

Abstract
The Sleeping Beauty transposon system, a non-viral, integrating vector that can deliver the alpha-L-iduronidase-encoding gene, is efficient in correcting mucopolysaccharidosis type I in NOD/SCID mice. However, in previous studies we failed to attain reliable long-term alpha-L-iduronidase expression in immunocompetent mice. Here, we focused on achieving sustained high-level expression in immunocompetent C57BL/6 mice. In our standard liver-directed treatment we hydrodynamically infuse mice with plasmids containing a SB transposon-encoding human alpha-L-iduronidase, along with a source of SB transposase. We sought to 1) minimize expression of the therapeutic enzyme in antigen-presenting cells, while avoiding promoter shutdown and gender bias, 2) increase transposition efficiency and 3) improve immunosuppression. By using a liver-specific promoter to drive IDUA expression, the SB100X hyperactive transposase and transient cyclophosphamide immunosuppression we achieved therapeutic-level (>100 wild-type) stabilized expression for 1 year in 50% of C57BL/6 mice. To gain insights into the causes of variability in transgene expression, we quantified the rates of alpha-L-iduronidase activity decay vis-a-vis transposition and transgene maintenance using the data obtained in this and previous studies. Our analyses showed that immune responses are the most important variable to control in order to prevent loss of transgene expression. Cumulatively, our results allow transition to pre-clinical studies of SB-mediated alpha-L-iduronidase expression and correction of mucopolysaccharidosis type I in animal models.
Introduction
The Sleeping Beauty (SB) transposon system [1] is a non-viral, integrating vector that is an alternative to viral vectors for gene therapy [2–5]. The SB system consists of two parts: the transposon, which is defined by inverted terminal repeats and which carries an expression cassette for the therapeutic gene, and a second expression cassette for the transposase enzyme (Figure 1A). When expressed, the transposase excises the transposon with its cargo from a plasmid and inserts it into the chromosomal DNA of the host. In practice, transposition results in stable integration of the transgene into a vertebrate host genome and prolongs expression of transgenic products such as Factor IX, β-glucuronidase and alpha-L-iduronidase (IDUA) [3,6–8] compared to most [9] non-integrating plasmid vectors.
Figure 1. Expression of IDUA from different promoters in NOD/SCID MPS I mice.

A. Vectors used in this study. The T2 SB transposon vectors (inverted arrowheads) contained either a “ubiquitous” CAGGS promoter or the liver-specific promoter (LSP) ApoEHCRhAAT. pKT2/ApoEHCRhAAT-hIDUA and pT2/miniCAGGS-hIDUA have different backbones and the selectable markers, kan and amp, respectively. B. Kinetics of hIDUA expression from LSP and CAGGS promoters in NOD.129(B6)-PrkdcscidIDUAtm1Clk/J (NOD/SCID) mice. Symbols: +SB11 females, filled red circles and solid lines; –SB females, open red circles and dotted lines; +SB11 males, filled blue squares and solid lines; –SB males, open blue squares and dotted lines. Each point represents the mean hIDUA activity ± SD, n=6. The CAGGS graph in panel B, is from Figure 1d of [8]. C. Comparison of transposition efficiency of SB11 and SB100X transposases in C57BL/6 mice by the excision product (EP) assay. 2μg of either pCMV-SB11 or pCMV-SB100X transposase-encoding plasmid was co-delivered with 15μg of pT2/CAGGS-GUSB [7] transposon plasmid. DNA for excision assay was isolated from livers 5 days p.i.
We have shown in immunodeficient NOD.129(B6)-Prkdcscid IDUAtm1Clk/J mice that the SB transposon system is efficacious in gene therapy of murine mucopolysaccharidosis type I (MPS I) [8], an inherited systemic disorder caused by IDUA deficiency [10]. However, the NOD/SCID MPS I mouse model (www.jaxmice.com), has drawbacks for studying both transposition and correction of MPS I because of impaired double-strand break repair that severely reduces transposition activity [11] and a shortened lifespan with mild clinical manifestations. However, evaluation of treatment effects in immunocompetent MPS I mice so far has not been feasible due to highly variable expression of IDUA following delivery of SB plasmids [7].
In our modified approach, we sought to minimize expression in antigen-presenting cells (APC), increase transposition efficiency by using the hyperactive transposase SB100X, and provide a more effective and less toxic cyclophosphamide regimen by transiently immunosuppressing mice at the time of vector administration. In addition, we aimed at reducing promoter shutdown and gender bias in IDUA expression associated with the CAGGS promoter [8]. As a result, we have attained sustained expression of IDUA in the liver at levels greater than 100-fold wildtype (WT) for one year in at least half of treated mice.
SB-mediated transgene expression is affected by many variables. The efficiency of transposition depends on the transposon and the transposase sequences, which are consistently being improved [2,5]. Since its first use as gene delivery vector [1], the SB transposase has been re-engineered from SB10 [12] through SB11 [13] and other intermediates [14] to the extremely hyperactive SB100X [15]. Likewise, the structure of the original transposon, T, has been re-engineered for greater activity, e.g. T2 [16] and other versions [17]. Besides the transposon system itself and its cargo, factors affecting expression are associated with the properties of expressed proteins, target cells or organs, delivery procedure, etc. [18]. Promoters regulating SB transposase and therapeutic genes can be either ubiquitously expressed or tissue-specific, either prone or resistant to silencing, and drive different levels of transgene expression. The expressed protein can either be restricted to the producer cell, e.g., luciferase [19], or it can escape the cell and be available for recapture by other cells as is typically the case with lysosomal hydrolases such as IDUA [10]. Host-related factors such as genetic background, gender and age can affect expression of the transposed gene. For example, we found that IDUA expression from the CAGGS promoter was silenced in female mice compared to male mice [8]. To gain insights into which variables affecting transgene expression lead to its loss and which facilitate stabilization of IDUA activity at therapeutic level, we examined decay rates in SB-mediated IDUA activity profiles obtained under a variety of conditions. We reasoned that these rates are by and large a reflection of various influences on transgene expression. We found that immune responses are the most important variable to control in order to prevent loss of transgene expression [20].
Results
SB transposon vectors support prolonged, high-level expression of human IDUA in immunocompetent mice
We designed a new set of human IDUA (hIDUA) expression cassettes to achieve reliable expression by meeting three criteria: 1) minimize expression of transgenic human IDUA in APCs to avoid anti-IDUA immune response; 2) provide high-level IDUA expression in both males and females; and 3) accommodate high-efficiency transposition. To achieve the first goal, we put the hIDUA cDNA under the regulation of a liver-specific promoter (LSP) ApoEHCRhAAT [21–23] instead of the ubiquitous CAGGS we used previously [7,8] (Figure 1A ). Transposons with and without a source of transposase were injected into WT C57BL/6 mice, which, although not IDUA-deficient, are immunogenically comparable to IDUA-deficient mice, because mice mount strong immune responses against human IDUA and innate responses to plasmids. Initial IDUA levels were equally high with both CAGGS and LSP promoters and gender bias was insignificant with the LSP in contrast to the 30-50-fold difference that we observed using the CAGGS promoter (Figure 1B ). Thus, LSP also satisfied our second requirement, i.e., lack of gender bias.
To measure the relative rates of transposition in liver tissue, we used the excision assay [24] in which plasmids that have lost the transposon by transposase-mediated excision (called excision products, EP) are quantified by qPCR. Comparison of transposition efficiency of SB11 [13] and SB100X [15] transposases by the excision assay in immunocompetent C57BL/6 mice showed that SB100X was about 15-fold more active in liver (Figure 1C ).
Expression of hIDUA in C57BL/6 mice over one year
Without immunosuppression, IDUA activity was lost in about 90% of IDUA vector-treated mice by 4-8 weeks post-injection (whether or not SB transposase-encoding plasmid was co-infused (±SB, Figures 2A and B). Cyclophosphamide (CP) treatment was used for immunosuppression in a regimen that consisted of four doses (120 mg/kg i.p.), -24, -6, +24 and +48 hrs relative to the hydrodynamic injection. The time-course of IDUA expression within each cohort of mice was not uniform (Figures 2C-F and Table 1 ). In four out of eight female mice treated with 5μg transposon-plasmid and in four out of six mice treated with the 25μg transposon-plasmid (+SB), IDUA activity was sustained for one year at more than 100-fold WT activity (Figures 2C and E), a level that is therapeutically corrective for MPS I mice [8]. Increasing the transposon dose from 5μg to 25μg DNA resulted in an initial approximately 5-fold increase in IDUA activity; however, the latter declined and stabilized by eight weeks post-infusion at levels that were not significantly higher than those attained with the 5μg dose (averages of 456- and 404-fold WT, respectively). In mice that did not receive the transposase plasmid (-SB), IDUA generally declined gradually over a one-year period, ending with an average activity level significantly below the therapeutic range (Figures 2D and F). Some mice appear to have been insufficiently immunosuppressed based on the rapid decline in IDUA expression after 4-36 weeks (Figures 2C, D and F , asterisks). Examples of gradual decline despite treatment +SB are indicated (Figures 2C and E, 1, 6 and 24, arrows).
Figure 2. Expression of transgenic hIDUA in C57BL/6 mice.

Mice aged 12 weeks were treated with 5μg or 25μg pKT2/LSP-IDUA ± pCMV-SB100X at 5:1 and 10:1 weight ratio, respectively. Plasma IDUA activity in untreated controls (WT) was 10±4 nmol/ml/hr. The dashed double-lines indicate the therapeutic level [8]. Arrows indicate expression profiles of +SB-treated mice with gradual or incomplete loss of IDUA activity. Asterisks indicate rapid decay in CP-immunosuppressed mice. Numbers indicate mouse IDs as listed in Table 1 .
Table 1. IDUA expression profiles and activity in plasma vis-à-vis transposition and transgene/plasmid maintenance in liver of immunosuppressed C57BL/6 mice one year post-treatment.
| Mouse ID | Profilea | Plasma IDUA, |
Frequency, DNA copy/cellb
|
||
|---|---|---|---|---|---|
| x WT c | IDUA | EP | SB | ||
| 5μg, +SB (Figure 2C ) | |||||
| 1 | G | 20 | 0.04 | bdld | 0.02 |
| 2 | S | 768 | 0.32 | 0.13 | 0.11 |
| 3 | S | 148 | 0.14 | 0.03 | 0.03 |
| 4 | R | 1 | bdl | bdl | Bdl |
| 5 | R | 1 | bdl | bdl | Bdl |
| 6 | R | 1 | bdl | bdl | bdl |
| 7 | S | 107 | 0.04 | 0.02 | 0.02 |
| 8 | S | 592 | 0.08 | 0.03 | 0.03 |
| eMean±SD for S | 404±330 | 0.15±0.12 | 0.05±0.05 | 0.05±0.04 | |
| 5μg, –SB (Figure 2D ) | |||||
| 9 | R | 1 | bdl | bdl | bdl |
| 10 | G | 17 | 0.58 | bdl | bdl |
| 11 | G | 34 | 0.45 | bdl | bdl |
| 12 | R | 2 | bdl | bdl | bdl |
| 13 | G | 25 | 0.09 | bdl | bdl |
| 14 | G | 19 | 0.37 | bdl | bdl |
| 15 | G | 8 | 0.15 | bdl | bdl |
| 16 | G | 22 | 0.52 | bdl | bdl |
| 17 | R | 3 | bdl | bdl | bdl |
| 18 | G | 10 | 0.21 | bdl | bdl |
| fMean±SD for G | 19.3±8.9 | 0.34±0.19 | bdl | bdl | |
| 25μg, +SB (Figure 2E ) | |||||
| 19 | S | 178 | 0.09 | 0.02 | bdl |
| 20 | S | 801 | 0.35 | 0.24 | 0.03 |
| 21 | R/S | 7 | bdl | bdl | bdl |
| 22 | S | 1051 | 0.59 | 0.22 | 0.03 |
| 23 | S | 164 | 0.08 | 0.02 | bdl |
| 24 | G/S | 84 | 0.07 | bdl | bdl |
| eMean±SD for S | 456±440 | 0.28±0.24 | 0.13±0.12 | 0.03 | |
| 25μg, –SB (Figure 2F ) | |||||
| 25 | G/R | 1 | bdl | bdl | bdl |
| 26 | G | 40 | 0.52 | bdl | bdl |
| 27 | G/R | 1 | bdl | bdl | bdl |
| 28 | G | 35 | 0.26 | bdl | bdl |
| fMean±SD for G | 37.5±3.5 | 0.4±0.2 | bdl | bdl | |
IDUA expression patterns are as defined in the Results: R-rapid decay; G-gradual decay; S-stable;R/S and G/R are hybrid profiles and G/S is very slow gradual decay; in R/S and G/S stabilization not confirmed;
qPCR determination of copy numbers of DNA sequences of human IDUA and SB100X (transposase) transgenes or excision/repair product plasmids;
IDUA activity was assayed in plasma; WT in C57BL/6 mice,10 nmol 4MU/ml/h;
bdl=below detection limit;
Mean±SD calculated for mice with stabilized expression;
Mean±SD calculated for mice with gradual expression.
Maintenance of transgenes and transposition in the liver
Whenever IDUA activity was rapidly lost (i.e., half-life of less than 3 days), neither transgenes nor plasmid sequences could be detected in samples of mouse liver by qPCR (Table 1 ). This suggested that in these cases, cells that initially took up and expressed the transgenic DNA were eliminated from the liver. In contrast, if the expression profile of transgenic IDUA was stable, or declined at a moderate rate, then we could always measure the IDUA transgene level by qPCR. Notably, plasmids that remained after transposition of the transposon cargo, i.e., EP, were detected only in mice with stabilized, high-level IDUA activity (Table 1 ). This observation supports our hypothesis that sustained, therapeutic-level transgene expression can be achieved after transposition. EP copy numbers per cell were not significantly different in mice dosed with either 5μg or 25μg of transposon (0.05±0.05 and 0.13±0.12, respectively); the variability in copy number may reflect episomal survival in liver cells one year post-injection. There was no evidence of EPs in mice whose IDUA activities gradually declined over a year (Table 1 ), although IDUA transgenes were detected at essentially the same frequency as in those with stabilized activity. Normalizing IDUA activity to IDUA transgene copy number for the non-saturating expression conditions of 5μg of transposon (404/0.15 = 2700 for +SB and 19/0.34 = 57 for –SB, Table 1) indicates that without SB transposase, expression of the transgenes was about 48-fold lower than with transposase (Figure 3A ). Transposition efficiency, reflected by the EP/IDUA ratio for both doses of transposon was 38±2% (n=8) in mice with sustained IDUA expression (Figure 3B ), i.e., about one-third of plasmids that survived as episomes in the liver after one year, had donated a transposon for integration. Our data from the 5μg transposon doses suggest that expression from episomes is about 130-fold (48/0.38) weaker than expression from transposed IDUA expression cassettes, i.e., expression of IDUA genes from episomes is about 1% that from chromosome.
Figure 3. IDUA transgene activity is enhanced when transposition is supported by SB transposase.
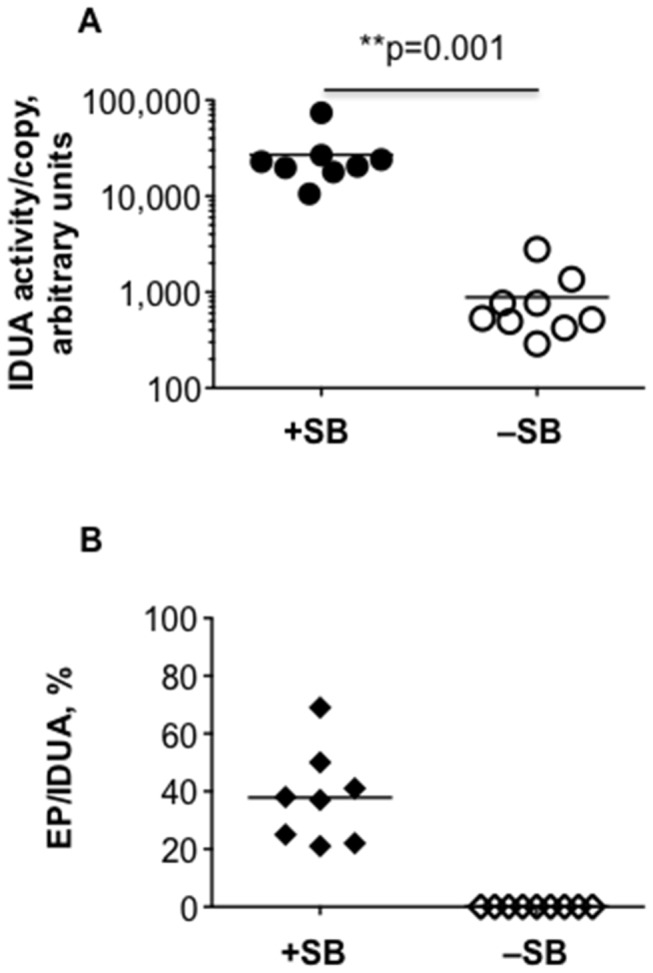
A. Plasma IDUA activities were normalized to IDUA copy per cell number. SB-treated mice with stabilized IDUA activity were compared with mice that were infused with only the transposon plasmids and did not lose IDUA activity in a rapid decay. The IDUA activity/copy number ratio, mean±SD, in arbitrary units, was +SB, 27000±19600 (n=8) and –SB, 900±800 -SB (n=9); **p=0.001. B. Transposition efficiency in mice with stabilized expression is calculated as the copy number ratio of pKT2/ApoEHCRhAAT-hIDUA plasmids that underwent transposon excision (EP, copy per cell) to the total number of pKT2/ApoEHCRhAAT-hIDUA (IDUA, copy per cell); mean EP/IDUA±SD =38±16%. Data from mice treated with 5μg and 25μg transposon doses were pooled.
Kinetics of loss of IDUA expression
Our inspection of transgene expression profiles under many different experimental conditions indicated that there are several components that account for loss of transgene expression. Quantified decay rates of LSP-IDUA expression from individual mice could be divided into three profiles: rapid decay, T1/2 =1.6±0.5 days (n=17) calculated from all mice treated without immunosuppression (Figure 4 , red solid line; data in Figures 2A and B); gradual decay, T1/2= 91±49 days (n=7), typical in CP-immunosuppressed mice that did not receive the transposase-encoding plasmid (Figure 4 , blue line; data in Figure 2D ); and sustained expression that was observed only in those immunosuppressed mice that received the SB-transposase-expressing plasmid (Figure 4 , black line; data in Figure 2C ). In those mice that lost expression at unpredictable times (Figure 4 , red dashed lines; mice 25 and 27 indicated by asterisks in Figure 2F ), the half-lives of expression were within the rapid decay range observed in animals that were not treated with CP.
Figure 4. Summary of quantitative evaluation of transgene expression in mice following hydrodynamic delivery to the liver.

Three decay rates can account for all of the profiles of expression that we have observed under about 128 different combinations of experimental variables. These decay rates are: initial (i), associated with promoter silencing [8], green line; rapid (ii), associated with loss of plasmids and transgenes, red line; and gradual (iii), blue line. Stabilized expression (iv), black line, at levels >100WTdeemed therapeutic [8] was attained only with LSP, transposition using SB 100X transposase and a transient 4-dose immunosuppression with CP administered intraperitoneally around the time of hydrodynamic delivery. We hypothesize that the delayed loss of expression (red dashed lines, mice 25 and 27 in Figure 2F), that has the same half-life of <3 days, is rapid as (ii).
Rapid decay at its earliest onset occurred between one and two weeks post-infusion, which is consistent with the onset of an adaptive immune response in the absence of immunosuppression by CP. Note that our T1/2 values have an inherent imprecision due to the infrequency of plasma IDUA measurements, which prohibits specific identification of the timing of initiation and termination of the decay process. We evaluated IDUA loss-of-expression rates obtained from our earlier studies using a CAGGS-IDUA expression cassette [7,8]. Compared to the LSP expression profiles, with CAGGS there was a gender-biased initial decay, steep in females and more gradual decay in males, during the first 3 days (Figures 1B and [8]). Thus, despite differences between CAGGS and LSP-expression due to gender bias and promoter silencing associated with CAGGS, the rates of final elimination of IDUA expression (T1/2=1-2 days) were similar for the LSP and CAGGS promoters, as was clearance of transgenes and excision products in those mice where all activity was lost.
Discussion
Achieving effective, life-long gene therapy is daunting because many variables can affect transgene expression. Such is the case with SB-mediated gene therapy for MPS I, where the goal is sustained, high-level IDUA expression in the liver.
In our previous work with the NOD/SCID MPS I model, IDUA expression profiles within a treatment group were consistent whereas high variability and inconsistency of expression were evident in immunocompetent MPS I mice even when immunosuppressed. This study, which employed delivery of immunogenic human IDUA-encoding transposons to immunocompetent mice, is a good model in which to examine the variable kinetics of transgene expression that one would expect in immunocompetent IDUA-deficient animals. Our achievement of sustained, high-level IDUA expression in 50% of C57BL/6 mice (Figures 2C and E) mimics what we have observed when treating MPS I mice. Our best results were obtained using the complete transposon system +SB100X transposase, a liver-specific promoter, and a single, four-dose regimen of CP immunosuppression around the time of transgene delivery. While the two former modifications to minimize immune responses are obvious, demonstration of SB-mediated effects on expression over one year has not been reported. That higher levels of SB activity were indeed attained is shown by quantitative measurements of excision circles (Figure 1C ), and the levels of plasma IDUA following SB transposition (e.g., Figure 2C ) are higher than those we have previously reported [7,8]. Our data suggest that transposition of the transgenic expression cassette into chromatin is necessary for long-term activity following plasmid delivery to the liver. However, stable transgene expression has been reported for minicircles [9,25–27] and mini-intronic plasmids [28], presumably because they either lack sequences that lead to transcriptional silencing [9,27] or are contained in an intron within the eukaryotic expression cassette [28]. Notably, in effectively immunosuppressed C57BL/6 mice, LSP-driven expression from episomes might continue for months, although we have never observed stabilized expression from episomes at >100 WT and the resulting episomal IDUA expression levels after one year never reached those deemed therapeutic for MPS I [8]. These observations are consistent with our calculations that expression from whole plasmids is about 1% that from transposed expression cassettes. They are also similar to what we saw upon Cre recombinase-induced inactivation of non-transposed erythropoietin transposons [29].
Prolonged persistence of low-level transgene expression from episomes may explain the absence of a clear-cut distinction in transgene activity between chromosomally integrated compared to episomal transposons in shorter-term experiments. By extending our studies out to one year, the long-term effects of using the complete SB system to achieve chromosomal integration of the IDUA expression cassette became obvious. Transposon doses of 5μg and 25μg yielded comparably high levels of stabilized transgenic IDUA activities (Figure 2C and E, Table 1 ). Although the increase of transposon+transposase dose from 5+1μg to 25+2.5μg, respectively, resulted in the expected approximately 5-fold increase in day-1 IDUA activity, there was a decline in average transgene expression in the 25+2.5μg dosed mice that ranged from 5 to 20-fold from peak activity at stabilization around 8 weeks p.i. (ibid). We interpret the gradual initial declines in +SB and –SB mice dosed with 25μg transposon-plasmid as being due to innate immune responses to a higher concentration of plasmid sequences, which can trigger transcriptional repression [9,25,27]. Thus, “more may be less” in the case of loading hepatocytes with transgenic constructs that remain as episomes.
Comparison of expression levels indicates the importance of carefully choosing the transgene promoter. IDUA activity from LSP was at therapeutic levels without the drawback of the initial fast shutdown of expression from CAGGS in females, which occured in both C57BL/6 and NOD/SCID mice [8] (Figures 1B and 4 ). However, the liver-specific promoter alone is not a solution against loss of expression, even with transposition. Although liver-specific (i.e., hepatocyte-specific) promoters minimize expression from APCs, overexpressed IDUA may not evade a robust immune system. Thus, while LSP-IDUA expression is not prone to silencing, LSP does not rescue expression from elimination by adaptive immune responses. Without immunosuppression, rapid decay, (Figures 2A and B) occurred at a time consistent with the onset of the adaptive immune response, 7 to 14 days post-infusion. Osborn et al. [28] demonstrated the presence of anti-IDUA IgG antibodies in the sera of mice treated with minicircle-encoded human IDUA without costimulatory blockade. However, it appears that cell-mediated responses are responsible for ultimately eliminating IDUA expression in both their experiments and in ours. These observations parallel ours in non-immunosuppressed mice that received CAGGS-IDUA; CTL-mediated immune responses removed transduced hepatocytes throughout the liver [7]. In this study, such rapid loss was evaded in over 70% of the mice treated with LSP-IDUA and transient CP regimen.
There are two plausible explanations for complete rapid loss of transgene expression in mice that were immunosuppressed: 1) some mice are resistant to immunosuppression by CP and 2) some intraperitoneal injections failed due to inadvertent placement of the injection other than intraperitoneal cavity [30,31]. Partial failure of immunosuppression with a 4-dose regimen could explain some of the outlier profiles of IDUA expression that we observed (Figures 2C and E, arrows). If loss of activity in the first six weeks in the 5μg and 25μg transposon-dosed groups was due to failed immunosuppression, then nearly 75% (8/11) of the truly immunosuppressed mice had stabilized expression of IDUA at therapeutic levels. If this is the case, our data suggest that improvement of immunosuppression will lead to sustained therapeutic effects in most mice.
In conclusion, in this and our previous studies [7,8,32] we have examined the levels of transgene expression for up to one year in transposon-injected mice under about 128 different combinations of SB transposases, transgene promoters, mouse genetic backgrounds, mouse gender, and cyclophosphamide immunosuppression. Broadly, two basic phenomena lead to the loss of transgene expression – presumably, transcriptional silencing and adaptive immune responses. Our quantitative analyses allowed us to discern the different kinetics underlying loss of transgene expression and better interpret experimental results. These causes can be divided into the ones that are feasible to control (e.g., choice of promoter, immunosuppression protocol, plasmid dose) and those that are poorly understood. We conclude that despite what looks like a large variation in patterns of transgene expression, many combinations of factors affecting transgene expression lead to only a limited number of post-treatment scenarios (Figure 4 ). The SB system is capable of providing effective gene therapy in mice, but improved methods of immunosuppression are needed to increase the percentage of treated animals that do not lose transgene expression.
Materials and Methods
Ethics statement
Mice were housed under specific pathogen-free conditions in AAALAC-accredited facilities. The Institutional Animal Care and Use Committee of the University of Minnesota approved all animal studies, protocol #1202A09921. Hydrodynamic injections were performed while the animals were under light anaesthesia to minimize suffering.
Plasmids
The source of the liver-specific promoter (LSP) ApoEHCRhAAT was pAAV-hFIX16 [23], a gift of Dr. Mark Kay (Stanford University, USA). The SB transposon pKT2/ApoEHCRhAAT-IDUA was engineered by subcloning an EcoRI-EcoRI fragment containing the full-length human α-L-iduronidase (hIDUA) cDNA into a unique EcoRI site of the L212-pKT2-ApoE-hAAT-BGintron plasmid. The ApoE-hAAT-BG intron was constructed by PCR amplifying the β-globin non-coding exon/mini intron from the mini-CAGGS vector and splicing it downstream of the ApoE-hAAT promoter. The PCR product was then TOPO-cloned (Invitrogen, Grand Island, NY, USA) and verified by sequencing. Subsequently, the ApoE-hAAT-BG was subcloned into the pKT2 vector containing the rabbit β-globin poly (A). The resulting plasmid, about 6.1 Kbp, had a kanamycin resistance gene, T2 inverted terminal repeats [16], the rabbit β-globin intron upstream and the rabbit β-globin polyadenylation site downstream the IDUA sequence, the latter was previously described [8]. The plasmid for transposase expression pCMV(CAT)T7-SB100, 4752 bp [15], was a gift from Dr. Zoltan Ivics (Max Delbruck Centre, Berlin, Germany). We refer to it in the text as pCMV-SB100X. As a negative control for transposition, the transposon plasmid was co-delivered with the pBluescript plasmid (Stratagene, La Jolla, CA, USA).
Animals and injections
C57BL/6 mice were purchased from Charles River (Wilmington, MA, USA) and housed under specific pathogen-free conditions in AAALAC-accredited facilities. Plasmids were prepared commercially (Aldevron, Fargo ND) and injected into mice using the hydrodynamics-based procedure as described [33]. Mice, in which complete dose failed to be injected or the injection time exceeded 7 sec, were eliminated from the study. Blood was collected at selected times by facial-vein phlebotomy without anesthesia. Animals were euthanized by carbon dioxide inhalation and perfused with saline. Livers were harvested and preserved for analyses as specified below. Cyclophosphamide (CP) at the dose of 120 mg/kg was injected intraperitoneally 24 h and 6 h before and 24 and 48 h after the hydrodynamic plasmid infusion.
IDUA enzyme assay
Liver and plasma specimens were stored at -80°C. Whole-liver specimens were pulverized by freezing in liquid nitrogen and grinding with mortar and pestle. Portions of each sample were extracted to assay for IDUA and plasmid excision product copy number by qPCR. IDUA activity was measured in plasma using the fluorometric assay with 4-MU-α-L-iduronide (Glycosynth, England) as a substrate, as described [34].
PCR analyses and copy number determinations
DNA was isolated from about 50 mg pulverized whole-liver specimens by phenol-chloroform extraction (www.molecularcloning.com). DNA copy number was determined by real-time PCR using an iCycler (Eppendorf), as previously described [7,8]. The mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) sequence, which served as an internal control of genomic DNA content, was amplified in a separate reaction. Standard curves for GAPDH, IDUA and SB genes and plasmid “excision products” (EP) were obtained as described (ibid). The mockEP used for preparation of EP standard curve was derived from the pKT2/ APOe-hAAT-BGintron plasmid in which the transposon was deleted between PvuII and DraI sites. The PCR primers for the excision assay were: FP-5’GCCTCGACGTTTCCCGTTGA and RP: 5’-GCGAGGAAGCGGAACAGATT. The primers to amplify both SB11 and SB100X were obtained from Dr. Vincent Keng (then at the University of Minnesota):
FP: 5’-ATGGGAAAATCAAAAGAAATCAGCC and
RP: 5’-CGCACCAAAGTACGTTCATCTCTA.
Statistical analysis
Data were analyzed using GraphPad Prizm 4.0 software (GraphPad Software Inc., San Diego, CA). The significance of differences between groups was determined based on exact two-tailed p values obtained with the Welch’s t-test. A value of p < 0.05 was considered statistically significant.
Acknowledgments
We thank Mark Kay for the gift of pAAV-hFIX 16 plasmid containinΥthe liver-specific promoter, and Vincent Keng for the gift of primers for SB11/100X transposases; Erik Olson for technical assistance; Michael Klug, Kelly Podetz-Pedersen, Jeff Reineke and Dan Wolf for helpful discussions.
Funding Statement
This project was supported by grants 1R01DK082516 and P01 HD32652 from the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ivics Z, Hackett PB, Plasterk RH, Izsvák Z (1997) Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 91: 501-510. doi: 10.1016/S0092-8674(00)80436-5. PubMed: 9390559. [DOI] [PubMed] [Google Scholar]
- 2. Izsvák Z, Hackett PB, Cooper LJ, Ivics Z (2010) Translating Sleeping Beauty transposition into cellular therapies: victories and challenges. Bioessays 32: 756-767. doi: 10.1002/bies.201000027. PubMed: 20652893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aronovich EL, McIvor RS, Hackett PB (2011) The Sleeping Beauty transposon system: a non-viral vector for gene therapy. Hum Mol Genet 20(R1): 14-20. doi: 10.1093/hmg/ddr140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hackett PB Jr, Aronovich EL, Hunter D, Urness M, Bell JB et al. (2011) Efficacy and safety of Sleeping Beauty transposon-mediated gene transfer in preclinical animal studies. Curr Gene Ther 11: 341-349. doi: 10.2174/156652311797415827. PubMed: 21888621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hackett PB, Largaespada DA, Cooper LJN (2010) A transposon and transposase system for human application. Mol Ther 18: 674-683. doi: 10.1038/mt.2010.2. PubMed: 20104209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yant SR, Meuse L, Chiu W, Ivics Z, Izsvak Z et al. (2000) Somatic integration and long-term transgene expression in normal and haemophilic mice using a DNA transposon system. Nat Genet 25: 35-41. doi: 10.1038/75568. PubMed: 10802653. [DOI] [PubMed] [Google Scholar]
- 7. Aronovich EL, Bell JB, Belur LR, Gunther R, Koniar B et al. (2007) Prolonged expression of a lysosomal enzyme in mouse liver after Sleeping Beauty transposon-mediated gene delivery: implications for non-viral gene therapy of mucopolysaccharidoses. J Gene Med 9: 403-415. doi: 10.1002/jgm.1028. PubMed: 17407189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aronovich EL, Bell JB, Khan SA, Belur LR, Gunther R et al. (2009) Systemic correction of storage disease in MPS I NOD/SCID mice using the Sleeping Beauty transposon system. Mol Ther 17: 1136-1144. doi: 10.1038/mt.2009.87. PubMed: 19384290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen ZY, He CY, Ehrhardt A, Kay MA (2003) Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo . Mol Ther 8: 495-500. doi: 10.1016/S1525-0016(03)00168-0. PubMed: 12946323. [DOI] [PubMed] [Google Scholar]
- 10. Neufeld EF, Muenzer J (2001) The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B. The Metabolic and Molecular Bases of Inherited Disease. Vol. III 8th edn. New York: McGraw-Hill; pp. 3427-3436. [Google Scholar]
- 11. Izsvák Z, Stüwe EE, Fiedler D, Katzer A, Jeggo PA et al. (2004) Healing the wounds inflicted by Sleeping Beauty transposition by double-strand break repair in mammalian somatic cells. Mol Cell 13: 279-290. doi: 10.1016/S1097-2765(03)00524-0. PubMed: 14759372. [DOI] [PubMed] [Google Scholar]
- 12. Plasterk RHA, Izsvák Z, Ivics Z (1999) Resident aliens: the Tc1/mariner superfamily of transposable elements. Trends Genet 15: 326-332. doi: 10.1016/S0168-9525(99)01777-1. PubMed: 10431195. [DOI] [PubMed] [Google Scholar]
- 13. Geurts AM, Yang Y, Clark KJ, Liu G, Cui Z et al. (2003) Gene transfer into genomes of human cells by the Sleeping Beauty transposon system. Mol Ther 8: 108-117. doi: 10.1016/S1525-0016(03)00099-6. PubMed: 12842434. [DOI] [PubMed] [Google Scholar]
- 14. Yant SR, Park J, Huang Y, Mikkelsen JG, Kay MA (2004) Mutational analysis of the N-terminal DNA-binding domain of Sleeping Beauty transposase: critical residues for DNA binding and hyperactivity in mammalian cells. Mol Cell Biol 24: 9239-9247. doi: 10.1128/MCB.24.20.9239-9247.2004. PubMed: 15456893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mátés L, Chuah MK, Belay E, Jerchow B, Manoj N et al. (2009) Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat Genet 41: 753-761. doi: 10.1038/ng.343. PubMed: 19412179. [DOI] [PubMed] [Google Scholar]
- 16. Cui Z, Geurts AM, Liu G, Kaufman CD, Hackett PB (2002) Structure-function analysis of the inverted terminal repeats of the Sleeping Beauty transposon. J Mol Biol 318: 1221-1235. doi: 10.1016/S0022-2836(02)00237-1. PubMed: 12083513. [DOI] [PubMed] [Google Scholar]
- 17. Zayed H, Izsvák Z, Walisko O, Ivics Z (2004) Development of hyperactive Sleeping Beauty transposon vectors by mutational analysis. Mol Ther 9: 292-304. doi: 10.1016/j.ymthe.2004.06.690. PubMed: 14759813. [DOI] [PubMed] [Google Scholar]
- 18. Ammar I, Izsvák Z, Ivics Z (2012) The Sleeping Beauty transposon toolbox. Methods Mol Biol 859: 229-240. doi: 10.1007/978-1-61779-603-6_13. PubMed: 22367875. [DOI] [PubMed] [Google Scholar]
- 19. Bell JB, Aronovich EL, Schreifels JM, Beadnell TC, Hackett PB (2010) Duration of expression and activity of Sleeping Beauty transposase in mouse liver following hydrodynamic DNA delivery. Mol Ther 18: 1796-1802. doi: 10.1038/mt.2010.152. PubMed: 20628359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arruda VR, Favaro P, Finn JD (2009) Strategies to modulate immune responses: a new frontier for gene therapy. Mol Ther 17: 1492-1503. doi: 10.1038/mt.2009.150. PubMed: 19584819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Okuyama T, Huber RM, Bowling W, Pearline R, Kennedy SC et al. (1996) Liver-directed gene therapy: a retroviral vector with a complete LTR and the ApoE enhancer-alpha 1-antitrypsin promoter dramatically increases expression of human alpha 1-antitrypsin in vivo. Hum Gene Ther 7: 637-645. doi: 10.1089/hum.1996.7.5-637. PubMed: 8845389. [DOI] [PubMed] [Google Scholar]
- 22. Miao CH, Ohashi K, Patijn GA, Meuse L, Ye X et al. (2000) Inclusion of the hepatic locus control region, an intron, and untranslated region increases and stabilizes hepatic factor IX gene expression in vivo but not in vitro . Mol Ther 1: 522-532. doi: 10.1006/mthe.2000.0075. PubMed: 10933977. [DOI] [PubMed] [Google Scholar]
- 23. Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M et al. (2006) Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med 12: 342-347. doi: 10.1038/nm1358. PubMed: 16474400. [DOI] [PubMed] [Google Scholar]
- 24. Liu G, Cui Z, Aronovich EL, Whitley CB, Hackett PB (2003) Excision of Sleeping Beauty transposons: parameters and applications to gene therapy. J Gene Med 6: 574-583. PubMed: 15133768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen ZY, Riu E, He CY, Xu H, Kay MA (2008) Silencing of episomal transgene expression in liver by plasmid bacterial backbone DNA is independent of CpG methylation. Mol Ther 16: 548-556. doi: 10.1038/sj.mt.6300399. PubMed: 18253155. [DOI] [PubMed] [Google Scholar]
- 26. Osborn MJ, McElmurry RT, Lees CJ, DeFeo AP, Chen ZY et al. (2011) Minicircle DNA-based gene therapy coupled with immune modulation permits long-term expression of α-L-iduronidase in mice with mucopolysaccharidosis type I. Mol Ther 19: 450-460. doi: 10.1038/mt.2010.249. PubMed: 21081900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gracey Maniar LE, Maniar JM, Chen ZY, Lu J, Fire AZ et al. (2013) Minicircle DNA vectors achieve sustained expression reflected by active chromatin and transcriptional level. Mol Ther 21: 131-138. doi: 10.1038/mt.2012.244. PubMed: 23183534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu J, Zhang F, Kay MA (2013) A Mini-intronic Plasmid (MIP): A Novel Robust Transgene Expression Vector In Vivo and In Vitro . Mol Ther 21: 954-963. doi: 10.1038/mt.2013.33. PubMed: 23459514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Score PR, Belur LR, Frandsen JL, Geurts JL, Yamaguchi T et al. (2006) Sleeping Beauty-mediated transposition and long-term expression in vivo: use of the Lox/Cre recombinase system to distinguish transposition-specific expression. Mol Ther 13: 617-624. doi: 10.1016/j.ymthe.2005.10.015. PubMed: 16356773. [DOI] [PubMed] [Google Scholar]
- 30. Miner NA, Koehler J, Greenaway L (1969) Intraperitoneal injection of mice. Appl Microbiol 17: 250-251. PubMed: 5775909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gaines Das R, North D (2007) Implications of experimental technique for analysis and interpretation of data from animal experiments: outliers and increased variability resulting from failure of intraperitoneal injection procedures. Lab Anim 41: 312-320. doi: 10.1258/002367707781282802. PubMed: 17640458. [DOI] [PubMed] [Google Scholar]
- 32. Podetz-Pedersen KM, Bell JB, Steele TW, Wilber A, Shier WT et al. (2010) Gene expression in lung and liver after intravenous infusion of polyethylenimine complexes of Sleeping Beauty transposons. Hum Gene Ther 21: 210-220. doi: 10.1089/hum.2009.128. PubMed: 19761403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bell JB, Podetz-Pedersen KM, Aronovich EL, Belur LR, McIvor RS et al. (2007) Preferential delivery of the Sleeping Beauty transposon system to livers of mice by hydrodynamic injection. Nat Protoc 2: 3153-3165. doi: 10.1038/nprot.2007.471. PubMed: 18079715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Garcia-Rivera MF, Colvin-Wanshura LE, Nelson MS, Nan Z, Khan SA et al. (2007) Characterization of an immunodeficient mouse model of mucopolysaccharidosis type 1 suitable for preclinical testing of human stem cell and gene therapy. Brain Res Bullet 74: 429-438. doi: 10.1016/j.brainresbull.2007.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]