

Abstract
Background
Subseafloor sediments accumulate large amounts of organic and inorganic materials that contain a highly diverse microbial ecosystem. The aim of this study was to survey the bacterial community of subseafloor sediments from the South China Sea.
Methodology/Principal Findings
Pyrosequencing of over 265,000 amplicons of the V3 hypervariable region of the 16S ribosomal RNA gene was performed on 16 sediment samples collected from multiple locations in the northern region of the South China Sea from depths ranging from 35 to 4000 m. A total of 9,726 operational taxonomic units (OTUs; between 695 and 2819 unique OTUs per sample) at 97% sequence similarity level were generated. In total, 40 bacterial phyla including 22 formally described phyla and 18 candidate phyla, with Proteobacteria, Firmicutes, Planctomycetes, Actinobacteria and Chloroflexi being most diverse, were identified. The most abundant phylotype, accounting for 42.6% of all sequences, belonged to Gammaproteobacteria, which possessed absolute predominance in the samples analyzed. Among the 18 candidate phyla, 12 were found for the first time in the South China Sea.
Conclusions
This study provided a novel insight into the composition of bacterial communities of the South China Sea subseafloor. Furthermore, abundances and community similarity analysis showed that the compositions of the bacterial communities are very similar at phylum level at different depths from 35-4000 m.
Introduction
Advances in high-throughput sequencing, also known as next-generation sequencing technology, including 454 pyrosequencing, Illumina sequencing etc., has significantly promoted microbial diversity and ecological studies. Deep sequencing makes it possible to precisely describe complicated microbial communities in several environments including marine, soil, animal or insects guts, which were all over 100 times more diverse than previously reported by traditional culture-dependent methods [1–4]. Marine microbial communities mediate biogeochemical ocean cycles including carbon, nitrogen and sulphur, and are probably play pivotal roles in maintaining marine ecosystem to prevent environmental changes such as warming and ocean acidification [5,6]. The ocean microbial community structure is influenced by pH, water temperature, salinity, silicate, seasonal shifts, and ocean currents as proved by surveys of the La Sal del Rey hypersaline lake located in southern Texas, USA [7], the western English Channel [8], and the western Arctic Ocean [9]. Phylotypic richness differed between summer and winter, but remarkable bacterial community structure stability was observed over time in the western Arctic Ocean [9]. Previously, the abundance of phylotypes in the oceanic microbial community was focused on abundant species because they were easily detectable. Thereafter, the 454 pyrosequencing technique revealed that most of the diversity of oceanic microbial communities is comprised of a high number of rare species and, in some cases, collectively comprise up to 75% of the abundance in their communities, named ‘‘the rare biosphere” [3,9,10]. The rare microbial biosphere together with abundant members play important roles in the functioning of the ocean ecosystem [11].
The South China Sea is one of the largest marginal seas and lies within the West Pacific marine. The detrital fluxes of sediments of South China Sea came from three of the largest rivers in the world (Mekong River, Red and Pearl rivers), and the monsoon activity plays an important role in the cycling of organic carbon and other biogenic component of sediments, which controlled the sea surface circulation. In summer, the subtropical waters are advected into the South China Sea through the southern straits and through the Taiwan Strait to exit the South China Sea. In winter, the cold and saline waters enter the South China Sea from the north as a reversed pattern [12].
It has an average water depth of 1200 m and a maximal depth of approximately 5380 m and has long been recognized as the global center of marine tropical biodiversity [13]. There are abundant organic matters in the deposited sediments of the seafloor [14]. Bacterial community richness estimated from rRNA sequences of ocean samples revealed hundreds to thousands of phylotypes [15,16]. To date, the South China Sea bacterial community distribution patterns remain unknown. Although a few surveys based on culture-dependent, denaturing gradient gel electrophoresis and constructed PCR product clone libraries methods analyzed the bacterial diversity, the obtained numbers of operational taxonomic units (OTUs) was lower than100 sequences for each sample [17–22]. These smaller datasets result in the underestimation of species richness and generally do not describe rare populations that might represent considerable diversity.
The goal of our study was to explore questions about bacterial diversity in subseafloor sediments of the South China Sea using sequences of the V3 region of the 16S rRNA gene as determined by 454 pyrosequencing. We examined 16 sediment samples obtained from the surface of shallow and deep-sea bottoms at depths from 35 to 4000 meters. We next compared the similarity of rare and abundant phylotypes of the communities. Furthermore, we described the taxonomic composition of bacterial communities from our samples.
Results and Discussion
To investigate bacterial diversity and provide an in-depth description of relative abundance in benthic regions of the South China Sea, 454 (Life Science, Branford, CT, USA) pyrosequencing technology was used to sequence 16 samples collected from different locations of the northern region of the South China Sea, with depths ranging from 35-4000 m (Table 1). We sequenced more than 265,000 PCR amplicons that span the V3 hypervariable region of rRNAs from our DNA preparations. Each sample produced from 13,000 to 20,000 reads. To eliminate random sequencing errors, reads were trimmed by removing the barcode and primers. Sequences with lengths less than 150bp or with ambiguous residues were also discarded. After removing potential erroneous sequences, data for the 16 samples was reduced from 21.8% to 10%, and on average data sizes were reduced by12.4%.
Table 1. Environmental DNA samples used for sequence analyses from the South China Sea.
| Sample ID | Lat °N, Long °W | Date | Depth, m |
|---|---|---|---|
| 1 | 111°23.243', 17°59.925' | 28/08/2011 | 1937.5 |
| 2 | 111°29.456', 17°58.927' | 28/08/2011 | 2026.6 |
| 8 | 112°08.124', 18°0.541' | 29/08/2011 | 2448.3 |
| 11 | 114°30.315', 18°1.841' | 31/08/2011 | 3563 |
| 12 | 117°2.913', 18°1.742' | 01/09/2011 | 3938 |
| 13 | 119°31.949', 18°2.114' | 02/09/2011 | 3023 |
| 15 | 119°44.263', 18°44.606' | 02/09/2011 | 3415 |
| 16 | 120°0.250', 20°22.971' | 04/09/2011 | 3536 |
| 17 | 119°19.896', 19°41.569' | 05/09/2011 | 2918 |
| 18 | 117°37.208', 21°23.202' | 06/09/2011 | 652 |
| 19 | 116°30.202', 22°29.355' | 06/09/2011 | 35 |
| 20 | 116°22.842', 20°38.176' | 07/09/2011 | 431 |
| 21 | 116°43.681', 20°14.109' | 07/09/2011 | 717 |
| 22 | 117°58.233', 19°0.368' | 08/09/2011 | 3739 |
| 25 | 115°17.868', 19°42.407' | 09/09/2011 | 1808 |
| 27 | 109°28.614', 18°13.299' | 26/08/2011 | 300 |
Lat, latitude, Long, longitude
Taxonomic richness of benthic bacterial communities of South China Sea
Based on a BLASTN search of trimmed 454 reads in the RefHVR_V3 database to identify the closest matches, sequence tags were clustered into groups by defining the variation from unique sequences to 10% differences. These clusters were calculated for OTUs, abundance-based coverage estimator (ACE), and the Chao1 estimator. The exponential Shannon index was calculated and the Simpson index at species, genus, and family levels were defined with the sequence similarity thresholds of 97%, 95%, and 90%, respectively. Rarefaction curves were generated based on a species level. In total, 9,726 unique OTUs at the 97% threshold were obtained from the 16 samples.
A ranges of 695 (sample 16) to 2819 OTUs (sample 19) were discovered in a total of 16 benthic sediment samples (Table 2). At the phylum level, all OTUs could be classified and belonged to 22 formally described bacterial phyla and 18 candidate phyla (Figure 1). Therefore, the overall known diversity in the South China Sea increases to 40 different bacterial phyla and candidate phyla, which was higher than the reported number of 35 phyla from other marine habitats, including the Arctic Ocean and the Western English Channel [8,11]. In this study, we used massively parallel signature sequencing technologies to obtain more than 265,000 sequences from 16 sediment samples from depths of 35 to 4000 m in a 1 million square kilometer area surrounded by the Sanyan Bay, Luzon Island, Shantou bay, and Paracel Islands to the west, north, east, and south. The rarefaction analysis of the OTUs indicated that the bacterial community diversity of sample 19 (depth <300 m) was significantly higher than other samples (depth >300 m; Figure 2). In addition, the similar result also was obtained from other sea area samples. For example, in a study to investigate the prokaryote diversity in the sub-seafloor biosphere (Accession: SRP001218), 79717 OTUs (97% genetic similarity) were obtained from three shallow sea sediments (~3.5 m) but only 46836 OTUs were obtained from three deep sea samples (depth 3860 m and 1326m) (Figure S1A). In another study (SRP001269) for investigating the microbial biodiversity of Indian Ocean region revealed the average amount of OTUs was 25367 for the shallow sea sediment samples, and the average amount of deep sea samples OTUs only reached 17556. This suggests that the bacterial community diversity of the shallow subseafloor near the coast was richer than the deep subseafloor area.
Table 2. Similarity-based OTUs and species richness estimates.
| Cluster distance |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.03 |
0.05 |
0.10 |
||||||||||||||
| Sample ID | reads | OUT | ACE | Chao | Shannon | Simpson | OUT | ACE | Chao | Shannon | Simpson | OUT | ACE | Chao | Shannon | Simpson |
| 1 | 15715 | 763 | 2403 | 1601 | 2.73 | 0.3279 | 527 | 1221 | 910 | 2.51 | 0.3586 | 277 | 381 | 396 | 2.21 | 0.3924 |
| 2 | 15536 | 708 | 2310 | 1526 | 2.33 | 0.4004 | 487 | 1130 | 837 | 2.14 | 0.4202 | 247 | 341 | 344 | 1.81 | 0.4562 |
| 8 | 14926 | 755 | 2400 | 1646 | 2.37 | 0.4047 | 533 | 1175 | 935 | 2.24 | 0.4078 | 271 | 341 | 337 | 1.96 | 0.4423 |
| 11 | 14290 | 854 | 3505 | 2081 | 3.59 | 0.1024 | 623 | 1734 | 1134 | 3.4 | 0.108 | 357 | 606 | 480 | 3.18 | 0.1158 |
| 12 | 13604 | 826 | 3087 | 1959 | 3.04 | 0.2487 | 596 | 1501 | 1198 | 2.83 | 0.2671 | 329 | 454 | 483 | 2.55 | 0.2916 |
| 13 | 14623 | 1002 | 4176 | 2475 | 4.16 | 0.0783 | 718 | 2252 | 1408 | 3.9 | 0.0855 | 416 | 832 | 650 | 3.55 | 0.095 |
| 15 | 14783 | 1264 | 4422 | 2872 | 4.32 | 0.061 | 950 | 2550 | 1807 | 4.01 | 0.0825 | 568 | 851 | 834 | 3.69 | 0.0966 |
| 16 | 12358 | 695 | 2597 | 1559 | 2.46 | 0.3775 | 485 | 1221 | 892 | 2.31 | 0.3801 | 242 | 330 | 323 | 1.97 | 0.4219 |
| 17 | 13978 | 799 | 2397 | 1717 | 2.52 | 0.3879 | 585 | 1339 | 1055 | 2.37 | 0.3947 | 349 | 483 | 481 | 2.08 | 0.4304 |
| 18 | 12139 | 705 | 2512 | 1768 | 2.59 | 0.3473 | 512 | 1293 | 1070 | 2.46 | 0.3501 | 268 | 366 | 355 | 2.16 | 0.3848 |
| 19 | 13287 | 2819 | 8559 | 5950 | 6.52 | 0.0056 | 2207 | 5358 | 4123 | 6.25 | 0.0067 | 1250 | 2201 | 1958 | 5.68 | 0.0088 |
| 20 | 12112 | 749 | 2446 | 1716 | 2.96 | 0.2785 | 530 | 1187 | 899 | 2.79 | 0.2884 | 293 | 383 | 372 | 2.52 | 0.3127 |
| 21 | 17768 | 1379 | 5367 | 3165 | 3.47 | 0.0976 | 1039 | 3151 | 2225 | 3.14 | 0.1234 | 642 | 1526 | 1188 | 2.85 | 0.1333 |
| 22 | 14999 | 908 | 3282 | 2139 | 3.03 | 0.283 | 645 | 1757 | 1202 | 2.88 | 0.2869 | 349 | 624 | 543 | 2.59 | 0.313 |
| 25 | 15750 | 1110 | 3782 | 2612 | 3.7 | 0.1163 | 839 | 2150 | 1538 | 3.51 | 0.1204 | 513 | 870 | 719 | 3.24 | 0.1311 |
| 27 | 16572 | 1138 | 3430 | 2506 | 2.52 | 0.407 | 843 | 2019 | 1561 | 2.38 | 0.4094 | 468 | 640 | 631 | 2.02 | 0.4532 |
Figure 1. Bacterial diversity richness and phylogenetic distribution on phylum (inner circle), class (middle circle), and order level (outer circle) (Based on 97% OTUs).

The shown phyla include formally described taxonomically phyla and candidate phyla. Phylum names are given in bold. Selected class and order groups are labeled. *represents unclassified groups.
Figure 2. Rarefaction analysis for the 16 sediment samples.

The curves were generated for 97% levels of OTU.
Abundance analysis showed that nine phyla account for over 95% of the total amplicons. The phyla include Proteobacteria, Firmicutes, Planctomycetes, Acidobacteria, Actinobacteria, Chloroflexi, Bacteroidetes, Gemmatimonadetes, and Nitrospirae (Figure 3A). Proteobacteria was the most abundant phylum in all samples and accounted for 37-80% of all bacterial amplicons. As the most dominant community in marine environments, Proteobacteria has also been described in the Arctic Ocean [11], marine sponges [1], and the benthic North Sea [23].
Figure 3. Bacterial community distribution in 16 sediment samples.

A, the relative abundances of different phyla; B, the relative abundances of different classes in Proteobacteria. The relative abundance is presented in terms of percentage in total effective bacterial sequences per sample.
The dominant communities of sample 19 were Proteobacteria (44.7%), Planctomycetes (15.5%), Acidobacteria (12.9%), Bacteroidetes (7.0%), and Actinobacteria (4.5%). In the remaining samples, Proteobacteria, Firmicutes, Actinobacteria, Chloroflexi and Bacteroidetes accounted for 11-42% of effective sequences. This suggests that in shallow sea sediments, the phyla Planctomycetes and Acidobacteria are the second and third dominant communities, accounting for 28.3% of tags. However, in deep-sea sediments, Firmicutes and Actinobacteria are the second and third most dominant communities, representing 8.9% to 45.2% of the tags. Planctomycetes and Acidobacteria only accounted for 0.4% to 14.8% of tags from overall deep sea sediment samples.
In Proteobacteria, Gammaproteobacteria were the most dominant class in all samples, accounting for 53.4% to 76.8%, and Deltaproteobacteria was the second most dominant class, accounting for 37.8% of tags in sample 19. However, Alphaproteobacteria and Betaproteobacteria were the second and the third most dominant classes in the deep-sea sediment samples (Figure 3B). Gammaproteobacteria, the predominant bacterial group, prevailed over other taxa identified in several deep-sea investigations, including the Eastern Mediterranean Sea [24] and Northeastern Pacific Ocean [25]. Sequences affiliated with Desulfobacterales, Myxococcales, and Sh765B-TzT-29, dominated the Deltaproteobacteria and their common role is to regulate the sulfur cycle. The ocean represents a major reservoir of sulfur on Earth and microbial transformation of sulfur compounds has had a profound effect on the properties of the biosphere and continues to affect geochemistry [26]. The types of sulfur-metabolizing microorganisms of Deltaproteobacteria include sulfate reducers, organic sulfur utilizers, and sulfur reducers [27].
Abundance analysis of bacterial community diversity comparing the South China Sea with other marine area sediment samples was performed at the phylum level and the class level for Proteobacteria. The bacterial communities of different marine area displayed similarity in dominant groups, which including Proteobacteria (Gammaproteobacteria, Deltaproteobacteria), Planctomycetes, Firmicutes, Actinobacteria, Acidobacteria, Bacteroidetes, and Chloroflexi (Figure S1). The phylum Proteobacteria, being most dominant group, was observed in a large proportion of shallow sea and deep-sea sediments. In the global overview, the dominant groups of bacterial communities of shallow sea sediments were similar with that of deep-sea sediments in the same sea area.
The benthic bacterial communities of the South China Sea showed similarity with other sea areas, but each area still has different characteristics in their dominant group’s proportions. Such as in samples from cluster CFU1 (Atlantic Ocean near Portugal), where more than 54.7% of the sequences belong to candidate phylum OP9; in a 5000 m depth benthic sample from the Indian Ocean, the class Alphaproteobacteria reached 93.4% in total sequences. The geographical location has a strong impact on microbial community composition, and explained 22.2% of the observed differences in benthic communities[16].
Principal-component analysis of the bacterial community of the South China Sea
To determine the distribution and biogeography of the bacterial community, the 454 data were analyzed in relation to sampling locations using principal-component analysis (Figure 4). The similarity of microbial communities among our 16 samples, collected from depths of 35-4000 m, was monitored with PCA at the phylum level and OTU0.03 levels. At the phylum level, samples collected from similar depths or locations did not contain more similar microbial communities to one another than to samples collected at other depths or locations (Figure 4A). For example, samples 16, 18, and 20, collected from depths of 3536 m, 652 m and 431 m, respectively, fell into a cluster, while samples 19, 20, and 21 were all collected from the Shantou bay but fell into different clusters. However, a large amount of deep-sea samples clustered into a group by PCA analysis at the OTU 0.03 levels (Figure 4B). The deep-sea samples exhibited a noticeable and regular separation from shallow sea in the first principal component (PC1), and 80.55% of the variation in the data explained in PC1. A large part of deep-sea samples corresponded to negative values and the shallow sea sample 19 had the highest value, which was more than 50. The shallow sea sample 19 had the lowest negative value in the second principal component (PC2), which represented 10.05% of the variation; a large amount of deep-sea samples fall in a range from -20 to +20.
Figure 4. Principal-component analysis of bacterial communities in the South China Sea.

A, in the level phylum; B, in the level OTU 0.03. The Number refers to the samples ID which is explained in Table 1.
In addition, the richness of bacterial diversity of the deep subseafloor was less than that of the shallow subseafloor (Figure 2). Because the nutrient-limited, low energy-flux, and high press environment of deep subseafloor leads to microbial abundance, activity and turnover rates in the deep subseafloor are extremely low relative to those in other global habitats [28].
Rare biosphere in South China Sea
Deep sequencing revealed that the rare biosphere accounts for tremendous diversity of marine bacterial communities. We defined the rare phylotypes as having a frequency <0.01% and abundant phylotypes at a frequency >1% within a sample, according to previous reports [11,29] .
The rare phylotype distribution in each sample was similar and within a range of 55-64% of OTUs but comprised <5% of the sequence abundance (Figure 5). Overall, 62 abundant phylotypes were counted in shallow sea and deep-sea sediment samples, covering 57-76% and 22% of the number of sequences, respectively. However, this only comprised <1.5% of the OTUs. In addition, 18 phylotypes of 62 abundant phylotypes were found to be abundant in some samples but rare in others. For example, the hydrocarbon-degrading Gammaproteobacteria, Cycloclasticus, a member of the rare biosphere, becomes an abundant member of the community when supplied appropriate conditions but returns to the rare biosphere by the loss of the supplied condition [30]. This suggests that the distribution of the bacterial rare biosphere is not obviously different in shallow and deep-sea sediments. The most abundant phylotype belonged to Gammaproteobacteria, accounting for 42.6% of total sequences and possessing absolute predominance in the South China Sea sediments. Interestingly, with the exception of the most abundant phylotype, 13 of 14 abundant phylotypes from sample 16 were rare or absent in other samples. Probably the difference of the sediments environment of deep and shallow sea such as temperature, pH, pressure, enrichment in reduce of compounds, and metals bring the variation of abundant phylotypes. In addition, the seasonal cycle brings environmental changes from the Pacific coast, which results in these populations dominating the assemblage during a period of time then declining in the abundant biosphere to the point where they become undetectable [31]. The majority of sequences from the community were occupied by abundant taxonomic groups. These groups were thought to be well adapted to the environment and to contribute the most to biomass production [32,33]. Conversely, the biomass of rare groups is negligible compared to that of the abundant members of the community, and their contribution to carbon flow is relatively small. However, some members of the rare biosphere that are actively growing can significantly contribute to particular elements such as nitrogen and sulfur cycling. For example, Desulfosporosinus account for <0.01% of the total cell count but could contribute to most of the sulfate reduction in peat [34,35].
Figure 5. Phylogenetic composition of rare bacterial phylotypes (≤0.01%) and richness of abundant phylotypes (≥1%frequency) for the 16 sediment samples.

Further analyses of community structure and function are needed to investigate the interaction between rare phylotypes and marine subseafloor habitats. The 454 pyrosequencing technique still has pitfalls and may affect our results for the rare biosphere. Even the largest published metagenomic investigations inadequately represent the full extent of microbial diversity, and primer efficiency for generating 16S rRNA gene fragments is limited [10,36].
Conclusions
Overall, this study is the first metagenomic analysis using pyrosequencing to characterize a more comprehensive overview of the bacterial community of the South China Sea. The massively parallel signature sequencing of 16 samples of subseafloor sediments and data analysis allowed novel insights into the complex composition of this microbial community. Detected diversity of bacterial communities increased to 40 different bacterial phyla and 18 candidate phyla, and the majority populations of the South China Sea at different depths from 35-4000 m showed a high similarity at the phyla level. However, the shallow subseafloor showed a higher bacterial diversity compared to deep subseafloor.
Materials and Methods
Sample collection and preparation for pyrosequencing
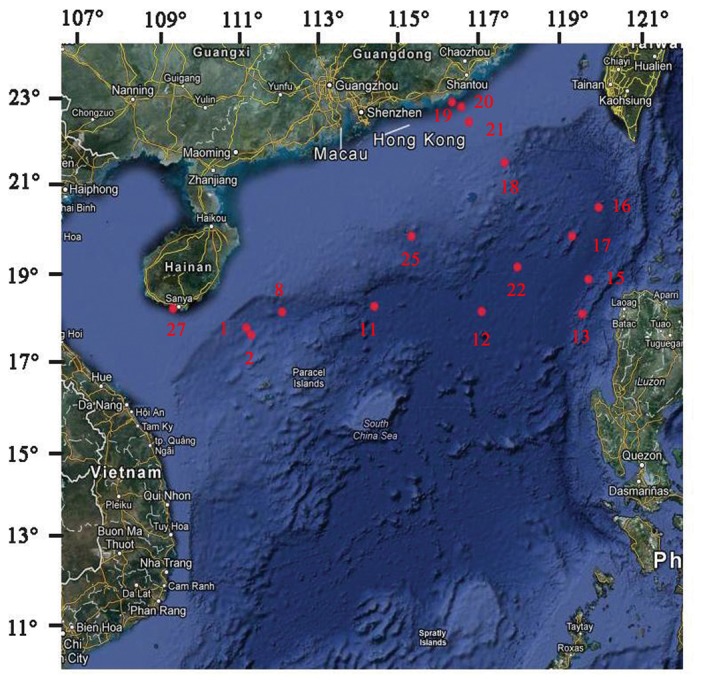
Samples were collected from the deposited sediment of benthic regions in 16 different locations of the northern region of South China Sea (between Lat °N, 111° 23.243' and 120° 0.250' to Long °W 17° 58.927' and 22° 29.355') at water depths ranging from 35-4000 m. Samples were transferred to sterilized plastic tubes and stored at -80°C (Figure 6). There are no specific permits required for the described sampling because collections did not involve endangered species and did not occur within a designated marine protected area, private reserve, or park.
Figure 6. Map of sampling locations in the South China Sea.
Total genomic DNA was extracted from 1g of the sediment samples using the EZgeneTM Soil gDNA Kit (Biomiga, San Diego, CA, USA) according to the manufacturer’s protocols. Bacterial 16S rRNA at the V3 hypervariable region were amplified using a set of primers designed by adding a 10-nucleotide barcode to the forward primer, 8F, (5’-AGAGTTTGATCCTGGCTCAG-3’) and reverse primer, 533R, (5’-TTACCGCGGCTGCTGGCAC-3’). The amplification reaction mixture contained 5 Units of Pfu Turbo DNA polymerase (Stratagene, La Jolla, CA, USA), 1×Pfu reaction buffer, 200 µM dNTPs (TaKaRa, Dalian, China), 0.2µM barcoded primer, and 20ng genomic DNA template for a total volume of 100 µl. PCR was performed with a thermal cycler (Bio-Rad, USA) under the following condition: 5 min at 94°C, 25 cycles of 30 s at 94°C plus 45 s at 55°C plus 30 s at 72°C, and finally 5 min at 72°C. The PCR products were purified by using a PCR Purification Kit (QIAGEN, Hilden, Germany). The 200 ng purified amplicons from each sample were mixed then pyrosequenced on the ROCHE 454 FLX Titanium platform (Roche, Basel, Switzerland) at Majorbio Bio Tech Co. Ltd (Shanghai, China). The results of the raw data have been deposited into the EMBL/GenBank/DDBJ Nucleotide Sequence Data Libraries under the Accession Number: DRA000705.
Sequence analysis
Raw sequence reads were filtered to eliminate the effect of random sequencing errors. The primer and barcode of each read were removed and trimmed. The sequences that (i) were shorter than 150 nucleotides, (ii) contained ambiguous bases (N), or (iii) contained homopolymer regions (>6 repetitions of the same base) were excluded.
Taxonomic identification of the reads (‘‘tags’’) were performed following the process described by Sogin et al [3]. All optimized reads were trimmed down to equal lengths (150bp), which contained the V3 hypervariable region. The tags were searched using the NCBI BLASTN tool in the reference database of hypervariable region tags (RefHVR_V3, http://vamps.mbl.edu/) based on the SILVA database, version 106 [37], and queried reads showing the minimum distance with the reference V3 tags were grouped and assigned the same phylotype. Taxonomy was assigned to each trimmed reference sequence (400bp) with Mothur 1.24.0 [38].
The reference ICoMM 454 bacterial 16S pyrotag dataset for comparing with data of this study were obtained from the internet (VAMPS website: http://vamps.mbl.edu, MICROBIS website: http://icomm.mbl.edu/microbis) and are provided in Table S1; sample descriptions were from Zinger, L. et al. [15].
Diversity and statistical analysis
The sequences were grouped into OTUs sharing ≥97%, ≥95%, or ≥90% similarity by DOTUR [39]. The bacterial community richness indices (non-parametric ACE and the Chao1) and diversity indices (Shannon and Simpson estimators) were calculated using Mothur and Shannon-ace-table.pl software programs (Majorbio, Shanghai, China). Rarefaction curves were calculated using Mothur and the software program Plot-rarefaction (Majorbio, Shanghai, China). Heat maps were drawn by hierarchal clustering performed in the R software environment (http://www.R-project.org) within the function “vegdist” in the Vegan Community Ecology Package.
Supporting Information
Bacterial community distribution in 17 ecosystems type. A, the relative abundances of different phyla in 17 ecosystems type which including 125 sediments samples; B, the relative abundances of different classes in Proteobacteria of 17 ecosystem type. The relative abundance is presented in terms of percentage in total effective bacterial sequences in an ecosystem type. NH1, sample 19 in this study, NH2, the other 15 samples except sample 19 in this study, other ecosystem type were described in Table S1.
(TIF)
Datasets, location and parameters of marine sediments samples.
(DOCX)
Funding Statement
This work was supported by a grant (Code: BK2012695) from the Natural Science Foundation of Jiangsu Province, China and by the grants from the National High Technology Research and Development Program of China (863 Program, 2012AA092104), as well as by the Project Startup Foundation for Distinguished Scholars of Jiangsu University (Code: 10JDG084), the Chinese Academy of Sciences for Key Topics in Innovation Engineering (KSCX2-EW-G-12) and Sponsored by the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Schmitt S, Tsai P, Bell J, Fromont J, Ilan M et al. (2012) Assessing the complex sponge microbiota: core, variable and species-specific bacterial communities in marine sponges. ISME J 6: 564-576. doi: 10.1038/ismej.2011.116. PubMed: 21993395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Godoy-Vitorino F, Goldfarb KC, Karaoz U, Leal S, Garcia-Amado MA et al. (2012) Comparative analyses of foregut and hindgut bacterial communities in hoatzins and cows. ISME J 6: 531-541. doi: 10.1038/ismej.2011.131. PubMed: 21938024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM et al. (2006) Microbial diversity in the deep sea and the underexplored "rare biosphere". Proc Natl Acad Sci U S A 103: 12115-12120. doi: 10.1073/pnas.0605127103. PubMed: 16880384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Teixeira LC, Peixoto RS, Cury JC, Sul WJ, Pellizari VH et al. (2010) Bacterial diversity in rhizosphere soil from Antarctic vascular plants of Admiralty Bay, maritime Antarctica. ISME J 4: 989-1001. doi: 10.1038/ismej.2010.35. PubMed: 20357834. [DOI] [PubMed] [Google Scholar]
- 5. Fuhrman JA (2009) Microbial community structure and its functional implications. Nature 459: 193-199. doi: 10.1038/nature08058. PubMed: 19444205. [DOI] [PubMed] [Google Scholar]
- 6. Graham DE, Wallenstein MD, Vishnivetskaya TA, Waldrop MP, Phelps TJ et al. (2012) Microbes in thawing permafrost: the unknown variable in the climate change equation. ISME J 6: 709-712. doi: 10.1038/ismej.2011.163. PubMed: 22094350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hollister EB, Engledow AS, Hammett AJ, Provin TL, Wilkinson HH et al. (2010) Shifts in microbial community structure along an ecological gradient of hypersaline soils and sediments. ISME J 4: 829-838. doi: 10.1038/ismej.2010.3. PubMed: 20130657. [DOI] [PubMed] [Google Scholar]
- 8. Gilbert JA, Field D, Swift P, Newbold L, Oliver A et al. (2009) The seasonal structure of microbial communities in the Western English Channel. Environ Microbiol 11: 3132-3139. doi: 10.1111/j.1462-2920.2009.02017.x. PubMed: 19659500. [DOI] [PubMed] [Google Scholar]
- 9. Kirchman DL, Cottrell MT, Lovejoy C (2010) The structure of bacterial communities in the western Arctic Ocean as revealed by pyrosequencing of 16S rRNA genes. Environ Microbiol 12: 1132-1143. doi: 10.1111/j.1462-2920.2010.02154.x. PubMed: 20132284. [DOI] [PubMed] [Google Scholar]
- 10. Huber JA, Mark Welch DB, Morrison HG, Huse SM, Neal PR et al. (2007) Microbial population structures in the deep marine biosphere. Science 318: 97-100. doi: 10.1126/science.1146689. PubMed: 17916733. [DOI] [PubMed] [Google Scholar]
- 11. Galand PE, Casamayor EO, Kirchman DL, Lovejoy C (2009) Ecology of the rare microbial biosphere of the Arctic Ocean. Proc Natl Acad Sci U S A 106: 22427-22432. doi: 10.1073/pnas.0908284106. PubMed: 20018741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tamburini F, Adatte T, Follmi K, Bernasconi SM, Steinmann P (2003) Investigating the history of East Asian monsoon and climate during the last glacial-interglacial period (0-140 000 years): mineralogy and geochemistry of ODP Sites 1143 and 1144, South China Sea. Mar Geol 201: 147-168. doi: 10.1016/S0025-3227(03)00214-7. [DOI] [Google Scholar]
- 13. Barber PH, Palumbi SR, Erdmann MV, Moosa MK (2000) Biogeography. A marine Wallace's line? Nature 406: 692-693. doi: 10.1038/35021135. PubMed: 10963585. [DOI] [PubMed] [Google Scholar]
- 14. Wang SH, Yan W, Magalhaes HV, Chen Z, Pinheiro ML et al. (2012) Calcium isotope fractionation and its controlling factors over authigenic carbonates in the cold seeps of the northern South China Sea. Chin Sci Bull 57: 1325-1332. doi: 10.1007/s11434-012-4990-9. [DOI] [Google Scholar]
- 15. Schauer R, Bienhold C, Ramette A, Harder J (2010) Bacterial diversity and biogeography in deep-sea surface sediments of the South Atlantic Ocean. Isme J 4: 159-170. doi: 10.1038/ismej.2009.106. PubMed: 19829317. [DOI] [PubMed] [Google Scholar]
- 16. Zinger L, Amaral-Zettler LA, Fuhrman JA, Horner-Devine MC, Huse SM et al. (2011) Global patterns of bacterial beta-diversity in seafloor and seawater ecosystems. PLOS ONE 6: e24570. doi: 10.1371/journal.pone.0024570. PubMed: 21931760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cao HL, Hong YG, Li M, Gu JD (2011) Diversity and abundance of ammonia-oxidizing prokaryotes in sediments from the coastal Pearl River estuary to the South China Sea. Antonie Van Leeuwenhoek Int J Gen Mol Microbiol 100: 545-556. doi: 10.1007/s10482-011-9610-1. PubMed: 21717206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liao L, Xu XW, Wang CS, Zhang DS, Wu M (2009) Bacterial and archaeal communities in the surface sediment from the northern slope of the South China Sea. J Zhejiang Univ-Sci B 10: 890-901. doi: 10.1631/jzus.B0920181. PubMed: 19946953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang GH, Dong JD, Li XA, Sun HM (2010) The bacterial diversity in surface sediment from the South China Sea. Acta Oceanol Sin 29: 98-105. doi: 10.1007/s13131-010-0028-9. [DOI] [Google Scholar]
- 20. Moisander PH, Beinart RA, Voss M, Zehr JP (2008) Diversity and abundance of diazotrophic microorganisms in the South China Sea during intermonsoon. Isme J 2: 954-967. doi: 10.1038/ismej.2008.51. PubMed: 18528417. [DOI] [PubMed] [Google Scholar]
- 21. Zhou MY, Chen XL, Zhao HL, Dang HY, Luan XW et al. (2009) Diversity of Both the Cultivable Protease-Producing Bacteria and Their Extracellular Proteases in the Sediments of the South China Sea. Microb Ecol 58: 582-590. doi: 10.1007/s00248-009-9506-z. PubMed: 19301066. [DOI] [PubMed] [Google Scholar]
- 22. Zhang Y, Jiao NZ, Sun ZY, Hu AY, Zheng QA (2011) Phylogenetic diversity of bacterial communities in South China Sea mesoscale cyclonic eddy perturbations. Res Microbiol 162: 320-329. doi: 10.1016/j.resmic.2010.12.006. PubMed: 21187147. [DOI] [PubMed] [Google Scholar]
- 23. Sapp M, Parker ER, Teal LR, Schratzberger M (2010) Advancing the understanding of biogeography-diversity relationships of benthic microorganisms in the North Sea. FEMS Microbiol Ecol 74: 410-429. doi: 10.1111/j.1574-6941.2010.00957.x. PubMed: 20735476. [DOI] [PubMed] [Google Scholar]
- 24. Polymenakou PN, Bertilsson S, Tselepides A, Stephanou EG (2005) Bacterial community composition in different sediments from the Eastern Mediterranean Sea: a comparison of four 16S ribosomal DNA clone libraries. Microb Ecol 50: 447-462. doi: 10.1007/s00248-005-0005-6. PubMed: 16328662. [DOI] [PubMed] [Google Scholar]
- 25. Kouridaki I, Polymenakou PN, Tselepides A, Mandalakis M, Smith KL Jr. (2010) Phylogenetic diversity of sediment bacteria from the deep Northeastern Pacific Ocean: a comparison with the deep Eastern Mediterranean Sea. Int Microbiol 13: 143-150. PubMed: 20890848. [DOI] [PubMed] [Google Scholar]
- 26. Klotz MG, Bryant DA, Hanson TE (2011) The microbial sulfur cycle. Front Microbiol 2: 241 PubMed: 22144979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sievert SM, Kiene R, Schulz H (2007) The sulfur cycle. J Oceanogr 20: 117–123. doi: 10.5670/oceanog.2007.55. [DOI] [Google Scholar]
- 28. Kobayashi T, Koide O, Mori K, Shimamura S, Matsuura T et al. (2008) Phylogenetic and enzymatic diversity of deep subseafloor aerobic microorganisms in organics- and methane-rich sediments off Shimokita Peninsula. Extremophiles 12: 519-527. doi: 10.1007/s00792-008-0157-7. PubMed: 18368287. [DOI] [PubMed] [Google Scholar]
- 29. Pedrós-Alió C (2006) Marine microbial diversity: can it be determined? Trends Microbiol 14: 257-263. doi: 10.1016/j.tim.2006.04.007. PubMed: 16679014. [DOI] [PubMed] [Google Scholar]
- 30. Teira E, Lekunberri I, Gasol JM, Nieto-Cid M, Alvarez-Salgado XA et al. (2007) Dynamics of the hydrocarbon-degrading Cycloclasticus bacteria during mesocosm-simulated oil spills. Environ Microbiol 9: 2551-2562. doi: 10.1111/j.1462-2920.2007.01373.x. PubMed: 17803779. [DOI] [PubMed] [Google Scholar]
- 31. Fuhrman JA, Hewson I, Schwalbach MS, Steele JA, Brown MV et al. (2006) Annually reoccurring bacterial communities are predictable from ocean conditions. Proc Natl Acad Sci U S A 103: 13104-13109. doi: 10.1073/pnas.0602399103. PubMed: 16938845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cottrell MT, Kirchman DL (2003) Contribution of major bacterial groups to bacterial biomass production (thymidine and leucine incorporation) in the Delaware estuary. Limnol Oceanogr 48: 168-178. doi: 10.4319/lo.2003.48.1.0168. [DOI] [Google Scholar]
- 33. Zhang Y, Jiao NZ, Cottrell MT, Kirchman DL (2006) Contribution of major bacterial groups to bacterial biomass production along a salinity gradient in the South China Sea. Aquat Microb Ecol 43: 233-241. doi: 10.3354/ame043233. [DOI] [Google Scholar]
- 34. Galli I, Teira G, Perlo F, Bonato P, Tisocco O et al. (2008) Animal performance and meat quality in cull cows with early weaned calves in Argentina. Meat Sci 79: 521-528. doi: 10.1016/j.meatsci.2007.10.007. PubMed: 22062912. [DOI] [PubMed] [Google Scholar]
- 35. Pester M, Bittner N, Deevong P, Wagner M, Loy A (2010) A 'rare biosphere' microorganism contributes to sulfate reduction in a peatland. ISME J 4: 1591-1602. doi: 10.1038/ismej.2010.75. PubMed: 20535221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang Y, Qian PY (2009) Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLOS ONE 4: e7401. doi: 10.1371/journal.pone.0007401. PubMed: 19816594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W et al. (2007) SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35: 7188-7196. doi: 10.1093/nar/gkm864. PubMed: 17947321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M et al. (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537-7541. doi: 10.1128/AEM.01541-09. PubMed: 19801464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schloss PD, Handelsman J (2005) Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71: 1501-1506. doi: 10.1128/AEM.71.3.1501-1506.2005. PubMed: 15746353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Bacterial community distribution in 17 ecosystems type. A, the relative abundances of different phyla in 17 ecosystems type which including 125 sediments samples; B, the relative abundances of different classes in Proteobacteria of 17 ecosystem type. The relative abundance is presented in terms of percentage in total effective bacterial sequences in an ecosystem type. NH1, sample 19 in this study, NH2, the other 15 samples except sample 19 in this study, other ecosystem type were described in Table S1.
(TIF)
Datasets, location and parameters of marine sediments samples.
(DOCX)