

Abstract
Synthesis of the 13C2-labelled analogues of the carcinogenic polycyclic aromatic hydrocarbon benzo[a]pyrene and its active metabolites are described. The method entails Pd-catalyzed Suzuki-Miyaura coupling of a naphthalene boronic acid with 2-bromobenzene-1,3-dialdehyde followed by Wittig reaction of the product with 13CH2=PPh3.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are ubiquitous environmental pollutants that are produced in combustion of fossil fuels and other organic matter.1–3 They occur commonly in auto and diesel engine exhaust,4 tobacco smoke,6–8 and smoked and charbroiled meats.1–3 PAHs have been designated as human carcinogens by the WHO,2 and they are implicated in the causation of human lung cancer.6,9,10
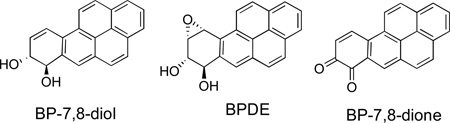
Benzo[a]pyrene (BP) is the prototype PAH carcinogen. It is enzymatically activated to metabolites that react with DNA leading to mutations. The most studied activation path involves cytochrome P-450 [CYP] mediated formation of a dihydrodiol (BP-7,8-diol) followed by its oxidation to a diol epoxide (BPDE).3,11 A second pathway entails aldo-keto reductase-mediated oxidation of BP-7,8-diol to a catechol that enters into a redox cycle with O2 to form a quinone (BP 7,8-dione) and reactive oxygen species (ROS) that attack DNA.12,13 A third pathway has also been proposed that entails peroxidase-mediated oxidation of BP to a radical-cation that reacts with DNA to form depurinated adducts.14,15 However, the relative importance of these pathways for human cancer is not certain.
Several advances have been made recently in understanding PAH activation in human cells susceptible to tumorigenesis. An investigation of metabolic activation of BP in human lung A549 cells has provided evidence that the AKR-mediated pathway to generate the BP quinone and ROS is operative in these cells.16 And human bronchoalveolar H358 cells were recently introduced as a model for study of PAH metabolism in normal human lung cells.17,18 In this connection, development of a stable isotope dilution liquid chromatography tandem mass spectrometric method for analysis of the metabolites of BP and its nucleoside adducts was also described.19 In order to improve the scope and sensitivity of this methodology, 13C-labelled analogues of BP and its active metabolites with at least two 13C-atoms in the aromatic ring system are needed as authentic standards.
Results and discussion
We now report synthesis of 13C2-benzo[a]pyrene (13C2-BP), 13C2-BP-7,8-diol, and 13C2-BP-7,8-dione required as standards for LC-MS/MS analysis of the patterns of BP metabolism and DNA adduct formation in human cells. Syntheses of all twelve of the mono-13C-labelled isomers of BP with a 13C-atom at each of the peripheral carbon atoms of the BP ring system were previously reported.20 However, the classical synthetic methods employed were not adaptable to preparation of the 13C2-labelled BP analogues.
Synthesis of 13C2-BP was carried out by a modified version of the new synthetic approach to BP recently reported.21b This method involves Pd-catalyzed Suzuki-Miyaura coupling of naphthalene 2-boronic acid (1a) with 2-bromobenzene-1,3-dialdehyde (2) to provide a dialdehyde product (3a) (Scheme 1). In the original approach the next step entailed double Wittig reaction of 3a with (methoxymethylene)triphenylphosphine (CH3OCH=PPh3)22 to provide the unlabelled di(methoxyvinyl compound (4b). However, the 13C-labelled methoxymethy halide (CH3O13CH2X) needed as the starting compound for preparation of CH3O13CH=PPh3 was unavailable. Fortunately, 13CH3I (99 atom %) was readily obtainable, and it was used for preparation of 13C-methylene)triphenylphosphine (13CH2=PPh3) by reaction with PPh3 followed by treatment of the resulting phosphonium salt with n-BuLi.23 Double Wittig reaction of 13CH2=PPh3 with 3a afforded a diolefin (4a) with 13C-atoms in both vinyl groups.24 Although the di(methoxyvinyl) analogue (4b) was found previously to undergo cyclization smoothly in the presence of an acid catalyst,21a analogous reaction of 4a gave principally polymeric products. This difficulty was avoided by conversion of 4a to the bis-epoxide (5a) by treatment with Oxone/acetone followed by acid-catalyzed cyclization and dehydration to provide 13C2-BP (6a).25 The 1H NMR spectrum of 6a was in good agreement with that of unlabelled BP. Its 13C NMR spectrum exhibited characteristic peaks at δ 122.1 and 128.1, confirming the presence of 13C atoms at the C-5 and C-11-positions of the BP ring system.
Scheme 1.

Because the yield of 6a was found to be dependent upon reaction conditions, a brief study of the influence of conditions on yield was undertaken using unlabelled 5a. The findings are summarized in Table 1. Significantly higher yields were obtained from reactions carried out at 70 °C than from those conducted at ambient temperature. The optimum yield of BP (94 %) was obtained from reaction of 5a at 70 °C with InCl3 (5%) as the catalyst. Good yields of BP were also obtained from reactions carried out with Hf(OTf)4 and methanesulfonic acid catalysts (84 % and 80 %, respectively).
Table 1.
Effects of reaction variables on cyclization of 5a to 6a.
| Catalysta | 25 °C | 70 °C | ||
|---|---|---|---|---|
| Time (h) |
Yield (%) |
Time (h) |
Yield (%) |
|
| Hf(OTf)4 | 12 | 52 | 1.5 | 84 |
| Cu(OTf)2 | 12 | <10 | 3.0 | 31 |
| In(OTf)3 | 12 | <10 | 3.0 | 46 |
| Ce(OTf)4 | 12 | 13 | 1.5 | 51 |
| InCl3 | 12 | <10 | 3.0 | 94 |
| BiOClO4 | 12 | <10 | 6.0 | <10 |
| ZnCl4 | 12 | <10 | 6.0 | <10 |
| MSA | 12 | <10 | 3.0 | 80 |
The molar ratio of the catalyst used was 5% of 5a. MSA = methanesulfonic acid.
13C2-8-Methoxy-BP (6b), needed as the starting compound for synthesis of the oxidized metabolites of BP, was prepared by an analogous sequence (Scheme 1). The dialdehyde 3b needed for this purpose was prepared by Pd-catalyzed Suzuki coupling of 6-methoxynaphthalene-2-boronic acid (1b) with 2, as previously described.21a Reaction of 3b with 13CH2=PPh3 furnished 13C2-2-methoxy-6-(2,6-divinylphenyl)naphthalene (4b). The 1H NMR spectrum of 4b was in good agreement with that of its unlabelled analogue 4a,21a and its 13C NMR spectrum exhibited a peak at δ 114.6, confirming the presence of 13C-atoms in the vinyl groups. Compound 4b was converted to the bis-epoxide (5b) by treatment with Oxone/acetone.25 Cyclization of 5b took place smoothly in the presence InCl3 at 70 °C to provide 13C2-8-MeO-BP (6b). The 1H NMR spectrum of 6b was in good agreement with this structural assignment.21a Its 13C NMR spectrum contained peaks at δ 122.0 and 128.1, closely similar to the signals found for 13C-BP with 13C in the C-5 and C-11-positions. Demethylation of 6b with HI/HOAc took place smoothly to furnish 13C2-8-HO-BP (6c) (97 %).26
The 13C2-labelled oxidized metabolites of 13C2-BP were synthesized from 6c via procedures analogous to those employed for synthesis of the unlabelled analogues (Scheme 2). Thus, oxidation of 6c with o-iodoxybenzoic acid (IBX) in DMF21a took place smoothly to furnish the o-quinone, 13C2-BP-7,8-dione (80 %).27 Reduction of 13C2-BP-7,8-dione with NaBH4/O221a, 28 furnished 13C2-trans-7,8-dihydro-7,8-dihydroxybenzo[a]pyrene (13C2-BP- 7,8-diol).29 This dihydrodiol may be readily converted to the 13C2-anti- and syn-diol epoxide isomers by the procedures previously described for synthesis of the corresponding unlabelled compounds.30
Scheme 2.

The syntheses described in preceding paragraphs provide convenient access to the 13C2-labelled analogues of BP and its presumed carcinogenic metabolites. Synthesis of the 13C2-labelled analogues of the potent carcinogenic PAH dibenzo[def,p]chrysene and its corresponding active metabolites by a different synthetic approach was recently reported by us.31 In principle, these methods are potentially applicable to synthesis of 13C2-labelled analogues of a wide range of other PAH carcinogens and their oxidized metabolites.
Acknowledgments
This investigation was supported by NIH Grants (P01 CA 92537, R01 CA 039504, R01 ES 015857, and P30 ES 013508)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.International Agency for Research on Cancer. Polynuclear Aromatic Compounds, Part 1, Chemical, Environmental and Experimental Data. Vol. 32. IARC: Lyon, France; 1983. Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans. [PubMed] [Google Scholar]
- 2.Straif K, Boan R, Grosse Y, Secretan B, Ghissassi FE, Cogliano V. Nature Oncology. 2005;6:931–932. doi: 10.1016/s1470-2045(05)70458-7. [DOI] [PubMed] [Google Scholar]
- 3.Harvey RG. Polycyclic Aromatic Hydrocarbons: Chemistry and Carcinogenicity. Cambridge, UK: Cambridge University Press; 1991. [Google Scholar]
- 4.Marr LC, et al. Environ. Sci. Technol. 1999;3:3091–3099. [Google Scholar]
- 5.International Agency for Research on Cancer. Tobacco Smoke and Involuntary Smoking. Vol. 83. IARC: Lyon, France; 2004. Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans. [Google Scholar]
- 6.Pfiefer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht S, Hainaut P. Oncogene. 2002;21:7435–7451. doi: 10.1038/sj.onc.1205803. [DOI] [PubMed] [Google Scholar]
- 7.World Health Organization. Tobacco or Health: A Global Status Report. WHO: Geneva; 1997. pp. 10–48. [Google Scholar]
- 8.Boffetta P, Jourenkova N, Gustavsson P. Cancer Causes and Control. 1997;8:444–472. doi: 10.1023/a:1018465507029. [DOI] [PubMed] [Google Scholar]
- 9.Armstrong B, Hutchinson E, Unwin J, Fletcher T. Environ. Health Perspect. 2004;112:970–978. doi: 10.1289/ehp.6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rubin H. Carcinogenesis. 2001;22:1903–1930. doi: 10.1093/carcin/22.12.1903. [DOI] [PubMed] [Google Scholar]
- 11.Baird WM, Hooven LA, Mahadevan B. Environ. Mol. Mutagen. 2005;45:106–114. doi: 10.1002/em.20095. [DOI] [PubMed] [Google Scholar]
- 12.Palackal NT, Burczynski ME, Harvey RG, Penning TM. Biochemistry. 2001;40:10901–10910. doi: 10.1021/bi010872t. [DOI] [PubMed] [Google Scholar]
- 13.Penning TM, Onishi ST, Onishi T, Harvey RG. Chem. Res. Toxicol. 1996;9:84–92. doi: 10.1021/tx950055s. [DOI] [PubMed] [Google Scholar]
- 14.Cavalieri EL, Rogan EG. Xenobiotica. 1995;25:677–688. doi: 10.3109/00498259509061885. [DOI] [PubMed] [Google Scholar]
- 15.Melendez-Colon VJ, Luch A, Seidel A, Baird WM. Carcinogenesis. 1999;20:1885–1891. doi: 10.1093/carcin/20.10.1885. [DOI] [PubMed] [Google Scholar]
- 16.Park JH, Mangal D, Tacka KA, Quinn A, Harvey RG, Blair IA, Penning TM. Proc. Natl. Acad. Sci. USA. 2008 doi: 10.1073/pnas.0802776105. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang H, Gelhaus SL, Mangal D, Harvey RG, Blair IA, Penning TM. Chem. Res. Toxicol. 2007;20:1331–1341. doi: 10.1021/tx700107z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruan Q, Gelhaus S, Penning TM, Harvey RG, Blair IA. Chem. Res. Toxicol. 2007;2:424–431. doi: 10.1021/tx060180b. [DOI] [PubMed] [Google Scholar]
- 19.Ruan Q, Kim HYH, Jiang H, Penning TM, Harvey RG, Blair IA. Rapid Commun. Mass Spectrom. 2006;20:1369–1380. doi: 10.1002/rcm.2457. [DOI] [PubMed] [Google Scholar]
- 20.Bodine RS, Hylarides M, Daub GH, VanderJagt DL. J. Org. Chem. 1978;43:4025–4028. [Google Scholar]; Simpson JE, Daub GH, VanderJagt DL. J. Labelled Compd. Radiopharm. 1980;17:895. [Google Scholar]; Klassen SE, Daub GH, VanderJagt DL. J. Org. Chem. 1983;48:4361–4366. [Google Scholar]; Bodine RS, Daub GH, VanderJagt DL. J. Labelled Compd. Radiopharm. 1984;22:475–485. [Google Scholar]
- 21.(a) Harvey RG, Dai Q, Ran C, Penning TM. J. Org. Chem. 2004;69:2024–2032. doi: 10.1021/jo030348n. [DOI] [PubMed] [Google Scholar]; (b) Harvey RG, Lim K, Dai Q. J. Org. Chem. 2004;69:1372–1373. doi: 10.1021/jo030313n. [DOI] [PubMed] [Google Scholar]
- 22.CH3OCH=PPh3 was prepared by reaction of CH3OCH2Br with PPh3 and treatment of the resulting phosphonium salt with t-BuOK (ref.21a).
- 23.13CH2=PPh3 was prepared via reaction of 13CH3I (99 atom %) with PPh3 and treatment of the phosphonium salt with n-BuLi. Thus, to a suspension of 13CH3PPh3I (2.0 g, 4.9 mmol) in anhydrous THF (20 mL) at −78 °C under argon was added a solution of n-BuLi (2.0 mL of a 2.5M solution in THF, 5.0 mmol). The solution was stirred at −78 °C for 20 min, then the cooling bath was removed, and stirring was continued for an additional 30 min.
- 24.13C2-2-(2,6-Divinylphenyl)naphthalene (4a) was synthesized by reaction of 3a with 13CH2=PPh3 prepared as described above. To the solution of 13CH2=PPh3 was added a solution of 3a (0.70 g, 2.45 mmol) in THF (10 mL), and the solution was stirred for 30 min when TLC indicated reaction to be complete. Conventional work up and flash chromatography on silica gel eluted with hexane/EtOAc (15:1) gave 4a (0.68g, 98%) as a colorless oil;the 1H NMR spectrum was in agreement with that for unlabelled 4a; 13 C NMR (CDCl3) δ 114.7. This compound was unstable and deteriorated on standing.
- 25.13C2-Benzo[a]pyrene (6a) was synthesized from 4a via conversion to the bisepoxide (5a) and acid-catalyzed cyclization. To a solution of 4a (0.29 g, 1.0 mmol) in EtOAc/acetone/H2O (20/10/10) was added NaHCO3 (0.63 g, 7.5 mmol), and to this solution was added dropwise a solution of Oxone (5.4 g, 9.0 mmol) in (30 mL) over a 4 h period. Stirring was continued for an additional 3 h, then the solvent was evaporated. The product was dissolved in EtOAc and purified by flash chromatography to provide 5a (290 mg). This was dissolved in CHCl3 (7.2 mL), InCl3 (11 mg) was added, and the mixture was heated at reflux for 12 h. Flash chromatography of the product provided 6a (70%); the 1H NMR spectrum was in generally good agreement with that of unlabelled BP; 13C NMR (CDCl3) δ 122.1 and 128.1.
- 26.A suspension of 6b (40 mg, 0.14 mmol) in 57 % HI (5 mL) and HOAc (5 mL) was stirred at 140 °C until TLC showed reaction to be complete (1.5 h). Then the solution was cooled to room temperature, and poured into ice water (50 mL) to afford 6c (37 mg, 97 %) which was used directly in the next step.
- 27.The 13C NMR spectrum of 13C2-BP-7,8-dione exhibited characteristic signals at δ 122.9 and 128.8, corresponding to the 13C-atoms in the C-5 and C-11-positions.
- 28.Sukumaran KB, Harvey RG. J. Org. Chem. 1980;45:4407–4413. [Google Scholar]
- 29.The 13C NMR spectrum of 13C2-BP-7,8-diol showed signals at δ 122.5 and 127.6 corresponding to the 13C-atoms in the C-5 and C-11-positions.
- 30.Harvey RG. In: Harvey RG, editor. Polycyclic Hydrocarbons and Carcinogenesis; ACS Symp. Series, No. 283; Washington DC: 1985. pp. 35–62. Edit. Amer. Chem. Soc. [Google Scholar]
- 31.Xu D, Duan Y, Blair IA, Penning TM, Harvey RG. Org. Lett. 2008;10:1058–1062. doi: 10.1021/ol7029323. [DOI] [PubMed] [Google Scholar]