

Abstract
Optical biosensors based on surface plasmon resonance (SPR) in metallic thin films are currently standard tools for measuring molecular binding kinetics and affinities – an important task for biophysical studies and pharmaceutical development. Motivated by recent progress in the design and fabrication of metallic nanostructures, such as nanoparticles or nanoholes of various shapes, researchers have been pursuing a new generation of biosensors harnessing tailored plasmonic effects in these engineered nanostructures. Nanoplasmonic devices, while demanding nanofabrication, offer tunability with respect to sensor dimension and physical properties, thereby enabling novel biological interfacing opportunities and extreme miniaturization. Here we provide an integrated overview of refractometric biosensing with nanoplasmonic devices and highlight some recent examples of nanoplasmonic sensors capable of unique functions that are difficult to accomplish with conventional SPR. For example, since the local field strength and spatial distribution can be readily tuned by varying the shape and arrangement of nanostructures, biomolecular interactions can be controlled to occur in regions of high field strength. This may improve signal-to-noise and also enable sensing a small number of molecules. Furthermore, the nanoscale plasmonic sensor elements may, in combination with nanofabrication and materials-selective surface-modifications, make it possible to merge affinity biosensing with nanofluidic liquid handling.
Keywords: Optical biosensors, refractometric sensors, surface plasmon resonance, plasmonics, figure of merit, single molecule detection, enzyme-linked biosensing, site-specific chemistry, supported lipid bilayer, pore-spanning lipid membrane, nanoparticle, nanohole, optofluidics
Introduction
Optical imaging, sensing, and trapping instruments have been among essential tools for life sciences and biotechnology, as illustrated in the examples of laser confocal scanning microscopy, fluorescence-activated cell sorters, optical tweezers, and surface plasmon resonance (SPR). With the completion of the Human Genome Project, there is a tremendous demand to catalogue proteins and to map their complex networks of interactions with other proteins, lipids, carbohydrates, nucleic acids, and drug molecules in a quantitative manner. SPR instruments are surface-based optical biosensors that can measure the kinetics and affinities of these diverse biological interactions in real-time by monitoring the interfacial refractive index changes caused by molecular interactions without having to label the molecules. Over the past two decades, SPR has become the “gold standard” in quantifying molecular binding kinetics – a task that is becoming increasingly important in the field of proteomics, systems biology, and drug discovery - and there is an increasing demand to improve its sensitivity, functionality, throughput and the information content in the measured response.
This review article will discuss how the emerging field of nanoplasmonics may further improve this unique and commercially important sensing technology. Focus is put on real-time measurements utilizing refractometric detection,1 i.e. changes in the far-field optical properties induced by the local change in refractive index (RI) on the surface upon molecular binding. The ultimate goal of SPR sensing is real-time measurements of binding kinetics between molecules. It should be emphasized that the overall sensor performance is determined not only by the sensitivity of the plasmonic nanostructure, but also depends heavily on the biorecognition elements employed. Therefore, rather than narrowly focusing on the physics of nanoplasmonic sensors, this review will provide an integrated view on refractometric SPR biosensing technologies, including plasmon resonances in nanostructures, biorecognition elements and surface modification strategies, mass transport effects, optical instrumentation and noise reduction techniques, and various performance metrics. Some emphasis will also be given to techniques for systems designed to interface biology. Our goal is to compare conventional SPR instruments with emerging nanoplasmonic sensors, in particular localized surface plasmon resonance (LSPR) and nanohole sensors based on the extraordinary optical transmission (EOT) effect. Finally, we show some recent highlights in using nanoplasmonics to demonstrate sensing tasks that are not easy or impossible to perform using conventional SPR.
Plasmonic Nanostructures
With the main focus of this review being recent advantages within nanoplasmonic biosensing, we will here give a shortened overview of the history, physics and biosensor applications of plasmon resonances in nanostructures. Appropriate references, especially to review articles and books, are provided for further reading. We also give a brief discussion on how the sensitivity of a given nanostructure is (or should be) evaluated.
Nanoparticle plasmons
The bright colors of noble metal nanoparticles have been utilized historically for decorative purposes e.g. in glass2 and on ceramics.3 In addition, colloidal gold has long been associated with various health benefits4 and is now beginning to appear in more sensible applications in modern medicine.5
The quantitative physical description of the localized plasmons in gold nanoparticles was presented by Mie slightly over 100 years ago.7 An extension from spherical to spheroidal particles was given shortly after.8 Although these theories are complete analytical solutions to Maxwell’s equations (for homogenous isotropic materials), it is common to present simplified equations based on electrostatics.9 For an arbitrary ellipsoid, the polarizability is then:10
| (1) |
Here V = 4πR1R2R3/3 is the ellipsoid volume, ε(ω) is the complex dielectric function of the metal (dispersive by dependence of angular frequency ω) and n is the refractive index (RI) of the environment. The far field extinction and scattering cross sections are acquired from σext = k Im(α) and σsca = k4|α|2/[6π],11 where k = 2πn/λ is the wave vector (λ is the vacuum wavelength). Notably, the scattering scales with the square of volume V and absorption of light is the primary decay mechanism in smaller particles (tens of nm). The parameter L(R1,R2,R3) in Equation 1 is the shape-determined factor associated with a given ellipsoidal axis, so that each axis has its own dipolar resonance.11 (Each L ranges from zero to one and L1 + L2 + L3 = 1.). Equation 1 fails to describe particles that are comparable in size to the wavelength of light (typically >50 nm) due to retardation, higher order modes and radiative damping.9 However, analytical approximations are available that describe the dipolar resonance contribution to the spectrum. The most commonly used is the modified long wavelength approximation, which defines a more accurate polarizability as a function of the quasistatic polarizability: 12,13
| (2) |
Here Rj is the radius of the ellipsoid dipole axis. Equation 2 gives excellent agreement with experimental data, even in absolute extinction magnitude, when used to model the spectrum of e.g. nanodisks14 (oblate spheroids). Further, there are analytical models that take into account the presence of a shell coating,11 which is particularly interesting for modeling the spectral changes induced by molecular binding in biosensing experiments. Also, the presence of a solid support, which is often included in biosensing applications, can be accounted for15 simply by modifying the value of L and making it depend on the RI of the solid support.16
Most plasmonic biosensors based on nanoparticles utilize spheres,17-20 rods,21-23 disks24 or shells25 and can thus be modeled analytically as described above. However, when dealing with particle shapes that deviate too much from ellipsoids, such as triangles,26 rings,27 octahedrons28 or “stars”,29 one is forced to use numerical approaches. This is also the case for more complex nanostructures containing particles in close proximity, in which case the individual resonances hybridize and produce new modes. There has been a recent interest in so-called Fano resonances appearing in such structures,30 not the least for refractometric sensing.31
In the beginning of the 1900’s, colloidal gold was used for diagnostics of cerebrospinal fluid.32,33 The first refractometric biosensing experiments with suspended nanoparticles were reported by Englebienne et al. in 1998.17 One year earlier, Mirkin’s group had showed the first assay based on plasmonic coupling between suspended nanoparticles.34 In the following years, many papers appeared where various nanoparticles on surface supports were used for refractometric detection, e.g. from the groups of van Duyne26 (triangular silver particles), Chilkoti18 (immobilized colloidal gold), Höök35 (quantification of LSPR response), and Rubinstein36 (gold “islands”).
Surface plasmons
The most important work for understanding the nature of surface plasmons is arguably what was presented in the middle of the 20th century by Ritchie,37 who used Maxwell’s equations to show that an electromagnetic wave (with transverse magnetic polarization) can exist and be confined to an interface between a metal and a dielectric. The concept of surface plasmons was used to explain the appearance of additional resonances (besides the metal bulk plasma frequency) in electron energy loss spectroscopy of thin metal foils.38
The fundamental difference between propagating surface plasmons and nanoparticle plasmons (often referred to as localized surface plasmons) is that they appear in a continuum of frequencies. Propagating surface waves carry a momentum and are described by a dispersion relation. For the simplest case with a single planar metal-dielectric interface (semi-infinite materials) one has:39
| (3) |
Here kx is the (complex) in-plane wavevector of the surface plasmon and c is the speed of light in vacuum. Excitation of surface plasmons by light requires matching both the frequency (energy) and wavevector(momentum) of incident photons, which is impossible under normal circumstances since the dispersions do not cross without additional photon momentum.40 The k of incident photons is normally increased either utilizing total internal reflection and a thin metal film (Kretchmann configuration for SPR; Figure 2) or by a grating on the metal surface.
Figure 2.
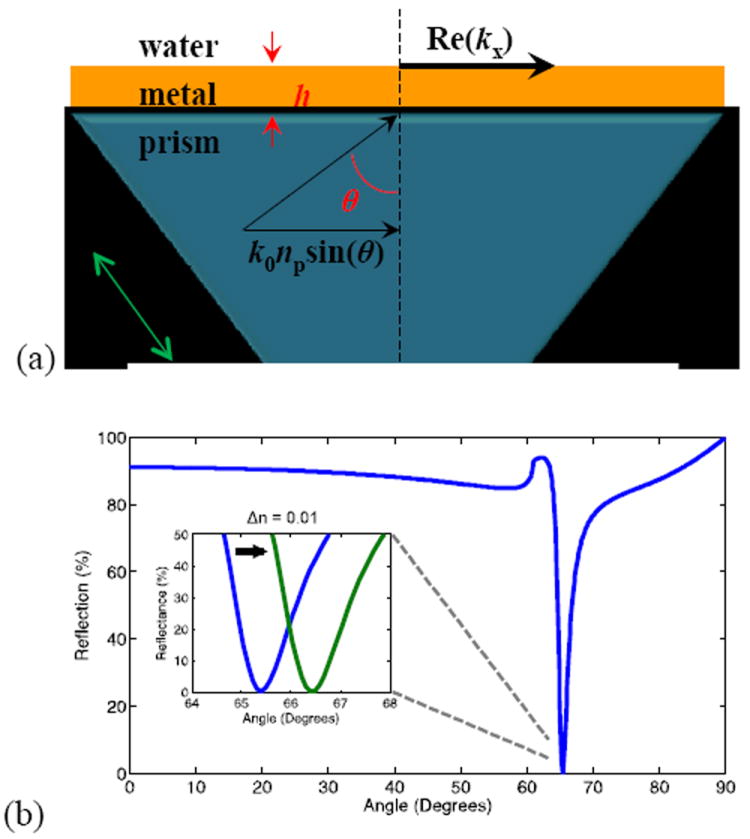
(a) A prism-coupler setup (also known as Kretschmann setup) for the excitation of SPR in thin metallic films. (b) Coupling to surface plasmons is detected by a sharp reduction in reflectance at a certain excitation angle. Shown here is the simulated reflection of a 50 nm-thick gold film on a glass substrate as a function of incident angle for 850 nm illumination. By changing the refractive index of the solution by Δn = 0.01, the resonance (dip) shifts by 1° in this example, corresponding to the sensitivity of 100°/(refractive index unit). From Lindquist et al.41
Importantly, since an optically thin film or a periodically patterned nanostructure is required for SPR, the dispersion relation in Equation 3 is always merely an approximation. A more accurate calculation of the resonance condition requires that the dispersion is modified such that it accounts for the fact that the metal film is finite in thickness (prism coupling in total internal reflection) or that scattering occurs at the structured surface (grating coupling). A convenient way to calculate the full spectrum in reflection spectroscopy is by Fresnel calculations using the transfer matrix method.1
The first utilization of surface plasmons in biosensing came in 1982 from Liedberg et al.42 and today surface plasmon resonance (SPR) is the primary established (and commercialized) method for quantitative real-time analysis of biomolecular interactions, generating thousands of papers every year.43 Lately, more sophisticated SPR sensors have been developed, based on phase detection44,45 or long-range surface plasmons.46,47
Nanohole arrays
The third of the most common type of nanostrucures used in plasmonic biosensors is nanoholes in thin metal films (Fig. 3).48-50 Considering first the case of a single hole, one can use electrostatic theory to show that a void in a metal should have a resonant polarizability quite similar to a nanoparticle (Fig. 3a).10 Further analogies between holes and particles were predicted already in 1954 by Bethe, who calculated the effective scattering cross section of a circular aperture in an infinitely thin perfect conductor.51 There seems to be a general consensus that single holes exhibit localized resonances similar to nanoparticles, i.e. high sensitivity to shape changes and polarization.52 The optical properties of arrays of nanoholes have been intensely studied since Ebbesen observed resonant transmission through square arrays in opaque gold and silver films.53
Figure 3.
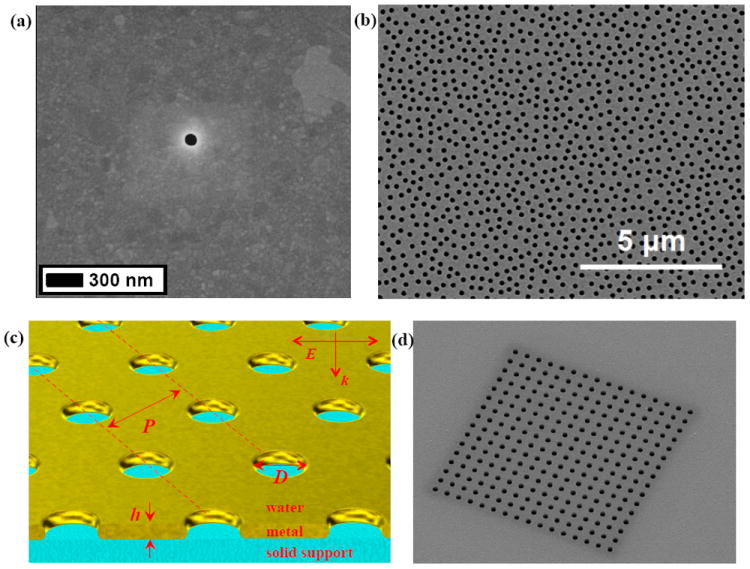
Metallic films perforated with nanoholes can support both propagating surface plasmons as well as LSPR-type resonances in the voids. (a) An isolated hole (100 nm in diameter) milled with FIB in a silver film. Image courtesy of Nathan Lindquist. (b) A random hole array in a metal film produced by colloidal lithography. (c) Schematic of periodic hole arrays in a metal film with a solid support. (d) A periodic array of nanoholes (200 nm diameter; 600 nm periodicity) milled with FIB in a gold film. Image courtesy of Hyungsoon Im.
In arrays of nanoholes (Fig. 3 b and d) one faces two principle types of resonances that couple to each other. One is the localized (particle-like) modes associated with individual holes and the other is surface plasmon excitation induced by the periodicity of holes in an array. Interestingly, short-range order in the arrays is sufficient for an SPR effect, i.e. structural correlation over longer distance (microns) is not necessary.54-56 For the case of coupling to surface plasmons at normal incidence using nanoholes, the resonance condition is simply:
| (4) |
This expression simply illustrates that the periodicity P of the array should equal a multiple of the surface plasmon wavelength. For short-range ordered arrays, P is simply the characteristic spacing between holes.55 The challenge lies in coming up with an accurate expression for the dispersion relation so that Equation 4 actually can be used to predict the resonance. In particular, one must account for the fact that the metal now contains holes. This leads to a redshift in the measured resonance, which can be understood as an effectively reduced plasma frequency (electron density) of the metal upon perforation.55 The situation is overall more complicated in optically thin films since they show significant “ordinary” transmission of light and strong hybridization between the surface plasmons at either interface.55,57 Also, a thin film prevents Fabry-Pérot type modes that propagate through individual holes (perpendicular to the surface).
Although there are well-known58,59 and extensive60,61 reviews on the optical properties of nanohole arrays, inconsistencies seem to remain in the literature still. For instance, in most work on >100 nm films the transmission maximum is associated with the surface plasmon, as originally suggested.53 However, it is also known that this approach does not provide accurate predictions of the resonance wavelength. There are theories that can explain this offset,62 but it has also been shown that the predicted resonance for various arrays agrees well with an extinction maximum.56 For the case of thinner films (10-50 nm) it seems quite clear that the extinction peak indeed corresponds to surface plasmon excitation.55,63 Even for single holes, some controversy on the optical properties remains since it has been suggested that they exhibit no localized resonances, but enable direct coupling to surface plasmons.57 Although a detailed physical description of the optical properties of nanohole arrays is not critical in many sensing situations, it becomes important when attempts are made to quantify the response in terms of bound mass, in the analysis of binding kinetics or if the performance of the sensor must be improved.
The first reports on biosensing through RI changes with nanohole arrays appeared in the early 2000’s. Brolo et al. utilized resonant transmission through long-range ordered arrays in thick (100 nm) Au films,64 while Dahlin et al. used short-range ordered nanoholes in thin (20 nm) Au films.65 The Käll group presented the first biosensing experiments on single holes.66
Sensitivity evaluation
How can one evaluate whether a plasmonic nanostructure, including a thin planar metal film for Kretschmann SPR, would be suitable for biosensing? This depends first of all on the transduction mechanism. For example, a sensor based on particle-particle coupling67 needs to have the biorecognition event cause a relatively large change in the distance between the metal particles. We focus here on how to evaluate the sensitivity in terms of refractometric detection, i.e. signal generation caused by a change in interfacial refractive index induced by a biomolecular recognition reaction.
The research field of nanoplasmonic sensors emerged in synergy with advances in nanofabrication.41 There was a great interest in testing new fabricating methods with the aim of finding “more sensitive” nanostructures, as exemplified above. This is still the case and now more sophisticated spectroscopy techniques such as phase detection are utilized as well.44,68 Naturally, one must consider carefully how the sensing capabilities should be evaluated, so that a sensor that has a higher “sensitivity” actually provides a better detection limit when employed for biomolecular sensing (although the actual resolution will, in the end, also depend on the instrumentation used for spectroscopy). Most early papers focused on the resonance shift per change in RI of the liquid environment. This is logical since our physical understanding of all refractometric plasmonic sensors is that the resonance shifts to lower energy (longer wavelength) when the RI of the environment is increased. Later, it became more common to use the “figure of merit (FOM)”, which (usually) means the peak-shift sensitivity divided by the peak width. While the FOM serves as a convenient metric to evaluate the performance of refractometric sensors, this parameter alone may not accurately predict the detection limit for real biomolecular sensing. For example, while a prism-based Kretschmann SPR instrument has a broader line width than optimized nanoplasmonic sensors, it still has a better detection limit in terms of molecular surface coverage (commercial Biacore™ instruments can go down to 0.01 ng/cm2) than nanoplasmonic sensors. With proper curve fitting algorithms and noise reduction schemes, a very small spectral shift (~0.001 nm) from a relatively broad SPR dip can be readily resolved.69 In terms of surface coverage, this corresponds to ~0.1 ng/cm2. On the other hand, if a sharp peak is obtained at the expense of reduced peak intensity, the overall performance may actually degrade. A paper from the Masson group suggested that the magnitude of the peak should be taken into account.70 For SPR kinetic measurements of rapid association or dissociation kinetics between molecules, one needs high temporal resolution and thus high photon flux, which also implies that the intensity of the peak or dip is of great importance. Rather than using the FOM defined above, one could argue that the minimum detectable RI change, or sensor resolution, may be a more realistic metric for SPR and nanoplasmonic sensors. Naturally, such a performance parameter also takes into account the quality of the optical components and the experimental setup (see below). However, a high resolution for bulk refractive index changes does not always translate into an improved detection limit for surface-bound molecules. For example, with a temperature control and mechanical stabilization, the resolution of prism-based SPR is approaching 10-7 refractive index units.71 While LSPR sensors cannot match such high refractive index resolution, they could show relatively similar performance for detecting surface-bound molecules.
As illustrated here, the discussion on how to evaluate sensitivity continues. Dahlin has presented detailed discussions on the topic,1,72 suggesting that the refractometric sensitivity is best defined in terms of relative intensity changes (e.g. extinction, transmission) per RI increase. In brief, the primary argument behind this conclusion is that a change in light intensity is the one and only thing that can be measured by a photodetector and thus it forms the basis for the sensor output. The same idea has been indicated in a few other papers that used a normalized intensity to evaluate the refractometric sensitivity.20,73 From this point of view, peaks that shift more, are narrow and strong in magnitude are preferable simply because they tend to give rise to higher intensity changes in the photodetector. Das et al. compared the signal-to-noise performance of different analysis methods for nanohole SPR sensors.74
Regardless of which parameter is chosen to represent refractometric sensing performance, one must also consider the extension of the plasmonic field into the liquid environment. It is widely accepted that nanoplasmonic sensors, especially those utilizing LSPR, have a shorter field extension (tens of nm) compared to Kretschmann SPR (hundreds of nm), making them more suitable for surface constructs consisting of biomolecular monolayers.75 However, in most SPR systems a 3D dextran matrix coating is used, which makes it possible to utilize the whole probing volume and enhance the signal.76 A recent study showed that for gold nanoislands, the field extension scaled approximately linearly with the liquid bulk sensitivity in terms of resonance shift per RI change.77 This suggests that as long as one works with biomolecular layers smaller than the field extension, the signal will be roughly the same regardless of the choice of nanoparticle.
Fabrication Techniques for Nanoplasmonic Sensors
In contrast to Kretschmann SPR, which works with simple flat gold films, nanoplasmonic substrates must be produced via top-down nanofabrication or bottom-up synthesis to pattern holes, particles, slits, etc. Recent advances in fabrication and synthesis techniques for nanoplasmonic structures have resulted in improved throughput, shape control, reproducibility and optical properties. For example, earlier work on studying EOT effects mainly relied on focused ion beam (FIB) milling53 to pattern periodic hole arrays typically at the scale of tens of microns, due to the low throughput of those serial writing techniques. While colloidal lithography has been used for large-area patterning of short-ranged ordered nanoholes,78 it is difficult, although possible, to make long-range periodic hole arrays.50,79 The growing interest in using metallic nanoholes for sensing and other applications has motivated researchers to develop practical routes to fabricate large-area (~mm to cm) periodic nanohole arrays, such as interference lithography80-82, nanoimprint lithography, or template stripping.83-85 Lindquist et al. summarized various top-down fabrication methods for making nanoplasmonic structures in a recent article.41
The optical performance of nanoplasmonic devices depends on various factors such as surface roughness, crystallinity,14 morphology (grain size and grain boundaries), and dielectric functions of the metal itself. One key advantage of nanoparticle sensors made with colloidal synthesis is that they are single-crystalline in nature, which reduces plasmon damping due to electron scattering at grain boundaries. In contrast, metal films deposited by conventional evaporation or sputtering are polycrystalline, and thus exhibit roughness. Unfortunately, tightly confined SP waves are not only sensitive to surface-bound analytes, but also to unwanted surface roughness, grain boundaries, and surface contaminants. Indeed it was shown than reducing the roughness from ~5 nm to sub-1-nm in a Ag film increased the plasmon propagation length by 3-5× at visible wavelengths.83 To minimize surface roughness and improve the dielectric functions of metal films, groups have shown techniques such as template stripping and the growth of single-crystalline metal films.86
Plasmon Spectroscopy for High-Resolution Biosensing
Experimental setup
What are the pros and cons of nanoplasmonics compared to SPR in terms of simplicity when performing spectroscopy? When the research field of nanoplasmonics started up, it was commonly claimed that one advantage is the possibility to be able to perform simpler spectroscopy, in transmission mode instead of the Kretschmann configuration.36,64 This has some truth because “ordinary” spectrophotometers, found in practically every molecular biology lab, can then be used in the sensing experiments.20,36,64,65 In terms of components needed for the experimental setup, the Kretschmann SPR is no more complicated. Homola’s group has presented simple and compact prism-based SPR sensors with high performance.87,88 However, performing transmission-mode spectroscopy (e.g. using nanohole arrays) in collinear setup offers some practical advantages, since it is more forgiving to a sample misalignment or tilt compared with the Kretschmann setup. Furthermore, as Tetz et al. pointed out,80 wide-field SPR imaging with a high-numerical-aperture (NA) lens, which is desirable to gather molecular binding kinetics from a dense microarray of sensing elements, is difficult to perform with the Kretschmann setup because of the limited depth of focus caused by the prism and oblique illumination. Wide-field, high-NA SPR imaging is considerably easier to perform with nanohole arrays,89 since it can operate with normal illumination and collection with high-NA imaging optics.
On the other hand, it should be noted that reflection-mode operation can also offer several advantages. Most notably, it simplifies measurements on opaque liquids and the top sensor surface is also freely accessible for integration with other analytical tools that may block optical paths in a transmission measurement setup. Recent work has now introduced reflection-mode measurements also for nanoplasmonic sensors.90,91 Using template-stripping methods, backside reflection-mode nanoplasmonic sensor has also been demonstrated,92 wherein the optical paths and fluidic paths are decoupled as with conventional SPR.
In summary, a key advantage of the prism-based Kretschmann setup is the fact that there is no need for lithographic patterning (the metal structure is just a thin film), while its oblique illumination optics puts some restrictions on the optical system. Nanoplasmonic sensors put more burdens on the chip fabrication, but have potential to simplify optical systems design and allows one to tailor the location and width of plasmon resonance peaks by patterned geometries. These pros and cons should be considered when choosing nanoplasmonics vs. conventional SPR for a given application.
Noise minimization
We will here mention some simple methods for eliminating the most common types of noise in optical spectroscopy. In any sensor, the limit of detection is eventually determined by not only the sensitivity of the plasmonic nanostructure, but also the instrumentation used for reducing noise. Judging from the literature, relatively little effort is spent on noise reduction compared to chasing high sensitivity. Obviously, the experimental setup must be mechanically stable, especially when measuring on smaller samples.24,93 Temperature control is normally more important for SPR because of the high sensitivity to changes in the liquid bulk RI. For instance, the RI of water decreases with 8×10-5 per K (at room temperature), suggesting that the temperature needs to be controlled almost down to 0.01K to maintain instrumental resolution. Although spectral changes can be induced by temperature changes in the dielectric environment, the temperature of the metal may be even more important for stability94 if it is strongly heated by the probing light.95 Another possible reason for instability is the light source. Quite high resolution (surface coverage of 0.1ng/cm2)) can be reached without the need of updating the reference spectrum,69 but continuous updates of the source intensity may be needed when pursuing even lower noise.44
For a stable experimental setup in a reasonably well-controlled environment, one can expect that most of the noise originates from the photodetector. There are three types of false electrons generated in the detector that do not originate from photons that have interacted with the sensor. First, thermal electrons are always present, but their contribution can be greatly reduced by cooling the detector. Second, there are electrons generated in the readout process. If significant, this noise can be reduced by gain in the detector. Third, there can be background light reaching the detector. (The solution is to work in the dark.) However, all these types of detector noise become insignificant if the probing intensity is high enough to dominate the photodetector response. It is then possible to reach the shot-noise limit, as can be verified by looking at the noise characteristics.24 Since shot-noise is part of the nature of light it cannot be truly eliminated, but its contribution to the overall noise can be reduced, again by operating at high intensity and by using photodetectors with high dynamic range.69,96
Multiplexed operation
It is often preferable to screen multiple biomolecular interactions or detect the presence of several analytes in a sample simultaneously. A small degree of multiplexing with particles in solution can be achieved by measuring the spectrum of a mixture of nanorods with different aspect ratio (L in Equation 1), so that the spectral peaks are reasonably well separated.21 However, normally multiplexing is achieved by operation in imaging mode on the surface, i.e. dispersive information is replaced with information on spatial intensity variations. If the full spectrum (not just one intensity value) needs to be acquired from several locations on the surface simultaneously, or from a linear array of parallel microfluidic channels, one can use 1D spectral imaging.97,98 Using some “tricks”, it is also possible to acquire the full scattering spectrum of well-separated individual nanoparticles (on a 2D surface) in parallel.99 For SPR, operation in imaging mode for multiplexing is an established technique40 and does not necessarily result in loss in resolution.87 Also for nanohole arrays82,89,100-102 and dense nanoparticle samples,103 imaging operation for multiplexed biosensing has been introduced.
Scattering spectroscopy
When performing spectroscopy in transmission or reflection mode, the intensity reaching the photodetector consists of light which has not coupled to any plasmon. An alternative technique is to only detect light which originates from radiative decay of plasmons, i.e. scattering spectroscopy. This requires a dark background and the technique is well suited for imaging plasmonic nanoparticles. Establishing dark-field illumination is relatively straightforward, but often requires that a significant amount of the excitation light is blocked in order to avoid direct transmission to the collection optics. One way to avoid this loss in intensity is to utilize total internal reflection for generating the dark field.104 Dahlin et al. have addressed the question in which situations scattering spectroscopy provides better signal to noise than operation in transmission mode,24 suggesting that scattering spectroscopy is preferable only when measuring on single nanoparticles.
Highlights in Nanoplasmonic Sensing
Single molecule resolution
One early major driving force for the development of nanoparticle-based LSPR sensors is the possibility to perform spectroscopy on single nanoparticles,19,22,23,25,29,105 which clearly represents an extremely small (~50 nm or below) sensor that cannot be easily constructed using diffraction-limited dielectric components. In fact, even a sensor based on propagating surface plasmons would never enable this degree of miniaturization due to limitations on how well light can be focused and the propagation length of the plasmons (several μm). It was shown in relatively early papers that the signal to noise in real-time measurements on light scattering from single particles was almost sufficient for resolving single molecules (typically a protein) binding to the particle.22,106 Enzymatic precipitation has been used to amplify the response post-binding, and it was argued that such an assay could provide single molecule detection although not in real-time.107 By enhancing the illumination intensity (white light laser in total internal reflection) and optimizing nanoparticle geometry, Sönnichsen’s group recently reported resolving single relatively large protein molecules (fibronectin, 450 kDa) adsorbing on a gold nanorod.104
The real-time detection of individual protein molecules binding to receptors on the nanoparticle surface, i.e. an operational biosensor, was recently shown by Orrit and coworkers.108 Not surprisingly, this required a significantly more sophisticated spectroscopy technique based on photothermal microscopy.109 In this method, two laser sources illuminate the nanoparticle. One beam is used for heating the nanoparticle (absorption, i.e. σabs) while the other beam is used as a probe for measuring the thermal signal. Upon biomolecular binding to the nanorod, the plasmon resonance shifts as usual. However, instead of attempting to detect the resonance shift directly through scattering spectroscopy, the fact that the absorption (and thus the temperature) is changed was utilized and single binding events could visualized in the thermal signal.108 As mentioned above, since absorption scales with the volume of the nanoparticle (while σsca scales with V2), thermal microscopy enables the use of very small nanoparticles109 (31×9 nm), which provide an extremely confined probing volume suitable for enhancing the response from a single molecule.
Although single-molecule resolution can be considered a highly impressive achievement by most standards, one should also ask the question in which sensing applications it would be relevant to see individual binding events. It is important to note that all molecules are not resolved individually unless they bind in the highly sensitive regions. Therefore, the surroundings must be perfectly passive and the molecules efficiently guided to the nanoparticle if the sensor should be useful in applications where the sample solution truly only contain a few target molecules.110 Further, it can be argued that this is a very rare scenario unless the content of single cells should be analyzed.111 In most samples of interest for biosensors, the number of molecules available is very high, even for what is normally considered low concentrations. This means that the challenge normally lies in detecting a low surface coverage rather than a low number of molecules and a high degree of sensor miniaturization tends to result in a worse detection limit in terms of surface coverage.111,112 However, the field has reached a state today which means that one can envision a device composed of an array of nanoplasmonic sensors that are imaged individually and in parallel, each of which capable of resolving single protein binding events. If the total number of nanoplasmonic particles is sufficiently large, the lowest detectable coverage could be extremely low, thus offering competitive limits of detection. One significant benefit of such a system is that one can envision, in analogy with single-molecule fluorescence imaging,113 that kinetic rate constants can be extracted at equilibrium binding conditions, i.e. without careful control of liquid injection and rinsing. This is possible because binding and dissociation rate constants can be obtained from the number of new binding events per time unite and the residence time of each binding event, respectively. With high time resolution, one would also be able to extract this type of information for weakly interacting components present at low concentrations, as also verified using fluorescence imaging.114
Site-Specific Surface Chemistry on Nanoplasmonic Sensors
In contrast to flat gold films used in conventional SPR sensors, patterned nanoplasmonic surfaces exhibit unique geometries (e.g. holes, tips, edges) and often a mixture of heterogeneous materials. This opens up some interesting options toward unique surface modification schemes that can enhance the utility of nanoplasmonic devices. Because refractometric sensors probe their immediate local refractive index irrespective of the identity of the molecules, the surface of the sensor must be prudently designed to detect only molecules of interest. In other words, refractometric sensors are inherently nonselective, and the analytical performance of an otherwise highly sensitive sensor can be significantly reduced by poor surface preparation. However, this requirement is not restricted to nanoplasmonic sensors; it holds for more traditional SPR approaches as well and surface-sensitive techniques in general. Most SPR and nanoplasmonic sensors employ noble metals, such as gold and silver, therefore the use of well-established thiol (-SH) chemistry to immobilize receptors is by far the most common method. When a gold or silver film is introduced into an thiol solution, the thiol groups form covalent bonds with the metal surface and after some time a well ordered self-assembled monolayer (SAM) is formed.115 A wide variety of thiols, with varying lengths, degrees of saturation and terminal groups are available commercially. Selecting a thiol with a terminal group compatible with further functionalization, such as a carboxylate or amine, allows the use of well-known conjugation schemes to immobilize proteins, peptides, small organic molecules, nucleotides and carbohydrates, preferably in plasmonically active zones.
As has been previously mentioned, plasmonic nanostructures exhibit a spatially nonuniform electromagnetic field distribution. One advantage afforded by nanoplasmonic sensors is the ability to selectively immobilize receptors at sites where the electromagnetic field is particularly strong; so-called “hot spots” such as inside nanogaps and at sharp tips and edges. In fact, when molecules bind to nanoplasmonic structures, most of the change in optical readout is due to binding at these hot spots, and the contribution to the signal change from binding elsewhere is lower. In sensing applications where equilibrium is not established, e.g. due to irreversible interactions, mass transport limitations or brief exposures to the sample solution, the sensor response can be enhanced by directing molecules to more sensitive regions.
One method to immobilize binding receptors at sensitive hot spots is through the use of thiol exchange chemistry. Beeram and Zamborini used a “place-exchange” strategy to selectively immobilize antibodies on the edges of triangular Au nanoplates, then used these functionalized nanoplates for LSPR sensing.116 The place-exchange process takes advantage of the fact that the thiols in a SAM located on the edges of the nanoplates are more readily exchanged for thiols in solution than are thiols located on the flat surfaces of the nanoplates due to decreased steric hindrance at high-curvature sites.117
The fabrication process for many nanoplasmonic sensors employs mixed materials, which can be exploited for site-selective surface chemistry. This is advantageous because varying material landscape leads to heterogeneous surface chemistry that can be exploited to selectively immobilize receptors at plasmonic hot spots. For example, Feuz and coworkers fabricated nanoholes in a TiO2/Au/TiO2 film on a glass substrate.118 The result was a nanohole array where the top surface was coated with TiO2, the hole sidewalls were composed of Au and the hole bottoms were coated with TiO2. Because poly-l-lysine-poly(ethylene glycol) (PLL-PEG) selectively adsorbs to the TiO2 surfaces, while thiolated PEG (HS-PEG) covalently bonds to the Au sidewalls, the functionalized nanohole arrays have heterogeneous chemical functionality. By employing a HS-PEG with a terminal biotin, it was possible to have avidin selectively bind to the hole sidewalls. In this sensing configuration the signal change per unit time for avidin to biotin was increased nearly 20-fold. Feuz and coworkers further showed that by using mixed materials to fabricate nanodisk pairs on a surface, they were able to direct binding of analytes to the region between the two nanodisks. This increased the signal per molecule bound by about a factor of 4 compared to binding on a single nanodisk.110 A similar, although not as robust, approach for selective blocking and functionalization in nanoholes was presented by Ferreira et al.119 Since new plasmonic architectures and fabrication methods using combinations of materials are constantly being developed, the use of site-selective chemistries to boost sensor performance seems promising for certain applications.120
Enzyme-Linked Nanoplasmonic Sensing
Enzymes are biological macromolecules (proteins) that function as catalysts in a wide variety of life-sustaining biochemical reactions. They also are used to amplify signals in a number of biochemical assays. Recently, groups have been harnessing the catalytic activity of enzymes to amplify the signals obtained from nanoplasmonic sensors with excellent results. In one example by Chen et al., circular Au nanodisks were fabricated on glass substrates by hole mask colloidal lithography.107 Then the nanodisks were functionalized with a biotin-terminated SAM. Then a streptavidin-conjugated enzyme (horseradish peroxidase, HRP) in various amounts was linked to the nanodisks. Then, in the presence of H2O2, HRP initiated the oxidation then precipitation of 3’-3-diaminobenzidine (DAB) in its polymerized form. The polymer film deposited on the nanodisks resulted in a red-shift of the LSPR spectrum. By lowering the concentration to the pM range one or at most a few HRP molecules were immobilized on each nanodisk. These single HRP molecules were then used to catalyze the precipitation of DAB polymer on the nanodisks, which resulted in a ~3 nm shift in the single particle LSPR spectrum. These results suggest that this method could push limits of detection toward the single molecule level for clinically-relevant biochemical assays.
The vast majority of sensing schemes rely on a larger signal corresponding to the presence of a higher concentration or larger amount of analyte. Using enzyme-coupled plasmonics however, Rodriguez-Lorenzo et al. have reversed this paradigm with an inverse sensitivity readout mode, where lower analyte concentrations lead to larger signals.121 In this work Au nanostars in solution function as the plasmonic sensing elements. An enzyme, glucose oxidase (GOx), was covalently linked to the nanostar surface, then upon addition of glucose, H2O2 was generated at the nanostar surface as a byproduct of glucose oxidation. Next Ag ions were introduced into solution, and H2O2 reduced the Ag ions, resulting in Ag deposition on the Au nanostars. (Figure 5) When a small amount of GOx was present on the nanostars the Ag reduction pathway lead to epitaxial deposition of a Ag layer on the nanostar surface, which resulted in a large blue-shift of the LSPR spectrum. But, when GOx was present in large amounts, the Ag reduction pathway favored nucleated deposition of Ag, which resulted in a much smaller spectral blue-shift. In short, less GOx on the nanostar surface results in a large change in the signal, while more GOx on the surface results in a small signal change, i.e. the calibration curve had a negative slope. This detection scheme was used to detect prostate-specific antigen (PSA), a biomarker for prostate cancer, with a sandwich immunoassay. Briefly, antibodies against PSA were immobilized on the nanostars then PSA was introduced. The anti-PSA antibodies captured the PSA, then a GOx-conjugated secondary antibody was introduced which bound to the captured PSA. The amount of GOx-conjugated antibody on the nanostars is directly proportional to the amount of captured PSA. When Ag ions were subsequently added, Ag was reduced and deposited epitaxially on the nanostars, in the case of low PSA concentrations, leading to large spectral shifts. At high PSA concentrations nucleated growth of Ag was observed, resulting in much smaller spectral shifts. The spectral shifts were linear as a function of PSA concentration between 10-18 and 10-13 g/mL, with the limit of detection being 10-18 g/mL. This represents an improvement of an order of magnitude over other advanced immunoassays for PSA. Because GOx can be conjugated to a wide variety of antibodies via well-known chemistries, this detection method is broadly applicable to many analytes. Moreover, the inverse sensitivity feature of this method is only possible due to the use of plasmonic nanostructures. These two examples of enzyme-linked plasmonic sensing have not shown real-time detection capabilities. However, their extreme sensitivity makes up for this shortcoming, at least when it is not critical to determine binding rate constants.
Figure 5.
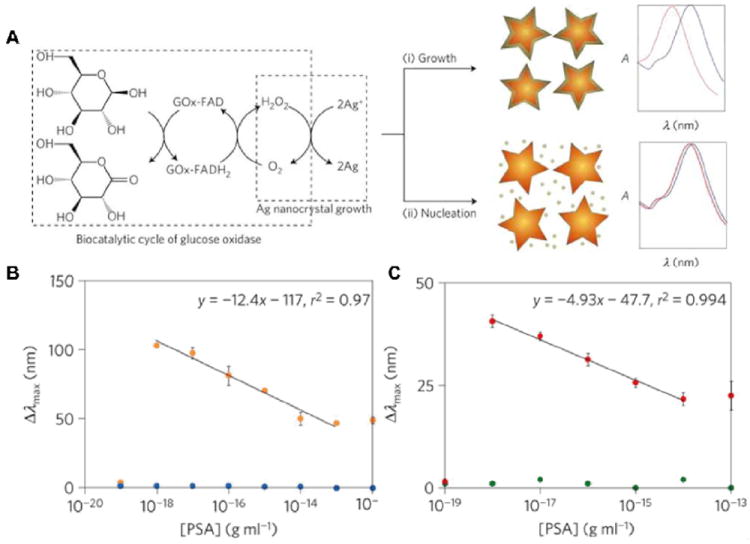
Inverse sensitivity sensing with enzyme-linked Au nanostars. (A) GOx generates hydrogen peroxide, which reduces silver ions to grow a silver coating around plasmonic nanosensors (Au nanostars); (i) at low concentrations of GOx the nucleation rate is slow, which favors the growth of a conformal silver coating that induces a large blueshift in the LSPR of the nanosensors; (ii) when GOx is present at high concentrations, the fast crystal growth conditions stimulate the nucleation of silver nanocrystals and less silver is deposited on the nanosensors, therefore generating a smaller variation of the LSPR. When the concentration of GOx is related to the concentration of a target molecule through immunoassay, this signal-generation step induces inverse sensitivity because condition (i) is fulfilled at low concentrations of analyte. (B, C) Blue shift of the LSPR absorbance band (Δλmax) as a function of the concentration of PSA (orange) and BSA (blue) in buffer (B) and of PSA (red) and BSA (green) spiked into whole serum (C). Reprinted with permission from ref. 121. Copyright 2012 Nature Publishing Group.
Combining Plasmonic Nanostructures with Lipid Bilayer Membranes
Lipid bilayer membranes are found throughout nature. They comprise the membranes that define the boundaries of cells and subcellular organelles. Their primary constituents are phospholipids, which are amphiphiles that self-assemble into ~3 nm-thick bilayer structures in the presence of aqueous solutions. Embedded in natural lipid bilayer membranes are many of the proteins that are necessary for normal cell function, such as receptors, transporters and ion channels. These membranes are also decorated with a wide variety of carbohydrates. Lipid membranes are of interest to the sensing community because of their importance in the drug development process. In fact, well over half of the top 100 selling pharmaceuticals target proteins that reside on or embedded in lipid membranes.122 Therefore, sensors that can successfully detect binding events on the surface of a lipid membrane are highly desirable. Schemes employing surface-sensitive fluorescence detection, such as total internal reflection fluorescence (TIRF) can be used,123 but they require the use of a fluorescent tag on the molecules of interest. Using a label-free detection method can eliminate the need to conjugate a fluorophore to the analyte of interest, thus plasmonic sensors are well suited to detecting molecular binding on lipid bilayer membrane.
Because natural cell membranes are difficult, though not impossible,127 to interface with plasmonic sensors, it is often convenient to use a model membrane system, such as liposomes or supported lipid bilayers (SLBs). Liposomes (also known as vesicles) are spherical lipid bilayers that can range from tens to hundreds of nanometers in diameter, while SLBs are planar lipid bilayers that are formed directly on a sensor surface and can be thought of as a 2-dimensional fluid because the lipids randomly diffuse in the SLB plane. Commercial SPR sensing systems employ specialized surface immobilization strategies to link liposomes to the surface of the sensor chip,122 even enabling sensing of membrane transport kinetics.128 An alternative strategy, though not generally commercially available, is to alter the surface of a sensor chip such that liposomes will rupture on the surface to form a planar SLB. This can be done by coating a thin (< 20 nm) layer of SiO2 over the metallic substrate,129 with the thinnest oxide layers typically deposited by atomic layer deposition.126 It should be noted, however, that addition of a thin oxide layer will shift the resonances of the sensor, so this must be taken into account in the sensor design process. Alternative strategies to form SLBs include the use of thiolated lipids to covalently tether the membrane to a bare metal film, or to form SLBs on a polymer cushion which lies upon the metal film.122 Plasmonic nanostructures can also be embedded in SLBs,130 or SLBs can be formed to envelop nanostructures, as will be discussed below. Figure 6 shows schematic illustrations of methods for interfacing membranes with plasmonic nanostructures.
Figure 6.
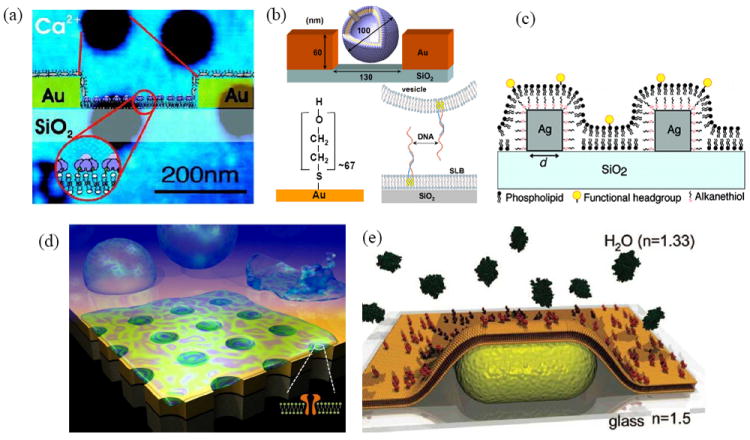
Integration of lipid bilayer membranes with plasmonic nanostructures. (a) Supported lipid bilayers formed on the SiO2 surface at the base of Au nanoholes. From Dahlin et al.65 (b) Intact vesicles tethered to SiO2 inside Au nanoholes via hybridization of complimentary DNA strands. From Dahlin et al.124 (c) Lipid bilayers formed over Ag nanocubes. The SAM on the nanocubes leads to a hybrid SAM-lipid bilayer that covers the nanocubes, while a lipid bilayer is formed on the glass between nanocubes. Adapted from Galush et al.125 (d) A lipid bilayer membrane suspended over nanopores in a free-standing Au film coated with an SiO2 shell. Adapted from Im et al.126 (e) A lipid bilayer membrane decorated with proteins covering a Au nanorod. Adapted from Baciu et al.23
One of the first examples of combining SLBs with nanoplasmonics was shown by Dahlin et al. In this work they fabricated random 110 nm-diameter holes in a Au film by colloidal lithography on a SiO2 coated substrate.65 The gold surface was then passivated, leaving the vesicles free to settle and rupture on the SiO2-coated nanohole bottoms, forming SLB patches containing lipid-conjugated biotin or single-stranded DNA or the lipid receptor GM1. With this setup the introduction of cholera toxin, a 56 kDa protein which binds to GM1, induced a large increase in the extinction of the nanohole array, which was monitored as a function of time. To demonstrate that this sensing approach worked for smaller molecules as well, they were able to detect the binding of a 15-base single-stranded DNA (5 kDa) to its complementary strand that was immobilized in the SLB patches. Single vesicles can also be immobilized inside nanoholes by tagging vesicles with single-stranded DNA, then functionalizing the bottom of nanoholes with the complementary sequence.124 Since the initial work by Dahlin et al., there have been a number of other examples where nanoholes are used to detect binding of a variety of different analytes to lipid bilayer membranes.
A challenge for combining SLBs that contain embedded proteins is that the presence of an underlying substrate can have negative effects on the protein, such as hindered diffusion and denaturation. One way to circumvent these effects is to partially remove the substrate, creating free-standing nanohole arrays that are surrounded on both sides by liquid. This allows the formation of a pore-spanning lipid membrane (PSLM) that reduces the influence of the underlying substrate on the lipids and proteins that comprise the membrane. PSLMs have been demonstrated on a variety of materials131,132 and been used for electrical biosensing studies,133 but examples where plasmonic sensing is used in conjunction with PSLMs are limited. In one example, Oh and coworkers formed PSLMs over SiO2-coated Au nanoholes.126 This allowed the insertion of a transmembrane protein, alpha-hemolysin, into the pore-spanning regions. The insertion process of the protein was followed by monitoring the transmission spectra in real-time. After protein insertion, it was possible to detect antibodies binding to alpha-hemolysin and calculate binding parameters, such as association and dissociation rate constants, as well as the dissociation constant.
Instead of nanoholes, surface immobilized nanoparticles can also be combined with lipid bilayer membranes. In one example, Galush et al. deposited Ag nanocubes on a glass surface then formed a SLB over them.125 By operating in transmission mode and monitoring the shift in the extinction maximum, binding of neutravidin to biotinylated lipids was characterized. This sensor was also used to observe unbinding of proteins from a specially functionalized lipid surface. SLB-covered Au nanorods have also been employed for lipid-protein binding assays.23 An alternative to completely covering nanostructures is to form a SLB around them, such as the work by Lohmuller and coworkers where SLBs were formed around bow tie nanoantennas.134 The large electromagnetic field between the antenna tips enhanced fluorescence emission, which allowed observation of individual molecules associated with the fluid 2-dimensional surface of the SLB diffusing through the tips.
Flow-through nanoplasmonic sensing
Mass transport limitations can plague all types of surface-based sensors. In order to record proper binding kinetics, an analyte must be delivered to the sensor surface at a rate that overcomes depletion at the surface due to binding. This is typically accomplished by optimizing the injection flow rate to a point where increasing flow rate does not increase the association rate constant. Unfortunately, when the sensor size shrinks, increasing the flow rate has diminishing return on improving the analyte delivery.112,135 Another way to overcome mass transport limitations is to force an analyte solution toward the active sensor surface. This can be achieved by using plasmonic nanoholes as nanofluidic channels.
These sensors generally rely upon free-standing open-ended nanoholes sandwiched between two microfluidic channels. In this configuration, solution approaches the nanoholes via a dead-ended microfluidic channel and then is forced through the nanoholes, where it then exits the sensing zone through a second outlet microfluidic channel (Figure 7). By forcing liquid through nanoholes, the rate of adsorption of molecules to the surface can be increased significantly compared to the case where solution simply flows over an array of dead-ended nanoholes.136-138
Figure 7.
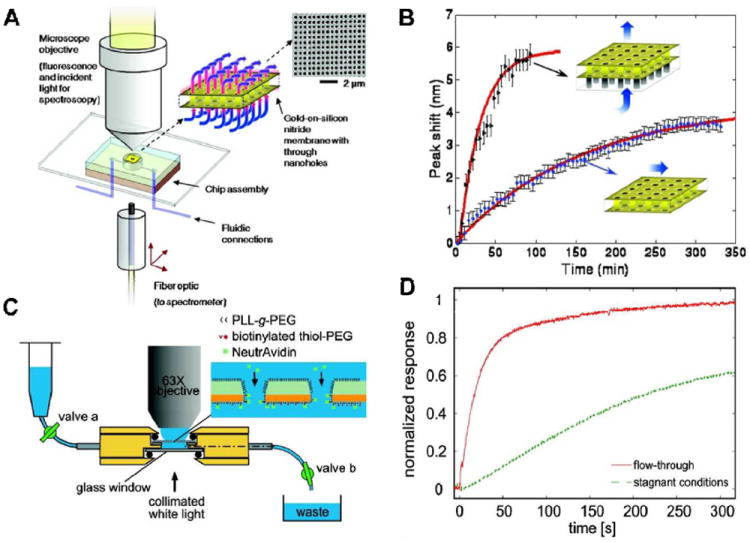
Flow-through nanoplasmonic sensing. (A, C) Schematic illustrations of experimental set-ups for flow-through sensing. (B) Kinetic curves showing SAM formation on a gold nanohole array in the flow-through configuration compared to the same SAM being formed using a flow-over configuration. (D) Comparison of avidin-biotin binding reactions carried out in stagnant solution vs. binding under flow-through conditions showing the drastically increased sensor response with flow-through sensing. Image A and B from Eftekhari et al.136; C and D from Jonsson et al.137
Flow-through plasmonic sensors are typically composed of a metallic layer supported by a thin film of silicon nitride, therefore it is possible to take advantage of the mixed materials to use material-specific chemistry to selectively functionalize only certain features of the nanoholes. In one example a suspended nanohole array composed of a Au layer on a Si3N4 thin film was functionalized with a biotinylated thiol, which is selectively immobilized on Au. Then the Si3N4 layer was passivated with a PEG layer.137 This results in the binding receptors for neutravidin being localized only to the Au surface residing inside the nanoholes. Although binding occurred also on the planar part of the gold film, which also possesses some sensitivity, the sensor response was a factor of 10 faster in the flow-through configuration than under conditions where mass transport is diffusion-limited. This means that capture-limited binding can be achieved at significantly lower flow rates, which drastically reduces sample consumption compared to standard SPR instruments. To avoid influence on the signal of biomolecular binding reactions on the less sensitive part of the gold film, Mazzotta et al. recently developed a fabrication protocol allowing discrete nanoplasmonic elements to be positioned inside the pores, thus being placed where the flow geometry is optimized for efficient binding.139
As these examples show, pressure driven flow through nanoholes can effectively overcome mass transport deficiencies that are associated with SPR sensing. Combining an electrophoretic concentration mechanism with a flow-though sensor architecture can further improve concentration of analytes at plasmonic nanoholes.140 It should be noted that care must be exercised when applying pressure to drive the flow through nanoholes. The Si3N4 membrane than supports the metallic nanoholes is usually only a few hundred nanometers thick and quite delicate. Application of excessive pressure can rupture the membrane rendering the sensor useless and the experiment failed. Despite this drawback, the advantages gained by using this design make flow-through sensors a worthwhile option when sample volume limitations hinder the use of flow-over nanoplasmonic sensors or traditional SPR sensors.
Conclusions
We have summarized the brief history and operating principles of SPR and nanoplasmonic sensors, and presented various metrics on how to evaluate the sensor performance. SPR still dominates in terms of low detection limits in surface coverage (e.g. Biacore instrument can detect ~0.01 ng/cm2), but we have shown some examples of recent nanoplasmonic sensors that truly can go where SPR cannot. The field of nanoplasmonic biosensing has been among the most multi-disciplinary research areas: The underlying physical principles of SPR and nanoplasmonic interactions have been seamlessly integrated with state-of-the-art nanofabrication, optical instrumentation techniques, a wide range of chemical surface modification techniques, microfluidics (and now toward nanofluidic “flow-through” schemes), and biological interfacing schemes involving soft matter and cellular membranes. There is still a barrier to commercial success of nanoplasmonic devices, but increasing demand for more advanced sensors will continue to motivate researchers to overcome those hurdles.
Figure 1.

(a) Metallic nanoparticles can support localized surface plasmon resonance (LSPR), collective oscillations of the conduction electrons excited by light. (b) A color photograph of single metallic nanoparticles illuminated with white light. (c) Scattered light spectra measured from nanoparticles shown in (b). Images (b) and (c) adapted from Schutz.6
Figure 4.
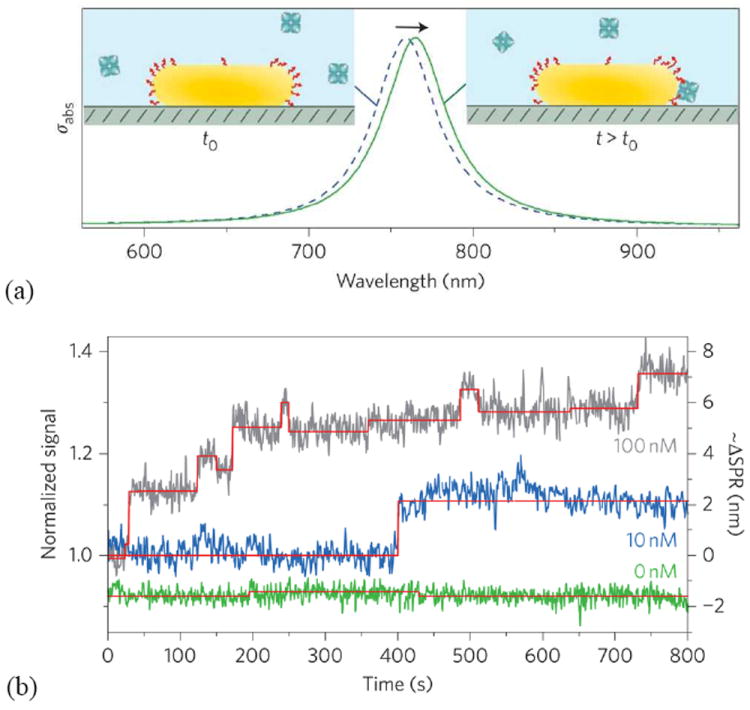
Binding of the analyte molecules to the receptors on a functionalized gold nanorod, shown in (a), shifts its plasmon resonance along the longitudinal direction. (b) Binding of streptavidin-R-phycoerythrin to a biotin-functionalized nanorod is monitored using photothermal microscopy. Adapted from Orrit et al.108
Contributor Information
Andreas B. Dahlin, Email: andreas.dahlin@chalmers.se.
Nathan J. Wittenberg, Email: witt0092@umn.edu.
Fredrik Höök, Email: fredrik.hook@chalmers.se.
Sang-Hyun Oh, Email: sang@umn.edu.
References
- 1.Dahlin AB. Plasmonic Biosensors: An Integrated View of Refractometric Detection. IOS Press; Washington, DC: 2012. [Google Scholar]
- 2.Freestone I, Meeks N, Sax M, Higgitt C. Gold Bull. 2007;40:270–277. [Google Scholar]
- 3.Bobin O, Schvoerer M, Miane JL, Fabre JF. J Non-Cryst Solids. 2003;332:28–34. [Google Scholar]
- 4.Brown CL, Bushell G, Whitehouse MW, Agrawal DS, Tupe SG, Paknikar KM, Tiekink ERT. Gold Bull. 2007;40:245–250. [Google Scholar]
- 5.Giljohann DA, Seferos DS, Daniel WL, Massich MD, Patel PC, Mirkin CA. Angew Chem Int Ed. 2010;49:3280–3294. doi: 10.1002/anie.200904359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schultz DA. Curr Opin Biotech. 2003;14:13–22. doi: 10.1016/s0958-1669(02)00015-0. [DOI] [PubMed] [Google Scholar]
- 7.Mie G. Annalen der Physik. 1908;25:377–445. [Google Scholar]
- 8.Gans R, Happel H. Annalen der Physik. 1909;29:277–300. [Google Scholar]
- 9.Myroshnychenko V, Rodriguez-Fernandez J, Pastoriza-Santos I, Funston AM, Novo C, Mulvaney P, Liz-Marzan LM, Garcia de Abajo FJ. Chem Soc Rev. 2008;37:1792–1805. doi: 10.1039/b711486a. [DOI] [PubMed] [Google Scholar]
- 10.Jackson JD. Classical Electrodynamics. 3. Wiley; 1998. [Google Scholar]
- 11.Bohren CF, Huffman DR. Absorption and Scattering of Light by Small Particles. Wiley; New York: 1983. [Google Scholar]
- 12.Kelly KL, Coronado E, Zhao LL, Schatz GC. J Phys Chem B. 2003;107:668–677. [Google Scholar]
- 13.Zeman EJ, Schatz GC. J Phys Chem. 1987;91:634–643. [Google Scholar]
- 14.Dahlin AB, Sannomiya T, Zahn R, Sotiriou GA, Vörös J. Nano Lett. 2011;11:1337–1343. doi: 10.1021/nl104424q. [DOI] [PubMed] [Google Scholar]
- 15.Dahlin AB, Zahn R, Vörös J. Nanoscale. 2012;4:2339–2351. doi: 10.1039/c2nr11950a. [DOI] [PubMed] [Google Scholar]
- 16.Bobbert PA, Vlieger J. Physica A. 1987;147:115–141. [Google Scholar]
- 17.Englebienne P. Analyst. 1998;123:1599–1603. doi: 10.1039/a804010i. [DOI] [PubMed] [Google Scholar]
- 18.Nath N, Chilkoti A. Anal Chem. 2002;74:504–509. doi: 10.1021/ac015657x. [DOI] [PubMed] [Google Scholar]
- 19.Raschke G, Kowarik S, Franzl T, Sonnichsen C, Klar TA, Feldmann J, Nichtl A, Kurzinger K. Nano Lett. 2003;3:935–938. [Google Scholar]
- 20.Nath N, Chilkoti A. Anal Chem. 2004;76:5370–5378. doi: 10.1021/ac049741z. [DOI] [PubMed] [Google Scholar]
- 21.Yu C, Irudayaraj J. Anal Chem. 2007;79:572–579. doi: 10.1021/ac061730d. [DOI] [PubMed] [Google Scholar]
- 22.Nusz GJ, Marinakos SM, Curry AC, Dahlin AB, Höök F, Wax A, Chilkoti A. Anal Chem. 2008;80:984–989. doi: 10.1021/ac7017348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baciu CL, Becker J, Janshoff A, Sönnichsen C. Nano Lett. 2008;8:1724–1728. doi: 10.1021/nl080805l. [DOI] [PubMed] [Google Scholar]
- 24.Dahlin AB, Chen S, Jonsson MP, Gunnarsson L, Käll M, Höök F. Anal Chem. 2009;81:6572–6580. doi: 10.1021/ac901175k. [DOI] [PubMed] [Google Scholar]
- 25.Raschke G, Brogl S, Susha AS, Rogach AL, Klar TA, Feldmann J, Fieres B, Petkov N, Bein T, Nichtl A, Kurzinger K. Nano Lett. 2004;4:1853–1857. [Google Scholar]
- 26.Haes AJ, Van Duyne RP. J Am Chem Soc. 2002;124:10596–10604. doi: 10.1021/ja020393x. [DOI] [PubMed] [Google Scholar]
- 27.Larsson EM, Prinetti A, Käll M, Sutherland DS. Nano Lett. 2007;7:1256–1263. doi: 10.1021/nl0701612. [DOI] [PubMed] [Google Scholar]
- 28.Lee S, Mayer KM, Hafner JH. Anal Chem. 2009;81:4450–4455. doi: 10.1021/ac900276n. [DOI] [PubMed] [Google Scholar]
- 29.Dondapati SK, Sau TK, Hrelescu C, Klar TA, Stefani FD, Feldmann J. ACS Nano. 2010;4:6318–6322. doi: 10.1021/nn100760f. [DOI] [PubMed] [Google Scholar]
- 30.Luk’yanchuk B, Zheludev NI, Maier SA, Halas NJ, Nordlander P, Giessen H, Chong CT. Nature Mater. 2010;9:707–715. doi: 10.1038/nmat2810. [DOI] [PubMed] [Google Scholar]
- 31.Verellen N, Van Dorpe P, Huang C, Lodewijks K, Vandenbosch GAE, Lagae L, Moshchalkov VV. Nano Lett. 2011;11:391–397. doi: 10.1021/nl102991v. [DOI] [PubMed] [Google Scholar]
- 32.Lange C. Berl klin Wohnschr. 1912;49:897. [Google Scholar]
- 33.Green F. Canadian Medical Association Journal. 1925;15:1139–1143. [PMC free article] [PubMed] [Google Scholar]
- 34.Elghanian R. Science. 1997;277:1078–1081. doi: 10.1126/science.277.5329.1078. [DOI] [PubMed] [Google Scholar]
- 35.Olofsson L, Rindzevicius T, Pfeiffer I, Käll M, Höök F. Langmuir. 2003;19:10414–10419. [Google Scholar]
- 36.Lahav M, Vaskevich A, Rubinstein I. Langmuir. 2004;20:7365–7367. doi: 10.1021/la0489054. [DOI] [PubMed] [Google Scholar]
- 37.Ritchie R. Phys Rev. 1957;106:874. [Google Scholar]
- 38.Powell C, Swan J. Phys Rev. 1959;115:869–875. [Google Scholar]
- 39.Homola J, Yee SS, Gauglitz G. Sens Actuators, B. 1999;54:3–15. [Google Scholar]
- 40.Homola J. Chem Rev. 2008;108:462–493. doi: 10.1021/cr068107d. [DOI] [PubMed] [Google Scholar]
- 41.Lindquist NC, Nagpal P, McPeak KM, Norris DJ, Oh S-H. Rep Prog Phys. 2012;75:036501. doi: 10.1088/0034-4885/75/3/036501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liedberg B, Nylander C, Lundstrom I. Sens Actuators. 1983;4:299–304. [Google Scholar]
- 43.Rich RL, Myszka DG. J Mol Recognit. 2011;24:892–914. doi: 10.1002/jmr.1138. [DOI] [PubMed] [Google Scholar]
- 44.Li Y-C, Chang Y-F, Su L-C, Chou C. Anal Chem. 2008;80:5590–5595. doi: 10.1021/ac800598c. [DOI] [PubMed] [Google Scholar]
- 45.Zhou W-J, Halpern AR, Seefeld TH, Corn RM. Anal Chem. 2012;84:440–445. doi: 10.1021/ac202863k. [DOI] [PubMed] [Google Scholar]
- 46.Wark AW, Lee HJ, Corn RM. Anal Chem. 2005;77:3904–3907. doi: 10.1021/ac050402v. [DOI] [PubMed] [Google Scholar]
- 47.Slavík R, Homola J. Sens Actuators, B. 2007;123:10–12. [Google Scholar]
- 48.Gordon R, Sinton D, Kavanagh KL, Brolo AG. Acc Chem Res. 2008;41:1049–1057. doi: 10.1021/ar800074d. [DOI] [PubMed] [Google Scholar]
- 49.Jonsson MP, Dahlin AB, Jönsson P, Höök F. Biointerphases. 2008;3:FD30–FD40. doi: 10.1116/1.3027483. [DOI] [PubMed] [Google Scholar]
- 50.Masson JF, Murray-Methot M-P, Live LS. Analyst. 2010;135:1483–1489. doi: 10.1039/c0an00053a. [DOI] [PubMed] [Google Scholar]
- 51.Bethe HA. Phys Rev. 1944;66:163–182. [Google Scholar]
- 52.Degiron A, Lezec HJ, Yamamoto N, Ebbesen TW. Opt Commun. 2004;239:61–66. [Google Scholar]
- 53.Ebbesen TW, Lezec HJ, Ghaemi HF, Thio T, Wolff P. Nature. 1998;391:667–669. [Google Scholar]
- 54.Przybilla F, Genet C, Ebbesen TW. Opt Express. 2012;20:4697–4709. doi: 10.1364/OE.20.004697. [DOI] [PubMed] [Google Scholar]
- 55.Sannomiya T, Scholder O, Jefimovs K, Hafner C, Dahlin AB. Small. 2011;7:1653–1663. doi: 10.1002/smll.201002228. [DOI] [PubMed] [Google Scholar]
- 56.Pacifici D, Lezec HJ, Sweatlock LA, Walters RJ, Atwater HA. Opt Express. 2008;16:9222–9238. doi: 10.1364/oe.16.009222. [DOI] [PubMed] [Google Scholar]
- 57.Park T-H, Mirin N, Lassiter JB, Nehl CL, Halas NJ, Nordlander P. ACS Nano. 2008;2:25–32. doi: 10.1021/nn700292y. [DOI] [PubMed] [Google Scholar]
- 58.Barnes WL, Dereux A, Ebbesen TW. Nature. 2003;424:824–830. doi: 10.1038/nature01937. [DOI] [PubMed] [Google Scholar]
- 59.Genet C, Ebbesen TW. Nature. 2007;445:39–46. doi: 10.1038/nature05350. [DOI] [PubMed] [Google Scholar]
- 60.de Ajajo FJG. Rev Mod Phys. 2007;79:1267. [Google Scholar]
- 61.García-Vidal FJ, Ebbesen TW, Kuipers L. Rev Mod Phys. 2010;82:729–787. [Google Scholar]
- 62.Genet C, Van Exter MP, Woerdman J. Opt Commun. 2003;225:331–336. [Google Scholar]
- 63.Braun J, Gompf B, Kobiela G, Dressel M. Phys Rev Lett. 2009;103 doi: 10.1103/PhysRevLett.103.203901. [DOI] [PubMed] [Google Scholar]
- 64.Brolo AG, Gordon R, Leathem B, Kavanagh KL. Langmuir. 2004;20:4813–4815. doi: 10.1021/la0493621. [DOI] [PubMed] [Google Scholar]
- 65.Dahlin AB, Zäch M, Rindzevicius T, Käll M, Sutherland DS, Höök F. J Am Chem Soc. 2005;127:5043–5048. doi: 10.1021/ja043672o. [DOI] [PubMed] [Google Scholar]
- 66.Rindzevicius T, Alaverdyan Y, Dahlin AB, Höök F, Sutherland DS, Käll M. Nano Lett. 2005;5:2335–2339. doi: 10.1021/nl0516355. [DOI] [PubMed] [Google Scholar]
- 67.Laromaine A, Koh L, Murugesan M, Ulijn RV, Stevens MM. J Am Chem Soc. 2007;129:4156–4157. doi: 10.1021/ja0706504. [DOI] [PubMed] [Google Scholar]
- 68.Lodewijks K, Van Roy W, Borghs G, Lagae L, Van Dorpe P. Nano Lett. 2012;12:1655–1659. doi: 10.1021/nl300044a. [DOI] [PubMed] [Google Scholar]
- 69.Dahlin AB, Tegenfeldt JO, Höök F. Anal Chem. 2006;78:4416–4423. doi: 10.1021/ac0601967. [DOI] [PubMed] [Google Scholar]
- 70.Murray-Methot M-P, Ratel M, Masson J-F. J Phys Chem C. 2010;114:8268–8275. [Google Scholar]
- 71.Piliarik M, Homola J. Opt Express. 2009;17:16505–16517. doi: 10.1364/OE.17.016505. [DOI] [PubMed] [Google Scholar]
- 72.Dahlin AB, Jonsson MP. In: Nanoplasmonic Sensors. Dmitriev A, editor. Springer New York; New York, NY: 2012. pp. 231–265. [Google Scholar]
- 73.Becker J, Trügler A, Jakab A, Hohenester U, Sönnichsen C. Plasmonics. 2010;5:161–167. [Google Scholar]
- 74.Das M, Hohertz D, Nirwan R, Brolo AG, Kavanagh KL, Gordon R. IEEE Photonics J. 2011;3:441–449. [Google Scholar]
- 75.Svedendahl M, Chen S, Dmitriev A, Käll M. Nano Lett. 2009;9:4428–4433. doi: 10.1021/nl902721z. [DOI] [PubMed] [Google Scholar]
- 76.Johnsson B, Löfås S, Lindquist G. Anal Biochem. 1991;198:268–277. doi: 10.1016/0003-2697(91)90424-r. [DOI] [PubMed] [Google Scholar]
- 77.Kedem O, Tesler AB, Vaskevich A, Rubinstein I. ACS Nano. 2011;5:748–760. doi: 10.1021/nn102617d. [DOI] [PubMed] [Google Scholar]
- 78.Prikulis J, Hanarp P, Olofsson L, Sutherland D, Käll M. Nano Lett. 2004;4:1003–1007. [Google Scholar]
- 79.Lee SH, Bantz KC, Lindquist NC, Oh S-H, Haynes CL. Langmuir. 2009;25:13685–13693. doi: 10.1021/la9020614. [DOI] [PubMed] [Google Scholar]
- 80.Tetz K, Pang L, Fainman Y. Opt Lett. 2006;31:1528–1530. doi: 10.1364/ol.31.001528. [DOI] [PubMed] [Google Scholar]
- 81.Henzie J, Lee MH, Odom TW. Nature Nanotech. 2007;2:549–554. doi: 10.1038/nnano.2007.252. [DOI] [PubMed] [Google Scholar]
- 82.Menezes JW, Ferreira J, Santos MJL, Cescato L, Brolo AG. Adv Funct Mater. 2010;20:3918–3924. [Google Scholar]
- 83.Nagpal P, Lindquist NC, Oh S-H, Norris DJ. Science. 2009;325:594–597. doi: 10.1126/science.1174655. [DOI] [PubMed] [Google Scholar]
- 84.Im H, Lee SH, Wittenberg NJ, Johnson TW, Lindquist NC, Nagpal P, Norris DJ, Oh S-H. ACS Nano. 2011;5:6244–6253. doi: 10.1021/nn202013v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lindquist NC, Johnson TW, Norris DJ, Oh S-H. Nano Lett. 2011;11:3526–3530. doi: 10.1021/nl2005737. [DOI] [PubMed] [Google Scholar]
- 86.Park JH, Ambwani P, Manno M, Lindquist NC, Nagpal P, Oh S-H, Leighton C, Norris DJ. Adv Mater. 2012;24:3988–3992. doi: 10.1002/adma.201200812. [DOI] [PubMed] [Google Scholar]
- 87.Piliarik M, Párová L, Homola J. Biosens Bioelectron. 2009;24:1399–1404. doi: 10.1016/j.bios.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 88.Vala M, Chadt K, Piliarik M, Homola J. Sens Actuators, B. 2010;148:544–549. [Google Scholar]
- 89.Lindquist NC, Lesuffleur A, Im H, Oh S-H. Lab Chip. 2009;9:382–387. doi: 10.1039/b816735d. [DOI] [PubMed] [Google Scholar]
- 90.Nakamoto K, Kurita R, Niwa O, Fujii T, Nishida M. Nanoscale. 2011;3:5067–5075. doi: 10.1039/c1nr10883b. [DOI] [PubMed] [Google Scholar]
- 91.Nakamoto K, Kurita R, Niwa O. Anal Chem. 2012;84:3187–3191. doi: 10.1021/ac203160r. [DOI] [PubMed] [Google Scholar]
- 92.Lindquist NC, Johnson TW, Jose J, Otto LM, Oh S-H. Annalen der Physik. 2012;524:687–696. doi: 10.1002/andp.201200144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stein B, Laluet J-Y, Devaux E, Genet C, Ebbesen TW. J Phys Chem C. 2012;116:6092–6096. [Google Scholar]
- 94.Moreira CS, Lima AMN, Neff H, Thirstrup C. Sens Actuators, B. 2008;134:854–862. [Google Scholar]
- 95.Baffou G, Quidant R. Laser & Photon Rev. 2012 [Google Scholar]
- 96.Im H, Sutherland JN, Maynard JA, Oh S-H. Anal Chem. 2012;84:1941–1947. doi: 10.1021/ac300070t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stewart ME, Mack NH, Malyarchuk V, Soares JANT, Lee TW, Gray SK, Nuzzo RG, Rogers JA. Proc Natl Acad Sci USA. 2006;103:17143–17148. doi: 10.1073/pnas.0606216103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lee SH, Lindquist NC, Wittenberg NJ, Jordan LR, Oh S-H. Lab Chip. 2012;12:3882–3890. doi: 10.1039/c2lc40455a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nusz GJ, Marinakos SM, Rangarajan S, Chilkoti A. Appl Opt. 2011;50:4198–4206. doi: 10.1364/AO.50.004198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lesuffleur A, Im H, Lindquist NC, Lim KS, Oh S-H. Opt Express. 2008;16:219–224. doi: 10.1364/oe.16.000219. [DOI] [PubMed] [Google Scholar]
- 101.Ji J, O’Connell JG, Carter DJD, Larson DN. Anal Chem. 2008;80:2491–2498. doi: 10.1021/ac7023206. [DOI] [PubMed] [Google Scholar]
- 102.Yang J-C, Ji J, Hogle JM, Larson DN. Nano Lett. 2008;8:2718–2724. doi: 10.1021/nl801043t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Endo T, Kerman K, Nagatani N, Hiepa HM, Kim DK, Yonezawa Y, Nakano K, Tamiya E. Anal Chem. 2006;78:6465–6475. doi: 10.1021/ac0608321. [DOI] [PubMed] [Google Scholar]
- 104.Ament I, Prasad J, Henkel A, Schmachtel S, Sönnichsen C. Nano Lett. 2012;12:1092–1095. doi: 10.1021/nl204496g. [DOI] [PubMed] [Google Scholar]
- 105.McFarland AD, Van Duyne RP. Nano Lett. 2003;3:1057–1062. [Google Scholar]
- 106.Nusz GJ, Curry AC, Marinakos SM, Wax A, Chilkoti A. ACS Nano. 2009;3:795–806. doi: 10.1021/nn8006465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen S, Svedendahl M, Van Duyne RP, Käll M. Nano Lett. 2011;11:1826–1830. doi: 10.1021/nl2006092. [DOI] [PubMed] [Google Scholar]
- 108.Zijlstra P, Paulo PMR, Orrit M. Nature Nanotech. 2012;7:379–382. doi: 10.1038/nnano.2012.51. [DOI] [PubMed] [Google Scholar]
- 109.Boyer D, Tamarat P, Maali A, Lounis B, Orrit M. Science. 2002;297:1160–1163. doi: 10.1126/science.1073765. [DOI] [PubMed] [Google Scholar]
- 110.Feuz L, Jonsson MP, Höök F. Nano Lett. 2012;12:873–879. doi: 10.1021/nl203917e. [DOI] [PubMed] [Google Scholar]
- 111.Dahlin AB. Sensors. 2012;12:3018–3036. doi: 10.3390/s120303018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Squires TM, Messinger RJ, Manalis SR. Nat Biotechnol. 2008;26:417–426. doi: 10.1038/nbt1388. [DOI] [PubMed] [Google Scholar]
- 113.Gunnarsson A, Jönsson P, Marie R, Tegenfeldt JO, Höök F. Nano Lett. 2008;8:183–188. doi: 10.1021/nl072401j. [DOI] [PubMed] [Google Scholar]
- 114.Bally M, Gunnarsson A, Svensson L, Larson G, Zhdanov V, Höök F. Phys Rev Lett. 2011;107:188103. doi: 10.1103/PhysRevLett.107.188103. [DOI] [PubMed] [Google Scholar]
- 115.Love JC, Estroff LA, Kriebel JK, Nuzzo RG, Whitesides GM. Chem Rev. 2005;105:1103–1170. doi: 10.1021/cr0300789. [DOI] [PubMed] [Google Scholar]
- 116.Beeram SR, Zamborini FP. J Am Chem Soc. 2009;131:11689–11691. doi: 10.1021/ja904387j. [DOI] [PubMed] [Google Scholar]
- 117.Hostetler MJ, Templeton AC, Murray RW. Langmuir. 1999;15:3782–3789. [Google Scholar]
- 118.Feuz L, Jönsson P, Jonsson MP, Höök F. ACS Nano. 2010;4:2167–2177. doi: 10.1021/nn901457f. [DOI] [PubMed] [Google Scholar]
- 119.Ferreira J, Santos MJL, Rahman MM, Brolo AG, Gordon R, Sinton D, Girotto EM. J Am Chem Soc. 2009;131:436–437. doi: 10.1021/ja807704v. [DOI] [PubMed] [Google Scholar]
- 120.Marie R, Dahlin AB, Tegenfeldt JO, Höök F. Biointerphases. 2007;2:49. doi: 10.1116/1.2717926. [DOI] [PubMed] [Google Scholar]
- 121.Rodríguez-Lorenzo L, la Rica de R, Álvarez-Puebla RA, Liz-Marzan LM, Stevens MM. Nature Mater. 2012;11:604–607. doi: 10.1038/nmat3337. [DOI] [PubMed] [Google Scholar]
- 122.Cooper MA. J Mol Recognit. 2004;17:286–315. doi: 10.1002/jmr.675. [DOI] [PubMed] [Google Scholar]
- 123.Shi J, Yang T, Kataoka S, Zhang Y, Diaz AJ, Cremer PS. J Am Chem Soc. 2007;129:5954–5961. doi: 10.1021/ja069375w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dahlin AB, Jonsson MP, Höök F. Adv Mater. 2008;20:1436–1442. [Google Scholar]
- 125.Galush WJ, Shelby SA, Mulvihill MJ, Tao A, Yang P, Groves JT. Nano Lett. 2009;9:2077–2082. doi: 10.1021/nl900513k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Im H, Wittenberg NJ, Lesuffleur A, Lindquist NC, Oh S-H. Chem Sci. 2010;1:688–696. doi: 10.1039/C0SC00365D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wittenberg NJ, Im H, Johnson TW, Xu X, Warrington AE, Rodriguez M, Oh S-H. ACS Nano. 2011;5:7555–7564. doi: 10.1021/nn202554t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Brändén M, Dahlin S, Höök F. ChemPhysChem. 2008;9:2480–2485. doi: 10.1002/cphc.200800614. [DOI] [PubMed] [Google Scholar]
- 129.Jonsson MP, Jönsson P, Dahlin AB, Höök F. Nano Lett. 2007;7:3462–3468. doi: 10.1021/nl072006t. [DOI] [PubMed] [Google Scholar]
- 130.Lohmüller T, Triffo S, O’Donoghue GP, Xu Q, Coyle MP, Groves JT. Nano Lett. 2011;11:4912–4918. doi: 10.1021/nl202847t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hennesthal C, Steinem C. J Am Chem Soc. 2000;122:8085–8086. [Google Scholar]
- 132.Lazzara TD, Carnarius C, Kocun M, Janshoff A, Steinem C. ACS Nano. 2011;5:6935–6944. doi: 10.1021/nn201266e. [DOI] [PubMed] [Google Scholar]
- 133.Janshoff A, Steinem C. Anal Bioanal Chem. 2006;385:433–451. doi: 10.1007/s00216-006-0305-9. [DOI] [PubMed] [Google Scholar]
- 134.Lohmüller T, Iversen L, Schmidt M, Rhodes C, Tu HL, Lin WC, Groves JT. Nano Lett. 2012;12:1717–1721. doi: 10.1021/nl300294b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sheehan PE, Whitman LJ. Nano Lett. 2005;5:803–807. doi: 10.1021/nl050298x. [DOI] [PubMed] [Google Scholar]
- 136.Eftekhari F, Escobedo C, Ferreira J, Duan X, Girotto EM, Brolo AG, Gordon R, Sinton D. Anal Chem. 2009;81:4308–4311. doi: 10.1021/ac900221y. [DOI] [PubMed] [Google Scholar]
- 137.Jonsson MP, Dahlin AB, Feuz L, Petronis S, Höök F. Anal Chem. 2010;82:2087–2094. doi: 10.1021/ac902925e. [DOI] [PubMed] [Google Scholar]
- 138.Escobedo C, Brolo AG, Gordon R, Sinton D. Anal Chem. 2010;82:10015–10020. doi: 10.1021/ac101654f. [DOI] [PubMed] [Google Scholar]
- 139.Mazzotta F, Höök F, Jonsson MP. Nanotechnology. 2012 doi: 10.1088/0957-4484/23/41/415304. [DOI] [PubMed] [Google Scholar]
- 140.Escobedo C, Brolo AG, Gordon R, Sinton D. Nano Lett. 2012;12:1592–1596. doi: 10.1021/nl204504s. [DOI] [PubMed] [Google Scholar]