

Abstract
In vivo, RNA molecules are constantly accompanied by RNA binding proteins (RBPs), which are intimately involved in every step of RNA biology, including transcription, editing, splicing, transport and localization, stability, and translation. RBPs therefore have opportunities to shape gene expression at multiple levels. This capacity is particularly important during development, when dynamic chemical and physical changes give rise to complex organs and tissues. This review discusses RBPs in the context of heart development. Since the targets and functions of most RBPs - in the heart and at large - are not fully understood, this review focuses on the expression and roles of RBPs that have been implicated in specific stages of heart development or developmental pathology. RBPs are involved in nearly every stage of cardiogenesis, including the formation, morphogenesis, and maturation of the heart. A fuller understanding of the roles and substrates of these proteins could ultimately provide attractive targets for the design of therapies for congenital heart defects, cardiovascular disease, or cardiac tissue repair.
Keywords: RNA binding protein, heart, development, morphogenesis, RNA processing, regulation
1. Introduction
Growing interest in the functional repertoire of RNA binding proteins (RBPs) has emerged as their potential to regulate gene expression has become more broadly appreciated. While the old paradigm of gene expression focused on the activation of transcriptional programs by DNA binding proteins, the roles of RBPs in post-transcriptional regulation have recently been given greater scrutiny. Post-transcriptional regulatory mechanisms have been identified at all levels of the life cycle of a transcript: regulation of pre-mRNA alternative splicing (Kelemen et al., 2013), mRNA editing (Chateigner-Boutin and Small, 2011), transcript stability (Schoenberg and Maquat, 2012), transcript localization (transport and sequestration) (Medioni et al., 2012), and regulation of translation (Kong and Lasko, 2012). RBPs are involved in the regulation of each of these processes [for an overview see (Glisovic et al., 2008)]; some specific examples are illustrated in Figure 1. Some of these functions are nuclear, while others are cytoplasmic, or take place within other organelles such as mitochondria. There are several different types of RNA binding domains, which divide RBPs into structurally distinct families. While many employ beta sheets as interaction surfaces to interface with client transcripts and utilize aromatic residues and base stacking interactions to achieve recognition of targets, a detailed understanding of the binding properties of many RBPs remain to be elucidated (Lunde et al., 2007). While their RNA binding domains can help catalog these proteins, RBP families are not characterized by unified functions. Individual RBPs may perform several functions within the same cell, and may have different functions in different cell types. The ability of RBPs to circumvent the transcription machinery allows them to quickly and selectively fine-tune expression, and this capacity has been recognized as especially important in developmental and pathological systems (Masuda et al., 2009; Misquitta et al., 2001; Siomi and Dreyfuss, 1997). For example, during early zygotic development, when maternal transcripts are translated but the transcription machinery is silent, RBPs provide robust mechanisms for regulating gene expression to direct processes such as pattern formation and cell-type specification (Lee and Schedl, 2006).
Figure 1. Mechanisms of RBP-mediated post-transcriptional regulation.

Schematic representations of mechanisms by which a number of proteins described in this review have been shown to regulate gene expression. Note that these are provided as examples; an exhaustive survey of RBP-mediated regulatory mechanisms is beyond the scope of this review. (1) RBFOX proteins regulate a variety of alternative splicing events by binding within introns flanking alternative exons. Binding upstream of an exon leads to skipping of that exon, while binding downstream of an exon leads to its inclusion (De Craene and Berx, 2013). (2) CELF2 directs the editing of a cytidine in the Apob transcript by binding to an AU-rich sequence element upstream of the editing site and recruiting ACF, a component of the editing machinery (Anant et al., 2001). (3) MBNL2 regulates the transport and localization of the Itga2 transcript to the plasma membrane by binding to a zipcode sequence in the 3′ UTR of the transcript (Adereth et al., 2005). (4) Multiple mechanisms have been proposed for how AUF1 regulates the stability of target transcripts, including the recruitment of the PARN deadenylase, leading to loss of the poly-A tail and rapid degradation of the RNA (White et al., 2013). (5) CELF1 enhances translation of the p21 transcript by antagonizing a regulatory protein, CRT, which normally blocks ribosome loading (Iakova et al., 2004).
The post-transcriptional regulation of gene expression by RBPs during development also has evolutionary consequences. Because of their generally small size and ability to rely on diffusion for tissue oxygenation, many invertebrate species lack a defined circulatory system. The invertebrate heart is typically a simple, beating tube or sac that moves fluid through the body via peristaltic contractions. Vertebrates, on the other hand, have closed circulatory systems with multi-chambered hearts. It has been proposed that vertebrates exhibit a greater degree of cellular and organismal complexity than invertebrates due in large part to expansion of the transcriptome (without proportional expansion of the genome) via an increase in alternative RNA processing, particularly pre-mRNA alternative splicing (Ast, 2004; Maniatis and Tasic, 2002). Consistent with this, several RBP families involved in alternative splicing regulation are differentially expanded in vertebrates compared to invertebrates, whereas basal splicing machinery proteins are generally invariant among all eukaryotes (Barbosa-Morais et al., 2006; Pascual et al., 2006).
The development of the heart is a complex and finely orchestrated process. RBPs have been shown to be involved in nearly every step of heart development, from the establishment of cardiac lineages to the maturation of the heart after birth (Figure 2). In addition, there are RBPs that are known to be expressed in the heart at some stage, but whose molecular and biological roles in heart development have not yet been determined. This review will focus on the expression and functions of RBPs that have been implicated in the formation, morphogenesis, and maturation of the heart (Figure 3). It should also be noted that a set of RBPs known as Argonaute proteins interact with microRNAs (miRNAs) within the RNA-induced silencing complex (RISC) to destabilize or inhibit the translation of target mRNAs that possess sequence complementarity (Amiel et al., 2012). Although miRNA-mediated gene expression changes are important in the heart during normal development and disease (Chen and Wang, 2012; Ono et al., 2011), we will not discuss Argonaute proteins further in this review, as it is generally the miRNAs rather than their associated RBPs that are developmentally regulated.
Figure 2. RNA binding proteins have been implicated in the formation, morphogenesis, and maturation of the heart.
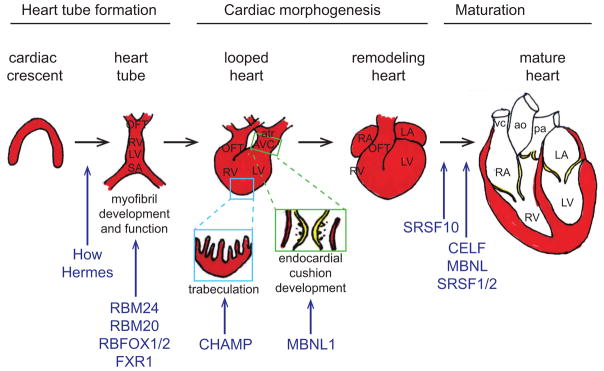
The primitive heart tube forms from precardiac mesoderm within the cardiac crescent. The heart tube undergoes extensive morphogenesis, including cardiac looping, endocardial cushion formation and remodeling, and myocardial trabeculation and compaction. Although the architecture of the heart is established during embryogenesis, maturation of the heart continues through postnatal life. RNA binding proteins that have been implicated in specific steps of heart development are indicated in blue. Abbreviations: OFT, outflow tract; RV, right ventricle; LV, left ventricle; SA, sinoatrial segment; atr, common atrium; AVC, atrioventricular canal; LA, left atrium; RA, right atrium; vc, vena cava; ao, aorta; pa, pulmonary artery.
Figure 3. Domain structure of RNA binding proteins implicated in regulation of heart development.

Schematic representations of the type and position of important domains within the RNA binding proteins described in this review are shown. RNA binding domains and other domains characteristic of these RNA binding protein families are shown in color; other conserved domains are shown in black and white. Proteins and domains are not drawn to scale. Molecular functions of these RNA binding proteins within the heart are indicated, if known; additional functions of these proteins that have been demonstrated in other tissue types are not shown. The domain structures of these proteins are highly conserved across vertebrate species, and the depicted structures represent all homologs described in the text. Note that the structure of QK/How is conserved from human to fly, whereas to date CHAMP/Csm has only been described in mammals.
2. Cardiac cell fate, heart tube formation, and differentiation
The heart is the first functional organ to form in the vertebrate embryo. Induction of cardiogenesis begins in the pregastrula embryo (Ladd et al., 1998; Yatskievych et al., 1997), and precardiac cells become specified within the nascent mesoderm as it arises during gastrulation (Brand, 2003). As presumptive cardiogenic cells migrate out of the primitive streak, the heart-forming regions are found on either side of the midline. Both positive and negative signals from nearby tissues act to induce cells in the heart-forming regions to migrate rostrally and medially to form the cardiac crescent (i.e., the primary heart field). A second cohort of cells (i.e., the secondary heart field) arising from splanchnic mesoderm at the arterial pole also contributes to the formation of the heart (Brand, 2003; Dunwoodie, 2007). Cell migration, along with flexion and folding of the embryo, bring the heart-forming regions together at the midline where they fuse to form the primitive heart tube: an outer, myocardial layer surrounding an inner, endocardial layer. The linear heart tube is arranged from the sinoatrial segment (inflow tract) at the posterior end to the presumptive left and right ventricles, and finally the conus arteriosus (outflow tract) at the anterior end. Completion of the heart tube coincides with the assembly of nascent myofibrils in the cardiac myocytes, and beating begins. In vertebrates, the primitive heart tube is later transformed into a multi-chambered heart.
The inductive signals that establish cardiac cell fates have been long studied, and include Wnt-βcatenin, fibroblast growth factor (FGF) and Hedgehog proteins, and members of the transforming growth factor β (TGFβ) superfamily such as the bone morphogenetic proteins (BMPs) (Brand, 2003; Dunwoodie, 2007). These growth factors initiate signaling cascades that activate important cardiogenic transcription factors of the NKX, TBX, GATA, and MEF2 families (Brand, 2003; Dunwoodie, 2007). Much less is known about the contributions of post-transcriptional programs to the establishment of cardiogenic cell lineages, but several RBPs have been shown to participate in heart tube formation or cardiac cell differentiation (Figure 2).
2.1. Formation of the heart tube
Unlike the multi-chambered vertebrate heart, the heart of the fruit fly Drosophila melanogaster remains a simple linear tube. The Drosophila heart tube is subdivided into an anterior “aorta” and a larger posterior cavity. Although the fly heart is structurally much simpler, gene expression programs important for cardiogenesis are largely conserved from flies to man. This conservation, along with the ease of performing large-scale genetic screens, has made Drosophila a powerful model system for the identification of important regulatory genes. For example, the importance of NK-class homeodomain-containing transcription factors (such as Nkx-2.5) in heart formation was first uncovered in studies of the fly paralog, tinman (Bodmer, 1993). Similarly, RBPs have been identified in flies that are important in the developing heart.
Held out wings (How) is a heterogeneous nuclear ribonucleoprotein (hnRNP) K-homology (KH)-domain protein (Figure 3) involved in muscle function (Baehrecke, 1997). It is most similar to the mouse Quaking (QK) and Caenorhabditis elegans GLD1 proteins, which belong to the signal transduction and activation of RNA (STAR) subfamily of KH-domain proteins. Members of this subfamily have been described to link signal transduction pathways to RNA metabolism in different tissues (Lasko, 2003). In flies, MAPK/ERK-dependent phosphorylation of How promotes its RNA binding activity, and How is phosphorylated in vivo in embryonic cardioblasts (Nir et al., 2012). A maternal how transcript is found broadly in the Drosophila zygote, although no detectable protein product is detected (Zaffran et al., 1997). A zygotic transcript is activated in later stages in the presumptive mesoderm, and then in cells of myogenic lineages and in epidermal muscle attachment cells (Baehrecke, 1997; Zaffran et al., 1997). Later in development, how transcripts are restricted to the heart and to muscle attachment sites. This expression pattern is maintained during and following morphogenesis, with the addition of expression in adult muscle precursor cells attached to the wing imaginal discs, resulting in the phenotype for which the gene is named (Baehrecke, 1997; Zaffran et al., 1997). Despite this broad early expression, histological analysis of mutants suggests that perturbation of how expression does not result in disruption of striated muscle formation, but rather leads to functional aberrations including reduced cardiac beat rate and weakened contraction (Zaffran et al., 1997). A recent study has suggested that How regulates the expression of sarcomeric proteins, although it is unclear whether this relationship is direct or indirect (Nir et al., 2012). How has also been shown to be part of a genetic pathway with the extracellular matrix protein Slit, its receptor Robo, and dystroglycan that controls formation of the cardiac lumen during heart tube morphogenesis (Medioni et al., 2008).
The mouse QK has nuclear and cytoplasmic isoforms generated by alternative splicing (Kondo et al., 1999). QK binds to consensus UACU(C/A)A hexanucleotide sequences (Ryder and Williamson, 2004), and regulates the alternative splicing of transcripts encoding proteins involved in myelination in the adult brain (Wu et al., 2002). An allelic series of mice bearing different Qk mutations, many of which are embryonic lethal, has revealed roles for QK in formation of the heart and vasculature (Justice and Hirschi, 2010). The specific targets of QK, and the specific functions of different QK isoforms, within the developing heart have yet to be elucidated. Another related STAR protein, Sam68, is also found in mouse heart, among a variety of other tissues, but its contributions to cardiac development or function have not been studied (Richard et al., 2005).
Heart and RRM expressed sequence (hermes), the homolog of the human RNA binding protein with multiple splicing type 1 (RBP-MS type 1), is an RNA recognition motif (RRM)-containing RBP (Figure 3) expressed in a variety of embryonic tissues including heart (Gerber et al., 1999). In Xenopus laevis, hermes is first detected in the developing heart concomitant with the first cardiac differentiation markers shortly before the coalescence of the linear heart tube, and remains high throughout morphogenesis (Gerber et al., 2002; Gerber et al., 1999). Likewise, in the chicken embryo HERMES is first detected in a crescent of Nkx-2.5-positive cardiac precursor cells, before expanding to encompass the entire myocardial layer of the heart tube (Wilmore et al., 2005). At later stages, however, its expression is curtailed in the outflow tract and persists primarily in the ventricles and atria (Wilmore et al., 2005). In the mouse, Hermes expression is absent in the early heart tube, but becomes widespread following looping, after which its expression becomes increasingly atrial (Gerber et al., 1999). Hermes likely regulates cardiac cell fate, as over-expression in the Xenopus embryo leads to dramatic reductions in the expression of cardiac markers, including cardiac α-actin and Nkx-2.5, and failure of heart tube formation (Gerber et al., 2002). This effect seems to be specific to the role of hermes in heart, since over-expression in somites does not result in aberrant skeletal muscle gene expression.
2.2. RBPs in cardiomyocyte differentiation and myofibril development
Recent studies investigating transcriptome-wide changes in alternative splicing during differentiation of pluripotent stem cells have highlighted the importance of post-transcriptional RNA processing in cell fate decisions and differentiation (Brandenberger et al., 2004; Cloonan et al., 2008; Pritsker et al., 2005; Salomonis et al., 2009; Salomonis et al., 2010; Yeo et al., 2007). In a comparison of human embryonic stem cells before and after differentiation into either cardiac or neural progenitors, Salomonis and colleagues identified alternative splicing events common to both differentiation pathways, but also events that were specific for the cardiac lineage (Salomonis et al., 2009). To what extent these alternative splicing events direct cardiomyocyte differentiation, and the RBPs responsible for their regulation, however, remain unknown. Knockdown/knockout experiments in fish, frogs, and rodents have identified several RBPs that are important during development for cardiomyocyte-specific gene expression, myofibril assembly, and myocardial function in vivo (Figure 2).
RNA binding motif (RBM) proteins are a subgroup of loosely related RRM-containing proteins (Figure 3) with varying functions, including splice site selection and nonsense-mediated RNA decay (Sutherland et al., 2005). RBM24 is up-regulated in both human and mouse embryonic stem cells upon differentiation into cardiomyocytes (Miller et al., 2008; Xu et al., 2009). Orthologs of RBM24 are expressed first in cardiac precursors, and then in the differentiated myocardium in zebrafish, frog, and mouse embryos (Fetka et al., 2000; Maragh et al., 2011; Miller et al., 2008; Xu et al., 2009). Knockdown of rbm24a/b in zebrafish embryos results in reductions in sarcomeric proteins, profound disorganization of the myofibril, looping defects, changes in heart rate, and reduced circulation (Maragh et al., 2011; Poon et al., 2012). A second RBM protein, Rbm20, has been shown to regulate the splicing of transcripts encoding titin, a large sarcomeric protein (Guo et al., 2012; Li et al., 2013). Mutations in RBM20 have been linked with dilated cardiomyopathy in humans, consistent with an important role in regulating myofibril structure and/or function (Brauch et al., 2009; Li et al., 2010; Refaat et al., 2012). Loss of another RBM protein, Rbm15, in mice leads to a variety of embryonic defects, including cardiac abnormalities and heart failure (Raffel et al., 2009), though its role in heart development remains poorly defined.
RNA binding protein, fox-1 homolog (RBFOX) proteins are RRM-containing proteins (Figure 3) that specifically bind to (U)GCAUG elements and regulate alternative splicing (Kuroyanagi, 2009). In addition to the RRM domain, RBFOX1 (also known as A2BP1) possesses two Sm domains, also found in small nuclear ribonucleoproteins, suggesting additional roles in RNA processing or transport (Shibata et al., 2000). RBFOX1 and RBFOX2 (also known as RBM9) are highly expressed in the developing heart, skeletal muscle, and brain, and (U)GCAUG motifs have been found to be enriched in transcripts that are alternatively spliced in these tissues (Bland et al., 2010; Brudno et al., 2001; Das et al., 2007; Gallagher et al., 2011; Jin et al., 2003; Sugnet et al., 2006; Zhang et al., 2008). The splicing activity of RBFOX proteins and splicing patterns of RBFOX targets in the heart are highly conserved across species (Gallagher et al., 2011; Jin et al., 2003; Venables et al., 2012). Although knockdown of individual Rbfox proteins in zebrafish does not result in gross defects, rbfox1/rbfox2 double morphant embryos exhibit severe skeletal and cardiac muscle defects, including myofibrillar disorganization, disrupted swimming, and reduced cardiac function (Gallagher et al., 2011). Expression of rbfox2 alone is sufficient to rescue wild type myocardial structure and function, suggesting that it can complement the functions of rbfox1 in cardiac myofibril development (Gallagher et al., 2011).
For proper assembly and maintenance of myofibrils, and for cardiomyocyte function, connections must be formed and maintained between apposing myofibrils, between myofibrils and the plasma membrane, and between neighboring cardiomyocytes. The RBP Fragile X mental retardation, autosomal homolog 1 (FXR1), has recently been shown to be involved in the proper regulation of these connections (Whitman et al., 2011). FXR1, a homolog of FMR1, which is lost in Fragile X Syndrome, contains two KH domains and an RGG box (Figure 3) (Burd and Dreyfuss, 1994; Siomi et al., 1995). A role in transport and translation regulation of mRNA targets has been proposed for fxr1 during somitogenesis in Xenopus (Huot et al., 2005). Fxr1-null mice have disorganized cardiac and skeletal muscle, and die within a few hours of birth, likely due to cardiac and respiratory insufficiency (Mientjes et al., 2004). Using these mice, Fxr1 was shown to regulate the expression of Talin2, which is found at the costameres (i.e., myofibril-membrane anchors), and Desmoplakin, which is found at the desmosomes (i.e., cell-cell junctions), in cardiac muscle at the level of translational repression (Whitman et al., 2011). Dysregulation of these proteins results in disruption of desmosome, costamere, and sarcomere structures at the microscopic level, and in overt cardiomyopathy and muscular dystrophy at the macroscopic level (Whitman et al., 2011). The expression and function of Fxr1 in the developing heart and skeletal muscle is conserved in zebrafish (Engels et al., 2004), where knockdown of fxr1 using antisense morpholinos likewise results in abnormal myotome formation and severe embryonic cardiomyopathy (Van’t Padje et al., 2009).
3. Cardiac morphogenesis
Following formation of the primitive heart tube, the heart undergoes extensive morphogenesis to transform from a simple tube into a multi-chambered pump that is capable of directing pulmonary and somatic circulation. Left-right asymmetry, established in the early embryo by a combination of asymmetrically deposited signaling molecules and the asymmetric activation of transcription programs, culminates in the heart in cardiac looping, in which right-handed bulging and twisting of the heart tube gives rise to a C-shaped tube with the inflow and outflow tracts both oriented rostrally (Brand, 2003). Differential proliferation distinguishes the “outer curvature” from the “inner curvature” of the heart, and contributes to subsequent expansion and definition of the chambers (Wagner and Siddiqui, 2007). RBPs have been implicated in the regulation of morphogenesis of the valves and septa that then divide the chambers, as well as morphogenesis of the developing myocardium (Figure 2).
3.1. Endocardial cushion development
The looped heart tube is subdivided into a four-chambered heart through the formation and remodeling of structures called endocardial cushions in the atrioventricular canal (AVC) and outflow tract (OFT) regions (DeLaughter et al., 2011; Person et al., 2005). In the AVC and OFT, the endocardium is pushed away from the myocardium by the localized expansion of extracellular matrix. The cushions are cellularized by the invasion of mesenchymal cells produced from a subpopulation of endocardial cells via epithelial-to-mesenchymal transition (EMT). Subsequent remodeling into the valves and septa involves condensation and differentiation of the cushion mesenchyme at post-EMT stages (Kirby, 2007). The AVC cushions give rise to the mitral and tricuspid valves, as well as to the atrioventricular septum, and contribute to the atrial and ventricular septa (Kirby, 2007). The OFT cushions give rise to the aortic and pulmonary valves, and transiently septate the OFT during early morphogenesis (Qayyum et al., 2001). Dysregulation of EMT in the heart can lead to valve and septal defects (Person et al., 2005). Although the tissue interactions, growth factors, signaling pathways, and transcription factors involved in regulating endocardial cushion EMT have been well studied (DeLaughter et al., 2011; Person et al., 2005), the roles of post-transcriptional RNA processing in regulating EMT and post-EMT remodeling are much less well understood.
A member of the muscleblind-like (MBNL) protein family, MBNL1, has been implicated in regulating endocardial cushion EMT (LeMasters et al., 2012; Vajda et al., 2009). MBNL proteins bind to RNA via two conserved pairs of zinc knuckle domains (Figure 3), and regulate pre-mRNA alternative splicing, RNA localization, and mRNA stability (Ho et al., 2004; Masuda et al., 2012; Wang et al., 2012). In the chicken embryo, MBNL1 expression is not detected in the linear heart tube prior to cardiac looping, but is detected in the looped heart prior to the formation of the endocardial cushions (LeMasters et al., 2012; Vajda et al., 2009). In the AVC and OFT, MBNL1 is strongly expressed in the endocardium (LeMasters et al., 2012; Vajda et al., 2009). Knockdown of MBNL1 in chick AVC or OFT explants induces enhanced EMT ex vivo, indicating that MBNL1 is a negative regulator of EMT in the endocardial cushions (LeMasters et al., 2012; Vajda et al., 2009). Active, secreted TGFβ3 levels are also elevated following MBNL1 knockdown (LeMasters et al., 2012). TGFβ proteins are important for inducing EMT (Arthur and Bamforth, 2011; Kruithof et al., 2012), suggesting MBNL1 may regulate EMT in part via modulation of EMT-inducing signals. MBNL1 preferentially binds to YGCY motifs in vitro and in vivo (Goers et al., 2010; Wang et al., 2012), and these motifs are highly enriched in known MBNL1-responsive transcripts (Gates et al., 2011; Ho et al., 2004; Wang et al., 2012). Strikingly, a recent study investigating alternative splicing in breast cancer cells found an enrichment of YGCY motifs near exons that exhibited decreased inclusion following EMT (Shapiro et al., 2011). This suggests MBNL1 may play a general role in regulating alternative splicing transitions during EMT.
A paralog of MBNL1, MBNL2, is also expressed in the embryonic heart (Fardaei et al., 2002; Fernandes et al., 2007; Kanadia et al., 2003b; Liu et al., 2008), but its temporal and spatial distribution within the developing heart, and whether it plays a role in EMT, have not yet been investigated. Knockdown of mbnl2 in zebrafish does result in cardiac abnormalities, however, including dilation and disorganization of the myofibril, and mis-regulated alternative splicing of tnnt2, a known target of MBNL proteins in the myocardium (Machuca-Tzili et al., 2011).
Additional RBPs have also been implicated in regulating alternative splicing during EMT. Epithelial splicing regulatory proteins (ESRP) 1 and 2 are RRM-containing RNA binding proteins that have been shown to regulate hundreds of alternative splicing events during EMT (Dittmar et al., 2012; Warzecha et al., 2009a; Warzecha et al., 2009b). Knockdown and over-expression studies have suggested that changes in ESRP expression are determinative for EMT and its converse, MET (Reinke et al., 2012; Shapiro et al., 2011; Warzecha et al., 2010), but in a recent study neither Esrp1 nor Esrp2 were detected in the embryonic heart (Revil and Jerome-Majewska, 2013). Motif analysis of intronic regions flanking EMT-regulated cassette exons not only suggests common regulatory roles for MBNL and ESRP proteins in EMT-specific alternative splicing, but also members of the polypyrimide tract binding protein (PTB), hnRNP, and RBFOX families (Shapiro et al., 2011). None of these have yet been interrogated for a role in endocardial cushion development.
3.2. Myocardial trabeculation and compaction
Myocardial cells are highly dynamic during cardiac morphogenesis, undergoing epithelialization, proliferation, and compaction as trabeculae form and then coalesce in the ventricular wall (Harvey and Rosenthal, 1999). The trabeculae consist of protrusions into the lumen of the heart, and are made up of poorly developed but strongly coupled cells, which contribute to the ventricular conduction system (Moorman and Christoffels, 2003). The trabeculae also increase the surface area of the thickening tissue, which is critical for oxygenation of the heart before the coronary arteries form. Although the anatomical changes that occur during trabeculation and compaction have been well described, the mechanisms that control these processes are not well understood.
Cardiac helicase activated by MEF2 protein (CHAMP) is an RNA helicase expressed specifically in the myocardium during prenatal and postnatal development (Liu et al., 2001). CHAMP is a member of the RNA helicase superfamily I, and shares several conserved motifs with helicases involved in DNA replication, transcription, and RNA processing (Figure 3) (Liu et al., 2001). Embryonic expression of CHAMP begins in the linear heart tube following the initiation of MEF2C expression. During trabeculation, CHAMP is strongly expressed in the non-proliferative trabecular cardiomyocytes, but not in the proliferative compact zone (Liu et al., 2001). This regional localization, as well as spatiotemporal similarities between the expression of CHAMP and both neurotrophin-3 and its receptor, Trk C, which regulate cardiomyocyte proliferation (Lin et al., 2000), led to speculation that CHAMP plays a role in repressing cardiomyocyte proliferation and growth during myocardial morphogenesis (Liu et al., 2001). Although this has not been experimentally confirmed in vivo, over-expression of CHAMP in primary neonatal cardiomyocytes inhibits cellular hypertrophy and leads to the up-regulation of the cell cycle inhibitor p21 (Liu and Olson, 2002).
Interestingly, CHAMP is a variant of the testis-specific helicase, MOV10 like-1 (MOV10l1), generated by alternative promoter usage, using a start codon within exon 14 (Liu et al., 2001). A second cardiac-specific helicase, cardiac-specific isoform of Mov10l1 (Csm), is also generated by alternative promoter usage from the MOV10l1 locus; its start codon lies within exon 16 (Ueyama et al., 2003). Despite their similar origins, the two helicases have distinct effects. While Csm potentiates cardiomyocyte hypertrophy induced by phenylephrine treatment (Ueyama et al., 2003), CHAMP over-expression is able to block this hypertrophy (Liu et al., 2001). The expression and role of Csm in embryonic heart development has not been investigated.
4. Postnatal maturation of the heart
Although the four-chambered architecture of the heart is established during embryogenesis, the rerouting of circulation to the lungs, switch from hyperplastic to hypertrophic growth, and increase in workload on the heart after birth prompts extensive molecular and cellular remodeling during early postnatal life. This remodeling involves changes in the expression of growth factors, cell cycle regulators, contractile and cytoskeletal proteins, and ion channels (Chen et al., 2004; Harrell et al., 2007; MacLellan and Schneider, 2000; Siedner et al., 2003), and is not limited to changes in transcription, but also includes changes in post-transcriptional RNA processing.
4.1. CELF- and MBNL-mediated alternative splicing programs
Many cardiac transcripts undergo fetal-to-adult changes in alternative splicing (Kalsotra et al., 2008; Park et al., 2011). Several families of RBPs have been implicated in fetal-to-adult reprogramming of alternative splicing in the heart (Figure 2). A study using splicing-sensitive microarrays identified fetal-to-adult splicing transitions in the developing mouse heart, many of which were conserved between mammals and birds (Kalsotra et al., 2008). Computational analyses identified several motifs that were highly enriched near the developmentally-regulated exons, including putative binding sites for hnRNP, PTB, STAR, RBFOX, MBNL, and CUGBP, Elav-like family (CELF) proteins (Kalsotra et al., 2008). Consistent with this, some of these RBPs were shown to be developmentally regulated. RBFOX1 transcript and protein levels are robustly up-regulated shortly after birth, whereas those of RBFOX2 decline slightly in the adult heart (Kalsotra et al., 2008). MBNL1 is up-regulated over the course of embryonic and postnatal heart development (Kalsotra et al., 2008; Terenzi and Ladd, 2010). CELF1 and CELF2 exhibit higher protein levels in embryonic than adult heart, and are down-regulated during early postnatal life (Kalsotra et al., 2008; Ladd et al., 2005a). Interestingly, the down-regulation of CELF proteins during heart development occurs without a change in CELF transcript levels, indicating that these RBPs are themselves post-transcriptionally regulated. Mechanisms include phosphorylation-driven changes in CELF1 protein stability and miRNA-mediated repression of CELF1 and CELF2 translation (Kalsotra et al., 2010; Kuyumcu-Martinez et al., 2007).
Strikingly, over half of the fetal-to-adult splicing transitions identified by Kalsotra and colleagues respond to over-expression of CELF1 or loss of MBNL1 in the hearts of genetically modified mice, suggesting these proteins are determinative for driving these developmental transitions (Kalsotra et al., 2008). A subset of these splicing events is regulated by both CELF1 and MBNL1 in an antagonistic manner. Although a cardiac phenotype has not yet been described for Mbnl1-null mice (Kanadia et al., 2003a), over-expression or repression of CELF proteins in the early postnatal myocardium leads to dysregulation of CELF-mediated alternative splicing and rapid onset of cardiac dysfunction in transgenic mice (Koshelev et al., 2010; Ladd et al., 2005b; Terenzi et al., 2009). The juvenile onset of cardiomyopathy, as well as the spontaneous recovery of cardiac function following maturity in a line of mice with mild repression of CELF activity (Terenzi et al., 2009), further supports a role for CELF-mediated alternative splicing programs specifically in postnatal remodeling.
CELF and MBNL proteins are found in the cytoplasm as well as the nucleus in the developing heart (Blech-Hermoni et al., 2013; Kalsotra et al., 2008; Ladd et al., 2005a), suggesting that the effects of these RBPs in postnatal maturation are likely not restricted to pre-mRNA alternative splicing. Although their cytoplasmic roles have not been investigated in the heart, in developing skeletal muscle CELF1 regulates the translation of Mef2A and p21, key regulators of muscle-specific gene expression and growth arrest (Timchenko et al., 2004), and both CELF1 and MBNL1 have been shown to regulate the stability of a large number of muscle transcripts (Masuda et al., 2012). Interestingly, CELF1 promotes decay of Mbnl1 transcripts and MBNL1 promotes decay of Celf1 transcripts, suggesting that these factors mutually contribute to their reciprocal patterns of expression during myogenesis (Masuda et al., 2012).
4.2. Roles of the SR protein family in the maturing heart
Members of the serine/arginine-rich (SR) protein family have also been implicated in fetal-to-adult cardiac reprogramming. There are twelve human SR proteins, SRSF1–12, each characterized by the presence of one or two RRMs and a carboxy-terminal RS domain enriched with arginine/serine dipeptides that functions as a protein:protein interaction domain (Figure 3) (Twyffels et al., 2011). SR proteins regulate splicing of both constitutive and alternative exons, mRNA export, stability, and translation (Shepard and Hertel, 2009; Twyffels et al., 2011). SR proteins also promote the processing of some miRNAs by facilitating cleavage by Drosha (Wu et al., 2010). The subcellular localization and activities of SR proteins are regulated in part through phosphorylation by SR protein-specific kinases (Zhou and Fu, 2013). Depletion of SRSF1 (formerly known as ASF/SF2) induces apoptosis in the DT40 chicken B-cell line (Li et al., 2005; Wang et al., 1996), and germline deletions of Srsf1, Srsf2 (formerly known as SC35), or Srsf3 (formerly known as SRp20), result in early embryonic lethality (Jumaa et al., 1999; Wang et al., 2001; Xu et al., 2005). Together, these studies have suggested essential, non-redundant roles for multiple SR proteins in cell viability. Cardiac-specific knockouts of two of these essential SR proteins, however, indicate that they have additional roles in regulating contractile function in developing heart muscle.
Cardiac-specific ablation of Srsf1 or Srsf2 was accomplished by crossing mice with floxed alleles with the MLC-2v-Cre transgenic line (Ding et al., 2004; Xu et al., 2005). Both cardiac-specific Srsf1- and Srsf2-null mice are healthy at birth, but develop early onset cardiomyopathy within the first four weeks of life (Ding et al., 2004; Xu et al., 2005), corresponding to the period of postnatal remodeling. Cardiomyocyte apoptosis was not observed in either model, but both displayed defects in excitation-contraction coupling (Ding et al., 2004; Xu et al., 2005). In cardiac-specific Srsf1-null mice this has been attributed at least in part to altered splicing of Ca2+/calmodulin-dependent kinase IIδ (CaMKIIδ), and transgenic over-expression of the inappropriate CaMKIIδ splice form was sufficient to phenocopy the defects in calcium handling in these mice (Xu et al., 2005).
SRSF10 (formerly known as SRp38) has also been implicated in regulating calcium handling in the developing heart. Unlike SRSF1, SRSF10 is not essential for viability in DT40 cells, although its loss does impair recovery from stress (Shin et al., 2004). Germline ablation of Srsf10 does not result in the early embryonic lethality seen in other SR protein gene knockouts, but nonetheless few homozygous Srsf10-null fetuses reach full term (Feng et al., 2009). Most Srsf10-null embryos die by embryonic day E15.5 and exhibit multiple cardiac abnormalities, including septal defects, thinning of the myocardium, and altered intracellular calcium handling in embryonic cardiomyocytes (Feng et al., 2009). It was suggested that the calcium handling defects may be due to changes in the level and alternative splicing of triadin (Feng et al., 2009), though this is unlikely to contribute to the other developmental defects in Srsf10-null mice as triadin-null mice are viable with no obvious cardiac malformations (Shen et al., 2007). The earlier onset of cardiac dysfunction and presence of additional defects in Srsf10-null versus cardiac-specific Srsf1- and Srsf2-null mice indicates that although multiple SR proteins are expressed in the heart, they play distinct roles in cardiac development.
5. Developmental dysregulation and disease
Dysregulation of developmental programs is often seen in disease states. Disruption of RBP function during embryogenesis can disrupt proper formation of the heart, leading to congenital heart disease. Dysregulation of developmental RBP pathways in the adult heart, however, can also perturb cardiac function and contribute to acquired heart disease.
5.1. RNA binding proteins and congenital heart defects
Several RBPs have been linked with syndromes characterized by congenital heart defects. Although a causal link has not been made in either case, CELF2 has been proposed as a candidate gene for congenital heart defects associated with two genetic disorders, partial monosomy 10p (Lichtner et al., 2002) and familial arrhythmogenic right ventricular dysplasia (Li et al., 2001). A single case report has linked a partial deletion of the RBFOX1 gene with complex heart defects (Lale et al., 2011), but further genetic evidence of RBFOX1 mutations causing developmental defects in humans is lacking.
Holt-Oram syndrome (HOS) is an autosomal dominant disorder characterized by upper limb abnormalities and a spectrum of cardiac birth defects, most typically septal and conduction defects (Mori and Bruneau, 2004). HOS is caused by a variety of mutations in the gene encoding the transcription factor TBX5, but the underlying mechanism of pathogenesis may not be limited to dysregulated transcription. TBX5 has been shown to form an RNA-dependent complex with the SR protein SRSF2, and affect constitutive and alternative splicing of reporter minigenes in cells (Fan et al., 2009). Strikingly, a severe mutation in TBX5 (G80R) associated with complete penetrance of cardiac defects strongly affects this splicing activity, whereas a less severe mutation (R237Q) associated with incomplete cardiac penetrance does not (Fan et al., 2009).
DiGeorge Syndrome (DGS) is caused in most cases by a deletion at the genomic locus 22q11.2, and is characterized by a constellation of birth defects attributable to disruption of neural crest cell development, including craniofacial and cardiac defects (Keyte and Hutson, 2012). The transcription factor gene TBX1 is a leading candidate for DGS pathogenesis, but Tbx1 is not expressed in cardiac neural crest cells in mice (Garg et al., 2001; Vitelli et al., 2002). The DiGeorge critical region 8 (DGCR8) gene also lies within the 22q11.2 region, and encodes a double-stranded RBP essential for miRNA biogenesis (Seitz and Zamore, 2006). Genetic inactivation of Dgcr8 specifically in neural crest cells in mice leads to cardiac malformations typical of DGS, including persistent truncus arteriosis, aortic arch malformations, and septal defects (Chapnik et al., 2012), supporting the idea that loss of this RBP contributes to the cardiac defects in DGS patients.
Mutations in RBM10 have been implicated in talipes equinovirus, atrial septal defect, Robin sequence (micrognathia, glossoptosis, and cleft palate), and persistence of the left superior vena cava (TARP), a rare X-linked disorder with severe congenital defects affecting multiple organs including the heart (Gripp et al., 2011; Johnston et al., 2010). Although the molecular functions of RBM10 are not well characterized, it was identified in a proteomic analysis of the human spliceosome (Rappsilber et al., 2002), and has been shown to regulate the stability of at least one mRNA (Mueller et al., 2009).
5.2. The recapitulation of fetal programs during adult heart disease
During heart disease, there is a partial reactivation of both transcriptional and post-transcriptional fetal gene expression programs. Many alternative splicing and alternative polyadenylation site choices found in the hypertrophic heart are more similar to those found at fetal stages than in healthy adults, indicating that patterns of fetal RNA processing are reestablished in response to pressure overload (Ames et al., 2013; Park et al., 2011). These differences are likely mediated by RBPs that become dysregulated during disease. For example, RBFOX1 is down-regulated while PTB is up-regulated during cardiac hypertrophy (Park et al., 2011). Alterations in alternative splicing are associated with heart disease in both mice and humans (Ames et al., 2013; Kong et al., 2010; Park et al., 2011; Song et al., 2012). Polymorphisms that affect alternative splicing of cardiac transcripts have also been linked with susceptibility to myocardial infarction and cardiac hypertrophy (Komamura et al., 2004; Mango et al., 2005).
Dysregulation of mRNA decay is another important driver of gene expression changes during cardiovascular disease. The levels of AUF1 (also known as hnRNP D), an RBP of the hnRNP family (Figure 3) that destabilizes transcripts via binding in the 3′ untranslated region (3′ UTR) (Misquitta et al., 2001), are elevated in cardiomyocytes in response to stress and in the hearts of human patients with heart failure (Glaser et al., 2006; Pende et al., 1996). The increase in AUF1 is strongly associated with a reduction in β-adrenergic receptor mRNA levels and impaired calcium handling in heart failure (Misquitta et al., 2006). AUF1 is expressed in the embryonic mouse heart (Gouble and Morello, 2000), but is normally undetectable in the adult heart (Lu and Schneider, 2004), suggesting that this may also represent the reactivation of a fetal decay program in adult heart disease.
Although the reemergence of fetal gene expression in the heart is generally thought to be compensatory, the reiteration of fetal programs in adult tissues can itself be pathogenic. In myotonic dystrophy (dystrophia myotonica, DM), the expression of mutant RNAs containing expanded CUG or CCUG repeats leads to a panoply of symptoms including electrocardiographic and echocardiographic anomalies, skeletal muscle myotonia, muscle wasting and weakness, neurological abnormalities, and endocrine dysfunction (Schoser and Timchenko, 2010). The expression of expanded repeat-containing RNAs disrupts a number of RBPs in DM cells, but pathogenesis is thought to be largely attributable to a gain of CELF1 function and loss of MBNL1 function that mirror embryonic expression patterns of these factors (Schoser and Timchenko, 2010). The reiteration of fetal alternative splicing patterns in adult DM tissues has been linked directly to patient symptoms (Charlet-B. et al., 2002; Savkur et al., 2001). Over-expression of CELF1 or deletion of MBNL1 in mice is sufficient to reestablish fetal CELF/MBNL-mediated alternative splicing patterns and mimic DM phenotypes (Ho et al., 2005; Kanadia et al., 2003a; Koshelev et al., 2010; Timchenko et al., 2004; Ward et al., 2010). Conversely, restoration of MBNL1 levels or repression of CELF activity rescues normal adult alternative splicing patterns and reduces pathogenesis in a DM mouse model (Berger and Ladd, 2012; Kanadia et al., 2006; Warf et al., 2009; Wheeler et al., 2009).
6. Concluding remarks
RNA binding proteins provide a robust and versatile mechanism for regulating gene expression. In eukaryotes, the regulation of alternative splicing of pre-mRNAs by RBPs underlies a great expansion in the proteome (Nilsen and Graveley, 2010), allowing for the production of multiple gene products from the majority of gene loci (Pan et al., 2008; Wang et al., 2008). RNA editing by RBPs can not only expand the proteome (Maas, 2010; Rosenthal and Seeburg, 2012), but is also important in the modification of non-coding RNAs (such as snoRNAs and miRNAs) and in viral attenuation (Mallela and Nishikura, 2012). Transport, localization, and regulation of RNA stability by RBPs allow for direct control of the spatial and temporal profiles of gene products (Pratt and Mowry, 2013; Weis et al., 2013), such as in the establishment of zygote polarity. Finally, regulation of translation by RBPs allows for the selective fine-tuning of protein production, and is particularly powerful in early zygotic events, during which maternal transcripts must be translated in the absence of a transcriptional apparatus (Lee and Schedl, 2006). All these mechanisms regulate gene expression without the need to transcribe new RNA, and thus make it possible to respond to external stimuli or developmental cues with remarkable speed and specificity.
This review describes the diverse RBP toolkit employed in the developing heart, which is involved in differentiation, morphogenesis, structure, and function. In contrast to other tissues, such as the nervous system (Boutz et al., 2007; Gao and Taylor, 2012; McKee et al., 2005; Okano and Darnell, 1997; Perrone-Bizzozero and Bolognani, 2002; Yano et al., 2010), the identities and functions of RBPs in the developing heart have been pursued with significantly less vigor. While the importance of the heart cannot be disputed, its early appearance and small size in the embryo make the ability to investigate this developing organ technically difficult. Fortunately, the ability to investigate RNA processing (such as RNA editing, alternative splicing, or transcript occupancy by RBPs) using the small amounts of tissue available from often microscopic embryos has recently become feasible thanks to the introduction of advanced methods such as laser-capture microdissection, high-throughput sequencing, and large-scale bioinformatic and computational analyses (Kishore et al., 2010; Wang et al., 2009).
A key challenge facing investigators as they adapt and apply these new tools is the identification of endogenous targets of the RBPs under study. For instance, while the splicing activity of regulators of alternative splicing has been investigated using artificial minigenes designed to mimic splicing substrates (Cooper, 2005), few bona fide targets have been identified for many of these regulators in vivo. At the same time, transcriptome analyses have indicated that the vast majority of genes (> 90%) are alternatively spliced (Pan et al., 2008; Wang et al., 2008), but have not linked these events to specific RBPs. Unlike DNA binding proteins, RBPs often do not have specific binding motifs. Instead, they bind with variable affinities to a range of sequences or secondary structures, with some preference for nucleotide content or morphology. This makes computational approaches to finding RBP binding sites challenging. Direct biochemical investigations of RBP binding have focused on identifying the highest affinity interactions, yet it is believed that most RBP:RNA interactions are not strong or are transient, and it is the relationships between RBPs and auxiliary factors that increase binding specificity (Burd and Dreyfuss, 1994; Lunde et al., 2007). The role of weak or suboptimal binding in RNA processing remains to be elucidated (Pickrell et al., 2010).
With a large number of potential targets for each RBP, another important question to address is how these specific targets contribute to developmental processes. In addition to directly affecting the expression of specific protein isoforms involved in cell fate or morphogenesis, RBPs can exert control over key regulatory proteins. For example, the activities of both transcription factors (e.g., Belaguli et al., 1999) and splicing factors (e.g., Terenzi and Ladd, 2010) have been shown to be regulated in the heart by alternative splicing. Thus the identification and elucidation of RBP programs at key developmental stages may shed light on how signaling molecules, their receptors, and their downstream mediators are controlled at multiple levels. The burgeoning appreciation for the importance of RBPs during normal development and in disease states, along with the rise of the technology necessary to properly interrogate their regulatory programs, will doubtless contribute important new insights into the formation and function of the developing heart. In time, RBPs and the transcripts they regulate during heart development may provide attractive targets for the design of treatments for congenital heart defects, cardiovascular disease, or cardiac tissue repair.
Acknowledgments
Research in the Ladd laboratory is supported by the National Institutes of Health (R01HL089376).
Abbreviations
- 3′ UTR
3′ untranslated region
- AVC
atrioventricular canal
- CELF
CUG-BP, Elav-like family
- CHAMP
cardiac helicase activated by MEF2 protein
- Csm
cardiac-specific isoform of Mov10l1
- DGCR8
DiGeorge Syndrome critical region gene 8
- DM
dystrophia myotonica (myotonic dystrophy)
- DGS
DiGeorge Syndrome
- EMT
epithelial-to-mesenchymal transition
- ESRP
epithelial splicing regulatory protein
- FXR1
Fragile X mental retardation autosomal homolog 1
- HERMES
heart and RRM expressed sequence
- hnRNP
heterogeneous nuclear ribonucleoprotein
- how
held out wings
- KH domain
hnRNP K homology domain
- MBNL
muscleblind-like
- MET
mesenchymal-to-epithelial transition
- miRNA
microRNA
- OFT
outflow tract
- PTB
polypyrimidine tract binding protein
- RBFOX
RNA binding Fox-1 homolog
- RBM
RNA binding motif
- RBP
RNA binding protein
- RISC
RNA-induced silencing complex
- RRM
RNA recognition motif
- RS domain
arginine/serine-rich domain
- SRSF
serine/arginine-rich splicing factor
- STAR
signal transduction and activation of RNA
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adereth Y, Dammai V, Kose N, Li R, Hsu T. RNA-dependent integrin alpha3 protein localization regulated by the Muscleblind-like protein MLP1. Nature Cell Biology. 2005;7:1240–7. doi: 10.1038/ncb1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames EG, Lawson MJ, Mackey AJ, Holmes JW. Sequencing of mRNA identifies re-expression of fetal splice variants in cardiac hypertrophy. Journal of Molecular and Cellular Cardiology. 2013;62C:99–107. doi: 10.1016/j.yjmcc.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiel J, de Pontual L, Henrion-Caude A. miRNA, development and disease. Advances in Genetics. 2012;80:1–36. doi: 10.1016/B978-0-12-404742-6.00001-6. [DOI] [PubMed] [Google Scholar]
- Anant S, Henderson J, Mukhopadhyay D, Navaratnam N, Kennedy S, Min J, et al. Novel role for RNA-binding protein CUGBP2 in mammalian RNA editing. Journal of Biological Chemistry. 2001;276:47338–51. doi: 10.1074/jbc.M104911200. [DOI] [PubMed] [Google Scholar]
- Arthur HM, Bamforth SD. TGFbeta signaling and congenital heart disease: Insights from mouse studies. Birth Defects Research Part A Clinical and Molecular Teratology. 2011;91:423–34. doi: 10.1002/bdra.20794. [DOI] [PubMed] [Google Scholar]
- Ast G. How did alternative splicing evolve? Nat Rev Genet. 2004;5:773–82. doi: 10.1038/nrg1451. [DOI] [PubMed] [Google Scholar]
- Baehrecke EH. who encodes a KH RNA binding protein that functions in muscle development. Development. 1997;124:1323–32. doi: 10.1242/dev.124.7.1323. [DOI] [PubMed] [Google Scholar]
- Barbosa-Morais NL, Carmo-Fonseca M, Aparicio S. Systematic genome-wide annotation of spliceosomal proteins reveals differential gene family expansion. Genome Research. 2006;16:66–77. doi: 10.1101/gr.3936206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belaguli NS, Zhou W, Trinh TH, Majesky MW, Schwartz RJ. Dominant negative murine serum response factor: alternative splicing within the activation domain inhibits transactivation of serum response factor binding targets. Molecular and Cellular Biology. 1999;19:4582–91. doi: 10.1128/mcb.19.7.4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger DS, Ladd AN. Repression of nuclear CELF activity can rescue CELF-regulated alternative splicing defects in skeletal muscle models of myotonic dystrophy. PLoS Currents. 2012;4:RRN1305. doi: 10.1371/currents.RRN1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bland CS, Wang ET, Vu A, David MP, Castle JC, Johnson JM, et al. Global regulation of alternative splicing during myogenic differentiation. Nucleic Acids Research. 2010;38:7651–64. doi: 10.1093/nar/gkq614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blech-Hermoni Y, Stillwagon SJ, Ladd AN. Diversity and conservation of CELF1 and CELF2 RNA and protein expression patterns during embryonic development. Developmental Dynamics. 2013;242:767–77. doi: 10.1002/dvdy.23959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodmer R. The gene tinman is required for specification of the heart and visceral muscles in Drosophila. Development. 1993;118:719–29. doi: 10.1242/dev.118.3.719. [DOI] [PubMed] [Google Scholar]
- Boutz PL, Stoilov P, Li Q, Lin CH, Chawla G, Ostrow K, et al. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes & Development. 2007;21:1636–52. doi: 10.1101/gad.1558107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand T. Heart development: molecular insights into cardiac specification and early morphogenesis. Developmental Biology. 2003;258:1–19. doi: 10.1016/s0012-1606(03)00112-x. [DOI] [PubMed] [Google Scholar]
- Brandenberger R, Wei H, Zhang S, Lei S, Murage J, Fisk GJ, et al. Transcriptome characterization elucidates signaling networks that control human ES cell growth and differentiation. Nature Biotechnology. 2004;22:707–16. doi: 10.1038/nbt971. [DOI] [PubMed] [Google Scholar]
- Brauch KM, Karst ML, Herron KJ, de Andrade M, Pellikka PA, Rodeheffer RJ, et al. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. Journal of the American College of Cardiology. 2009;54:930–41. doi: 10.1016/j.jacc.2009.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudno M, Gelfand M, Spengler S, Zorn M, Dubchak I, Conboy J. Computational analysis of candidate intron regulatory elements for tissue-specific alternative pre-mRNA splicing. Nucleic Acids Research. 2001;29:2338–48. doi: 10.1093/nar/29.11.2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd C, Dreyfuss G. Conserved structures and diversity of functions of RNA-binding proteins. Science. 1994;265:615–21. doi: 10.1126/science.8036511. [DOI] [PubMed] [Google Scholar]
- Chapnik E, Sasson V, Blelloch R, Hornstein E. Dgcr8 controls neural crest cells survival in cardiovascular development. Developmental Biology. 2012;362:50–6. doi: 10.1016/j.ydbio.2011.11.008. [DOI] [PubMed] [Google Scholar]
- Charlet-B N, Savkur R, Singh G, Philips A, Grice E, Cooper T. Loss of the muscle-specific chloride channel in type I myotonic dystrophy lead to misregulated alternative splicing. Molecular Cell. 2002;10:45–53. doi: 10.1016/s1097-2765(02)00572-5. [DOI] [PubMed] [Google Scholar]
- Chateigner-Boutin AL, Small I. Organellar RNA editing. Wiley Interdisciplinary Reviews RNA. 2011;2:493–506. doi: 10.1002/wrna.72. [DOI] [PubMed] [Google Scholar]
- Chen HW, Yu SL, Chen WJ, Yang PC, Chien CT, Chou HY, et al. Dynamic changes of gene expression profiles during postnatal development of the heart in mice. Heart. 2004;90:927–34. doi: 10.1136/hrt.2002.006734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Wang DZ. microRNAs in cardiovascular development. Journal of Molecular and Cellular Cardiology. 2012;52:949–57. doi: 10.1016/j.yjmcc.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloonan N, Forrest AR, Kolle G, Gardiner BB, Faulkner GJ, Brown MK, et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nature Methods. 2008;5:613–9. doi: 10.1038/nmeth.1223. [DOI] [PubMed] [Google Scholar]
- Cooper TA. Use of minigene systems to dissect alternative splicing elements. Methods. 2005;37:331–40. doi: 10.1016/j.ymeth.2005.07.015. [DOI] [PubMed] [Google Scholar]
- Das D, Clark TA, Schweitzer A, Yamamoto M, Marr H, Arribere J, et al. A correlation with exon expression approach to identify cis-regulatory elements for tissue-specific alternative splicing. Nucleic Acids Research. 2007;35:4845–57. doi: 10.1093/nar/gkm485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nature Reviews Cancer. 2013;13:97–110. doi: 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- DeLaughter DM, Saint-Jean L, Baldwin HS, Barnett JV. What chick and mouse models have taught us about the role of the endocardium in congenital heart disease. Birth Defects Research Part A Clinical and Molecular Teratology. 2011;91:511–25. doi: 10.1002/bdra.20809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J-H, Xu X, Yang D, Chu P-H, Dalton N, Ye Z, et al. Dilated cardiomyopathy caused by tissue-specific ablation of SC35 in heart. EMBO Journal. 2004;23:885–96. doi: 10.1038/sj.emboj.7600054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmar KA, Jiang P, Park JW, Amirikian K, Wan J, Shen S, et al. Genome-wide determination of a broad ESRP-regulated posttranscriptional network by high-throughput sequencing. Molecular and Cellular Biology. 2012;32:1468–82. doi: 10.1128/MCB.06536-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwoodie SL. Combinatorial signaling in the heart orchestrates cardiac induction, lineage specification and chamber formation. Seminars in Cell & Developmental Biology. 2007;18:54–66. doi: 10.1016/j.semcdb.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Engels B, van ‘t Padje S, Blonden L, Severijnen LA, Oostra BA, Willemsen R. Characterization of Fxr1 in Danio rerio; a simple vertebrate model to study costamere development. Journal of Experimental Biology. 2004;207:3329–38. doi: 10.1242/jeb.01146. [DOI] [PubMed] [Google Scholar]
- Fan C, Chen Q, Wang QK. Functional role of transcriptional factor TBX5 in pre-mRNA splicing and Holt-Oram syndrome via association with SC35. The Journal of Biological Chemistry. 2009;284:25653–63. doi: 10.1074/jbc.M109.041368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fardaei M, Rogers M, Thorpe H, Larkin K, Hamshere M, Harper P, et al. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Human Molecular Genetics. 2002;11:805–14. doi: 10.1093/hmg/11.7.805. [DOI] [PubMed] [Google Scholar]
- Feng Y, Valley MT, Lazar J, Yang AL, Bronson RT, Firestein S, et al. SRp38 regulates alternative splicing and is required for Ca(2+) handling in the embryonic heart. Developmental Cell. 2009;16:528–38. doi: 10.1016/j.devcel.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes JM, Kinghorn JR, Johnston IA. Characterization of two paralogous muscleblind-like genes from the tiger pufferfish (Takifugu rubripes) Comparative Biochemistry and Physiology Part B, Biochemistry & Molecular Biology. 2007;146:180–6. doi: 10.1016/j.cbpb.2006.10.100. [DOI] [PubMed] [Google Scholar]
- Fetka I, Radeghieri A, Bouwmeester T. Expression of the RNA recognition motif-containing protein SEB-4 during Xenopus embryonic development. Mechanisms of Development. 2000;94:283–6. doi: 10.1016/s0925-4773(00)00284-7. [DOI] [PubMed] [Google Scholar]
- Gallagher TL, Arribere JA, Geurts PA, Exner CR, McDonald KL, Dill KK, et al. Rbfox-regulated alternative splicing is critical for zebrafish cardiac and skeletal muscle functions. Developmental Biology. 2011;359:251–61. doi: 10.1016/j.ydbio.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao FB, Taylor JP. RNA-binding proteins in neurological disease. Brain Research. 2012;1462:1–2. doi: 10.1016/j.brainres.2012.05.038. [DOI] [PubMed] [Google Scholar]
- Garg V, Yamagishi C, Hu T, Kathiriya IS, Yamagishi H, Srivastava D. Tbx1, a DiGeorge syndrome candidate gene, is regulated by sonic hedgehog during pharyngeal arch development. Developmental Biology. 2001;235:62–73. doi: 10.1006/dbio.2001.0283. [DOI] [PubMed] [Google Scholar]
- Gates DP, Coonrod LA, Berglund JA. Autoregulated splicing of muscleblind-like 1 (MBNL1) Pre-mRNA. The Journal of Biological Chemistry. 2011;286:34224–33. doi: 10.1074/jbc.M111.236547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber WV, Vokes SA, Zearfoss NR, Krieg PA. A role for the RNA-binding protein, hermes, in the regulation of heart development. Developmental Biology. 2002;247:116–26. doi: 10.1006/dbio.2002.0678. [DOI] [PubMed] [Google Scholar]
- Gerber WV, Yatskievych TA, Antin PB, Correia KM, Conlon RA, Krieg PA. The RNA-binding protein gene, hermes, is expressed at high levels in the developing heart. Mechanisms of Development. 1999;80:77–86. doi: 10.1016/s0925-4773(98)00195-6. [DOI] [PubMed] [Google Scholar]
- Glaser ND, Lukyanenko YO, Wang Y, Wilson GM, Rogers TB. JNK activation decreases PP2A regulatory subunit B56alpha expression and mRNA stability and increases AUF1 expression in cardiomyocytes. American Journal of Physiology Heart and Circulatory Physiology. 2006;291:H1183–92. doi: 10.1152/ajpheart.01162.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glisovic T, Bachorik JL, Yong J, Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Letters. 2008;582:1977–86. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goers ES, Purcell J, Voelker RB, Gates DP, Berglund JA. MBNL1 binds GC motifs embedded in pyrimidines to regulate alternative splicing. Nucleic Acids Research. 2010;38:2467–84. doi: 10.1093/nar/gkp1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouble A, Morello D. Synchronous and regulated expression of two AU-binding proteins, AUF1 and HuR, throughout murine development. Oncogene. 2000;19:5377–84. doi: 10.1038/sj.onc.1203910. [DOI] [PubMed] [Google Scholar]
- Gripp KW, Hopkins E, Johnston JJ, Krause C, Dobyns WB, Biesecker LG. Long-term survival in TARP syndrome and confirmation of RBM10 as the disease-causing gene. American Journal of Medical Genetics Part A. 2011;155A:2516–20. doi: 10.1002/ajmg.a.34190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Schafer S, Greaser ML, Radke MH, Liss M, Govindarajan T, et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nature Medicine. 2012;18:766–73. doi: 10.1038/nm.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrell MD, Harbi S, Hoffman JF, Zavadil J, Coetzee WA. Large-scale analysis of ion channel gene expression in the mouse heart during perinatal development. Physiological Genomics. 2007;28:273–83. doi: 10.1152/physiolgenomics.00163.2006. [DOI] [PubMed] [Google Scholar]
- Harvey R, Rosenthal N. Heart Development. San Diego: Academic Press, Inc; 1999. p. 530. [Google Scholar]
- Ho T, Charlet-B N, Poulos M, Singh G, Swanson M, Cooper T. Muscleblind proteins regulate alternative splicing. EMBO Journal. 2004;23:3103–12. doi: 10.1038/sj.emboj.7600300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TH, Bundman D, Armstrong DL, Cooper TA. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Human Molecular Genetics. 2005;14:1539–47. doi: 10.1093/hmg/ddi162. [DOI] [PubMed] [Google Scholar]
- Huot ME, Bisson N, Davidovic L, Mazroui R, Labelle Y, Moss T, et al. The RNA-binding protein fragile X-related 1 regulates somite formation in Xenopus laevis. Molecular Biology of the Cell. 2005;16:4350–61. doi: 10.1091/mbc.E05-04-0304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iakova P, Wang GL, Timchenko L, Michalak M, Pereira-Smith OM, Smith JR, et al. Competition of CUGBP1 and calreticulin for the regulation of p21 translation determines cell fate. Embo Journal. 2004;23:406–17. doi: 10.1038/sj.emboj.7600052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Suzuki H, Maegawa S, Endo H, Sugano S, Hashimoto K, et al. A vertebrate RNA-binding protein Fox-1 regulates tissue-specific splicing via the pentanucleotide GCAUG. The EMBO Journal. 2003;22:905–12. doi: 10.1093/emboj/cdg089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JJ, Teer JK, Cherukuri PF, Hansen NF, Loftus SK, Chong K, et al. Massively parallel sequencing of exons on the X chromosome identifies RBM10 as the gene that causes a syndromic form of cleft palate. American Journal of Human Genetics. 2010;86:743–8. doi: 10.1016/j.ajhg.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumaa H, Wei G, Nielsen PJ. Blastocyst formation is blocked in mouse embryos lacking the splicing factor SRp20. Current Biology. 1999;9:899–902. doi: 10.1016/s0960-9822(99)80394-7. [DOI] [PubMed] [Google Scholar]
- Justice MJ, Hirschi KK. The role of quaking in mammalian embryonic development. Advances in Experimental Medicine and Biology. 2010;693:82–92. doi: 10.1007/978-1-4419-7005-3_6. [DOI] [PubMed] [Google Scholar]
- Kalsotra A, Wang K, Li PF, Cooper TA. MicroRNAs coordinate an alternative splicing network during mouse postnatal heart development. Genes & Development. 2010 doi: 10.1101/gad.1894310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB, et al. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proceedings of the National Academy of Sciences U S A. 2008;105:20333–8. doi: 10.1073/pnas.0809045105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, et al. A muscleblind knockout model for myotonic dystrophy. Science. 2003a;302:1978–80. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- Kanadia RN, Shin J, Yuan Y, Beattie SG, Wheeler TM, Thornton CA, et al. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proceedings of the National Academy of Sciences U S A. 2006;103:11748–53. doi: 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanadia RN, Urbinati CR, Crusselle VJ, Luo D, Lee YJ, Harrison JK, et al. Developmental expression of mouse muscleblind genes Mbnl1, Mbnl2 and Mbnl3. Gene Expression Patterns. 2003b;3:459–62. doi: 10.1016/s1567-133x(03)00064-4. [DOI] [PubMed] [Google Scholar]
- Kelemen O, Convertini P, Zhang Z, Wen Y, Shen M, Falaleeva M, et al. Function of alternative splicing. Gene. 2013;514:1–30. doi: 10.1016/j.gene.2012.07.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyte A, Hutson MR. The neural crest in cardiac congenital anomalies. Differentiation. 2012;84:25–40. doi: 10.1016/j.diff.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby ML. Cardiac Development. New York: Oxford University Press; 2007. [Google Scholar]
- Kishore S, Luber S, Zavolan M. Deciphering the role of RNA-binding proteins in the post-transcriptional control of gene expression. Brief Functional Genomics. 2010;9:391–404. doi: 10.1093/bfgp/elq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komamura K, Iwai N, Kokame K, Yasumura Y, Kim J, Yamagishi M, et al. The role of a common TNNT2 polymorphism in cardiac hypertrophy. Journal of Human Genetics. 2004;49:129–33. doi: 10.1007/s10038-003-0121-4. [DOI] [PubMed] [Google Scholar]
- Kondo T, Furuta T, Mitsunaga K, Ebersole TA, Shichiri M, Wu J, et al. Genomic organization and expression analysis of the mouse qkI locus. Mammalian Genome. 1999;10:662–9. doi: 10.1007/s003359901068. [DOI] [PubMed] [Google Scholar]
- Kong J, Lasko P. Translational control in cellular and developmental processes. Nature Reviews Genetics. 2012;13:383–94. doi: 10.1038/nrg3184. [DOI] [PubMed] [Google Scholar]
- Kong SW, Hu YW, Ho JW, Ikeda S, Polster S, John R, et al. Heart failure-associated changes in RNA splicing of sarcomere genes. Circulation: Cardiovascular Genetics. 2010;3:138–46. doi: 10.1161/CIRCGENETICS.109.904698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshelev M, Sarma S, Price RE, Wehrens XH, Cooper TA. Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Human Molecular Genetics. 2010;19:1066–75. doi: 10.1093/hmg/ddp570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruithof BP, Duim SN, Moerkamp AT, Goumans MJ. TGFbeta and BMP signaling in cardiac cushion formation: Lessons from mice and chicken. Differentiation. 2012;84:89–102. doi: 10.1016/j.diff.2012.04.003. [DOI] [PubMed] [Google Scholar]
- Kuroyanagi H. Fox-1 family of RNA-binding proteins. Cellular and Molecular Life Sciences. 2009;66:3895–907. doi: 10.1007/s00018-009-0120-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Molecular Cell. 2007;28:68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladd A, Stenberg M, Swanson M, Cooper T. A dynamic balance between activation and repression regulates pre-mRNA alternative splicing during heart development. Developmental Dynamics. 2005a;233:783–93. doi: 10.1002/dvdy.20382. [DOI] [PubMed] [Google Scholar]
- Ladd A, Taffet G, Hartley C, Kearney D, Cooper T. Cardiac-specific repression of CELF activity disrupts alternative splicing and causes cardiomyopathy. Molecular and Cellular Biology. 2005b;25:6267–78. doi: 10.1128/MCB.25.14.6267-6278.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladd AN, Yatskievych TA, Antin PB. Regulation of avian cardiac myogenesis by activin/TGFbeta and bone morphogenetic proteins. Developmental Biology. 1998;204:407–19. doi: 10.1006/dbio.1998.9094. [DOI] [PubMed] [Google Scholar]
- Lale S, Yu S, Ahmed A. Complex congenital heart defects in association with maternal diabetes and partial deletion of the A2BP1 gene. Fetal and Pediatric Pathology. 2011;30:161–6. doi: 10.3109/15513815.2010.547555. [DOI] [PubMed] [Google Scholar]
- Lasko P. Gene regulation at the RNA layer: RNA binding proteins in intercellular signaling networks. Science Signal Transduction Knowledge Environment. 2003:RE6. doi: 10.1126/stke.2003.179.re6. [DOI] [PubMed] [Google Scholar]
- Lee MH, Schedl T. WormBook. 2006. RNA-binding proteins; pp. 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeMasters KE, Blech-Hermoni Y, Stillwagon SJ, Vajda NA, Ladd AN. Loss of muscleblind-like 1 promotes invasive mesenchyme formation in endocardial cushions by stimulating autocrine TGFbeta3. BMC Developmental Biology. 2012;12:22. doi: 10.1186/1471-213X-12-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Bachinski L, Roberts R. Genomic organization and isoform-specific tissue expression of human NAPOR (CUGBP2) as a candidate gene for familial arrhythmogenic right ventricular dysplasia. Genomics. 2001;74:396–401. doi: 10.1006/geno.2001.6558. [DOI] [PubMed] [Google Scholar]
- Li D, Morales A, Gonzalez-Quintana J, Norton N, Siegfried JD, Hofmeyer M, et al. Identification of novel mutations in RBM20 in patients with dilated cardiomyopathy. Clinical and Translational Science. 2010;3:90–7. doi: 10.1111/j.1752-8062.2010.00198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Guo W, Dewey CN, Greaser ML. Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Research. 2013;41:2659–72. doi: 10.1093/nar/gks1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang J, Manley JL. Loss of splicing factor ASF/SF2 induces G2 cell cycle arrest and apoptosis, but inhibits internucleosomal DNA fragmentation. Genes & Development. 2005;19:2705–14. doi: 10.1101/gad.1359305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtner P, Attié-Bitach T, Schuffenhauer S, Henwood J, Bouvagnet P, Scambler P, et al. Expression and mutation analysis of Brunol3, a candidate gene for heart and thymus developmental defects associated with partial monosomy 10p. Journal of Molecular Medicine. 2002;80:431–42. doi: 10.1007/s00109-002-0331-9. [DOI] [PubMed] [Google Scholar]
- Lin MI, Das I, Schwartz GM, Tsoulfas P, Mikawa T, Hempstead BL. Trk C receptor signaling regulates cardiac myocyte proliferation during early heart development in vivo. Developmental Biology. 2000;226:180–91. doi: 10.1006/dbio.2000.9850. [DOI] [PubMed] [Google Scholar]
- Liu YF, Liu HY, Tu LC, Lin CW, Hsiao KM, Pan H. Zebrafish muscleblind-like genes: identification, structural features and expression. Comparative Biochemistry and Physiology Part B Biochemistry and Molecular Biology. 2008;151:118–24. doi: 10.1016/j.cbpb.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Liu ZP, Nakagawa O, Nakagawa M, Yanagisawa H, Passier R, Richardson JA, et al. CHAMP, a novel cardiac-specific helicase regulated by MEF2C. Developmental Biology. 2001;234:497–509. doi: 10.1006/dbio.2001.0277. [DOI] [PubMed] [Google Scholar]
- Liu ZP, Olson EN. Suppression of proliferation and cardiomyocyte hypertrophy by CHAMP, a cardiac-specific RNA helicase. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:2043–8. doi: 10.1073/pnas.261708699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JY, Schneider RJ. Tissue distribution of AU-rich mRNA-binding proteins involved in regulation of mRNA decay. The Journal of Biological Dhemistry. 2004;279:12974–9. doi: 10.1074/jbc.M310433200. [DOI] [PubMed] [Google Scholar]
- Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nature Reviews Molecular Cell Biology. 2007;8:479–90. doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas S. Gene regulation through RNA editing. Discovery Medicine. 2010;10:379–86. [PubMed] [Google Scholar]
- Machuca-Tzili LE, Buxton S, Thorpe A, Timson CM, Wigmore P, Luther PK, et al. Zebrafish deficient for Muscleblind-like 2 exhibit features of myotonic dystrophy. Disease Models & Mechanisms. 2011;4:381–92. doi: 10.1242/dmm.004150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLellan WR, Schneider MD. Genetic dissection of cardiac growth control pathways. Annual Review of Physiology. 2000;62:289–319. doi: 10.1146/annurev.physiol.62.1.289. [DOI] [PubMed] [Google Scholar]
- Mallela A, Nishikura K. A-to-I editing of protein coding and noncoding RNAs. Critical Reviews in Biochemistry and Molecular Biology. 2012;47:493–501. doi: 10.3109/10409238.2012.714350. [DOI] [PubMed] [Google Scholar]
- Mango R, Biocca S, del Vecchio F, Clementi F, Sangiuolo F, Amati F, et al. In vivo and in vitro studies support that a new splicing isoform of OLR1 gene is protective against acute myocardial infarction. Circulation Research. 2005;97:152–8. doi: 10.1161/01.RES.0000174563.62625.8e. [DOI] [PubMed] [Google Scholar]
- Maniatis T, Tasic B. Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature. 2002;418:236–43. doi: 10.1038/418236a. [DOI] [PubMed] [Google Scholar]
- Maragh S, Miller RA, Bessling SL, McGaughey DM, Wessels MW, de Graaf B, et al. Identification of RNA binding motif proteins essential for cardiovascular development. BMC Developmental Biology. 2011;11:62. doi: 10.1186/1471-213X-11-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda A, Andersen HS, Doktor TK, Okamoto T, Ito M, Andresen BS, et al. CUGBP1 and MBNL1 preferentially bind to 3′ UTRs and facilitate mRNA decay. Scientific Reports. 2012;2:209. doi: 10.1038/srep00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda K, Marasa B, Martindale JL, Halushka MK, Gorospe M. Tissue- and age-dependent expression of RNA-binding proteins that influence mRNA turnover and translation. Aging. 2009;1:681–98. doi: 10.18632/aging.100073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AE, Minet E, Stern C, Riahi S, Stiles CD, Silver PA. A genome-wide in situ hybridization map of RNA-binding proteins reveals anatomically restricted expression in the developing mouse brain. BMC Developmental Biology. 2005;5:14. doi: 10.1186/1471-213X-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medioni C, Astier M, Zmojdzian M, Jagla K, Semeriva M. Genetic control of cell morphogenesis during Drosophila melanogaster cardiac tube formation. The Journal of Cell Biology. 2008;182:249–61. doi: 10.1083/jcb.200801100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medioni C, Mowry K, Besse F. Principles and roles of mRNA localization in animal development. Development. 2012;139:3263–76. doi: 10.1242/dev.078626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mientjes EJ, Willemsen R, Kirkpatrick LL, Nieuwenhuizen IM, Hoogeveen-Westerveld M, Verweij M, et al. Fxr1 knockout mice show a striated muscle phenotype: implications for Fxr1p function in vivo. Human Molecular Genetics. 2004;13:1291–302. doi: 10.1093/hmg/ddh150. [DOI] [PubMed] [Google Scholar]
- Miller RA, Christoforou N, Pevsner J, McCallion AS, Gearhart JD. Efficient array-based identification of novel cardiac genes through differentiation of mouse ESCs. PLoS One. 2008;3:e2176. doi: 10.1371/journal.pone.0002176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misquitta CM, Chen T, Grover AK. Control of protein expression through mRNA stability in calcium signalling. Cell Calcium. 2006;40:329–46. doi: 10.1016/j.ceca.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Misquitta CM, Iyer VR, Werstiuk ES, Grover AK. The role of 3′-untranslated region (3′-UTR) mediated mRNA stability in cardiovascular pathophysiology. Molecular and Cellular Biochemistry. 2001;224:53–67. doi: 10.1023/a:1011982932645. [DOI] [PubMed] [Google Scholar]
- Moorman AF, Christoffels VM. Cardiac chamber formation: development, genes, and evolution. Physiological Reviews. 2003;83:1223–67. doi: 10.1152/physrev.00006.2003. [DOI] [PubMed] [Google Scholar]
- Mori AD, Bruneau BG. TBX5 mutations and congenital heart disease: Holt-Oram syndrome revealed. Current Opinion in Cardiology. 2004;19:211–5. doi: 10.1097/00001573-200405000-00004. [DOI] [PubMed] [Google Scholar]
- Mueller CF, Berger A, Zimmer S, Tiyerili V, Nickenig G. The heterogenous nuclear riboprotein S1-1 regulates AT1 receptor gene expression via transcriptional and posttranscriptional mechanisms. Archives of Biochemistry and Biophysics. 2009;488:76–82. doi: 10.1016/j.abb.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–63. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nir R, Grossman R, Paroush Z, Volk T. Phosphorylation of the Drosophila melanogaster RNA-binding protein HOW by MAPK/ERK enhances its dimerization and activity. PLoS Genetics. 2012;8:e1002632. doi: 10.1371/journal.pgen.1002632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano HJ, Darnell RB. A hierarchy of Hu RNA binding proteins in developing and adult neurons. Journal of Neuroscience. 1997;17:3024–37. doi: 10.1523/JNEUROSCI.17-09-03024.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K, Kuwabara Y, Han J. MicroRNAs and cardiovascular diseases. FEBS Journal. 2011;278:1619–33. doi: 10.1111/j.1742-4658.2011.08090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nature Genetics. 2008;40:1413–5. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- Park JY, Li W, Zheng D, Zhai P, Zhao Y, Matsuda T, et al. Comparative analysis of mRNA isoform expression in cardiac hypertrophy and development reveals multiple post-transcriptional regulatory modules. PLoS One. 2011;6:e22391. doi: 10.1371/journal.pone.0022391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual M, Vicente M, Monferrer L, Artero R. The Muscleblind family of proteins: an emerging class of regulators of developmentally programmed alternative splicing. Differentiation. 2006;74:65–80. doi: 10.1111/j.1432-0436.2006.00060.x. [DOI] [PubMed] [Google Scholar]
- Pende A, Tremmel KD, DeMaria CT, Blaxall BC, Minobe WA, Sherman JA, et al. Regulation of the mRNA-binding protein AUF1 by activation of the beta-adrenergic receptor signal transduction pathway. The Journal of Biological Chemistry. 1996;271:8493–501. doi: 10.1074/jbc.271.14.8493. [DOI] [PubMed] [Google Scholar]
- Perrone-Bizzozero N, Bolognani F. Role of HuD and other RNA-binding proteins in neural development and plasticity. Journal of Neuroscience Research. 2002;68:121–6. doi: 10.1002/jnr.10175. [DOI] [PubMed] [Google Scholar]
- Person AD, Klewer SE, Runyan RB. Cell biology of cardiac cushion development. International Review of Cytology. 2005;243:287–335. doi: 10.1016/S0074-7696(05)43005-3. [DOI] [PubMed] [Google Scholar]
- Pickrell JK, Pai AA, Gilad Y, Pritchard JK. Noisy splicing drives mRNA isoform diversity in human cells. PLoS Genetics. 2010;6:e1001236. doi: 10.1371/journal.pgen.1001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon KL, Tan KT, Wei YY, Ng CP, Colman A, Korzh V, et al. RNA-binding protein RBM24 is required for sarcomere assembly and heart contractility. Cardiovascular Research. 2012;94:418–27. doi: 10.1093/cvr/cvs095. [DOI] [PubMed] [Google Scholar]
- Pratt CA, Mowry KL. Taking a cellular road-trip: mRNA transport and anchoring. Current Opinion in Cell Biology. 2013;25:99–106. doi: 10.1016/j.ceb.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritsker M, Doniger TT, Kramer LC, Westcot SE, Lemischka IR. Diversification of stem cell molecular repertoire by alternative splicing. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:14290–5. doi: 10.1073/pnas.0502132102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qayyum SR, Webb S, Anderson RH, Verbeek FJ, Brown NA, Richardson MK. Septation and valvar formation in the outflow tract of the embryonic chick heart. The Anatomical Record. 2001;264:273–83. doi: 10.1002/ar.1162. [DOI] [PubMed] [Google Scholar]
- Raffel GD, Chu GC, Jesneck JL, Cullen DE, Bronson RT, Bernard OA, et al. Ott1 (Rbm15) is essential for placental vascular branching morphogenesis and embryonic development of the heart and spleen. Molecular and Cellular Biology. 2009;29:333–41. doi: 10.1128/MCB.00370-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Ryder U, Lamond AI, Mann M. Large-scale proteomic analysis of the human spliceosome. Genome Research. 2002;12:1231–45. doi: 10.1101/gr.473902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refaat MM, Lubitz SA, Makino S, Islam Z, Frangiskakis JM, Mehdi H, et al. Genetic variation in the alternative splicing regulator RBM20 is associated with dilated cardiomyopathy. Heart Rhythm. 2012;9:390–6. doi: 10.1016/j.hrthm.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke LM, Xu Y, Cheng C. Snail represses the splicing regulator epithelial splicing regulatory protein 1 to promote epithelial-mesenchymal transition. The Journal of Biological Chemistry. 2012;287:36435–42. doi: 10.1074/jbc.M112.397125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revil T, Jerome-Majewska LA. During embryogenesis, esrp1 expression is restricted to a subset of epithelial cells and is associated with splicing of a number of developmentally important genes. Developmental Dynamics. 2013;242:281–90. doi: 10.1002/dvdy.23918. [DOI] [PubMed] [Google Scholar]
- Richard S, Torabi N, Franco GV, Tremblay GA, Chen T, Vogel G, et al. Ablation of the Sam68 RNA binding protein protects mice from age-related bone loss. PLoS Genetics. 2005;1:e74. doi: 10.1371/journal.pgen.0010074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal JJ, Seeburg PH. A-to-I RNA editing: effects on proteins key to neural excitability. Neuron. 2012;74:432–9. doi: 10.1016/j.neuron.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryder SP, Williamson JR. Specificity of the STAR/GSG domain protein Qk1: implications for the regulation of myelination. RNA. 2004;10:1449–58. doi: 10.1261/rna.7780504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomonis N, Nelson B, Vranizan K, Pico AR, Hanspers K, Kuchinsky A, et al. Alternative splicing in the differentiation of human embryonic stem cells into cardiac precursors. PLoS Computational Biology. 2009;5:e1000553. doi: 10.1371/journal.pcbi.1000553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomonis N, Schlieve CR, Pereira L, Wahlquist C, Colas A, Zambon AC, et al. Alternative splicing regulates mouse embryonic stem cell pluripotency and differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:10514–9. doi: 10.1073/pnas.0912260107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savkur R, Phillips A, Cooper T. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nature Genetics. 2001;29:40–7. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- Schoenberg DR, Maquat LE. Regulation of cytoplasmic mRNA decay. Nature Reviews Genetics. 2012;13:246–59. doi: 10.1038/nrg3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoser B, Timchenko L. Myotonic dystrophies 1 and 2: complex diseases with complex mechanisms. Current Genomics. 2010;11:77–90. doi: 10.2174/138920210790886844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz H, Zamore PD. Rethinking the microprocessor. Cell. 2006;125:827–9. doi: 10.1016/j.cell.2006.05.018. [DOI] [PubMed] [Google Scholar]
- Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, et al. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genetics. 2011;7:e1002218. doi: 10.1371/journal.pgen.1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Franzini-Armstrong C, Lopez JR, Jones LR, Kobayashi YM, Wang Y, et al. Triadins modulate intracellular Ca(2+) homeostasis but are not essential for excitation-contraction coupling in skeletal muscle. The Journal of Biological Chemistry. 2007;282:37864–74. doi: 10.1074/jbc.M705702200. [DOI] [PubMed] [Google Scholar]
- Shepard PJ, Hertel KJ. The SR protein family. Genome Biology. 2009;10:242. doi: 10.1186/gb-2009-10-10-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata H, Huynh DP, Pulst SM. A novel protein with RNA-binding motifs interacts with ataxin-2. Human Molecular Genetics. 2000;9:1303–13. doi: 10.1093/hmg/9.9.1303. [DOI] [PubMed] [Google Scholar]
- Shin C, Feng Y, Manley JL. Dephosphorylated SRp38 acts as a splicing repressor in response to heat shock. Nature. 2004;427:553–8. doi: 10.1038/nature02288. [DOI] [PubMed] [Google Scholar]
- Siedner S, Kruger M, Schroeter M, Metzler D, Roell W, Fleischmann BK, et al. Developmental changes in contractility and sarcomeric proteins from the early embryonic to the adult stage in the mouse heart. J Physiol. 2003;548:493–505. doi: 10.1113/jphysiol.2002.036509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi H, Dreyfuss G. RNA-binding proteins as regulators of gene expression. Current Opinion in Genetics & Development. 1997;7:345–53. doi: 10.1016/s0959-437x(97)80148-7. [DOI] [PubMed] [Google Scholar]
- Siomi MC, Siomi H, Sauer WH, Srinivasan S, Nussbaum RL, Dreyfuss G. FXR1, an autosomal homolog of the fragile X mental retardation gene. The EMBO Journal. 1995;14:2401–8. doi: 10.1002/j.1460-2075.1995.tb07237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song HK, Hong SE, Kim T, Kim do H. Deep RNA sequencing reveals novel cardiac transcriptomic signatures for physiological and pathological hypertrophy. PLoS One. 2012;7:e35552. doi: 10.1371/journal.pone.0035552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugnet CW, Srinivasan K, Clark TA, O’Brien G, Cline MS, Wang H, et al. Unusual intron conservation near tissue-regulated exons found by splicing microarrays. PLoS Computational Biology. 2006;2:e4. doi: 10.1371/journal.pcbi.0020004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland LC, Rintala-Maki ND, White RD, Morin CD. RNA binding motif (RBM) proteins: a novel family of apoptosis modulators? Journal of Cellular Biochemistry. 2005;94:5–24. doi: 10.1002/jcb.20204. [DOI] [PubMed] [Google Scholar]
- Terenzi F, Brimacombe KR, Penn MS, Ladd AN. CELF-mediated alternative splicing is required for cardiac function during early, but not later, postnatal life. Journal of Molecular and Cellular Cardiology. 2009;46:395–404. doi: 10.1016/j.yjmcc.2008.10.030. [DOI] [PubMed] [Google Scholar]
- Terenzi F, Ladd AN. Conserved developmental alternative splicing of muscleblind-like (MBNL) transcripts regulates MBNL localization and activity. RNA Biology. 2010;7:43–55. doi: 10.4161/rna.7.1.10401. [DOI] [PubMed] [Google Scholar]
- Timchenko NA, Patel R, Iakova P, Cai ZJ, Quan L, Timchenko LT. Overexpression of CUG triplet repeat-binding protein, CUGBP1, in mice inhibits myogenesis. Journal of Biological Chemistry. 2004;279:13129–39. doi: 10.1074/jbc.M312923200. [DOI] [PubMed] [Google Scholar]
- Twyffels L, Gueydan C, Kruys V. Shuttling SR proteins: more than splicing factors. FEBS Journal. 2011;278:3246–55. doi: 10.1111/j.1742-4658.2011.08274.x. [DOI] [PubMed] [Google Scholar]
- Ueyama T, Kasahara H, Ishiwata T, Yamasaki N, Izumo S. Csm, a cardiac-specific isoform of the RNA helicase Mov10l1, is regulated by Nkx2.5 in embryonic heart. The Journal of Biological Chemistry. 2003;278:28750–7. doi: 10.1074/jbc.M300014200. [DOI] [PubMed] [Google Scholar]
- Vajda NA, Brimacombe KR, LeMasters KE, Ladd AN. Muscleblind-like 1 is a negative regulator of TGF-beta-dependent epithelial-mesenchymal transition of atrioventricular canal endocardial cells. Developmental Dynamics. 2009;238:3266–72. doi: 10.1002/dvdy.22155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van’t Padje S, Chaudhry B, Severijnen LA, van der Linde HC, Mientjes EJ, Oostra BA, et al. Reduction in fragile X related 1 protein causes cardiomyopathy and muscular dystrophy in zebrafish. Journal of Experimental Biology. 2009;212:2564–70. doi: 10.1242/jeb.032532. [DOI] [PubMed] [Google Scholar]
- Venables JP, Vignal E, Baghdiguian S, Fort P, Tazi J. Tissue-specific alternative splicing of Tak1 is conserved in deuterostomes. Molecular Biology and Evolution. 2012;29:261–9. doi: 10.1093/molbev/msr193. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Morishima M, Taddei I, Lindsay EA, Baldini A. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Human Molecular Genetics. 2002;11:915–22. doi: 10.1093/hmg/11.8.915. [DOI] [PubMed] [Google Scholar]
- Wagner M, Siddiqui MA. Signal transduction in early heart development (II): ventricular chamber specification, trabeculation, and heart valve formation. Experimental Biology and Medicine. 2007;232:866–80. [PubMed] [Google Scholar]
- Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, et al. Transcriptome-wide Regulation of Pre-mRNA Splicing and mRNA Localization by Muscleblind Proteins. Cell. 2012;150:710–24. doi: 10.1016/j.cell.2012.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–6. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Xu X, Ding JH, Bermingham JR, Jr, Fu XD. SC35 plays a role in T cell development and alternative splicing of CD45. Molecular Cell. 2001;7:331–42. doi: 10.1016/s1097-2765(01)00181-2. [DOI] [PubMed] [Google Scholar]
- Wang J, Takagaki Y, Manley JL. Targeted disruption of an essential vertebrate gene: ASF/SF2 is required for cell viability. Genes & Development. 1996;10:2588–99. doi: 10.1101/gad.10.20.2588. [DOI] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward AJ, Rimer M, Killian JM, Dowling JJ, Cooper TA. CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Human Molecular Genetics. 2010;19:3614–22. doi: 10.1093/hmg/ddq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warf MB, Nakamori M, Matthys CM, Thornton CA, Berglund JA. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:18551–6. doi: 10.1073/pnas.0903234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha CC, Jiang P, Amirikian K, Dittmar KA, Lu H, Shen S, et al. An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition. The EMBO Journal. 2010;29:3286–300. doi: 10.1038/emboj.2010.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Molecular Cell. 2009a;33:591–601. doi: 10.1016/j.molcel.2009.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha CC, Shen S, Xing Y, Carstens RP. The epithelial splicing factors ESRP1 and ESRP2 positively and negatively regulate diverse types of alternative splicing events. RNA Biology. 2009b;6:546–62. doi: 10.4161/rna.6.5.9606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis BL, Schleiff E, Zerges W. Protein targeting to subcellular organelles via MRNA localization. Biochimica et Biophysica Acta. 2013;1833:260–73. doi: 10.1016/j.bbamcr.2012.04.004. [DOI] [PubMed] [Google Scholar]
- Wheeler TM, Sobczak K, Lueck JD, Osborne RJ, Lin X, Dirksen RT, et al. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science. 2009;325:336–9. doi: 10.1126/science.1173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White EJ, Brewer G, Wilson GM. Post-transcriptional control of gene expression by AUF1: mechanisms, physiological targets, and regulation. Biochimica et Biophysica Acta. 2013;1829:680–8. doi: 10.1016/j.bbagrm.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman SA, Cover C, Yu L, Nelson DL, Zarnescu DC, Gregorio CC. Desmoplakin and talin2 are novel mRNA targets of fragile X-related protein-1 in cardiac muscle. Circulation Research. 2011;109:262–71. doi: 10.1161/CIRCRESAHA.111.244244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmore HP, McClive PJ, Smith CA, Sinclair AH. Expression profile of the RNA-binding protein gene hermes during chicken embryonic development. Developmental Dynamics. 2005;233:1045–51. doi: 10.1002/dvdy.20392. [DOI] [PubMed] [Google Scholar]
- Wu H, Sun S, Tu K, Gao Y, Xie B, Krainer AR, et al. A splicing-independent function of SF2/ASF in microRNA processing. Molecular Cell. 2010;38:67–77. doi: 10.1016/j.molcel.2010.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JI, Reed RB, Grabowski PJ, Artzt K. Function of quaking in myelination: regulation of alternative splicing. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:4233–8. doi: 10.1073/pnas.072090399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Yang D, Ding JH, Wang W, Chu PH, Dalton ND, et al. ASF/SF2-regulated CaMKIIdelta alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell. 2005;120:59–72. doi: 10.1016/j.cell.2004.11.036. [DOI] [PubMed] [Google Scholar]
- Xu XQ, Soo SY, Sun W, Zweigerdt R. Global expression profile of highly enriched cardiomyocytes derived from human embryonic stem cells. Stem Cells. 2009;27:2163–74. doi: 10.1002/stem.166. [DOI] [PubMed] [Google Scholar]
- Yano M, Hayakawa-Yano Y, Mele A, Darnell RB. Nova2 regulates neuronal migration through an RNA switch in disabled-1 signaling. Neuron. 2010;66:848–58. doi: 10.1016/j.neuron.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatskievych TA, Ladd AN, Antin PB. Induction of cardiac myogenesis in avian pregastrula epiblast: the role of the hypoblast and activin. Development. 1997;124:2561–70. doi: 10.1242/dev.124.13.2561. [DOI] [PubMed] [Google Scholar]
- Yeo GW, Xu X, Liang TY, Muotri AR, Carson CT, Coufal NG, et al. Alternative splicing events identified in human embryonic stem cells and neural progenitors. PLoS Computational Biology. 2007;3:1951–67. doi: 10.1371/journal.pcbi.0030196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaffran S, Astier M, Gratecos D, Semeriva M. The held out wings (how) Drosophila gene encodes a putative RNA-binding protein involved in the control of muscular and cardiac activity. Development. 1997;124:2087–98. doi: 10.1242/dev.124.10.2087. [DOI] [PubMed] [Google Scholar]
- Zhang C, Zhang Z, Castle J, Sun S, Johnson J, Krainer AR, et al. Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes & Development. 2008;22:2550–63. doi: 10.1101/gad.1703108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Fu XD. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma. 2013;122:191–207. doi: 10.1007/s00412-013-0407-z. [DOI] [PMC free article] [PubMed] [Google Scholar]