

Abstract
Resveratrol (3,5,4′-trihydroxylstilbene) has been proposed to elicit a variety of positive health effects including protection against cancer and cardiovascular disease. The highest affinity target of resveratrol identified so far is the oxidoreductase enzyme quinone reductase 2 (QR2), which is believed to function in metabolic reduction and detoxification processes; however, evidence exists linking QR2 to the metabolic activation of quinones, which can lead to cell toxicity. Therefore, inhibition of QR2 by resveratrol may protect cells against reactive intermediates and eventually cancer. With the aim of identifying novel inhibitors of QR2, we designed, synthesized, and tested two generations of resveratrol analogue libraries for inhibition of QR2. In addition, X-ray crystal structures of six of the resveratrol analogues in the active site of QR2 were determined. Several novel inhibitors of QR2 were successfully identified as well as a compound that inhibits QR2 with a novel binding orientation.
1. Introduction
Resveratrol (3,5,4′-trihydroxystilbene, Figure 1) is a naturally occurring phytoalexin that was discovered in 1940, when it was isolated from the roots of white hellebore.1-3 Resveratrol occurs in nature as both the cis- and trans-isomers and it can be found in a variety of dietary sources including peanuts, pistachios, and berries.4,5 Of the more common dietary sources of resveratrol, the skins and seeds of grapes are the most notable with red wine being the most heavily consumed form.5,6
Figure 1. Trans- and cis-resveratrol.

Interest in resveratrol increased dramatically in 1992, when it was hypothesized to explain the cardioprotective effects of red wine and the “French paradox,” the observation of reduced incidence of cardiovascular disease in regions of France where red wine and saturated fats are consumed in greater quantities than in the US.7,8 Since then, numerous studies have demonstrated the ability of resveratrol to prevent or slow the progression of various disease states including cancer and cardiovascular disease.9,10 Resveratrol has even been shown to increase the lifespans of several organisms including yeast, worms, fruit flies and fish.11-13
A number of direct targets for resveratrol have been discovered in vitro, including cyclooxygenase-1 (COX1), cyclooxygenase-2 (COX2), and the transcription factor NF-κB.14-16 The highest affinity target of resveratrol identified to date is quinone reductase 2 (QR2), a FAD-dependent cytosolic enzyme that catalyzes the 1-, 2-, or 4-electron reduction of quinones and other compounds using N-alkyl- and N-ribosylnicotinamides.17,18 QR2 is an oxidoreductase thought to function in metabolic reduction and detoxification; however, the true physiological role of QR2 is currently unknown.19 Evidence exists that QR2 is capable of catalyzing the metabolic activation of quinones and anti-tumor drugs, leading to cell toxicity.20-22 Thus, in some cases, inhibition of QR2 by resveratrol may guard cells against these reactive species that induce DNA damage, which may subsequently lead to cancer.23
Resveratrol has been found to bind tightly to the oxidized, FAD-form of QR2 and it acts as a competitive inhibitor against N-methyldihydronicotinamide (NMeH) with a Ki value of 88 ± 20 nM, determined by steady-state kinetic studies, and a Kd value of 54 ± 0.6 nM, determined by isothermal titration calorimetry17,24 Additionally, plasma levels of resveratrol are able to reach concentrations of 500 nM, which suggests that significant inhibition of QR2 by resveratrol in vivo may be achievable24 Taken together, these data suggest that the amount of resveratrol consumed from dietary sources may be sufficient for effective inhibition of QR2. However, circulating resveratrol is rapidly metabolized in the liver and gut by sulfation and glucuronation to its 3- and 4′-O-sulfate and 3-O-glucuronide conjugates24-27 These primary metabolites of resveratrol have been shown to have far lower affinity for QR2.24
The present work was undertaken in an attempt to identify novel analogues of resveratrol that could potently inhibit QR2 with increased affinity and to serve as leads for the development of future QR2 inhibitors as cancer chemopreventive or anticancer drugs. To do this, we first tested a library of 78 previously synthesized resveratrol analogues designed to investigate the effects of different steric and electronic substituents on both the aryl rings and central olefin resveratrol.28 Based on the inhibition of QR2 by the most active of these compounds, we set out to determine what effect functionalization of the central olefin of resveratrol with electron withdrawing substituents would have on inhibition of QR2 by creating a series of olefin-substituted and benzanilide resveratrol analogues. In addition, to circumvent inhibitory inactivation of resveratrol by its rapid metabolism, identification of effective resveratrol analogues that lacked the 3- and 4′-hydroxyl groups required for sulfation and glucuronation was of interest.
2. Results and discussion
2.1. Inhibition of QR2 by a first-generation resveratrol analogue library
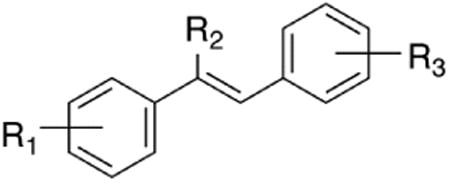
The first-generation library of resveratrol analogues was designed to investigate the effects of substitution on each of the two aryl rings and central alkene of resveratrol. Therefore, to explore the electronic and steric demands of each of the aryl rings, electron-donating (OH, OMe, and NMe2) and electron-withdrawing (F, CF3 and NO2) and naphthyl substitutents were selected. Four substituents were chosen to determine the effect of sterics and electronics on the central olefin (H, Me, Et, CF3). The synthesis of this first generation library of 78 resveratrol has been previously reported.28 Twenty-four of the seventy-eight resveratrol analogues were found to effectively inhibit QR2. These results are displayed in Table 1.
Table 1. Inhibition of QR2 by first generation resveratrol analogue library.
| ||||
|---|---|---|---|---|
|
| ||||
| Analogue | R1 | R2 | R3 | IC50 (μM)a |
| resveratrol | 4-OH | H | 3,5-(OH)2 | 11.5 ± 3.2 |
| 1a | 4-OH | H | 3,4-(OH)2 | 6.0 ± 1.2 |
| 1b | 4-OH | H | 3,5-(OMe)2 | 5.1 ± 1.2 |
| 1c | 3,4-(OH)2 | H | 4-OMe | 9.3 ± 3.7 |
| 1d | 3,4-(OH)2 | H | 3-F | 20.8 ± 8.6 |
| 1e | 3,4-(OH)2 | H | 4-CF3 | <14.2 ± 4.3 |
| 1f | 4-OMe | H | 3,5-(OMe)2 | 14.6 ± 4.3 |
| 1g | 3,5-(OMe)2 | H | 3,4-(OMe)2 | 37.1 ± 6.8 |
| 1h | 3,5-(OMe)2 | H | 4-F | 4.6 ± 1.0 |
| 1i | 3,5-(OMe)2 | H | 2-Naphthyl | 0.73 ± 0.12 |
| 1j | 3,4-(OMe)2 | H | 3-OMe | 6.8 ± 1.2 |
| 1k | 3,4-(OMe)2 | H | 4-NMe2 | 13.0 ± 3.5 |
| 1l | 3,4-(OMe)2 | H | 2-naphthyl | 5.5 ± 1.3 |
| 1m | 4-OH | Me | 3,5-(OH)2 | 39.0 ± 17.2 |
| 1n | 4-OH | Me | 4-CF3 | 2.8 ± 1.2 |
| 1o | 3,5-(OH)2 | Me | 4-OH | 4.8 ± 1.2 |
| 1p | 4-OMe | Me | 3,5-(OH)2 | 12.0 ± 8.4 |
| 1q | 4-OMe | Me | 3,5-(OMe)2 | 20.1 ± 0.1 |
| 1r | 4-OMe | Me | 3,4-(OMe)2 | 6.7 ± 3.0 |
| 1s | 3,4-(OMe)2 | Me | 3,5-(OH)2 | 33.7 ± 32.4 |
| 1t | 3,4-(OMe)2 | Me | 3,5-(OMe)2 | 9.9 ± 4.0 |
| 1u | 3,5-(OMe)2 | CF3 | 4-OMe | 13.0 ± 3.1 |
| 1v | 3-CF3 | CF3 | 3,4-(OMe)2 | 0.18 ± 0.3 |
| 1w | 3,5-(OH)2 | Et | 4-NMe2 | 10.9 ± 2.2 |
| 1x | 3,4-(OMe)2 | Et | 3,5-(OMe)2 | 16.3 ± 7.0 |
The lowest IC50 values are highlighted in bold
Of the 78 resveratrol analogues tested, twenty-four were found to actively inhibit QR2. Of the twenty-four active analogues, ten were more potent inhibitors of QR2 than resveratrol, five of which lacked both the 3- and 4′-hydroxyl substituents that undergo metabolic sulfation and glucuronation (1h, 1i, 1j, 1r, and 1v). Interestingly, two of the analogues found to be more active inhibitors of QR2 than resveratrol had a naphthyl substituted for an aryl ring (1i and 1l), possibly allowing for a greater pi-stacking interaction with the oxidized isoalloxazine ring of the FAD in the active site of QR2. The most potent inhibitor of QR2 identified from the first generation library was compound 1v, which has both a highly electron-deficient aryl ring and central olefin as a result of the trifluoromethyl substituents. Unfortunately, attempts at obtaining an X-ray crystal structure of our most active compound 1v in the active site of QR2 were unsuccessful; however, as a result we were determined to further investigate the effect of substitution at the central olefin. Therefore, we set out to synthesize a second-generation library comprised of analogues with electron-withdrawing substituents on the central olefin as well as a set of resveratrol analogues where the central olefin had been replaced by an amide.
2.2. Synthesis of olefin-substituted resveratrol analogues
To investigate the effect of substitution of both the central olefin and the phenols, as well as the conformational effect of E/Z-isomerism of resveratrol on its inhibition of QR2, a small library of 21 olefin-substituted resveratrol analogues was synthesized. The synthesis of (Z)-cyano resveratrol analogues 4a, 4b, and 4c was accomplished by condensation of the appropriate aldehyde 2 with the appropriate 2-phenylacetonitrile 3, as shown in Scheme 1.29,30 With these easily functionalized nitriles in hand, the remaining eighteen resveratrol structural analogues could be synthesized. Deprotection of 4b with boron tribromide yielded (Z)-nitrile analogue 4d. Protection of the phenols of 4d as tert-butyldimethylsilyl ethers followed by the reduction of the nitrile using diisobuytlaluminum hydride yielded a mixture of (E)- and (Z)-aldehydes 5. Treatment of 5 with tetra-n-butylammonium fluoride resulted in the isomerically pure (E)-acrylaldehyde 4e, where the (Z)-isomer was not detected. Further reduction of 5 with diisobutylaluminum followed by deprotection of the hydroxyls with dilute hydrochloric acid in methanol yielded the isomerically pure (Z)-alcohol 4f (Scheme 1).
Scheme 1. Synthesis of olefin-substituted resveratrol analogues.

Reduction of (Z)-nitriles 4b and 4c using diisobutylaluminum hydride resulted in (Z)-aldehyde analogues 4g and 4h in moderate to good yields. Further reduction of the aldehydes 4g and 4h with sodium borohydride yielded mixtures of both (Z)- and (E)- alcohols, which could be separated by flash chromatography to give the four analogues 4i, 4j, 4k, and 4l. The (E)-methyl ester 4m was obtained by oxidation of aldehyde 4h using manganese dioxide and potassium cyanide in methanol (Scheme 2).31
Scheme 2. Synthesis of olefin-substituted resveratrol analogues 4g-4m.

A Pinnick-type oxidation of aldehyde 4h by sodium chlorite-hydrogen peroxide yielded an inseparable mixture of the desired (Z)-carboxylic acid 6 and chlorinated byproduct 7, formed by reaction with hypochlorite generated in situ.32 Fischer esterification of the acids 6 and 7 yielded the methyl esters (Z)-4n and (Z)-4o, which could be separated by flash chromatography. Finally, deprotection of 4o using boron tribromide yielded the desired (Z)-ester analogue 4p (Scheme 3).
Scheme 3. Synthesis of olefin-substituted resveratrol analogues 4o, 4n, & 4p.

Palladium-catalyzed hydration of nitrile 4b yielded the (Z)-amide 4q, which upon treatment with boron tribromide gave (Z)-amide analogue 4r in good yield, as shown in Scheme 4.33 Concomitant reduction of the nitrile and olefin of 4b was accomplished using sodium borohydride and cobalt chloride hexahydrate, resulting in amine analogue 4s.34 The (Z)-methyl carbamate 4t was obtained in good yield by oxidative Hofmann rearrangement of 4q using (diacetoxyiodo)benzene. Treatment of the carbamate 4t with boron tribromide deprotected the methyl carbamate, which upon aqueous workup afforded the imine analogue 4u (Scheme 4).35 The structures of all olefin-substituted resveratrol analogues are displayed in Figure 2.
Scheme 4. Synthesis of olefin-substituted resveratrol analogues 4q, 4r, 4s, 4t, & 4u.

Figure 2. (Top) Benzanilide resveratrol analogue library, (Bottom) Olefin-substituted resveratrol analogue library.

2.3. Synthesis of benzanilides
A separate modification of the central alkene of resveratrol was accomplished by replacing the alkene with an amide bond; consequently, a small library of 26 substituted benzanilides (Figure 2) was synthesized using standard amide bond forming reactions. Construction of the library was accomplished by reaction of the appropriately activated benzoic acid derivative with the corresponding substituted aniline. In most cases, benzoyl chlorides were suitable for the coupling reaction; however, when acid-sensitive substrates were used, EDC activation was employed instead. N-methyl benzanilide derivatives were made by treatment of the appropriate benzanilide with sodium hydride and methyl iodide. The imidate 8v (Figure 2) was obtained by treatment of TBS-protected 8r with thionyl chloride and methanol.
The relationship between resveratrol and its benzanilide analogues' bioactivity was directly compared using analogues 8a, 8b, 8c, 8d, and 8e where the methylation of the amide and the phenols, and the position of the resorcinol ring relative to the nitrogen of the amide were varied. An additional 21 benzanilide resveratrol analogues were synthesized to probe the effect of different substitutions on bioactivity (Figure 2, top).
2.4. Inhibition of QR2 by olefin-substituted and benzanilide resveratrol analogues
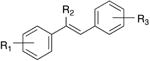
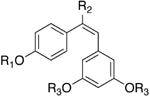
The ability of the second-generation analogues to inhibit QR2 is outlined in Table 2. Of the forty-seven second generation resveratrol analogues that were synthesized, nine were found to inhibit QR2. Two analogues, 4d and 4f, inhibited QR2 with IC50s comparable to resveratrol, while the remaining seven were slightly weaker inhibitors of QR2 as compared to resveratrol (6.9 ± 0.4 μM). Ten analogues showed weak inhibition of QR2 (between 20-50% at 100 μM) and twenty-nine of the forty-seven analogues showed minimal inhibition of QR2 (<20% at 100 μM). This result could be a consequence of substitution at the central olefin perturbing the binding orientation of the analogue within the active site of QR2. Each of the analogues that inhibited QR2 strongly had free hydroxyl groups, with the exception of analogues 4b, 4g, and 4k, which had methyl ethers. In general, inhibitory activity decreased as steric bulk associated with the aryl rings increased. For example, the nitrile analogue 4d was found to nhibit QR2 with an IC50 of 5.9 ± 0.3 μM and 88.8% inhibition, while the nitrile analogue 4b, which differs from 4d only in that the hydroxyl groups have been replaced with methyl ethers, was found to inhibit QR2 with a six-fold increase in IC50 of 31.6 ± 4.3 μM and 57.4% inhibition. Furthermore, the nitrile analogues 4a and 4c, where the methyl ethers of 4b have been replaced with bulkier isopropyl ethers, did not show inhibition of resveratrol over 20% at a concentration of 100 μM. This result may be a consequence of the bulky ethers reducing the ability of the molecule to form stabilizing hydrogen bonds to ordered QR2 active site water molecules (vide infra). Previous reports have demonstrated that methoxy groups and even acetates at the 2-, 3-, 4-, and 5-positions of resveratrol can be accommodated by the QR2 active site when the 4′-position is substituted with an amine.36,37
Table 2. Inhibition of QR2 by olefin-substituted and benzanilide resveratrol analogues.
| Structure | Analogue | R1 | R2 | R3 | IC50 (μM) |
|---|---|---|---|---|---|
|
resveratrol | 4-OH | H | 3,5-(OH)2 | 6.9 ± 0.4 |
| 4b | 4-OMe | CN | 3,5-(OMe)2 | 31.6 ± 4.3 | |
| 4d | 4-OH | CN | 3,5-(OH)2 | 5.9 ± 0.3 | |
| 4f | 4-OH | CH2OH | 3,5-(OH)2 | 5.1 ± 0.3 | |
| 4g | 4-OMe | CHO | 3,5-(OMe)2 | 24.2 ± 1.3 | |
| 4r | 4-OH | CONH2 | 3,5-(OH)2 | 9.3 ± 1.9 | |
|
| |||||
|
4e | H | CHO | H | 25.5 ± 3.5 |
| 4k | Me | CH2OH | Me | 27.5 ± 4.9 | |
|
| |||||
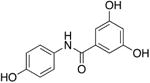
|
8a | - | - | - | 27.6 ± 4.6 |
|
| |||||
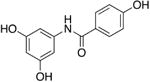
|
8d | - | - | - | 16.8 ± 1.3 |
Two of the olefin-substituted resveratrol analogues found to inhibit QR2, 4e and 4k, were of the E geometry, which more closely resembles cis-resveratrol. Interestingly, the (E)-alcohol 4k was found to effectively inhibit QR2 with an IC50 of 27.5 ± 4.9 μM and 57.2% inhibition at 100 μM, whereas its (Z)-isomer 4i only had an average inhibition of 27.7% at 100 μM. Furthermore, the analogue 4f, which only differs from 4i in that the methoxy substituents have been replaced with hydroxyl groups, was found to be the most potent inhibitor in our library, having an IC50 of 5.1 ± 0.3 μM and 87.4% average inhibition at 100 μM of QR2. This supports our observation that substitution of sterically bulky ethers for the hydroxyl groups of resveratrol decreases inhibitory ability of a given compound; however, this data suggests that this trend may be negated when taking into consideration the effect of conformation. To the best of our knowledge, this is the first report of an isomerically pure (E)-resveratrol analogue inhibiting QR2 and having greater inhibition of QR2 than its corresponding (Z)-conformation (Figure 3).
Figure 3. Effect of substitution and alkene geometry on QR2 inhibition.

Two of the twenty-six benzanilide resveratrol analogues were found to inhibit QR2, 8a and 8d. Examination of the IC50 data shows that the position of the amide nitrogen relative to the resorcinol ring has a measurable effect on the ability of the benzanilide analogue to inhibit QR2: when the nitrogen is directly bound to the resorcinol ring the IC50 is decreased by roughly 40% (16.8 ± 1.3 μM vs. 27.6 ± 4.6 μM). Additionally, only hydroxyl substituents on the aryl rings are tolerated for good inhibition of QR2 by the benzanilide analogues and even then, only at the same positions found in resveratrol itself. Comparison of 8a, which has free hydroxyl groups at the 3-, 5-, and 4′-positions, to 8c, where the 3- and 5-hydroxyls are replaced by methyl ethers, shows the average percent inhibition at 100 μM of QR2 decreases from 59.7% to 43.2%, respectively. Additionally, moving one hydroxyl substituent from the 3- to the 4-position on the resorcinol ring, as observed in the comparison of 8a and 8t, decreases the analogue's ability to inhibit QR2 from 59.7% to 49.9% at 100 μM. Replacement of the 4′-hydroxyl group by a 4′-dimethylamino group eliminates inhibition of QR2, which is seen in both the comparison of 8a to 8f and 8d to 8k. This is significant since previous reports have shown that replacement of the hydroxyl group at the 4′-position of resveratrol by an amine can increase its inhibitory activity.36,37
Of the eight synthesized naphthyl-substituted benzanilide analogues, only 8z showed weak inhibition of QR2 (30.5% average inhibition at 100 μM). In contrast, the first generation analogue 1l, which differs from 8z only in that 8z has an amide in place of the central alkene, was a potent inhibitor of QR2, having an IC50 of 5.5 μM. Interestingly, second generation benzanilide analogue 8p showed no inhibition of QR2 at 100 μM, while compound 1i was the second most active analogue in the first generation library, having an IC50 of 0.7 μM.
2.5. X-ray crystallographic analysis of QR2 in complex with active olefin-substituted and benzanilide resveratrol analogues
To elucidate the structural factors underlying the inhibitory trends observed in the kinetic data, and to answer questions such as why does the (E)-geometry 4k analogue lead to a more potent inhibitor of QR2 than its (Z)-isomer 4i, a series of X-ray crystal structures were determined for the most potent olefin-substituted and benzanilide QR2 inhibitors. Resveratrol binds in the in the active site of QR2 fitting into a hydrophobic cleft that is approximately 17 Å long and 7 Å wide (Figure 4, A and B). Three sides of the QR2 binding cavity are composed of hydrophobic amino acids, while the fourth side is solvent exposed. The ends of the cavity contain residues that are available for hydrogen bond formation. The planar structure of resveratrol allows it to fit into the QR2 binding cleft in an orientation coplanar to the isoalloxazine ring of the FAD cofactor.17 The narrow width of the QR2 binding site dictates that it can only accommodate molecules such as resveratrol that can adopt essentially planar conformations. Additionally, molecules that bind tightly to QR2 are generally planar and polyaromatic with the ability to stack with the isoalloxazine ring of FAD, possibly utilizing a pi-stacking interaction with the isoalloxazine for binding (Figure 4, A and B)38,39
Figure 4.

Resveratrol-QR2 complex (PDB: 1SG0). Resveratrol is shown in green, colored by atom type, and shown in ball and stick representation. Water molecules are shown as red spheres. Hydrogen bonds are shown as grey dashes labeled with the distance in Angstroms (Å). (A) X-ray structure of QR2 in complex with resveratrol displaying hydrogen bond net Å k, (B) Resveratrol in binding cavity of QR2, where QR2 is shown in grey surface representation and colored according to atom type.17
Complete X-ray crystallographic data sets of QR2 in complex with inhibitors 4d, 4f, 4k, 4r, 8a, and 8d were collected and the structures for each complex were determined. The final data collection and refinement statistics are summarized in Table 3. With the exception of 4k, all of the olefin-substituted and benzanilide resveratrol analogues bind in the same orientation as resveratrol, coplanar to the oxidized isoalloxazine ring of the bound cofactor FAD (Figure 5). The binding of analogs 4d, 4f, 4k, 4r, 8a, and 8d does not significantly alter the conformation of the active site residues compared to the resveratrol-bound structure. As with resveratrol, the (Z)-analogues show water mediated hydrogen bonds from the 4′-hydroxyl group of the ligand to the hydroxyl group of Thr71, a tight hydrogen bond from the 5-hydroxyl to Asn161, and a hydrogen bond from the 3-hydroxyl to the carbonyl oxygen of Gly174.17 The 5-hydroxyls of the (Z)-configured analogues also form a hydrogen bond to an ordered active site water molecule. In some cases, such as 8a (Figure 5, E) and 8d (Figure 5, F), a network of water molecules within the active site can extend to the substitution at the central olefin.
Table 3. X-Ray Data Collection and Refinement Statistics.
| Data Collection | 4d | 4r | 4f | 8a | 8d | 4k | 4k |
|---|---|---|---|---|---|---|---|
| Detector | MarMosaic 300mm | MarMosaic 300mm | MarMosaic 300mm | MarMosaic 225mm | MarMosaic 225mm | MarMosaic 225mm | MarMosaic 225mm |
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 |
| a,b,c (Å) | 56.90, 83.36, 106.53 | 56.94, 83.39, 106.64 | 56.65, 83.39, 106.57 | 56.69, 83.15, 106.68 | 57.10, 83.51, 106.47 | 56.33, 83.59, 106.43 | 56.26, 83.58, 106.34 |
| α=β=γ (degrees) | 90 | 90 | 90 | 90 | 90 | 90 | 90 |
| Resolution (Å) | 50-1.40 | 50-1.45 | 50-1.45 | 50-1.55 | 50-1.63 | 50-1.45 | 50-1.40 |
| Reflections Observed | 436515 | 402271 | 389780 | 349634 | 287051 | 437831 | 441401 |
| Unique Reflections | 85793 | 90632 | 84618 | 72933 | 62952 | 86788 | 98717 |
| Rmerge (%) | 58.7 (5.1) | 54.4 (5.6) | 50.2 (5.2) | 53.0 (6.6) | 54.9 (7.8) | 46.1 (5.7) | 50.3 (5.2) |
| I/σ | 33.1 (2.5) | 28.0 (2.9) | 28.3 (2.9) | 27.1 (2.6) | 17.4 (2.0) | 31.7 (3.0) | 31.4 (2.5) |
| % | 85.9 | 99.8 | 94.0 | 98.9 | 98.4 | 96.8 | 99.4 |
| Completeness | (85.8) | (99.8) | (98.7) | (98.1) | (97.6) | (99.8) | (98.6) |
| Refinement | |||||||
| Resolution range (Å) | (1.40-1.42) | (1.45-1.48) | (1.45-1.48) | (1.55-1.58) | (1.63-1.66) | (1.45-1.48) | (1.40-1.42) |
| Reflections in Working Set | 81481 | 86029 | 80331 | 69206 | 59715 | 82382 | 93724 |
| Reflections in Test Set | 5532 | 5778 | 5840 | 3672 | 3968 | 5772 | 6413 |
| Rwork (%) | 16.3 | 15.4 | 16.7 | 16.8 | 16.4 | 16.4 | 16.6 |
| Rfree (%) | 19.0 | 18.0 | 19.2 | 18.9 | 19.3 | 18.7 | 18.8 |
| Average B-factor (Å2) | 14.0 | 13.8 | 13.8 | 12.9 | 11.7 | 14.0 | 14.1 |
Figure 5.

X-ray crystal structures of (A) 4d, (B) 4f, (C) 4k, (D) 4r, (E) 8a, and (F) 8d in complex with QR2. Resveratrol analogues are shown in green, colored according to atom type, and shown in ball and stick representation. Water molecules are shown as red spheres. Hydrogen bonds are shown as grey dashes with distances (Å) labeled. The FAD cofactor is shown in grey lines and colored according to atom. Electron density omit maps (Fo - Fc) are shown in white mesh and contoured to 3.0 σ around the ligand only. The binding orientation of each resveratrol analogue was the same in both active sites of the QR2 dimer, therefore only one active site is shown for clarity.
Examination of the X-ray crystal structures of the (Z)-configured resveratrol analogues show that they bind in identical conformations. The substitution of methyl ethers for phenols, such as in analogue 4b, could change this preferred binding orientation. In support of this, we did not observe strong electron density of 4b in the QR2 active site, suggesting that the ligand was only present at partial occupancy. This also supports our observations regarding the series of analogues 4f, 4i, and 4k (Figure 3). Our most active inhibitor, 4f, has a Z geometry and has hydroxyl substituents that hydrogen-bond key residues and structural waters in the QR2 active site. The inhibitor 4i, which differs from 4f only by substitution of methyl ethers for the hydroxyl groups of 4f, did not show potent enough inhibition of QR2 to make determination of an IC50 feasible, suggesting that increasing the size of the 4′-, 3-, and 5-substituents is not readily accommodated by the QR2 active site.
Most surprisingly, the E isomer of 4i, 4k, is a potent inhibitor of QR2 and binds in a completely different orientation than the other analogues where only one aryl ring occupies the same molecular space as the other Z analogues (Figure 5, E). Examination of the binding orientation of 4k shows that it does not form a direct hydrogen bond to the network of active site water molecules that hydrogen bond to the side chain of Thr71. Instead, an additional water molecule is present in the active site and is positioned where the 4′-hydroxyl group of the (Z)-configured resveratrol analogues usually resides, forming a water-mediated hydrogen bond to the side chain of Asn161. Taking together the structure of 4k in the active site and the partial occupancy observed for the 4b analogue, we hypothesize that, for the QR2 active site to accommodate resveratrol analogues with bulkier substituents, the molecules must rotate 90° from their preferred binding orientation to allow at least one aryl ring to stack on the isoalloxazine ring (Figure 6, A). Whether this rotation will be accommodated by the QR2 active site depends on the other substituents on the analogs and whether water molecules can aid in the binding process.
Figure 6.

(A) Crystal structure of inhibitor 4k bound in the QR2 active site. QR2 is shown in grey surface representation and colored according to atom type; 4k is shown in green ball and stick and colored according to atom. FAD and active site residue Asn161 are shown in grey ball and stick and colored according to atom. (B) Superposition of X-ray structures of benzanilides 8a (shown in orange ball and stick) and 8d (shown in green ball and stick) in complex with QR2, where active site waters are shown as non-bonding spheres, are colored according to ligand and are identical between the two compounds.
The two most active benzanilide analogues, 8a and 8d (Figure 5E and 5F, respectively), orient their hydroxyl substituents in identical locations (Figure 6B), taking advantage of the same hydrogen bond network. The carbonyl groups of the amide core of each analogue are positioned to interact with the solvent exposed region of the active site, avoiding the hydrophobic interior, again both utilizing almost identical water molecules.
3. Conclusions
In conclusion, two sets of resveratrol analogue libraries were designed, synthesized and tested for inhibition of QR2. Twenty-four of the seventy-eight first generation resveratrol analogues were found to potently inhibit QR2 and, of those twenty-four analogues, ten were more potent QR2 inhibits than resveratrol. Our goal of identifying analogues that were both more potent than resveratrol and lacking the metabolically sensitive 3- and 4′-hydroxyl groups was achieved with the identification of five analogues --1h, 1i, 1j, 1r, and 1v -- from the first-generation library. A second-generation library of 47 resveratrol analogues was designed to probe the effects of substitution on the central alkene. Nine of the second-generation resveratrol analogues were found to inhibit QR2. Two of the analogues, 4d and 4f, had comparable IC50 values to that of resveratrol while the remaining seven analogues were slightly weaker inhibitors of QR2 in comparison to resveratrol. Because of the unique substitutions made at the central alkene, X-ray crystal structures of six of the nine inhibitors in complex with QR2 were determined. From the series of crystal structures obtained, we learned that all of the Z-configured resveratrol analogues share a common binding orientation within the active site of QR2. Additionally, the analogues all utilize the same ordered, active site water molecules to form water-mediated hydrogen bonds to the same residues within the QR2 active site, Asn161, Gly174 and Thr71. We also identified a novel binding orientation of the E-configured resveratrol analogue 4k, which is the first example of an E-configured resveratrol analogue bound in the QR2 active site. Future investigations will focus on the identification of additional E-configured resveratrol analogues that lack the metabolically sensitive 3- and 4′-hydroxyl groups, but also have more potent inhibition of QR2.
4. Experimental
4.1. General Procedures
All reagents (chemicals) were purchased from Sigma or Acros and used without further purification. Infrared spectra were obtained using a ThermoNicolet Nexus 470 FT-IR spectrometer with OMNIC software package. Mass spectral analyses were performed using a Waters Micromass ZQ with ESI-MS injection port utilizing MassLynx V4.1 software package. Analytical thin-layer chromatography was performed on Sorbent Technologies Glass Backed Silica Gel HL TLC Plates w/UV254. Flash chromatography was performed with Sorbent Technologies 200-400 mesh silica gel. 1H NMR spectra were obtained at 300 or 400 MHz and 13C NMR spectra were obtained at 75 or 100 MHz, using Varian Inova300 and Bruker ARX400 spectrometers, respectively. E/Z isomerism was determined by Nuclear Overhauser Effect experiments. All compounds were purified by flash chromatography to >90% purity by 1H NMR (see Supporting Information).
4.2. Synthesis
4.2.1. 3,5-diisopropoxybenzaldehyde (2)
A mixture of 3,5-dihydroxybenzoic acid (1.00 g, 6.49 mmol), potassium carbonate (4.04 g, 29.2 mmol) and isopropyl bromide (3.65 mL, 38.93 mmol) in 12 mL dimethylformamide was heated to reflux for 4 days. After cooling to room temperature, 6 mL water and 6 mL 2M hydrochloric acid were added to dissolve the carbonate and acidify the reaction mixture. The aqueous layer was extracted with ethyl acetate (3 × 20 mL) and the combined organic layers were dried over magnesium sulfate, filtered and concentrated to yield 3,5-diisopropoxybenzoic acid as a clear oil that was used without further purification.
A 0.54 M solution of DIBAL-H in dichloromethane (4.66 mL, 2.52 mmol) was added to a solution of 3,5-diisopropoxybenzoic acid (282 mg, 1.01 mmol) in toluene (5 mL) at −78 °C. The reaction mixture was allowed to stir for 45 minutes at that temperature before quenching with 5% aqueous hydrochloric acid (1 mL). After warming room temperature, the reaction mixture was diluted with water and the aqueous layer was extracted with ethyl acetate (3 × 5 mL), the combined organic layers were dried over magnesium sulfate, filtered and concentrated to yield a mixture of 3,5-diisopropoxybenzaldehyde and (3,5-diisopropoxyphenyl)methanol in 97% combined yield. 3,5-Diisopropoxybenzaldehyde: 1H-NMR (400 MHz, CDCl3) δ (ppm): 9.77 (1H, s), 6.86 (2H, d, J = 2.31), 6.57 (1H, t, J = 2.31), 4.48 (2H, m, J = 6.15), 1.24 (6H, d, J = 6.16). 13C NMR (100 MHz, CDCl3) δ (ppm): 191.72, 159.44, 110.10, 108.33, 70.03, 21.72.
4.2.2. 2-(4-Methoxyphenyl)acetonitrile (3)
Triphenyl phosphine (1.14 g, 4.34 mmol) was added portion-wise to a solution of (4-methoxyphenyl)methanol (300 mg, 2.17 mmol) and carbon tetrabromide (1.22 g, 3.69 mmol) in anhydrous dichloromethane (7.2 mL) at 0 °C. The reaction mixture was stirred at that temperature for 20 minutes then concentrated to yield a viscous oil which was dissolved in 10 mL of 50% hexanes 50% ethyl acetate. The precipitate was filtered off and the filtrate concentrated. The residue was taken up in dimethylformamide (7.2 mL) and potassium cyanide (0.99 g, 15.2 mmol) was added. The reaction mixture was allowed to stir for 90 minutes before quenching with saturated aqueous sodium bicarbonate (10 mL). The aqueous layer was extracted with ethyl acetate (3 × 10 mL) and the combined organic layers were washed with brine, dried over magnesium sulfate, filtered and concentrated. The crude product was purified by flash chromatography using 20% ethyl acetate 80% hexanes to yield the pure product 3 in 45% yield. 1H-NMR (300 MHz, CDCl3) δ (ppm): 7.19 (2H, d, J = 8.38), 6.86 (2H, d, J = 8.38), 3.74 (3H, s), 3.61 (2H, s). 13C NMR (75 MHz, CDCl3) δ (ppm): 159.15, 129.01, 121.89, 118.42, 114.35, 55.18, 22.50.
4.2.3. General Procedure for Aldol Condensation (4a-4c)
Aqueous 40% potassium hydroxide (0.23 mL/mmol nitrile) was diluted with absolute ethanol (0.46 mL/mmol nitrile) and added at room temperature to a solution of the appropriate aldehyde (1.1 equivalents) and nitrile (1.0 equivalent) in absolute ethanol (0.35 mL/mmol nitrile). The reaction was then allowed to stir at room temperature for 12-24 hours before concentrating. The resulting yellow residue was taken up in water and ethyl acetate and dilute hydrochloric acid was added to neutralize the aqueous layer. The aqueous layer was then extracted with ethyl acetate and the combined organic layers were washed with brine, dried over magnesium sulfate, filtered and concentrated to yield the crude product. Purification by flash chromatography yielded the pure nitrile products in 80-95% yield.
4.2.4. (Z)-3-(3,5-diisopropoxyphenyl)-2-(4-hydroxyphenyl)acrylonitrile (4a)
General procedure for Aldol condensation was used in combination with 2 and 2-(4-((tert-butyldimethylsilyl)oxy)phenyl)acetonitrile (28%). 1H-NMR (300 MHz, CDCl3) δ (ppm): 7.56 (2H, d, J = 8.71), 7.31 (1H, s), 6.98 (2H, d, J = 2.15), 6.90 (2H, d, J = 8.71), 6.50 (1H, t, J = 2.15), 5.06 (1H, s), 4.58 (2H, m, J = 6.05), 1.36 (12H, d, J = 2.05). ESIMS(+) m/z 338 (M + H), 360 (M + Na), 376 (M + K).
4.2.5. (Z)-3-(3,5-dimethoxyphenyl)-2-(4-methoxyphenyl)acrylonitrile (4b)
General procedure for Aldol condensation was used in combination with 3,5-dimethoxybenzaldehyde and 3 (95%). 1H-NMR (300 MHz, CDCl3) δ (ppm): 7.61 (2H, d, J = 8.91), 7.34 (1H, s), 7.04 (2H, d, J = 2.13), 6.96 (2H, d, J = 8.91), 6.53 (1H, t, 2.13), 3.85 (9H, s). 13C NMR (75 MHz, CDCl3) δ (ppm): 160.73, 160.32, 139.83, 135.47, 127.16, 126.59, 118.11, 114.27, 111.14, 106.71, 102.65, 55.28. ESIMS(+) m/z 318 (M + Na).
4.2.6. (Z)-3-(3,5-diisopropoxyphenyl)-2-(4-methoxyphenyl)acrylonitrile (4c)
General procedure for Aldol condensation was used in combination with 2 and 3 (92%). 1H-NMR (300 MHz, CDCl3) δ (ppm): 7.56 (2H, d, J = 8.83), 7.32 (1H, s), 7.01 (2H, d, J = 1.86), 6.94 (2H, d, J = 8.83), 6.51 (1H, t, 1.86), 4.58 (2H, m, J = 6.04), 3.81 (3H, s), 1.37 (12H, d, J = 6.04). 13C NMR (75 MHz, CDCl3) δ (ppm): 160.31, 159.15, 140.24, 135.45, 127.19, 126.79, 118.08, 114.30, 110.98, 108.30, 108.24, 106.13, 70.07, 70.01, 55.25, 21.96. ESIMS(+) m/z 352 (M + H), 374 (M + Na), 390 (M + K).
4.2.7. (Z)-3-(3,5-dihydroxyphenyl)-2-(4-hydroxyphenyl)acrylonitrile (4d)
A 1.0 M solution of boron tribromide in dichloromethane (2.34 mL, 2.34 mmol) was added slowly to a solution of 4b (138 mg, 0.46 mmol) in dichloromethane (2.3 mL) at −78 °C under nitrogen atmosphere. The reaction was allowed to stir at room temperature for four hours before cooling to −78 °C and quenching with an aqueous solution of saturated sodium bicarbonate (2 mL). The reaction mixture was warmed to room temperature and the aqueous layer was extracted with ethyl acetate (3 × 2 mL). The combined organic layers were washed with saturated sodium bicarbonate (3 × 5 mL), water (1 × 5 mL), and brine (1 × 5 mL) before being dried over magnesium sulfate, filtered, and concentrated. The pure product was obtained in 80% yield after purification by flash chromatography using 50% ethyl acetate 50% hexanes. 1H-NMR (400 MHz, CD3OD) δ (ppm): 7.52 (2H, d, J = 8.70), 7.42 (1H, s), 6.85 (2H, d, J = 8.70), 6.82 (2H, d, J = 2.02), 6.33 (1H, t, J = 2.02). 13C NMR (100 MHz, CD3OD) δ (ppm): 159.85, 141.34, 137.32, 128.35, 127.07, 119.17, 116.86, 111.91, 108.56, 105.58. IR vmax 3216 (OH), 2227 (CN) cm−1.
4.2.8. (E)-3-(3,5-dihydroxyphenyl)-2-(4-hydroxyphenyl)acrylaldehyde (4e)
A 1.0 M solution of tetrabutylammonium fluoride solution in THF (0.15 mL, 0.15 mmol) was added to a solution of 5 (29 mg, 0.05 mmol) in THF (0.25 mL) at 0 °C. The reaction was allowed to stir overnight before quenching with saturated ammonium chloride. The aqueous layer was extracted with ethyl acetate (3 × 3 mL) and the combined organic layers were washed with brine, dried over magnesium sulfate, filtered and concentrated. The pure product 4e was obtained in 15% yield after flash chromatography using 50% ethyl acetate 50% hexanes. 1H-NMR (400 MHz, CD3OD:CDCl3 50:50) δ (ppm): 9.44 (1H, s), 7.03 (1H, s), 6.81 (2H, d, J = 8.59), 6.63 (2H, d, J = 8.59), 6.07 (2H, s). 13C NMR (100 MHz, CD3OD) δ (ppm): 195.25, 157.48, 157.33, 151.12, 140.98, 135.65, 130.39, 123.78, 115.27, 109.10, 104.54. ESIMS(+) m/z 257 (M + H).
4.2.9. 3-(3,5-bis((tert-butyldimethylsilyl)oxy)phenyl)-2-(4-((tert-butyldimethylsilyl)oxy)phenyl)- acrylaldehyde (5)
Tert-Butyldimethylsilyl chloride (202 mg, 1.34 mmol) and imidazole (228 mg, 3.35 mmol) were added to a stirred solution of 4d (94 mg, 0.37 mmol) in dimethylformamide (2 mL) at 0 °C. The reaction mixture was warmed to room temperature. After stirring overnight, the reaction mixture was diluted with water and extracted with ethyl acetate (3 × 5 mL). The combined organic layers were then washed with brine, dried over magnesium sulfate, filtered and concentrated to yield the protected nitrile in 52% yield after flash chromatography using 5% ethyl acetate 95% hexanes.
A 0.54 M solution of DIBAL-H in hexanes (0.31 mmol, 0.57 mL) was added slowly to the protected nitrile (155 mg, 0.26 mmol) in toluene (1.3 mL) at 0 °C under nitrogen atmosphere. After 40 minutes the reaction was quenched by addition of methanol (0.32 mL) and water (0.31 mL), warmed to room temperature and stirred for an additional 3 hours. The reaction mixture was diluted with water and the aqueous phase was extracted with ethyl acetate (3 × 5 mL). The combined organic layers were washed with brine, dried over magnesium sulfate, filtered and concentrated. Flash chromatography using 5% ethyl acetate 95% hexanes yielded the pure aldehyde product 5 in 80% yield. (Z)-5: 1H-NMR (300 MHz, CDCl3) δ (ppm): 9.71 (1H, s), 7.20 (1H, s), 7.05 (2H, d, J = 8.41), 6.85 (2H, s, J =8.41), 6.39 (2H, d, J = 2.18), 6.29 (1H, t, J = 2.18), 1.00 (12H, s), 0.92 (18H, s), 0.23 (6H, s), 0.08 (9H, s). (E)-5: 1H-NMR (300 MHz, CDCl3) δ (ppm): 10.10 (1H, 1), 7.70 (1H, s), 7.39 (2H, d, J = 8.38), 6.94 (2H, d, J = 8.38), 6.50 (2H, d, J = 2.07), 6.41 (1H, t, J =2.07), 1.00 (12H, s), 0.92 (18H, s), 0.23 (6H, s), 0.08 (9H, s). 13C NMR (75 MHz, CDCl3) δ (ppm): 192.46, 156.47, 145.57, 140.63, 135.76, 129.80, 128.96, 127.20, 119.85, 115.32, 113.12, 25.56, 18.13.
4.2.10. (Z)-5-(3-hydroxy-2-(4-hydroxyphenyl)prop-1-en-1-yl)benzene-1,3-diol (4f)
A 0.54 M solution of DIBAL-H in hexanes (0.7 mmol, 0.12 mL) was added slowly to 5 (40 mg, 0.07 mmol) in toluene (.4 mL) at −78 °C under nitrogen atmosphere. The reaction was allowed to warm to room temperature and stirred for 4 hours. The reaction was quenched by addition of methanol (0.12 mL) and 0.1 N HCl (0.12 mL) at −78 °C. The reaction mixture was diluted with water and the aqueous phase was extracted with ethyl acetate (3 × 5 mL). The combined organic layers were washed with brine, dried over magnesium sulfate, filtered and concentrated. Flash chromatography using 5% ethyl acetate 95% hexanes yielded the pure protected alcohol in 55% yield.
The protected alcohol (15 mg, 0.04 mmol) was dissolved in methanol (0.5 mL) and a catalytic amount of hydrochloric acid was added. The reaction was allowed to stir until all of the starting material had disappeared from TLC. The reaction mixture was then concentrated. The residue was taken up in ethyl acetate and water, and the aqueous phase was extracted with ethyl acetate (3 ×). The combined organic layers were washed with brine, dried over magnesium sulfate, filtered and concentrated. The product 4f was obtained in 35% yield after purification by flash chromatography using 70% ethyl acetate 30% hexanes. 1H-NMR (400 MHz, CD3OD) δ (ppm): 7.43 (2H, d, J = 8.64), 6.78 (2H, d, J = 8.64), 6.73 (1H, s), 6.37 (2H, d, J = 2.13), 6.19 (1H, t, J = 2.13), 4.59 (2H, s). 13C NMR (100 MHz, CD3OD) δ (ppm): 159.33, 158.05, 141.01, 140.74, 133.78, 130.22, 128.76, 116.09, 109.11, 108.50, 102.42, 101.57, 84.02, 60.22.
4.2.11. General Procedure for Nitrile Reduction (4g, 4h)
A 0.54 M solution of DIBAL-H in hexanes (2.0 equivalents) was added slowly to a solution of the appropriate nitrile (1.0 equivalent) in toluene (0.2 M) at −78 °C. The reaction mixture was stirred at that temperature for four hours before quenching with water and ethyl acetate. The organic layer was washed (3 ×) with dilute hydrochloric acid. The combined aqueous layers were extracted with ethyl acetate (3 ×) and the combined organic layers were washed with brine, dried over magnesium sulfate, filtered and concentrated. The desired aldehyde products were obtained in 67-80% yield after flash chromatography.
4.2.12. (Z)-3-(3,5-dimethoxyphenyl)-2-(4-methoxyphenyl)acrylaldehyde (4g)
General procedure for nitrile reduction was used starting from 4b (67%). 1H-NMR (400 MHz, CDCl3) δ (ppm): 9.75 (1H, s), 7.28 (1H, s), 7.15 (2H, d, J = 8.71), 6.96 (2H, d, J = 8.71), 6.41 (3H, s), 3.83 (3H, s), 3.59 (6H, s). 13C NMR (100 MHz, CDCl3) δ (ppm): 194.16, 160.36, 159.56, 149.81, 141.68, 135.80, 130.70, 125.30, 114.23, 108.37, 102.83, 55.23, 55.06.
4.2.13. (Z)-3-(3,5-diisopropoxyphenyl)-2-(4-methoxyphenyl)acrylaldehyde (4h)
General procedure for nitrile reduction was used starting from 4c (80%). 1H-NMR (300 MHz, CDCl3) δ (ppm): 10.13 (1H, s), 7.73 (1H, s), 7.37 (2H, d, J = 8.77), 6.94 (2H, d, J = 8.77), 6.49 (3H), 4.54 (2H, m, J = 6.04), 3.84 (3H, s), 1.35 (12H, d, J = 6.04). 13C NMR (75 MHz, CDCl3) δ (ppm): 192.58, 159.69, 158.88, 145.96, 140.65, 135.82, 129.89, 128.43, 113.72, 109.81, 104.68, 70.07, 55.22, 21.94.
4.2.14. General Procedure for Aldehyde Reduction (4i-4l)
Sodium borohydride (1.2 equivalents) was added portion-wise to a stirred solution of the appropriate aldehyde (1.0 equivalent) in methanol (0.2 M) at 0 °C. The reaction mixture was concentrated under reduced pressure after 30 min. The residue was dissolved in CH2Cl2 and extracted with H2O, and the aqueous layer was washed with CH2Cl2 (3×). The combined organic layers were washed with H2O (3×) and brine, dried over MgSO4, filtered and concentrated. Purification by flash chromatography afforded the pure alcohol products in 13-44% yield.
4.2.15. (Z)-3-(3,5-dimethoxyphenyl)-2-(4-methoxyphenyl)prop-2-en-1-ol (4i)
General procedure for aldehyde reduction as used from 4g (13%). 1H-NMR (300 MHz, CDCl3) δ (ppm): 7.53 (2H, d, J = 8.82), 6.94 (2H, d, J = 8.82), 6.85 (1H, s), 6.57 (2H, d, J = 2.16), 6.42 (1H, t, J = 2.16), 4.70 (2H, s), 3.84 (3H, s), 3.82 (6H, s). 13C NMR (100 MHz, CDCl3) δ (ppm): 160.63, 159.32, 139.94, 139.00, 132.70, 129.62, 127.68, 114.01, 106.80, 99.44, 60.39, 55.29.
4.2.16. (Z)-3-(3,5-diisopropoxyphenyl)-2-(4-methoxyphenyl)prop-2-en-1-ol (4j)
General procedure for aldehyde reduction was used from 4h (27%). 1H-NMR (400 MHz, CDCl3) δ (ppm): 7.52 (2H, d, J = 8.71), 6.93 (2H, d, J = 8.71), 6.82 (1H, s), 6.53 (2H, d, J = 2.16), 6.39 (1H, t, J = 2.16), 4.70 (2H, s), 4.54 (2H, m, J = 6.07), 3.84 (3H, s), 1.34 (12H, d, J = 6.07). 13C NMR (100 MHz, CDCl3) δ (ppm): 159.25, 158.92, 139.60, 138.86, 132.80, 129.84, 127.66, 113.98, 108.49, 102.90, 69.87, 60.39, 55.25, 22.03.
4.2.17. (E)-3-(3,5-dimethoxyphenyl)-2-(4-methoxyphenyl)prop-2-en-1-ol (4k)
General procedure for aldehyde reduction was used from 4g (35%). 1H-NMR (300 MHz, CDCl3) δ (ppm): 7.17 (2H, d, J = 8.77), 6.88 (2H, d, J = 8.77), 6.59 (1H, s), 6.24 (1H, t, J = 2.23), 6.20 (2H, d, J = 2.23), 4.43 (2H, s), 3.80 (3H, s), 3.56 (6H, s). 13C NMR (75 MHz, CDCl3) δ (ppm): 160.05, 158.97, 141.52, 138.37, 130.47, 129.86, 126.01, 114.09, 106.98, 99.50, 68.36, 55.17, 54.88.
4.2.18. (E)-3-(3,5-diisopropoxyphenyl)-2-(4-methoxyphenyl)prop-2-en-1-ol (4l)
General procedure for aldehyde reduction was used from 4h (44%). 1H-NMR (300 MHz, CDCl3) δ (ppm): 7.17 (2H, d, J = 8.63), 6.88 (2H, d, J = 8.63), 6.56 (1H, s), 6.21 (1H, t, J = 2.02), 6.16 (2H, d, J = 2.02), 4.41 (2H, s), 4.19 (2H, m, J = 6.05), 1.17 (12H, d, J = 6.05).
4.2.19. (E)-methyl 3-(3,5-diisopropoxyphenyl)-2-(4-methoxyphenyl)acrylate (4m)
To a stirred solution of 4h (64 mg, 0.18 mmol) in methanol (1.8 mL) was added potassium cyanide (59 mg, 0.91 mmol) and manganese dioxide (317 mg, 3.65 mmol). After six days, the reaction mixture was diluted with dichloromethane and filtered through a celite pad. The combined organic layers were washed with brine, dried over magnesium sulfate, filtered and concentrated. Flash chromatography using 10% ethyl acetate 90% hexanes afforded the pure product in 17% yield. 1H-NMR (300 MHz, CDCl3) δ (ppm): 7.72 (1H, s), 7.16 (2H, d, J = 8.70), 6.92 (2H, d, J = 8.70), 6.29 (1H, t, J = 2.22), 6.20 (2H, d, J = 2.22), 4.16 (2H, m, J = 6.06), 3.81 (3H, s), 3.79 (3H, s), 1.17 (12H, d, J = 6.06).
4.2.20. (Z)-methyl 3-(3,5-diisopropoxyphenyl)-2-(4-methoxyphenyl)acrylate (4n)
See procedure for 4o (34%). 1H-NMR (400 MHz, CDCl3) δ (ppm): 7.37 (2H, d, 8.86), 6.91 (2H, d, J = 8.86), 6.85 (1H, s), 6.49 (2H, d, J = 2.11), 6.38 (1H, t, J = 2.11), 4.50 (2H, m, J = 6.07), 3.83 (3H, s), 3.80 (3H, s), 1.33 (12H, d, J = 6.07). 13C NMR (100 MHz, CDCl3) δ (ppm): 170.24, 159.71, 159.02, 137.48, 134.37, 129.44, 129.23, 127.52, 114.06, 107.71, 104.14, 69.92, 55.26, 52.17, 22.03, 21.90.
4.2.21. (Z)-methyl 3-(2-chloro-3,5-diisopropoxyphenyl)-2-(4-methoxyphenyl)acrylate (4o)
Sodium chlorite (66 mg, 0.73 mmol) in water (0.73 mL) was added drop-wise to a mixture of 4h (185 mg, 0.52 mmol) in acetonitrile (0.54 mL), monosodium phosphate (108 mg, 0.78 mmol) in water (0.25 mL) and 30% hydrogen peroxide (0.07 mL) at 0 °C. The reaction was then allowed to stir for 48 hours at room temperature before addition of a saturated solution of sodium thiosulfate. The aqueous phase was then extracted with ethyl acetate (3 × 5 mL). The combined organic layers were washed with brine, dried over magnesium sulfate, filtered and concentrated to afford an inseparable mixture of (Z)-3-(3,5-diisopropoxyphenyl)-2-(4-methoxyphenyl)acrylic acid and (Z)-3-(2-chloro-3,5-diisopropoxyphenyl)-2-(4-methoxyphenyl)acrylic acid in 81-88% yield.
The above mixture of acids was dissolved in methanol (2.33 mL) and a catalytic amount of concentrated sulfuric acid was added. The reaction mixture was then refluxed for 48 hours, cooled to room temperature and concentrated. The residue was taken up in ethyl acetate and water, and the aqueous layer was extracted with ethyl acetate (3 × 8 mL). The combined organic layers were washed with brine, dried over magnesium sulfate, filtered and concentrated to yield a mixture of 4o and 4n, which were separated by flash chromatography using 49% petroleum ether, 49% toluene, 1% t-butanol to yield 4o in 34% yield and 4n in 14% yield. 1H-NMR (400 MHz, CDCl3) δ (ppm): 7.42 (2H, d, J = 8.84), 7.15 (1H, s), 6.92 (2H, d, J = 8.84), 6.55 (1H, d, J = 2.63), 6.47 (1H, d, J = 2.63), 4.51 (1H, m, J = 6.03), 4.46 (1H, m, J = 6.05), 3.83 (3H, s), 3.71 (3H, s), 1.39 (6H, d, J = 6.05), 1.33 (6H, d, J = 6.03). 13C NMR (100 MHz, CDCl3) δ (ppm): 169.70, 159.88, 156.63, 154.38, 136.10, 135.85, 128.97, 127.97, 127.73, 115.50, 114.03, 106.95, 104.71, 71.99, 70.39, 55.27, 52.04, 21.97. ESIMS m/z 441 (M + Na). IR vmax 1726 (CO) cm-1.
4.2.22. (Z)-methyl 3-(2-chloro-3,5-dihydroxyphenyl)-2-(4-hydroxyphenyl)acrylate (4p)
A 1.0 M solution of boron tribromide in dichloromethane ((0.60 mL, 0.60 mmol) was added slowly to a solution of 4o (50 mg, 0.12 mmol) in dry dicholormethane (0.60 mL) at −78 °C. The reaction was gradually allowed to warm to room temperature. After six hours the reaction mixture was cooled to −78 °C and quenched with saturated sodium bicarbonate. The aqueous layer was extracted with ethyl acetate (3 × 5 mL) and the combined organic layers were washed with brine, dried over magnesium sulfate, filtered and concentrated. The pure product was obtained in 10% yield after flash chromatography using 50% ethyl acetate 50% hexanes. 1H-NMR (400 MHz, CD3OD) δ (ppm): 7.80 (1H, s), 6.94 (2H, d, J = 8.67), 6.69 (2H, d, J = 8.67), 6.26 (1H, d, J = 2.70), 5.84 (1H, d, J = 2.70). 13C NMR (100 MHz, CD3OD) δ (ppm): 170.02, 158.35, 157.17, 155.04, 137.90, 136.51, 135.48, 132.47, 128.87, 115.98, 112.88, 110.09, 104.76, 52.77.
4.2.23. (Z)-3-(3,5-dimethoxyphenyl)-2-(4-methoxyphenyl)acrylamide (4q)
A mixture of 2b (45 mg, 0.15 mmol), acetaldoxime (0.02 mL, 0.30 mmol), palladium acetate (3 mg, 0.02 mmol), and triphenylphosphine (8 mg, 0.03 mmol) in aqueous ethanol (0.5 mL, 1:4 water:ethanol) was heated to reflux for three hours under nitrogen atmosphere. The reaction mixture was diluted with ethanol, filtered through a celite pad, washed with dichloromethane, and the combined organic layers were concentrated. The pure product was obtained in 92% yield after purification by flash chromatography using 40% ethyl acetate 60% hexanes. 1H-NMR (300 MHz, CDCl3) δ (ppm): 7.34 (2H, d, J = 8.91), 6.79 (2H, d, J = 8.91), 6.70 (1H, s), 6.57 (2H, d, J = 2.06), 6.28 (1H, t, J = 2.06), 3.86 (2H, s), 3.71 (3H, s), 3.66 (6H, s). 13C NMR (100 MHz, CDCl3) δ (ppm): 173.13, 160.51, 159.62, 137.32, 137.24, 129.21, 127.30, 126.65, 113.92, 106.17, 100.24, 55.05.
4.2.24. (Z)-3-(3,5-dihydroxyphenyl)-2-(4-hydroxyphenyl)acrylamide (4r)
A 1.0 M solution of boron tribromide in dichloromethane ((0.94 mL, 0.94 mmol) was added slowly to a solution of 4q (50 mg, 0.13 mmol) in dry dicholormethane (0.60 mL) at −78 °C. The reaction was gradually allowed to warm to room temperature. After four hours the reaction mixture was cooled to −78 °C and quenched with saturated sodium bicarbonate. The aqueous layer was extracted with ethyl acetate (3 × 5 mL) and the combined organic layers were washed with brine, dried over magnesium sulfate, filtered and concentrated. The pure product was obtained in quantitative yield after flash chromatography using 60% ethyl acetate 40% hexanes. 1H-NMR (400 MHz, CD3OD) δ (ppm): 7.38(2H, d, J = 8.66), 6.79 (2H d, J = 8.66), 6.70 (1H, s), 6.52 (2H, s), 6.19 (1H, s). 13C NMR (100 MHz, CD3OD) δ (ppm): 175.86, 159.43, 158.82, 139.17, 138.47, 130.03, 128.45, 126.91, 116.43, 108.12, 103.31.
4.2.25. 3-(3,5-dimethoxyphenyl)-2-(4-methoxyphenyl)propan-1-amine (4s)
To a mixture of 4b (20 mg, 0.07 mmol) cobalt chloride hexahydrate (32 mg, 0.14 mmol) in anhydrous methanol (0.5 mL) was added sodium borohydride (26 mg, 0.68 mmol) portionwise. After stirring overnight a second portion of sodium borohydride was added. After one hour, an aqueous solution of 3N HCl was added to the reaction mixture until it because clear in appearance. The aqueous layer was then extracted with ether (3 ×) to remove any remaining nitrile. The aqueous layer was then made alkaline using ammonium hydroxide (pH = 10) and extracted with ethyl acetate (3 × 5 mL). The combined organic layers were washed with brine, dried over magnesium sulfate, filtered and concentrated. The pure product was obtained in 64% yield after purification by flash chromatography using 5% methanol 95% dichloromethane. 1H-NMR (400 MHz, CDCl3) δ (ppm): 7.09 (2H, d, J = 8.57), 6.84 (2H, d, J = 8.57), 6.26 (1H, t, J = 2.17), 6.21 (2H, d, J = 2.17), 3.78 (3H, s), 3.71 (6H, s), 2.96-2.81 (5H, m). 13C NMR (100 MHz, CDCl3) δ (ppm): 160.42, 158.22, 142.46, 134.57, 128.82, 113.89, 107.01, 97.93, 55.11, 49.88, 46.82, 41.15.
4.2.26. (Z)-methyl (2-(3,5-dimethoxyphenyl)-1-(4-methoxyphenyl)vinyl)carbamate (4t)
Potassium hydroxide (14 mg, 0.26 mmol) was added to 4q (32 mg, 0.10 mmol) in anhydrous methanol (1.25 mL) at room temperature under nitrogen atmosphere. Once a clear reaction mixture was obtained, (diacetoxyiodo)benzene (33 mg, 0.10 mmol) was added in one portion. The reaction mixture was concentrated after three hours and the crude residue as taken up in water and dichloromethane. The aqueous phase was extracted with dichloromethane (3 × 5 mL) and the combined organic layers were washed with brine, dried over magnesium sulfate, filtered and concentrated. The pure product was obtained in 86% yield after purification by flash chromatography using 30% ethyl acetate 70% hexanes. 1H-NMR (400 MHz, CDCl3) δ (ppm): 7.44 (2H, d, J = 8.80), 6.91 (2H, d, J = 8.80), 6.58 (2H, d, J = 2.20), 6.37 (1H, t, J = 2.20), 6.20 (1H, s), 3.83 (3H, s), 3.78 (6H, s), 3.67 (3H, s). 13C NMR (100 MHz, CDCl3) δ (ppm): 160.84, 159.85, 154.67, 137.66, 135.13, 130.56, 127.38, 118.19, 113.72, 106.30, 99.55, 55.20, 52.51.
4.2.27. 2-(3,5-dimethoxyphenyl)-1-(4-methoxyphenyl)ethanimine (4u)
A 1.0M solution of boron tribromide in dichloromethane (0.66 mL, 0.66 mmol) was added slowly to a solution of 4t (30 mg, 0.09 mmol) in dichloromethane (0.5 mL) at −78 °C under nitrogen atmosphere. The reaction was allowed to stir at room temperature for four hours before cooling to −78 °C and quenching with an aqueous solution of saturated sodium bicarbonate (2 mL). The reaction mixture was warmed to room temperature and the aqueous layer was extracted with ethyl acetate (3 × 2 mL). The combined organic layers were washed with saturated sodium bicarbonate (3 × 5 mL), water (1 × 5 mL), and brine (1 × 5 mL) before being dried over magnesium sulfate, filtered, and concentrated. The pure product was obtained in 77% yield after purification by flash chromatography using 30% ethyl acetate 70% hexanes. 1H-NMR (400 MHz, CDCl3) δ (ppm): 7.99 (2H, d, J = 8.97), 6.92 (2H, d, J = 8.97), 6.43 (2H, d, J = 2.26), 6.34 (1H, t, J = 2.26), 4.15 (2H, s), 3.85 (3H, s), 3.76 (6H, s). 13C NMR (100 MHz, CDCl3) δ (ppm): 195.94, 163.45, 160.83, 137.07, 130.90, 129.50, 113.70, 107.32, 98.76, 55.37, 55.20, 45.53.
4.2.28. General Benzanilide Synthesis, Method A
The benzoyl chloride was prepared by stirring benzoic acid (1.0 mmol) with SOCl2 (0.363 mL, 5.0 mmol) and DMF (1 drop) in CH2Cl2 (5 ml) for overnight and evaporating the volatile materials under reduced pressure. To the solution of benzoyl chloride in CH2Cl2 (5 mL) were added an aniline (1.0 mmol) and DIEA (0.871 mL, 5.0 mmol), and the reaction was stirred until no starting material by TLC (1~24 h) at room temperature. After dilution with CH2Cl2 (30 mL), the organic layer was washed with brine (40 mL), dried with anhydrous MgSO4, and evaporated to dryness under reduced pressure. The residue was purified by flash column chromatography to obtain a benzanilide.
4.2.29. General Benzanilide Synthesis, Method B
To the solution of benzoic acid (1.0 mmol) in CH2Cl2 (5 ml) were added EDC (192 mg, 1.0 mmol) and DIEA (0.523 mL, 3.0 mmol), and stirred for 5 min at room temperature. To the solution of activated benzoic acid was added an aniline (1.0 mmol) and the reaction was stirred for overnight at room temperature. After dilution with CH2Cl2 (35 mL), the organic layer was washed with brine (40 mL), dried with anhydrous MgSO4, and evaporated to dryness under reduced pressure. The residue was purified by flash column chromatography to obtain a benzanilide.
4.2.30. General Benzanilide Synthesis, Method C
To the solution of secondary amide (0.5 mmol) in THF (5 ml) were added NaH (25mg, 60 % in oil, 0.6 mmol) and stirred for 30 min at 0°C. MeI (0.062 mL, 1 mmol) was added dropwise to the solution, and the reaction was stirred for overnight at room temperature. After dilution with CH2Cl2 (40 mL), the organic layer was washed with brine (40 mL), dried with anhydrous MgSO4, and evaporated to dryness under reduced pressure. The residue was purified by flash column chromatography to obtain a benzanilide.
4.2.31. General Benzanilide Deprotection, Method D
If R1 or R2 were protected by TBS, that were deprotected before final column purification by TBAF treatment; the product was dissolved in THF (5mL), and TBAF (1.0 mmol per protecting group) was added and stirred for 20 min. After dilution with CH2Cl2 (30 mL), the organic layer was washed with brine (40 mL), dried with anhydrous MgSO4, and evaporated to dryness under reduced pressure.
4.2.32.7V-(3,5-Dihydroxyphenyl)-4-hydroxybenzamide (8a)
Method B and D were used in combination with 3,5-bis(tert-butyldimethylsilyloxy)benzoic acid and 4-(tert-butyldimethylsilyloxy)aniline (79 %). 1H-NMR (300 MHz, (CD3)2CO) δ (ppm): 7.69 (2H, d, J = 8.7), 7.04 (2H, d, J = 1.8), 6.91 (2H, d, J = 8.7), 6.62 (1H, t, J = 1.8). 13C NMR (75 MHz, (CD3)2CO) δ (ppm): 206.88, 166.03, 158.68, 154.13, 137.68, 131.11, 122.52, 115.25, 106.12. ESIMS(+) m/z 246 (M + H), 268 (M + Na), 284 (M + K).
4.2.33. 3,5-Dihydroxy-7V-(4-hydroxyphenyl)-7V-methylbenzamide (8b)
Method C and D were used starting from TBS protected form of 8a (38 %). 1H-NMR (300 MHz, (CD3)2CO) δ (ppm): 7.05 (2H, d, J = 8.1), 6.80 (2H, d, J = 8.1), 6.38 (2H, s), 6.32 (1H, s), 3.40 (3H, s). 13C NMR (75 MHz, (CD3)2CO) δ (ppm): 205.98, 157.95, 155.98, 138.69, 136.84, 128.13, 115.67, 107.13, 103.57, 37.96. ESIMS(+) m/z 260 (M + H), 282 (M + Na).
4.2.34. N-(4-Hydroxyphenyl)-3,5-dimethoxybenzamide (8c)
Method A and D were used in combination with 3,5-dimethoxybenzoic acid and 4-(tert-butyldimethylsilyloxy)aniline (90 %). 1H-NMR (300 MHz, (CD3)2CO) δ (ppm): 7.70 (2H, d, J = 8.7), 7.20 (2H, d, J = 2.4), 6.91 (2H, d, J = 8.7), 6.72 (1H, t, J = 2.4), 3.89 (6H, s). 13C NMR (75 MHz, (CD3)2CO) δ (ppm): 165.05, 161.01, 154.07, 137.68, 131.25, 122.32, 115.14, 105.31, 103.23, 55.06. ESIMS(+) m/z 274 (M + H), 296 (M + Na), 312 (M + K).
4.2.35. N-(3,5-Dihydroxyphenyl)-4-hydroxybenzamide (8d)
Method B and D were used in combination with 4-(tert-butyldimethylsilyloxy)benzoic acid and 3,5-bis(tert-butyldimethylsilyloxy)aniline (41 %). 1H-NMR (300 MHz, CD3OD) δ (ppm): 7.86 (2H, d, J = 9.0), 6.93 (2H, d, J = 9.0), 6.78 (2H, d, J = 1.8), 6.16 (1H, t, J = 1.8). 13C NMR (75 MHz, CD3OD) δ (ppm): 167.40, 160.89, 158.28, 140.20, 129.34, 125.78, 114.77, 99.75, 98.54. ESIMS(+) m/z 246 (M + H), 268 (M + Na), 284 (M + K).
4.2.36. N-(3,5-dihydroxyphenyl)-4-hydroxy-N-methylbenzamide (8e)
Method B and D were used in combination with 4-(tert-butyldimethylsilyloxy)benzoic acid and N-methyl 3,5-bis(tert-butyldimethylsilyloxy) aniline (11 %). 1H-NMR (300 MHz, CD3OD) δ (ppm): 7.31 (2H, d, J = 9.0), 6.69 (2H, d, J = 9.0), 6.19 (1H, t, J = 1.8), 6.12 (2H, d, J = 1.8), 3.45 (3H, s). 13C NMR (75 MHz, CD3OD) δ (ppm): 171.34, 159.24, 158.82, 146.61, 130.44, 126.39, 114.12, 105.48, 100.87, 37.74. ESIMS(+) m/z 260 (M + H), 282 (M + Na), 298 (M + K).
4.2.37. N-(4-(dimethylamino)phenyl)-4-hydroxybenzamide (8f)
Method B and D were used in combination with 4-(tert-butyldimethylsilyloxy)benzoic acid and 4-(N,N-dimethylamino)aniline (46 %). 1H-NMR (300 MHz, CD3OD) δ (ppm): 7.87 (2H, d, J = 9.6), 7.53 (2H, d, J = 9.6), 6.92 (2H, d, J = 8.7), 6.87 (2H, d, J = 8.7), 2.98 (6H, s). 13C NMR (75 MHz, CD3OD) δ (ppm): 167.18, 161.34, 148.35, 129.17, 128.64, 125.35, 122.72, 114.94, 113.09, 40.04. ESIMS(+) m/z 257 (M + H), 279 (M + Na), 295 (M + K).
4.2.38. N-(4-(dimethylamino)phenyl)-4-methoxy-N-methylbenzamide (8g)
Method C was used starting from 8f (12 %). 1H-NMR (300 MHz, CDCl3) δ (ppm): 7.34 (2H, d, J = 9), 6.96 (2H, d, J = 8.7), 6.73 (2H, d, J = 8.7), 6.61 (2H, d, J = 9), 3.78 (3H, s), 3.48 (3H, s), 2.96 (6H, s). 13C NMR (75 MHz, CDCl3) δ (ppm): 170.30, 160.28, 148.85, 134.51, 130.88, 128.47, 127.60, 112.89, 112.61, 55.20, 40.57, 38.94. ESIMS(+) m/z 285 (M + H), 307 (M + Na).
4.2.39. N-(4-(dimethylamino)phenyl)-3,5-dimethoxybenzamide (8h)
Method A was used in combination with 3,5-hydroxybenzoic acid and 4-(N,N-dimethylamino)aniline (9 %). 1H-NMR (300 MHz, (CD3)2SO) δ (ppm): 9.87 (1H, s), 9.60 (2H, s), 7.62 (2H, d, J = 8.7), 6.82 (2H, s), 6.76 (2H, d, J = 8.7), 6.41 (1H, s), 2.91 (6H, s). 13C NMR (75 MHz, (CD3)2SO) δ (ppm): 165.56, 158.79, 147.73, 138.02, 129.57, 122.18, 112.97, 106.18, 105.62, 40.26. ESIMS(+) m/z 273 (M + H), 295 (M + Na), 311 (M + K).
4.2.40. N-(4-(dimethylamino)phenyl)-3,5-dimethoxybenzamide (8i)
Method A was used in combination with 3,5-dimethoxybenzoic acid and 4-(N,N-dimethylamino)aniline (83 %). 1H-NMR (300 MHz, CDCl3) δ (ppm): 8.51 (1H, s), 7.54 (2H, d, J = 8.7), 7.03 (2H, d, J = 2.4), 6.71 (2H, d, J = 8.7), 6.58 (1H, t, J = 2.4), 3.77 (6H, s), 2.95 (6H, s). 13C NMR (75 MHz, CDCl3) δ (ppm): 165.78, 160.78, 148.09, 137.51, 127.97, 122.38, 112.95, 105.03, 103.71, 55.51, 40.91. ESIMS(+) m/z 301 (M + H), 323 (M + Na), 339 (M + K).
4.2.41. N-(4-(dimethylamino)phenyl)-3,5-dimethoxy-N-methylbenzamide (8j)
Method C was used from 8i (67 %). 1H-NMR (300 MHz, CDCl3) δ (ppm): 6.97 (2H, d, J = 9.0), 6.61 (2H, d, J = 9.0), 6.54 (2H, d, J = 1.8), 6.38 (1H, t, J = 1.8), 3.70 (6H, s), 3.49 (3H, s), 2.96 (6H, s). 13C NMR (75 MHz, CDCl3) δ (ppm): 170.44, 159.97, 149.10, 138.21, 133.94, 127.52, 112.64, 106.71, 102.28, 55.37, 40.60, 38.66. ESIMS(+) m/z 315 (M + H), 337 (M + Na), 353 (M + K).
4.2.42. N-(3,5-dihydroxyphenyl)-4-(dimethylamino)benzamide (8k)
Method A and D were used in combination with 4-(N,N-dimethylamino)benzoic acid and 3,5-bis(tert-butyldimethylsilyloxy)aniline (54 %). 1H-NMR (300 MHz, (CD3)2SO) δ (ppm): 7.88 (2H, d, J = 8.7), 6.80 (2H, s), 6.78 (2H, d, J = 8.7), 5.98 (1H, s), 3.04 (6H, s). 13C NMR (75 MHz, (CD3)2SO) δ (ppm): 165.56, 158.62, 152.78, 141.55, 129.62, 121.93, 111.23, 99.19, 98.29, 40.26. ESIMS(+) m/z 273 (M + H), 295 (M + Na), 311 (M + K).
4.2.43. N-(3,5-dihydroxyphenyl)-4-(dimethylamino)-N-methylbenzamide (8l)
Method A and D were used in combination with 4-(N,N-dimethylamino)benzoic acid and N-methyl-3,5-bis(tert-butyldimethylsilyloxy)aniline (69 %). 1H-NMR (300 MHz, CD3OD) δ (ppm): 7.34 (2H, d, J = 8.7), 6.53 (2H, d, J = 8.7), 6.21 (1H, t, J = 2.4), 6.16 (2H, d, J = 2.4), 3.42 (3H, s), 2.91 (6H, s). 13C NMR (75 MHz, CD3OD) δ (ppm): 171.82, 158.82, 151.80, 147.14, 130.41, 121.70, 110.33, 105.53, 100.82, 38.92, 38.07. ESIMS(+) m/z 287 (M + H), 309 (M + Na), 325 (M + K).
4.2.44. 4-(Dimethylamino)-N-(4-hydroxyphenyl)benzamide (8m)
Method A and D were used in combination with 4-(N,N-dimethylamino)benzoic acid and 4-(tert-butyldimethylsilyloxy)aniline (81 %). 1H-NMR (300 MHz, (CD3)2SO) δ (ppm): 7.91 (2H, d, J = 8.7), 7.59 (2H, d, J = 8.1), 6.80 (4H, d, J = 8.7), 3.05 (6H, s). 13C NMR (75 MHz, (CD3)2SO) δ (ppm): 165.24, 153.79, 152.70, 131.73, 129.45, 122.60, 121.93, 115.39, 111.29, 40.26. ESIMS(+) m/z 257 (M + H), 279 (M + Na), 295 (M + K).
4.2.45. 4-(Dimethylamino)-N-(4-hydroxyphenyl)-N-methylbenzamide (8n)
Method C and D was used from TBS protected 8m (51 %). 1H-NMR (300 MHz, (CD3)2SO) δ (ppm): 7.16 (2H, d, J = 8.7), 6.98 (2H, d, J = 9.0), 6.70 (2H, d, J = 8.7), 6.52 (2H, d, J = 9.0), 3.13 (3H, s), 2.92 (6H, s). 13C NMR (75 MHz, (CD3)2SO) δ (ppm): 169.85, 155.93, 151.21, 137.65, 130.86, 128.53, 122.97, 116.17, 110.76, 40.60, 40.32.
4.2.46. 3,5-dihydroxy-N-(naphthalen-2-yl)benzamide (8o)
Method A and D was used in combination with 3,5-bis(tert-butyldimethylsilyloxy)benzoic acid and 2-naphthyl amine (43 %). 1H-NMR (300 MHz, (CD3)2CO) δ (ppm): 8.57 (1H, s), 7.94-7.85 (4H, m), 7.52-7.40 (4H, m), 7.05 (2H, d, J = 1.8), 6.63 (1H, t, J = 1.8). 13C NMR (75 MHz, (CD3)2CO) δ (ppm): 166.28, 158.99, 137.90, 137.28, 134.25, 130.86, 128.55, 127.80, 126.62, 125.02, 120.97, 116.88, 106.38, 105.98. ESIMS(+) m/z 280 (M + H), 302 (M + Na), 318 (M + K).
4.2.47. 3,5-dimethoxy-N-(naphthalen-2-yl)benzamide (8p)
Method A was used in combination with 3,5-dimethoxybenzoic acid and 2-naphthlamine (87 %). 1H-NMR (300 MHz, CDCl3) δ (ppm): 8.31 (1H, s), 7.78-7.62 (4H, m), 7.46-7.38 (2H, m), 7.02 (2H, d, J = 2.4), 6.55 (1H, t, J = 2.4), 3.74 (6H, s). 13C NMR (75 MHz, CDCl3) δ (ppm): 137.15, 135.57, 133.86, 130.83, 128.75, 127.80, 127.63, 126.51, 125.19, 120.50, 117.44, 108.20, 107.13, 105.17, 103.91, 55.56. ESIMS(+) m/z 308 (M + H), 330 (M + Na), 346 (M + K).
4.2.48. 3,5-dimethoxy-N-methyl-N-(naphthalen-2-yl)benzamide (8q)
Method A was used in combination with 3,5-dimethoxybenzoic acid and N-methyl 2-naphthlamine (91 %). 1H-NMR (300 MHz, CDCl3) δ (ppm): 7.81-7.12 (3H, m), 7.56 (1H, d, J = 1.8), 7.49-7.46 (2H, m), 7.26 (1H, dd, J = 1.8, 8.7), 6.58 (2H, d, J = 2.4), 6.33 (1H, t, J = 2.4), 3.61 (3H, s), 3.57 (6H, s). 13C NMR (75 MHz, CDCl3) δ (ppm): 170.50, 160.14, 142.34, 137.71, 133.47, 131.64, 129.20, 127.74, 126.76, 126.34, 125.21, 124.74, 106.83, 102.39, 55.25, 38.66. ESIMS(+) m/z 322 (M + H), 344 (M + Na), 360 (M + K).
4.2.49. N-(3,5-dihydroxyphenyl)-2-naphthamide (8r)
Method A and D were used in combination with 2-naphtoic acid and 3,5-bis(tert-butyldimethylsilyloxy)aniline (61 %). 1H-NMR (300 MHz, CD3OD) δ (ppm): 8.51 (1H, s), 8.09-7.99 (4H, m), 7.66 (2H, m), 6.87 (2H, d, J = 2.1), 6.19 (1H, t, J = 2.1). 13C NMR (75 MHz, CD3OD) δ (ppm): 167.66, 158.42, 140.06, 134.96, 132.65, 132.32, 128.84, 128.11, 127.85, 127.66, 127.52, 126.59, 123.89, 99.92, 99.11. ESIMS(+) m/z 280 (M + H), 302 (M + Na), 318 (M + K).
4.2.50. N-(3,5-dihydroxyphenyl)-N-methyl-2-naphthamide (8s)
Method A and D were used in combination with 2-naphtoic acid and N-methyl 3,5-bis(tert-butyldimethylsilyloxy) aniline (32 %). 1H-NMR (300 MHz, (CD3)2CO) δ (ppm): 8.09 (1H, s), 7.91-7.87 (2H, m), 7.80 (1H, d, J = 8.7), 7.61-7.52 (3H, m), 6.32 (3H, s), 3.51 (3H, s). 13C NMR (75 MHz, (CD3)2CO) δ (ppm): 170.13, 158.99, 146.69, 134.09, 133.75, 132.54, 128.75, 128.64, 127.71, 127.29, 127.24, 126.56, 125.49, 106.07, 101.35, 37.88. ESIMS(+) m/z 294 (M + H), 316 (M + Na), 332 (M + K).
4.2.51. 3,4-Dihydroxy-N-(4-hydroxyphenyl)benzamide (8t)
Method B and D were used in combination with 3,4-bis(tert-butyldimethylsilyloxy)benzoic acid and 4-(tert-butyldimethylsilyloxy)aniline (26 %). 1H-NMR (300 MHz, CD3OD) δ (ppm): 7.47 (2H, d, J = 8.7), 7.45 (1H, d, J = 2.4), 7.22 (1H, dd, J = 2.4, 8.4), 6.91 (1H, d, J = 8.4), 6.85 (2H, d, J = 8.7). 13C NMR (75 MHz, CD3OD) δ (ppm): 167.41, 154.21, 148.94, 145.01, 130.38, 127.38, 126.22, 123.16, 119.54, 114.85, 114.63. ESIMS(+) m/z 246 (M + H), 268 (M + Na), 284 (M + K).
4.2.52. 3,4-Dihydroxy-N-(4-hydroxyphenyl)-N-methylbenzamide (8u)
Method C and D were used starting from 8t (58 %). 1H-NMR (300 MHz, CD3OD) δ (ppm): 6.99 (2H, d, J = 8.7), 6.86 (1H, d, J = 1.8), 6.75 (2H, d, J = 8.7), 6.71-6.61 (2H, m), 3.44 (3H, s). 13C NMR (75 MHz, CD3OD) δ (ppm): 171.87, 156.01, 147.00, 144.33, 136.78, 127.80, 127.01, 121.12, 115.89, 115.44, 113.98, 37.99. ESIMS(+) m/z 260 (M + H), 282 (M + Na).
4.2.53. (Z)-methyl N-(3,5-dihydroxyphenyl)-2-naphthimidate (8v)
Compound 8v was prepared by O-methylation of 8r; 8r (200 mg, 0.39 mmol) was stirred with SOCl2 (5 mL) for 3 h, and volatile materials were removed by evaporation under reduced pressure. MeOH (5 mL) was added to the residue and continued the stirring for 1 h. After evaporation, method D was used to deprotect TBS group and purified by flash column chromatography (ethyl acetate only) to obtain a benzanilide (41 mg, 36 %). 1H-NMR (300 MHz, (CD3)2SO) δ (ppm): 8.59 (1H, s), 8.15-8.02 (4H, m), 7.71-7.67 (2H, m), 6.87 (2H, d, J = 2.1), 6.05 (1H, t, J = 2.1), 3.42 (3H, s). 13C NMR (75 MHz, (CD3)2SO) δ (ppm): 207.05, 165.98, 158.79, 141.10, 134.70, 133.10, 132.57, 129.45, 128.39, 128.16, 127.32, 125.07, 99.33, 98.91, 31.22. ESIMS(+) m/z 332 (M + K).
4.2.54. N-(3,5-Dihydroxyphenyl)-3,4-dimethoxybenzamide (8w)
Method A and D were used in combination with 3,4-dimethoxy-aniline and 3,5-bis(tert-butyldimethylsilyloxy)benzoic acid (71 %). 1H-NMR (300 MHz, (CD3)2CO) δ (ppm): 7.65 (1H, dd, J = 1.8, 8.4), 7.62 (1H, d, J = 1.8), 7.08 (1H, d, J = 8.4), 7.03 (2H, d, J = 2.4), 6.22 (1H, t, J = 2.4), 3.93 (6H, s). 13C NMR (75 MHz, (CD3)2CO) δ (ppm): 165.30, 158.62, 152.30, 149.10, 141.13, 127.77, 120.72, 111.06, 110.78, 99.02, 98.32, 55.31. ESIMS(+) m/z 290 (M + H), 312 (M + Na), 328 (M + K).
4.2.55. N-(3,5-Dihydroxyphenyl)-3,4-dimethoxy-N-methylbenzamide (8x)
Method A and D were used in combination with 3,4-dimethoxy-benzoic acid and N-methyl 3,5-bis(tert-butyldimethylsilyloxy)aniline (64 %). 1H-NMR (300 MHz, CD3OD) δ (ppm): 7.14 (1H, d), 7.01 (1H, d), 6.86 (1H, d), 6.21 (1H, s), 6.15 (2H, s), 3.83 (3H, s), 3.70 (3H, s), 3.46 (3H, s). 13C NMR (75 MHz, CD3 OD) δ (ppm): 170.80, 159.04, 150.70, 147.95, 146.61, 127.71, 122.32, 112.22, 110.22, 105.51, 101.01, 54.92, 37.65. ESIMS(+) m/z 304 (M + H), 326 (M + Na), 342 (M + K).
4.2.56. 3,4-Dihydroxy-N-(naphthalen-2-yl)benzamide (8y)
Method B and D were used in combination with 3,4-bis(tert-butyldimethylsilyloxy)benzoic acid and 2-naphtyl amine (77 %). 1H-NMR (300 MHz, (CD3)2SO) δ (ppm): 8.30 (1H, d, J = 1.8), 7.88-7.45 (4H, m), 7.55-7.43 (4H, m), 6.96 (1H, d, J =8.1). 13C NMR (75 MHz, CD3OD) δ (ppm): 167.72, 149.24, 145.12, 136.22, 133.89, 130.86, 128.08, 127.26, 126.17, 126.06, 124.74, 120.95, 119.85, 117.63, 114.83, 114.63. ESIMS(+) m/z 318 (M + K).
4.2.57. 3,4-Dimethoxy-N-(naphthalen-2-yl)benzamide (8z)
Method A was used in combination with 3,4-dimethoxybenzoic acid and 2-naphtyl amine (99 %). 1H-NMR (300 MHz, CDCl3) δ (ppm): 7.76 (3H, m), 7.70 (1H, m), 7.55 (2H, m), 7.45 (2H, m), 6.65 (1H, d), 3.79 (3H, s), 3.76 (3H, s). 13C NMR (75 MHz, CDCl3) δ (ppm): 166.23, 151.94, 148.88, 135.94, 133.83, 130.74, 128.64, 127.71, 127.60, 127.26, 126.48, 125.13, 120.89, 120.33, 117.66, 110.70, 110.25, 55.87, 55.79.
4.3. Expression and purification of human QR2
Human QR2 was purified from 4L of Escherichia coli BL21(DE3) grown in Luria-Bertani medium supplemented with 100μg/mL ampicillin as previously described.23 The concentration of purified hQR2 was determined using BioRad Protein assay. QR2 was concentrated to 4mg/mL for all kinetic and crystallization assays.
4.4. Steady-state kinetic assays and IC50 value determination
The enzymatic activity of QR2 at steady-state was determined using two assays, where both utilized NMeH (N-methyldihydronicotinamide) as the co-substrate.23,35,36 In the first assay, menadione was used as the quinone substrate and QR2 activity was monitored by the decrease in absorbance of NMeH at 360 nm. The second assay utilized MTT ((3-(4,5-Dimethylthiazol-2-yl)-2,5-dipenyltetrazolium bromide)) as a quinone substrate and QR2 activity was monitored by the increase of the formazan product at 612 nm. Both assays were performed at 25°C on a BioTek Synergy H1 multimode microplate reader. Reactions were carried out in a 96-well plate to a final volume of 200 μL. Each assay mixture contained 12 nM QR2, NMeH (100μM for the first assay and 17.5 μM on the second assay), either 30 μM menadione in the first assay or 200 μM MTT in the second assay, in a reaction buffer containing 100 mM NaCl, 50 mM Tris (pH 7.5) and 0.1% Triton-X-100. Substrates, inhibitor and buffer were added to the wells, and the plate was shaken for 5 seconds using a Thermo LabSystems WellMix microtiter plate shaker at a setting of 9 to 10 to mix the assay components. With the microtiter plate still on the shaker (not shaking), the enzyme, and associated control with no enzyme, were rapidly pipetted into the appropriate wells using a multi-channel pipette, and the plate was immediately shaken for 5 s to mix the enzyme with reagents. The plate was immediately transferred to the plate reader and the change in absorbance was monitored until the reaction reached completion. Initial slopes of the reaction (ΔAbs/Δtime) were measured and were used to calculate initial rates of the reaction using a value of 7,060 M−1cm−1 as an extinction coefficient for NMeH and 11,300 M−1cm−1 as an extinction coefficient for MTT.
Inhibitors were tested at 100 μM and compounds that showed greater than 50% inhibition at 100 μM were assayed for their IC50 determination. The inhibition of QR2 activity by resveratrol analogs were determined using the same assay conditions described above except that inhibitor concentrations were varied from 0.2 to 100 μM. Assays at each inhibitor concentration were performed in triplicate and the average and standard deviations in the rates were used to determine the final IC50 values by calculating the percent inhibition at each inhibitor concentration verses the negative control with zero inhibitor. The percent inhibition data were then plotted as a function of inhibitor concentration [I] and the data were fit to the following equation using nonlinear regression:
All data were fit to the equation using the Enzyme Kinetics Module of the program SigmaPlot from SPSS Scientific. IC50 values are reported along with their standard error.
4.5. Crystallization and X-ray structure determination of human QR2 in complex with inhibitors
QR2 was crystallized using our previously described methods.23,35,36 Briefly, the hanging-drop, vapor-diffusion method was used by setting up drops adding 1 μL purified QR2 with 1 μL of reservoir solution that contained between 1.3 to 1.7 M ammonium sulfate, 0.1 M Bis-Tris buffer pH between 6.0 to 7.0, 0.1 M NaCl, 5 mM DTT and 12 μM FAD. Crystals were transferred from hanging-drops to a 10 μL drop of prepared with 9 μL reservoir solution and 1 μL stock solution of inhibitor (10 mM in 100% DMSO). Crystals were soaked for 24 hours, retrieved with a nylon loop, which was then swiped through the same mother-liquor solution supplemented with 20% glycerol. The crystals were flash-frozen by plunging into liquid nitrogen. Crystals were stored in shipping dewars containing liquid nitrogen until X-ray data collection.
All diffraction data were collected at 100K at the Life Sciences Collaborative Access Team (LS-CAT) at the Advanced Photon Source (APS) at Argonne National Laboratories. Crystals were transferred from shipping dewars and mounted on a goniostat under a stream of dry N2. X-ray data sets were collected using both MarMosaic 300 mm and 225 mm CCD detectors. QR2-inhibitor complexes were processed and scaled using the program HKL2000. The crystals belonged to the primitive orthorhombic space group P212121 with a dimer in the asymmetric unit.
The initial phases for QR2 in complex with the inhibitors were determined by molecular replacement using PHASER in the CCP4 suite.40 The search model used was the structure of human QR2 in complex with resveratrol (PDB 1SG0).17 Molecular library files and coordinates for the inhibitors were built using SKETCHER in CCP4. The inhibitors were manually built into density using the program COOT. Fourier maps were calculated and visualized using the program COOT,41 and the structures were refined using the program REFMAC. Water molecules were added manually to 2Fo-Fc density peaks that were greater than 3.0 σ. Iterative rounds of refinement were continued until Rwork and Rfree values reached their lowest values.42 At this point, TLS refinement was used by submitting the coordinates to the TLS server,43-45 and then two rounds of TLS and restrained refinement were performed in REFMAC to arrive at the final models, which were validated using MolProbity.46 Electron density maps presented in the figures were calculated using CCP4 and the figures were generated using the program PyMol.
Supplementary Material
Acknowledgments
Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). K.C.J. acknowledges support by the National Institute of Health Biophysics Training grant T32 GM008296 (NIH/GM). This research was supported in part by grants from the Walther Cancer Foundation (to A.D.M) and the NIH (NCI CA48112 to A.D.M.) A.D.M. and K.C.J. also acknowledge support from the Macromolecular Crystallography Shared Research Facility via the Purdue University Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Takaoka MJ. J Faculty Sci Hokkaido Imperial University. 1940;3:1. [Google Scholar]
- 2.Langcake P, Pryce RJ. Phytochemistry. 1977;16:1193. [Google Scholar]
- 3.Langcake P, Pryce RJ. Physiological Plant Pathology. 1976;9:77. [Google Scholar]
- 4.Burns J, Yokota T, Ashihara H, Lean MEJ, Crozier A. Journal of Agricultural and Food Chemistry. 2002;50:3337. doi: 10.1021/jf0112973. [DOI] [PubMed] [Google Scholar]
- 5.Baur JA, Sinclair DA. Nat Rev Drug Discov. 2006;5:493. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- 6.Fernandez-Mar MI, Mateos R, Garcia-Parrilla MC, Puertas B, Cantos-Villar E. Food Chem. 2012;130:797. [Google Scholar]
- 7.Siemann EH, Creasy LL. American Journal of Enology and Viticulture. 1992;43:49. [Google Scholar]
- 8.Renaud S, Delorgeril M. Lancet. 1992;339:1523. doi: 10.1016/0140-6736(92)91277-f. [DOI] [PubMed] [Google Scholar]
- 9.Jang MS, et al. Science. 1997;275:218. [Google Scholar]
- 10.Bradamante S, Barenghi L, Villa A. Cardiovasc Drug Rev. 2004;22:169. doi: 10.1111/j.1527-3466.2004.tb00139.x. [DOI] [PubMed] [Google Scholar]
- 11.Howitz KT, et al. Nature. 2003;425:191. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 12.Valenzano DR, et al. Curr Biol. 2006;16:296. doi: 10.1016/j.cub.2005.12.038. [DOI] [PubMed] [Google Scholar]
- 13.Wood JG, et al. Nature. 2004;430:686. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- 14.Jang M, et al. Science. 1997;275:218. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- 15.Subbaramaiah K, et al. In: Cancer Prevention: Novel Nutrient and Pharmaceutical Developments. Bradlow HL, Fishman J, Osborne MP, editors. Vol. 889. New York Acad Sciences; New York: 1999. pp. 214–223. [Google Scholar]
- 16.Holmes-McNary M, Baldwin AS. Cancer Res. 2000;60:3477. [PubMed] [Google Scholar]
- 17.Buryanovskyy L, et al. Biochemistry. 2004;43:11417. doi: 10.1021/bi049162o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Winger JA, Hantschel O, Superti-Furga G, Kuriyan J. BMC structural biology. 2009;9:7. doi: 10.1186/1472-6807-9-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vella F, Gilles FA, Delagrange P, Boutin JA. Biochem Pharmacol. 2005;71:1. doi: 10.1016/j.bcp.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 20.Celli CM, Tran N, Knox R, Jaiswal AK. Biochem Pharmacol. 2006;72:366. doi: 10.1016/j.bcp.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 21.Jamieson D, Tung ATY, Knox RJ, Boddy AV. Br J Cancer. 2006;95:1229. doi: 10.1038/sj.bjc.6603414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knox RJ, et al. Cancer Res. 2000;60:4179. [PubMed] [Google Scholar]
- 23.Maiti A, et al. SJournal of Medicinal Chemistry. 2009;52:1873. doi: 10.1021/jm801335z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calamini B, et al. Biochem J. 2010;429:273. doi: 10.1042/BJ20091857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boocock J, et al. J Chromatogr B. 2007;848:182. doi: 10.1016/j.jchromb.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Santi C, Pietrabissa A, Mosca F, Pacifici GM. Xenobiotica. 2000;30:1047. doi: 10.1080/00498250010002487. [DOI] [PubMed] [Google Scholar]
- 27.De Santi C, Pietrabissa A, Spisni R, Mosca F, Pacifici GM. Xenobiotica. 2000;30:857. doi: 10.1080/004982500433282. [DOI] [PubMed] [Google Scholar]
- 28.Kang SS, et al. Bioorganic & Medicinal Chemistry. 2009;17:1044. doi: 10.1016/j.bmc.2008.04.031. [DOI] [PubMed] [Google Scholar]
- 29.Meyers MJ, et al. Journal of Medicinal Chemistry. 2001;44:4230. doi: 10.1021/jm010254a. [DOI] [PubMed] [Google Scholar]
- 30.Knoevenagel E. Berichte der deutschen chemischen Gesellschaft. 1898;31:2596. [Google Scholar]
- 31.Lai G, Anderson WK. Synthetic Communications. 1997;27:1281. [Google Scholar]
- 32.Dalcanale E, Montanari F. The Journal of Organic Chemistry. 1986;51:567. [Google Scholar]
- 33.Kim ES, Lee HS, Kim JN. Tetrahedron Letters. 2009;50:6286. [Google Scholar]
- 34.Satoh T, Suzuki S, Suzuki Y, Miyaji Y, Imai Z. Tetrahedron Letters. 1969;10:4555. [Google Scholar]
- 35.Butler DCD, Alper H. Chemical Communications. 1998:2575. [Google Scholar]
- 36.Kondratyuk TP, et al. Molecular Nutrition & Food Research. 2011;55:1249. doi: 10.1002/mnfr.201100122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun B, et al. Bioorganic & Medicinal Chemistry. 2012;18:5352. doi: 10.1016/j.bmc.2010.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foster CE, Bianchet MA, Talalay P, Faig M, Amzel LM. Free Radical Biology and Medicine. 2000;29:241. doi: 10.1016/s0891-5849(00)00299-9. [DOI] [PubMed] [Google Scholar]
- 39.Zhou Z, et al. Biochemistry. 2003;42:1985. [Google Scholar]
- 40.C. C. P. N. CCP4. The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. 1994;D50:760. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 41.Emsley P, Cowtan K. Acta Crystallogr D Biol Crystallogr. 2004;60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 42.Brunger AT. Nature. 1992;355:472. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 43.Painter J, Merritt EA. Acta Crystallogr D Biol Crystallogr. 2005;61:465. doi: 10.1107/S0907444905001897. [DOI] [PubMed] [Google Scholar]
- 44.Painter J, Merritt EA. Acta Crystallogr D Biol Crystallogr. 2006;62:439. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 45.Painter J, Merritt EA. Journal of Applied Crystallography. 2006;39:109. [Google Scholar]
- 46.Chen VB, et al. Acta crystallographica Section D, Biological crystallography. 2010;66:12. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.