activated T cells and thereby regulates the ability of the cells to attain full effector status.
Figure 4. The Translational Defect in Cytokine Production Evident in the Absence of Aerobic Glycolysis Is Marked by Enhanced GAPDH Binding to IFN-γ mRNA.

(A) Polysome analysis of T cells that were activated with anti-CD3/28 for 3 days in media containing glucose and then differentially cultured in media with either glucose (Glc) or galactose (Gal) for an additional day. The 40, 60, and 80S ribosomal subunits and polysomes were fractionated and monitored with continuous A254 measurements. A representative polysome profile of cells in Glc (red) and in Gal (blue) is shown in the upper panel. Total RNA was extracted from each fraction, and IFN-γ (middle) and β-actin (bottom) expression was measured by qPCR and calculated as a percentage of total RNA collected in all fractions. The proportion of each mRNA between nontranslated (1–7) and translated (8–14) fractions is plotted. Data are representative results from two independent experiments.
(B) GAPDH-specific antibodies were used to immunoprecipitate GAPDH from extracts of activated T cells differentially cultured in media with either Glc or Gal. Bound GAPDH and associated mRNA were analyzed by western blot (top) and qRT-PCR (bottom). Total input per immunoprecipitation is shown (top). Data show results from six independent experiments as mean ± SEM (error bar) and are normalized to Glc cells (*p = 0.025).
(C) IFN-γ mRNA transcripts and housekeeping gene transcripts (GAPDH mRNA) that bind to GAPDH protein in cells cultured in galactose (Gal) versus cells cultured in glucose (Glc). Relative levels were calculated by subtracting background binding (no primary antibody control) from primary antibody-specific binding and then normalizing to transcripts levels bound to GAPDH in Glc cells.
Mean ± SEM (error bar) of three independent experiments. See also Figure S4.
The Defect in IFN-γ Translation Evident in the Absence of Aerobic Glycolysis Is Marked by Enhanced GAPDH Binding to IFN-γ mRNA
We next investigated how engaging aerobic glycolysis could specifically alter IFN-γ mRNA translation. Given that cells grown on galactose do not engage aerobic glycolysis (Figures 1G and 2F)—nor do they robustly metabolize galactose into the glycolysis pathway (Figure S2)—we hypothesized that, when cells are grown in galactose, their glycolysis enzymes are not engaged to the same extent as they are in cells grown in glucose and therefore become available to perform other functions. Besides its metabolic function, the glycolysis enzyme GAPDH has several additional roles, including acting as a mRNA-binding protein (Singh and Green, 1993) that regulates mRNA translation (Bonafé et al., 2005; Kondo et al., 2011; Nagy et al., 2000; Rodríguez-Pascual et al., 2008; Zhou et al., 2008). Specifically, GAPDH has been shown to bind to adenylate-uridylate (AU)-rich elements in the 3′ UTRs of IFN-γ and IL-2 mRNAs (Nagy and Rigby, 1995). Furthermore, it is well established that 3′ UTR-dependent mechanisms limit the translation of cytokine mRNA (Anderson, 2010; Garcia-Sanz and Lenig, 1996; Villarino et al., 2011). To investigate whether aerobic glycolysis regulates the ability of GAPDH protein to bind cytokine mRNA, we immunoprecipitated GAPDH from activated T cells cultured in glucose or galactose and assessed its association with IFN-γ transcripts. We found a >10-fold increase in GAPDH-associated IFN-γ transcripts in cells cultured in galactose as compared to those cultured in glucose (Figure 4B). This enhanced binding of GAPDH to IFN-γ mRNA in galactose-cultured cells correlated with their defect in IFN-γ production (Figures 3A, 3B, S3A, and S3B). Similar binding of GAPDH mRNA to GAPDH protein was observed between both cell types, indicating that GAPDH protein preferentially binds IFN-γ transcripts—rather than housekeeping gene transcripts—only in cells cultured in galactose (Figure 4C). We also found that IL-2 mRNA was associated with GAPDH to a greater extent when cells were cultured in galactose rather than in glucose (Figure S4). Together, these results suggest that the engagement of aerobic glycolysis permits effector cytokine production by preventing GAPDH from binding to and inhibiting the translation of cytokine transcripts.
A 3′ UTR-Dependent Mechanism Limits IFN-γ Expression in Galactose-Cultured CD4 T Cells
It has been previously shown that 3′ UTR-dependent mechanisms limit the translation of cytokine mRNA (Villarino et al., 2011). We sought to establish whether a 3′ UTR-dependent mechanism limited the production of IFN-γ in CD4 T cells cultured in galactose. To accomplish this, we used a series of previously reported UTR sensors, where the IFN-γ 3′ UTR or a control 3′ UTR were fused to green fluorescent protein (GFP) in a retroviral vector, thus allowing for protein expression to be measured by GFP fluorescence (Villarino et al., 2011). We found that, in CD4 T cells transduced with a mutant in which specific residues of the AU-rich elements were mutated (ARE*), GFP expression was greatly increased over that of cells transduced with the full-length IFN-γ 3′ UTR construct (Figure 5A), showing that translation of mRNA is limited by the AU-rich region of the IFN-γ 3′ UTR. We also transduced cells with two other IFN-γ 3′ UTR mutants—one that lacks the AU-rich region and one that contains the AU-region but is truncated in a different region of the promoter—and found that they either enhance or limit GFP expression, respectively. As a control, we transduced cells with a construct containing the GAPDH 3′ UTR fused to GFP. As expected, the GAPDH 3′ UTR, which lacks an AU-rich region, does not limit GFP expression (Figure 5A). These results confirm that the AU-rich region of the IFN-γ 3′ UTR limits IFN-γ production in galactose-cultured cells.
Figure 5. GAPDH Associates with the AU-Rich Region of the IFN-γ 3′ UTR and Regulates IFN-γ Production.

(A) T cells cultured in glucose (Glc) were transduced with UTR-sensor constructs for 2 days and then recultured in galactose (Gal) for 1 more day before GFP fluorescence was measured by flow cytometry. The percentage of GFP+ cells is depicted on the top left, and the MFI of those GFP+ events is shown vertically as indicated.
(B) GAPDH-specific antibodies were used to immunoprecipitate GAPDH from extracts of indicated UTR-sensor construct-transduced T cells cultured in media with Gal. Associated GFP mRNA bound to GAPDH was analyzed by qRT-PCR. Data show results from one experiment that is normalized to the ARE* mutant IFN-γ 3′ UTR construct-transduced cells.
(C) T cells were transfected with Scrambled or GAPDH siRNA. Cells were incubated for 24 hr before analyzing IFN-γ by flow cytometry. Shown vertically is the MFI of the events from IFN-γ-producing cells. Data show a representative result from seven independent experiments.
(D) GAPDH expression was determined by qRT-PCR. Relative expression of GAPDH mRNA after Scrambled or GAPDH siRNA transfection is normalized to the GAPDH expression of Scrambled siRNA samples, p = 0.0049. Bar graph shown as the mean ± SEM (error bar) is generated from data from four independent experiments.
(E and F) T cells were transduced with GAPDH (GAPDH EX) and empty vector control retrovirus (EV), and intracellular IFN-γ (E) and IFN-γ secretion by ELISA (F) were measured after restimulation. Dot plots show IFN-γ MFI from CD4+ T cells and are representative of three independent experiments; supernatant was collected from one experiment (mean ± SEM).
(G) Activated T cells were exposed to varying concentrations of glyceraldehyde 3-phosphate (G3P) in 0.01% saponin on ice for 10 min prior to restimulation. Data represent similar results from three independent experiments. See also Figure S5.
GAPDH Binds the AU-Rich Region of the IFN-γ 3′ UTR in Galactose-Cultured CD4 T Cells
The finding that GAPDH binds to the AU-rich region of IFN-γ mRNA in T cells has been previously reported (Nagy and Rigby, 1995), so we sought to establish whether GAPDH protein binds to the AU-rich region of the IFN-γ 3′ UTR in our system. Therefore, we transduced galactose-cultured CD4 Th1 cells with either the full-length IFN-γ 3′ UTR or the ARE* mutant constructs, immunoprecipitated GAPDH, and assessed bound GFP mRNA transcripts by qRT-PCR. We found that there was ~10 times more GFP mRNA associated with GAPDH in the cells transduced with the full-length IFN-γ 3′ UTR construct as compared to those expressing the ARE* mutant (Figure 5B). Together, these results confirm that GAPDH protein binds to the AU-rich region of the IFN-γ promoter in CD4 T cells cultured in galactose.
GAPDH Directly Regulates IFN-γ Protein Expression in CD4 T Cells
To assess whether GAPDH directly regulates IFN-γ production, we used a small interfering RNA (siRNA) approach to decrease GAPDH expression in CD4 T cells. We found a small but statistically significant increase in IFN-γ mean fluorescence intensity (~10% increase, where p = 0.00009 and n = 7) in cells expressing GAPDH siRNA compared to those expressing scrambled siRNA (Figure 5C). This result correlates with the modest siRNA-mediated decrease in gene expression of this highly abundant transcript (Figure 5D). To further examine how modulating GAPDH expression could influence IFN-γ production, we created a retroviral construct to enforce the expression of GAPDH in activated T cells. We found that retroviral expression of GAPDH decreased IFN-γ production when compared to T cells transduced with the empty vector (EV) alone (Figures 5E and 5F). Together, these results indicate that total levels of GAPDH in activated T cells can influence cytokine production.
To more firmly establish a direct link between GAPDH and IFN-γ production in CD4 T cells and to test our idea that, in addition to total levels of GAPDH protein, the engagement of GAPDH in the glycolysis pathway can also influence cytokine production, we took a different approach and introduced the GAPDH substrate glyceraldehyde 3-phosphate (G3P) into activated T cells and assessed its influence on cytokine expression. We reasoned that, because the catalytic site for the enzymatic activity of GAPDH is the same site where mRNA has been reported to bind (Nagy et al., 2000), we could essentially force GAPDH to “engage” in the glycolysis pathway by loading T cells with G3P. We found that galactose-cultured T cells increased IFN-γ production after G3P introduction, whereas glucose-cultured cells did not (Figure 5G). We speculate that G3P is already available in T cells cultured in glucose due to endogenous glycolytic flux. We also found that introduction of G3P promoted the expression of IL-2 in galactose-cultured cells (Figure S5A). Taken together, these results support that GAPDH directly regulates IFN-γ production in T cells and extend our findings to show that levels of GAPDH expression, in addition to its preoccupation in the glycolysis pathway, can influence cytokine production.
It has been previously shown that GAPDH can exist as part of a complex of proteins that drives transcript selective translation in myeloid cells (Mukhopadhyay et al., 2009). The formation of this complex, referred to as the GAIT (gamma interferon (IFN-γ)-activated inhibitor of translation) complex, is induced by IFN-γ signals (Mukhopadhyay et al., 2009). Therefore, we investigated whether GAPDH exists as part of the GAIT complex in T cells. We first assessed whether IFN-γ signaling, the regulator of GAIT, influences IFN-γ expression in T cells. Neither culturing T cells with an exogenous concentration of IFN-γ that is known to induce GAIT in myeloid cells (Arif et al., 2012), nor the addition of neutralizing antibodies against IFN-γ, had any effect on IFN-γ expression by T cells (Figure S5B). We also immunoprecipitated GAPDH from glucose- and galactose-cultured cells and assessed the binding of other GAIT complex proteins to GAPDH by western blot. We found no evidence that the GAIT complex proteins EPRS, NSAP1, or L13a bind to GAPDH to any appreciable extent in these T cells, whether cultured in glucose or galactose (Figure S5C). Although these observations do not preclude that GAPDH might exist as part of GAIT in T cells, they indicate that this is not a major mechanism of how GAPDH is functioning in our system.
GAPDH Expression Inversely Correlates with IFN-γ Production in CD4 T Cells In Vivo after Infection
To investigate whether the total level of GAPDH can influence IFN-γ production in vivo, we infected mice with Listeria monocytogenes and analyzed CD4 T cells 7 days later at the peak of the effector T cell response (Figure 6A) to this pathogen. We measured GAPDH expression in CD4 T cells and found that T cells expressing a higher level of GAPDH produce much less IFN-γ than cells expressing a lower level of GAPDH (Figure 6B), showing that the level of GAPDH expression determines effector cytokine production in T cells after infection. Given that PD-1 is a negative regulator of T cell function and that its expression is often linked with the inability of T cells to produce optimal levels of cytokine (termed T cell exhaustion) in many disease states (Barber et al., 2006), we also assessed the expression of PD-1 on cells with high and low levels of GAPDH. We found that T cells expressing the highest level of GAPDH, which have the lowest cytokine production, also have the highest expression of PD-1 when compared to cells expressing low levels of GAPDH (Figure 6C). These data suggest a link between PD-1 expression and the engagement of aerobic glycolysis.
Figure 6. Expression of GAPDH in CD4 T Cells Determines IFN-γ Production after Listeria monocytogenes Infection In Vivo.
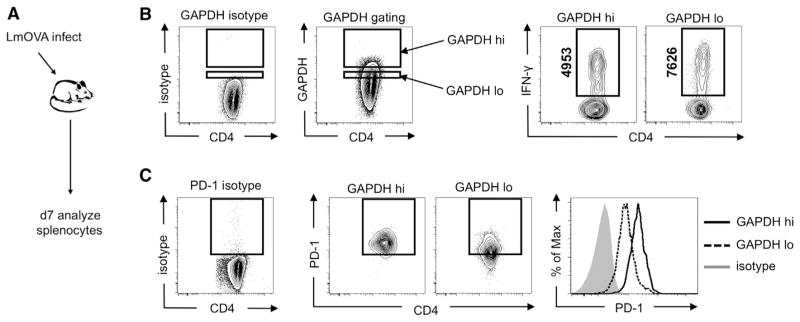
(A–C) A schematic is depicted (A) showing that mice were infected with L. monocytogenes, and splenocytes were analyzed 7 days later for GAPDH (B) and (C) and IFN-γ (B) and PD-1 (C) expression. Gating for high (GAPDH hi) and low (GAPDH lo) levels of GAPDH expression among GAPDH-positive cells, or PD-1 expression, was determined according to isotype controls. The mean fluorescence intensity of IFN-γ+ events is shown vertically as indicated. Data (B) and (C) are representative of at least two independent experiments.
Metabolic Phenotype Determines PD-1 Expression on CD4 T Cells
To further explore the relationship between PD-1 and metabolism, we measured PD-1 expression on CD4 T cells cultured in glucose and galactose and found that galactose-cultured cells express higher levels of PD-1 (Figure 7A), which correlated with their defect in cytokine production. We next assessed whether cells could regain the ability to produce cytokine after forced respiration and found that, when cells using aerobic glycolysis (glucose) are forced to respire (galactose), they develop a defect in IFN-γ production that is immediately reversed upon re-exposure to glucose and engagement of aerobic glycolysis (Figures 7B and 7C). We also found that, as CD4 T cells were switched between glucose and galactose, the level of PD-1 expression directly correlated with, and was preceded by, the metabolic adaptation as dictated by substrate utilization (Figures 7D and 7E). Together, these results indicate that cellular metabolism can influence the level of PD-1 expression in CD4 T cells.
Figure 7. Tumor Cells Impose a Nutrient Restriction on T Cells that Dampens IFN-γ Production.

(A) T cells were activated with anti-CD3/28 in media containing glucose (Glc) or galactose (Gal), and PD-1 expression was measured.
(B) Activated T cells were cultured in media containing Glc and then differentially cultured in either Glc or Gal (Glc→Gal) for 1 additional day. Cells cultured in Gal were then recultured in Glc for 1 more day (Glc→Gal→Glc). IFN-γ expression was measured 5 days postactivation. Frequencies of IFN-γ producing cells are shown, and plots are representative of three independent experiments.
(C and D) The OCR over ECAR ratio (C) and PD-1 expression (D) of activated T cells cultured in indicated media as described in (B) were assessed.
(E) Activated T cells were cultured in media containing Glc and then differentially cultured in either Glc or Gal for 1 additional day (Glc→Gal (1d)) or 2 days (Glc→ Gal (2d)). Cells cultured in Gal were then recultured in Glc for 1 more day (Glc→Gal→Glc). PD-1 expression was measured 5 days postactivation. Data are representative of at least two independent experiments (A), (B), and (D), and data are presented as mean ± SEM (error bar) from at least three independent experiments (C) and (E). *p = 0.0001.
(F and G) (F) T cells were activated with anti-CD3/28 under Th1 polarizing conditions for 3 days. Cells were subsequently incubated overnight with equal numbers of Th1 cells alone or in a 50/50 mix of EL4 lymphoma cells: Th1 cells. Cells were restimulated (restim) and IFN-γ production (F) and glucose concentration in the media (G) were measured. At the time of restimulation, either nothing (no change), 10 mM glucose, or new media were added back to the cells. Data (F) and (G) are representative of three independent experiments. Data (G) are presented as mean ± SEM (error bar).
(H) The MFI of PD-1 expression on T cells from the indicated coculture ratios of EL-4 lymphoma cells:Th1 cells are shown.
Data are presented as mean ± SEM (error bar) from one experiment (n = 6). *p = 0.0423; **p = 0.0001. See also Figure S6.
Tumor Cells Impose a Reversible Nutrient Restriction on T Cells that Dampens Cytokine Production
To further substantiate how metabolic adaptions imposed by nutrient restriction can influence T cell effector function, we used a tumor cell:CD4 T cell coculture system. We incubated overnight equal numbers of Th1 cells alone or in a 50/50 mix of EL-4 lymphoma cells:Th1 cells, and then after a 4 hr restimulation, we measured cytokine production. During the restimulation, we either added back nothing (no change), 10 mM glucose, or new media. We found that, as tumor cell density increased, CD4 T cells reduced IFN-γ production to a greater degree than if cultured with T cells alone (Figure 7F, top row). We found that this defect in IFN-γ production could be rescued simply by the addition of glucose (Figure 7A, middle row) and even further enhanced by the addition of new media (Figure 7F, bottom row). Importantly, we confirmed that glucose concentrations in the media were in fact lower when T cells were cocultured with tumor cells than when T cells were cultured alone (Figure 7G) and that media alone contain much higher concentrations of glucose than either of these conditions (Figure S6). We also found that the level of PD-1 increased on CD4 T cells as numbers of tumor cells increased in the coculture (Figure 7H). These results suggest that lymphoma cells impose a nutrient deprivation on T cells that limits their ability to produce effector cytokine.
DISCUSSION
The regulation of metabolic pathways in proliferating cells, as well as how aerobic glycolysis contributes to metabolically regulated signaling mechanisms, remains poorly understood (Loca-sale and Cantley, 2011; Lunt and Vander Heiden, 2011). We show that OXPHOS and aerobic glycolysis interchangeably fuel T cell proliferation and survival, but only aerobic glycolysis can facilitate full effector status. Specifically, engagement/disengagement of aerobic glycolysis allows the posttranscriptional regulation of IFN-γ production in T cells. We speculate that the advantage of this regulatory mechanism is that it offers a metabolic checkpoint for allowing T cell proliferation with or without accompanying cytokine production. This level of regulation is important because T cells are required to undergo both homeostatic proliferation, when IFN-γ production is neither required nor desirable, and antigen-driven proliferation during an immune response, when effector cytokine production is essential. In this model, the engagement of CD28 by costimulatory molecules on pathogen-activated antigen-presenting cells drives aerobic glycolysis and allows T cells to attain full effector status (Frauwirth et al., 2002).
Culturing cells with galactose rather than glucose allowed us to enforce respiration while providing all the known critical factors for T cell activation and cytokine production—i.e., T cell receptor ligation, costimulation, and growth factors. By changing only the sugar substrate provided, we were able to unravel that aerobic glycolysis is a critical regulator of cytokine production in T cells. In addition to the regulation of cytokine production, we also found that PD-1 expression was directly influenced by the metabolism of the T cells. This finding hints at the intriguing idea that differing states of T cell exhaustion, which, in many disease states, is characterized by a loss of effector function and enhanced PD-1 expression, is fundamentally regulated by metabolic constraints, rather than chronic antigen stimulation per se (Barber et al., 2006). It is also worth noting that culturing cells in galactose where they can use only OXPHOS might model physiological conditions in which T cells’ access to or acquisition of nutrients is altered. This could result from a lack of appropriate costimulation, a lack of growth factor signals, or direct nutrient limitations enforced by the presence of other rapidly proliferating cells, such as might be expected to occur within a tumor micro-environment; our data showing that, in coculture, tumor cells impose nutrient restrictions that dampen the ability of T cells to produce cytokines illustrate this possibility. Although our experiments specifically address the impact of tumor-cell-mediated glucose deprivation on T cell cytokine production, it is clear that new complete media can enhance IFN-γ production even further than the addition of glucose alone in our T cell: tumor cell coculture model. This could be a result of other nutrients in the media affecting T cell cytokine production. For example, replenishing glutamine in addition to glucose may provide additional substrates to the T cells and allow a further increase in cytokine expression. If this were the case, it might suggest that T cells are severely nutrient impaired, beyond the simple depletion of glucose, in a tumor microenvironment. However, it is also possible that tumor cells produce inhibitory factors that dampen T cell cytokine production and that replacing the media removes or dilutes these factors, allowing T cells to produce cytokines. This may also apply if there is a buildup of metabolites, or byproducts of metabolism, such as lactate, that are able to dampen cytokine production (Fischer et al., 2007). How these types of metabolic restrictions might manifest in vivo is currently under investigation.
Our results show that the glycolysis enzyme GAPDH posttranscriptionally regulates T cell effector function by binding to the AU-rich region in the 3′ UTR of cytokine mRNA and reducing protein translation. Our data indicate that GAPDH-mediated inhibition of effector function in T cells may be controlled at several levels, such as whether or not the enzyme is occupied by its metabolic function or by its expression level within cells. It is also known that the activity of GAPDH is heavily influenced by the redox state of a cell and that redox-mediated changes can alter its function and even location within different cellular compartments (Colell et al., 2007; Tristan et al., 2011). It is likely that all of these processes contribute to how GAPDH controls cytokine expression in vivo. Although GAPDH clearly plays an important role in cytokine translation in T cells, our study does not preclude, and may even suggest, the possibility that other metabolic enzymes control effector cytokine translation or even other translational programs within cells. This will remain a focus of further investigation.
Looking beyond any one specific metabolic enzyme, our results more broadly implicate aerobic glycolysis in general as a target to control T cell effector functions specifically, rather than simply controlling the survival and proliferation of responding T cells. In this light, it is possible that anticancer drugs developed to inhibit aerobic glycolysis in tumors could be repurposed to target autoreactive T cells. This approach may allow the inhibition of cytokine production by differentiated effector T cells without affecting survival, maintenance, or proliferation of the broader T cell compartment. Furthermore, it is appealing to speculate that some tumors adopt Warburg metabolism not for proliferation per se, but for function, and even perhaps for the optimal expression of factors that facilitate their survival or modulate the immune response. Regardless of the applications of these findings, they unexpectedly reveal that a purpose of engaging aerobic glycolysis is to posttranscriptionally regulate specific cellular functions.
EXPERIMENTAL PROCEDURES
Mice and Immunizations
C57BL/6 mice were used for all of the experiments. All animals were cared for according to the Animal Care Guidelines of Washington University School of Medicine in St. Louis. An attenuated strain of recombinant Listeria monocytogenes deleted for actA (LmOVA) was used for immunizations. Mice were infected intraperitoneally with a sublethal dose of 1 × 107 colony-forming units (CFU). Naive (CD44loCD62Lhi) and effector (CD44hiCD62Llo) CD4+ T cells were isolated and sorted from spleens and lymph nodes.
Cell Cultures
Naive CD4+ T cells were purified by MACS (Miltenyi Biotech) and used in all experiments. T cells were activated with plate-bound anti-CD3 (5.0 μg/ml) and soluble anti-CD28 (0.5 μg/ml) antibodies for 3 to 4 days in either nonpolarizing (10 U/ml of IL-2) or in T helper type 1 (Th1)-polarizing conditions (10 U/ml of IL-2, 10 ng/ml of IL-12, and 10 μg/ml anti-IL-4 antibody) as indicated. For EL4/CD4 cell coculture, activated T cells were cocultured with or without EL4 cells in varying ratios of cell number as indicated. For cell culture experiments, cells were cultured with RPMI medium (no glucose) containing 10% dialyzed serum, 1 mM sodium pyruvate, and 2 mM L-glutamine, along with either 10 mM glucose or 10 mM galactose. For in vitro survival assays, cells were plated in a 96-well plate in the presence of the indicated inhibitors, and live cells were assessed daily by 7-AAD exclusion. Cell size/volume were measured on a Moxi Z cell counter.
Flow Cytometry and Intracellular Cytokine Staining
All antibodies were purchased from BD PharMingen or eBioscience. To assess proliferation, cells were fluorescein-labeled with 1 μM CFSE for 8 min at room temperature. For intracellular cytokine staining, cells were stimulated at 37°C for 4 hr in either glucose or galacose medium supplemented, with or without indicated inhibitors, in the presence of GolgiStop (BD PharMingen), PMA (Sigma), and ionomycin (Sigma). For the introduction of glyceraldehyde 3-phosphate (G3P) into cells, T cells were permeabilized on ice for 10 min with 0.01% saponin in glucose- or galactose-containing media. Cells were then washed and put in media in the presence of G3P (a 30 min recovery time at 37°C before restimulation) for restimulation prior to measuring intracellular cytokines. Representative plots from flow cytometry were all gated on live cells, either by 7-AAD exclusion or fixable aqua dead cell staining (Invitrogen).
Extracellular Flux Analysis
Oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) were measured in XF media (nonbuffered RPMI 1640 containing either 10 mM or 25 mM glucose or galactose, 2 mM L-glutamine, and 1 mM sodium pyruvate) under basal conditions and in response to mitochondrial inhibitors, 1 μM oligomycin, and/or 100 nM rotenone + 1 μM antimycin A (Sigma) on the XF-24 or XF-96 Extracellular Flux Analyzers (Seahorse Bioscience).
RT-PCR and Western Blotting
Total RNA was isolated with the RNeasy kit (QIAGEN), and cDNA was synthesized with the High Capacity cDNA Reverse Transcription Kit (Applied Bio-systems). All quantitative RT-PCR was performed by the Taqman method, except for GFP mRNA, which was assayed by the SYBR green method. The expression of mRNA for genes of interest was normalized to the expression of β-actin. Cell lysate preparation, SDS–PAGE, electrophoretic transfer, immunoblotting, and development were accomplished as previously described (Pearce et al., 2009). Antibodies for western analysis were purchased from Cell Signaling. Total protein concentration was determined according to the BCA method (Thermo Scientific Pierce). Sypro Ruby (Molecular Probes, Inc.) staining of the gel was performed according to the protocol.
Ribosome Analysis
Cells were treated with cycloheximide (Sigma) for 5 min before being harvested. Equal numbers of cells were lysed, and cytosolic extracts were separated over 7%–47% sucrose gradients by ultracentrifugation as previously described (Miceli et al., 2012). Fractions were collected with constant monitoring of absorbance at 254 nm using a density gradient system (Teledyne ISCO, Lincoln, NE). RNA was extracted from monosome (40 and 60S), disome (80S), and polysome fractions using RNAsolv (Omega Bio-Tek) according to the manufacturer’s specifications. Reverse transcription and qPCR were performed as described above. Numbers of IFN-γ and IL-2 transcripts were calculated from a standard curve generated from serial dilutions of a known quantity of subcloned cDNAs. mRNA distribution was calculated as a percentage of the total number of transcripts in all collected fractions.
RNA Immunoprecipitation
Cells were freshly harvested and then crosslinked by treating with 1% formaldehyde for 10 min. Cells were resuspended and lysed in polysome lysis buffer. The lysate was precleared and immunoprecipitated overnight with a rabbit anti-GAPDH polyclonal antibody (Sigma). Immune complexes were recovered by Protein A Agarose (Invitrogen), were washed five times with washing buffers, and were eluted with 1% SDS and 0.1 M NaHCO3. Total lysate and 1% of the eluted fraction were used for western blot analysis. For crosslinking reversal, the eluted fraction was incubated at 42°C for 1 hr, followed by an additional 2 hr at 65°C in the presence of 200 mM NaCl and 20 μg proteinase K. RNA was then extracted from the eluted fraction using RNAsolv, and mRNA expression was measured by qPCR as described above.
Supplementary Material
{kind=link}
Acknowledgments
We thank Andrey Shaw, Ken Murphy, Skip Virgin, Abul Abbas, Eyal Amiel, Rong Zeng, Qiongyu Chen, Monika Vig, Georgia Perona-Wright, Bart Everts, and the Goodman Cancer Research Centre Metabolomics Core Facility at McGIll University. This work was supported in part by grants from the NIH (AI091965 and CA158823 to E.L.P. and CA164062 to E.J.P.), CIHR (MOP-93799 to R.G.J.), The Arthritis Society of Canada (R.G.J.), NWO (G.J.W.v.d.W.), and the Emerald Foundation Young Investigator Award (E.L.P.). C.-H.C., L.B.M., J.D.W., A.V.V., E.J.P., R.G.J., and E.L.P. designed the research. C.C., J.D.C., S.C.-C.H., G.J.W.v.d.W, D.O., J.B., B.F., J.Q., and E.L.P. analyzed data. C.C., J.D.C., L.B.M., B.F., S.C.-C.H., D.O., G.J.W.v.d.W., J.Q., and E.L.P. performed experiments. C.C., E.J.P., and E.L.P. contributed to the preparation of the manuscript. C.C. and E.L.P. wrote the manuscript.
Footnotes
Supplemental Information includes Extended Experimental Procedures and six figures and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2013.05.016.
References
- Anderson P. Post-transcriptional regulons coordinate the initiation and resolution of inflammation. Nat Rev Immunol. 2010;10:24–35. doi: 10.1038/nri2685. [DOI] [PubMed] [Google Scholar]
- Arif A, Chatterjee P, Moodt RA, Fox PL. Heterotrimeric GAIT complex drives transcript-selective translation inhibition in murine macrophages. Mol Cell Biol. 2012;32:5046–5055. doi: 10.1128/MCB.01168-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- Bonafé N, Gilmore-Hebert M, Folk NL, Azodi M, Zhou Y, Chambers SK. Glyceraldehyde-3-phosphate dehydrogenase binds to the AU-Rich 3′ untranslated region of colony-stimulating factor-1 (CSF-1) messenger RNA in human ovarian cancer cells: possible role in CSF-1 posttranscriptional regulation and tumor phenotype. Cancer Res. 2005;65:3762–3771. doi: 10.1158/0008-5472.CAN-04-3954. [DOI] [PubMed] [Google Scholar]
- Bustamente E, Morris HP, Pedersen PL. Hexokinase: the direct link between mitochondrial and glycolytic reactions in rapidly growing cancer cells. Adv Exp Med Biol. 1977;92:363–380. doi: 10.1007/978-1-4615-8852-8_15. [DOI] [PubMed] [Google Scholar]
- Cham CM, Gajewski TF. Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. J Immunol. 2005;174:4670–4677. doi: 10.4049/jimmunol.174.8.4670. [DOI] [PubMed] [Google Scholar]
- Cham CM, Driessens G, O’Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol. 2008;38:2438–2450. doi: 10.1002/eji.200838289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colell A, Ricci JE, Tait S, Milasta S, Maurer U, Bouchier-Hayes L, Fitz-gerald P, Guio-Carrion A, Waterhouse NJ, Li CW, et al. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell. 2007;129:983–997. doi: 10.1016/j.cell.2007.03.045. [DOI] [PubMed] [Google Scholar]
- Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109:3812–3819. doi: 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. 2005;5:844–852. doi: 10.1038/nri1710. [DOI] [PubMed] [Google Scholar]
- Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH, Thompson CB. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769–777. doi: 10.1016/s1074-7613(02)00323-0. [DOI] [PubMed] [Google Scholar]
- Garcia-Sanz JA, Lenig D. Translational control of interleukin 2 messenger RNA as a molecular mechanism of T cell anergy. J Exp Med. 1996;184:159–164. doi: 10.1084/jem.184.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatza E, Wahl DR, Opipari AW, Sundberg TB, Reddy P, Liu C, Glick GD, Ferrara JL. Manipulating the bioenergetics of alloreactive T cells causes their selective apoptosis and arrests graft-versus-host disease. Sci Transl Med. 2011;3:ra8. doi: 10.1126/scitranslmed.3001975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerriets VA, Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol. 2012;33:168–173. doi: 10.1016/j.it.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohil VM, Sheth SA, Nilsson R, Wojtovich AP, Lee JH, Perocchi F, Chen W, Clish CB, Ayata C, Brookes PS, Mootha VK. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat Biotechnol. 2010;28:249–255. doi: 10.1038/nbt.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RG, Thompson CB. Revving the engine: signal transduction fuels T cell activation. Immunity. 2007;27:173–178. doi: 10.1016/j.immuni.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Kondo S, Kubota S, Mukudai Y, Nishida T, Yoshihama Y, Shirota T, Shintani S, Takigawa M. Binding of glyceraldehyde-3-phosphate dehydrogenase to the cis-acting element of structure-anchored repression in ccn2 mRNA. Biochem Biophys Res Commun. 2011;405:382–387. doi: 10.1016/j.bbrc.2011.01.034. [DOI] [PubMed] [Google Scholar]
- Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, Pearce EJ. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115:4742–4749. doi: 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Goffe C, Vallette G, Jarry A, Bou-Hanna C, Laboisse CL. The in vitro manipulation of carbohydrate metabolism: a new strategy for deciphering the cellular defence mechanisms against nitric oxide attack. Biochem J. 1999;344:643–648. doi: 10.1042/0264-6021:3440643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale JW, Cantley LC. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011;14:443–451. doi: 10.1016/j.cmet.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miceli AP, Saporita AJ, Weber JD. Hypergrowth mTORC1 signals translationally activate the ARF tumor suppressor checkpoint. Mol Cell Biol. 2012;32:348–364. doi: 10.1128/MCB.06030-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186:3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay R, Jia J, Arif A, Ray PS, Fox PL. The GAIT system: a gatekeeper of inflammatory gene expression. Trends Biochem Sci. 2009;34:324–331. doi: 10.1016/j.tibs.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy E, Rigby WF. Glyceraldehyde-3-phosphate dehydrogenase selectively binds AU-rich RNA in the NAD(+)-binding region (Rossmann fold) J Biol Chem. 1995;270:2755–2763. doi: 10.1074/jbc.270.6.2755. [DOI] [PubMed] [Google Scholar]
- Nagy E, Henics T, Eckert M, Miseta A, Lightowlers RN, Kellermayer M. Identification of the NAD(+)-binding fold of glyceralde-hyde-3-phosphate dehydrogenase as a novel RNA-binding domain. Biochem Biophys Res Commun. 2000;275:253–260. doi: 10.1006/bbrc.2000.3246. [DOI] [PubMed] [Google Scholar]
- Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem. 1979;254:2669–2676. [PubMed] [Google Scholar]
- Rodríguez-Pascual F, Redondo-Horcajo M, Magán-Marchal N, Lagares D, Martínez-Ruiz A, Kleinert H, Lamas S. Glyceraldehyde-3-phosphate dehydrogenase regulates endothelin-1 expression by a novel, redox-sensitive mechanism involving mRNA stability. Mol Cell Biol. 2008;28:7139–7155. doi: 10.1128/MCB.01145-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, Capaldi RA. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004;64:985–993. doi: 10.1158/0008-5472.can-03-1101. [DOI] [PubMed] [Google Scholar]
- Scheu S, Stetson DB, Reinhardt RL, Leber JH, Mohrs M, Locksley RM. Activation of the integrated stress response during T helper cell differentiation. Nat Immunol. 2006;7:644–651. doi: 10.1038/ni1338. [DOI] [PubMed] [Google Scholar]
- Singh R, Green MR. Sequence-specific binding of transfer RNA by glyceraldehyde-3-phosphate dehydrogenase. Science. 1993;259:365–368. doi: 10.1126/science.8420004. [DOI] [PubMed] [Google Scholar]
- Tripmacher R, Gaber T, Dziurla R, Häupl T, Erekul K, Grützkau A, Tschirschmann M, Scheffold A, Radbruch A, Burmester GR, Buttgereit F. Human CD4(+) T cells maintain specific functions even under conditions of extremely restricted ATP production. Eur J Immunol. 2008;38:1631–1642. doi: 10.1002/eji.200738047. [DOI] [PubMed] [Google Scholar]
- Tristan C, Shahani N, Sedlak TW, Sawa A. The diverse functions of GAPDH: views from different subcellular compartments. Cell Signal. 2011;23:317–323. doi: 10.1016/j.cellsig.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Windt GJ, Pearce EL. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol Rev. 2012;249:27–42. doi: 10.1111/j.1600-065X.2012.01150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarino AV, Katzman SD, Gallo E, Miller O, Jiang S, McManus MT, Abbas AK. Posttranscriptional silencing of effector cytokine mRNA underlies the anergic phenotype of self-reactive T cells. Immunity. 2011;34:50–60. doi: 10.1016/j.immuni.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, Green DR. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Yi X, Stoffer JB, Bonafe N, Gilmore-Hebert M, McAlpine J, Chambers SK. The multifunctional protein glyceraldehyde-3-phosphate dehydrogenase is both regulated and controls colony-stimulating factor-1 messenger RNA stability in ovarian cancer. Mol Cancer Res. 2008;6:1375–1384. doi: 10.1158/1541-7786.MCR-07-2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.