

Abstract
Chronic hepatitis C virus (HCV) infection is responsible for the development of liver cirrhosis and hepatocellular carcinoma. It has been a tremendous burden on global health care systems. With the advent of a number of new direct-acting and host-targeting antiviral agents, current interferon-α- and ribavirin-based HCV therapy has started to move towards an interferon-sparing or even interferon-free strategy. In this regard, a recently identified NS5A inhibitor, daclatasvir, showed a great promise in clinical trials as another new class of direct-acting anti-HCV therapeutics, with a distinct mechanism of action. In this review, a variety of preclinical as well as clinical proof-of-concept studies of daclatasvir, including the studies of its discovery, mechanism of action, viral resistance, and host polymorphism profiles are reviewed. In addition, a role of daclatasvir in the future therapy for HCV patients is discussed briefly.
Keywords: hepatitis C virus, nonstructural protein 5A, NS5A inhibitor, hepatitis C treatment
Introduction
The global estimation of hepatitis C virus (HCV)-infected patients is around 170 million, with 4 million new infections reported annually.1,2 There has been a solid cause-and-effect relationship between persistent HCV infection and the development of chronic and often deadly liver diseases, including chronic hepatitis, cirrhosis, and hepatocellular carcinoma.3,4 HCV infection is accountable for more than two-thirds of all cases of liver cancers and transplantations performed in the US.5 In spite of the introduction of two recently approved direct-acting antiviral agents (DAAs), boceprevir and telaprevir, specifically targeting an NS3 viral protease,6 the current standard of care for HCV patients still relies on the combined treatment of a weekly injection of pegylated (PEG) interferon-α (IFNα) and daily administration of the nucleoside analog, ribavirin (RBV). However, this IFNα-based combination therapy has been associated with undesirable side effects, including flu-like symptom, hemolytic anemia, depression, and suicidal thoughts.7 Pulmonary and metabolic complications, including pneumonitis and diabetic ketoacidosis, were also found as IFNα-induced side effects of high severity.8–10 In addition, its unsatisfactory efficacy, which is less than 50% for genotype (GT) 1 and 4 patients has been severe clinical problems.11,12 This emphasizes an urgent need to develop an IFNα-sparing or even an IFNα-free anti-HCV regimen. In this regard, one of the NS5A inhibitors, daclatasvir (DCV), recently developed by Bristol-Myers Squibb and currently progressing through the last stage of clinical trials, has ignited a great excitement as a new and promising component of combination therapy. This review discusses the relevant preclinical as well as clinical data regarding DCV, in order to predict its potential role in HCV treatment in the near future.
Classification and organization of HCV
HCV belongs to the Flaviviridae family of viruses with a single-stranded ribonucleic acid (RNA) of a positive polarity as its viral genome. Following entry into a host liver cell, HCV delivers its RNA genome inside the target cell. Then, internal ribosome entry site (IRES)-assisted translation of its RNA genome results in the production of a polyprotein composed of around 3000 amino acids. This polyprotein subsequently cleaves into ten different viral proteins by virtue of host and virally-encoded proteases.13,14 The first three viral proteins freed from the original polyprotein are called structural proteins. They include core capsid protein and two envelope glycoproteins, E1 and E2.15 They serve as the structural components of a mature virus particle. The remaining seven viral nonstructural (NS) proteins, including p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B, represent the components of a functional replication complex in charge of viral RNA genome amplification on endoplasmic reticulum membranes.16–19 DCV has been shown to abrogate HCV replication by specifically targeting the critical functions of an NS5A protein in the replication complex.20
Role of NS5A in the HCV life cycle
NS5A is a 447 amino acid–long phosphoprotein with an RNA-binding activity.21 In spite of lacking an enzymatic activity, NS5A has been shown to play a major role in HCV RNA replication as well as in infectious particle assembly.22 It consists of an amino terminal amphipathic helix plus three structurally distinct domains. An amino terminal amphipathic helix was demonstrated to be required for the endoplasmic reticulum membrane association, to support HCV RNA replication.23 Domain I was shown to form a dimer, to accommodate a single-strand RNA molecule.24 Replication-enhancing adaptive mutations were found around domain II, suggesting its possible role in HCV RNA replication.25 In addition, the essential role of domain III in HCV infectious particle assembly was also characterized.26 Based on results of these domain mapping studies, NS5A was proposed to play a role in fine-tuning both viral replication as well as particle production in overall HCV life cycle.22
Discovery of DCV and its preclinical studies
DCV was initially discovered by means of a chemical genetic strategy.27,28 In this strategy, a large number of chemically diverse compounds are first screened based on their effect on HCV replication, without any knowledge of their mechanism of action. Then, an error-prone HCV RNA-dependent RNA polymerase (RDRP) generates mutant HCV genomes, which are resistant to identified compounds. Analysis of these resistant mutant genomes leads to the identification of their potential viral targets. Unlike the traditional bottom-up screening approach, which requires the definition of their molecular targets in the first place, this top-down strategy enjoys a freedom of targets selection as long as they are key elements of HCV replication. The discovery of DCV as an NS5A inhibitor proves the usefulness and applicability of this bias-free approach.
By taking advantage of this strategy, Lemm et al conducted a cell-based high-throughput HCV replicon screening.29 In this screening, HCV replication was measured indirectly by monitoring an NS3 protease activity, coupled with fluorescence resonance energy transfer (FRET). Following successful screening, a number of compounds with a thiazolidinone core were identified as inhibitors of HCV replication. Among them, BMS-824 demonstrated a half-maximum effective concentration (EC50) of 5 nM and a half-maximum cytotoxic concentration of more than 50 μM, with a therapeutic index of more than 10,000 in the GT 1b replicon.29 However, BMS-824 showed a relatively poor activity against the GT 1a replicon (> 10 μM). After realizing symmetry as an important contributor to their antiviral activity in GT 1a, BMS-790052 (a previous name of DCV), with a symmetric structure and greater potency against both the GT 1a and 1b replicons, was identified and developed as a candidate for advancement into clinical trials (Figure 1).27 DCV has been the most potent HCV replication inhibitor reported so far, with a picomolar EC50 towards the GT 1 replicons (50 pM against GT 1a, 9 pM against GT 1b), which represent the majority of HCV patients.27 In addition, DCV also turned out to be an inhibitor of the GT 2a JFH1 replicon and cell culture infectious J6/JFH1 virus, with EC50 values of 46.8 pM and 16.1 pM, respectively20 A study of hybrid HCV replicons harboring either GT 3a or 4 NS5A genes also confirmed their susceptibilities to DCV with EC50 values ranging from 120–870 pM for GT 3a and from 7–13 pM for GT 4.30,31 In addition to its potent antiviral activity alone, the additive or synergistic effects of combining NS3 protease or NS5B RNA polymerase inhibitors with DCV on replicon inhibition and clearance were also demonstrated in an in vitro replicon study32 In addition, GT 1a replicon elimination was shown to be markedly enhanced by the combination of PEG-IFNα and DCV33 These data strongly suggested DCV as a prime candidate for a new component of an IFN-sparing or IFN-free anti-HCV regimen in the future.32
Figure 1.
Structure of daclatasvir.
Mechanism of action for DCV
In order to gain an insight into its potential mechanism of action, the HCV replicon of GT 1b was exposed to DCV to generate mutant HCV replicon variants resistant to DCV. After successful sequencing of these variants, a DCV sensitivity domain was found to map to the N terminus of NS5A.29 Identification of NS5A as a possible target of DCV was more or less unexpected, given the ability of DCV to achieve such an extraordinary antiviral potency by targeting a viral protein without any known enzymatic activity. Specifically, DCV was shown to be able to target a specific function of NS5A, which involves downregulation of the hyperphosphorylation of NS5A (Figure 2).29 By exploring this unique phosphorylation-regulating property of DCV, two distinct functions of NS5A in HCV RNA replication were also discovered. These include a cis-acting function of basally phosphorylated NS5A in maintaining HCV replication complex and a trans-acting function of hyperphosphorylated NS5A in modulating HCV assembly and particle formation. DCV seems to target both the cis- and trans-acting functions of NS5A (Figure 2).20 When the effect of DCV on polyprotein processing was further examined, DCV was shown to promote the accumulation of an HCV polyprotein intermediate.34 In line with this evidence, the disruption of polyprotein processing by an NS3 protease inhibitor also led to downregulation of the hyperphosphorylation of NS5A, further emphasizing an intimate relationship between polyprotein processing and the phosphorylation status of NS5A.29 In addition, hyperphosphorylated NS5A proteins were demonstrated to separate into different membrane fractions during discontinuous sucrose gradient centrifugation, upon DCV treatment.34 A localization pattern of only hyperphosphorylated NS5A proteins was shown to be disrupted specifically by DCV.34 This finding was further supported by the demonstration of altered biochemical fractionation and the subcellular localization of specific populations of NS5A proteins by DCV.35,36 Based on these observations, DCV seems to perturb the function of new HCV replication complexes, rather than acting on preformed complexes, by modulating the NS5A phosphorylation status (Figure 2).36 In addition, one modeling study37 even suggested the possibility of the existence of two modes of action for DCV, due to the multiple roles of NS5A in HCV replication and particle production. These involved the DCV-dependent blockage of two distinct stages of the viral life cycle – viral RNA synthesis and virion assembly/secretion. This study also yielded a more precise estimate of the serum HCV half-life, which was calculated to be around 45 minutes.
Figure 2.
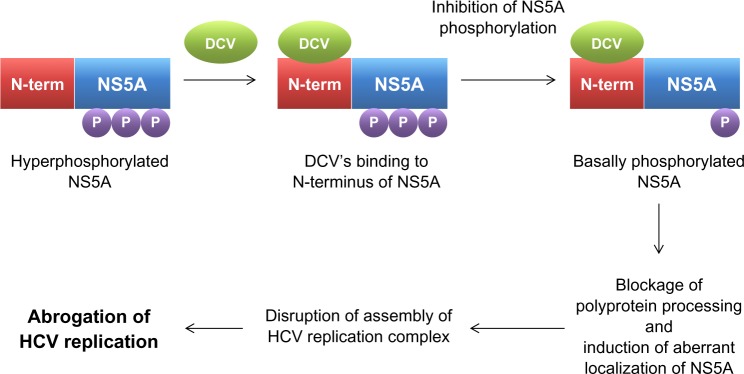
Mechanism of action for daclatasvir.
Abbreviations: DCV, daclatasvir; HCV, hepatitis C virus; NS, nonstructural viral protein; N-term, N terminal; P, phosphate.
In vitro and in vivo HCV resistance to DCV
The analysis of the resistance profiles of HCV variants against DCV is critical in order to predict the potential synergistic efficacy of combination therapy with other anti-HCV agents. Nonoverlapping resistance profiles among different anti-HCV drugs usually lead to a more favorable combination treatment outcome. A majority of DCV-resistant variants turned out to include the L31V, P32L, Q54L, and Y93H mutations of NS5A in the GT 1b replicon, and the M28T, Q30H, Q30R, L31M, L31V, P32L, and Y93C mutations of NS5A in the GT 1a replicon.27,29,38 Specifically, the Y93H mutation displayed the maximum fold resistance phenotype, in spite of its negative impact on fitness of the virus.20 Nevertheless, DCV was reported to be able to maintain subnanomolar potency against all the variants from GT 1b with single amino acid substitutions.38 This suggests a requirement of multiple mutations for the development of significant in vivo resistance in this genetic background.38 Fortunately, most of the DCV-resistant variants remained fully sensitive to IFNα and the small-molecule inhibitors of HCV protease and polymerase.33,38 Most importantly, the resistant variants observed in this in vitro study were very similar to those observed in a multiple ascending dose monotherapy clinical trial of DCV.27,39 These data further validate an in vitro HCV replicon study as an invaluable model to predict clinical efficacy and resistance profile.33
In the following paragraphs, each DCV-related clinical trial data is discussed in depth. In order to conduct a systemic analysis of each DCV-related clinical trial data, all the treatment regimens used in the DCV clinical studies were classified based on the presence or absence of IFN, as studies of DCV alone, studies of IFN-containing regimens, and studies of IFN-free regimens. The characteristics of each clinical study are summarized in Table 1.
Table 1.
Summary of the clinical trial results of daclatasvir
| Study | Reference | Number of patients | GT | Treatment | Note |
|---|---|---|---|---|---|
| DCV-alone regimens | |||||
| Phase I | Gao et al27 | 6 | 1 | Single dose of 1, 10, and 100 mg |
aCmin>bEC90 cT1/2 10–14 hr MCAE: headache |
| 3.3 log10 reduction at 24 hr postdose with 10 and 100 mg of DCV | |||||
| A sustained reduction for an additional 120 hr in two GT 1b patients | |||||
| A more sustained decline for GT 1b than for GT 1a patients | |||||
| Phase II | Nettles et al39 | 30 | 1 | Once daily dose of 1, 10, 30, 60, or 100 mg or twice daily dose of 30 mg for 14 days |
cT1/2 12–15 hr MCAE: headache |
| A 2.8 to 4.1 log10 of mean maximum decline | |||||
| Viral rebound on or before day 7 | |||||
| A greater antiviral response for GT 1b than for GT 1a patients | |||||
| IFN-containing regimens | |||||
| Phase IIa | Pol et al28 | 48 | 1 | Once daily dose of 3, 10, and 60 mg of DCV with PEG-IFNα and RBV for 48 weeks | PEP: eRVR |
| 8%, 42%, 83%, and 75% of eRVR achieved for patients treated with 0, 3, 10, and 60 mg of DCV with PEG-IFNα and RBV, respectively | |||||
| Phase IIa | Lok et al43 | 21 (all null responders to previous PEG-IFNα and RBV treatment) | 1 | Once daily dose of 60 mg of DCV and 600 mg of ASV with or without PEG-IFNα and RBV for 24 weeks | PEP: SVR12 MCAE: diarrhea |
| 36% and 90% of SVR12 achieved for patients treated with PEG-IFNα and RBV alone or together with 60 mg of DCV and 600 mg of ASV, respectively | |||||
| IFN-free regimens | |||||
| Phase IIa | Sulkowski et al45 | 170 | 1, 2, 3 | Once daily dose of 60 mg of DCV and 400 mg of SOF with or without RBV for 24 weeks | PEP: SVR24 |
| Cotreatment of DCV and SOF with or without RBV achieved SVR24 in more than 93% of patients with HCV GT 1, 2, or 3 | |||||
| Phase IIa | Sulkowski et al44 | 41 (all null responders to previous telaprevir or boceprevir plus PEG-IFNα and RBV treatment) | 1 | Once daily dose of 60 mg of DCV and 400 mg of SOF with or without RBV for 24 weeks | PEP: SVR12 |
| The all-oral, once-daily combination of DCV and SOF with or without RBV achieved SVR12 in all 41 HCV GT 1–infected patients who failed prior treatment | |||||
| Phase IIa | Chayama et al46 | 9 (all null responders to previous PEG-IFNα and RBV treatment) | 1b | Once daily dose of 60 mg of DCV and twice daily dose of 600 mg of ASV for 24 weeks | PEP: SVR12 MCAEs: diarrhea and headache |
| 100% of SVR12 achieved, for patients treated with 60 mg of DCV and 600 mg of ASV | |||||
| No viral breakthrough or relapse observed during or after treatment | |||||
| Phase IIa | Suzuki et al47 | 21 (all null responders to previous PEG-IFNα and RBV treatment) 22 (all ineligible/intolerant for PEG-IFNα and RBV treatment) |
1b | Once daily dose of 60 mg of DCV and twice daily dose of 200 mg of ASV for 24 weeks | PEP: SVR12 |
| 90.5% and 63.6% of SVR12 achieved, for null responders and ineligible/intolerant patients, respectively | |||||
| Viral breakthrough for three ineligible/intolerant patients | |||||
Notes:
Minimal plasma concentration
protein binding–adjusted 90% median effective concentration
mean plasma elimination half-life.
Abbreviations: ASV, asunaprevir; DCV, daclatasvir; eRVR, extended rapid virologic response at 4 and 12 weeks after start of treatment; GT, genotype; PEP, primary end point; MCAE, most common adverse event; PEG-IFNα, pegylated interferon-α; RBV, ribavirin; SOF, sofosbuvir; SVR12, sustained virologic response at 12 weeks after end of treatment; SVR24, sustained virologic response at 24 weeks after end of treatment; hr, hours.
DCV-alone regimens
Phase I clinical study by Gao et al27
In order to test the clinical efficacy of DCV against HCV infection, a randomized, double-blind, placebo-controlled, single ascending dose study was initiated.27 First, six subjects with GT 1 chronic HCV were treated with doses of 1, 10, and 100 mg of DCV as an oral solution. The mean plasma elimination half-life of DCV was reported to range from 10 to 14 hours by a pharmacokinetic study. After a single oral dose of 10–100 mg of DCV, the 24-hour plasma concentration turned out to be above tenfold the protein-binding-adjusted 90% median effective concentration values (EC90) for HCV GT 1a and 1b. In regards to its antiviral efficacy, the administration of a single 100 mg dose of DCV resulted in a 3.3 log10 reduction in the mean viral load measured 24 hours postdose. This reduction was further sustained for an additional 120 hours in two patients infected with GT 1b virus. Interestingly, a greater and more sustained decline in HCV RNA was detected for subjects infected with GT 1b than for subjects infected with GT 1a.27 The genotypic analysis of clinical samples showed that the major observed HCV variants had substitutions at amino acid positions identified using the in vitro replicon system (M28T, Q30H/R, and L31M mutations for GT 1a and Y93H mutation for GT 1b).27 Headache was the most frequent adverse event, as reported by four subjects after the administration of DCV. This study was the first clinical validation of an the vivo efficacy of the HCV NS5A inhibitor DCV.
Phase I clinical study by Nettles et al39
In the Phase I clinical trial by Nettles et al, a double-blind, placebo-controlled, sequential panel, a multiple ascending dose study was conducted in thirty patients with chronic HCV GT 1 infection.39 The patients were treated with DCV (1, 10, 30, 60, or 100 mg once daily or 30 mg twice daily) or placebo for 14 days. The mean maximum decline from baseline in HCV RNA was observed to be around 2.8–4.1 log10 IU/mL. Inconsistent with a previous result, those who were infected with HCV GT 1b generally demonstrated greater antiviral responses than those who were infected with HCV GT 1a. Specifically, undetectable HCV RNA levels were detected at day 14 in four of the seven patients with GT 1b infection versus none of the 17 patients with GT 1a infection. Viral rebound was also observed on or before day 7 of treatment. Once-daily dosing yielded median peak plasma concentrations at 1–2 hours postdose and a mean terminal half-life of 12–15 hours. A steady state was also achieved following 3–4 days of daily dosing. The identified resistant substitutions included M28, Q30, L31, and Y93 for GT 1a and L31 and Y93 for GT 1b. After 14 days of dosing of 1–100 mg DCV, all patients were able to achieve maximal plasma concentration values above the replicon (EC90) of 0.283 and 0.0362 ng/mL for GT 1a and GT 1b, respectively. This was a first proof-of-concept clinical study with multiple doses of an HCV NS5A inhibitor, DCV.
In vitro and in vivo correlation study by Fridell et al40
An in vitro and in vivo correlation study was conducted for the genotypic and phenotypic analysis of variants resistant to DCV in humans.40 In this study, a sequence analysis was performed on viral complementary DNA isolated from serum specimens, collected at baseline and days 1, 2, 4, 7, and 14 postdosing after a single ascending dose study and a 14-day multiple ascending dose monotherapy study. HCV RNA was reported to remain detectable in all GT 1a–infected patients, and viral breakthrough (VBT) was observed during the course of treatment in the majority of these patients. Due to no cross-resistance observed with other DAAs, DCV was regarded as an excellent candidate for combination therapy. The resistant mutation analysis also found the linkage of L31V/M with Y93H mutations in GT 1b. DCV appeared to have a low genetic barrier for resistance since the plasma minimum concentrations of DCV monotherapy at the dose range used for the multiple ascending dose study were not sufficient to prevent all VBTs because of the emergence of resistant variants.
Single-dose pharmacokinetic study of DCV by Bifano et al41
DCV was shown to be eliminated by hepatic metabolism and direct biliary excretion.41 DCV was also known for its high protein binding. Consistent with its daily dosing, DCV demonstrated a mean plasma half-life of 12 to 15 hours in patients with chronic HCV infection.39 Due to the varying degrees of hepatic impairment, the DCV target population exhibited different levels of drug exposure. In order to examine the influence of hepatic impairment on the singledose pharmacokinetics of DCV, in this open-label, parallel-group study, subjects without active HCV who had mild, moderate, or severe hepatic impairment and controls with normal hepatic function received a single oral dose of 30 mg DCV, and a plasma analysis was performed predose and postdose for 72 hours. DCV turned out to be well tolerated in hepatically impaired patients. The time to reach maximal plasma concentration in the hepatically impaired subjects was similar to that in the controls, and the plasma half-life across the study groups was consistent with the observations in HCV-infected patients.39 In addition, exposure to active and unbound DCV was found to be comparable between the controls and subjects with moderate or severe hepatic impairment. Therefore, DCV dose adjustments in subjects with hepatic impairment would not be recommended.
IFN-containing regimens
Phase IIa clinical study by Pol et al28
In this Phase IIa trial, a randomized parallel-group, double-blind placebo-controlled dose-finding study was conducted for those patients with previously untreated chronic HCV GT 1 infection without cirrhosis.28 The patients were randomly assigned to receive PEG-IFNα (180 μg per week) and RBV (1000–1200 mg daily) plus placebo or 3 mg, 10 mg, or 60 mg of DCV taken once daily, for 48 weeks. The primary efficacy end point was undetectable HCV RNA at 4 weeks and 12 weeks after the start of treatment (extended rapid virologic response [eRVR]). A total of 48 patients were randomly assigned to make 12 per group. Although 15 patients discontinued treatment before week 48, five of 12 patients (42%) who received 3 mg DCV ten of 12 (83%) who received 10 mg DCV nine of 12 (75%) who received 60 mg DCV and one of 12 (8%) who received placebo achieved eRVR. Eleven virologic failures were found including three cases of VBT, four cases of virologic relapse, and four cases of detectable HCV RNA at the end of 48 weeks of therapy. This study suggested a further development of regimens containing 60 mg of DCV for the treatment of chronic GT 1 HCV infection.
Relevant case report by Fontana et al42
In this study, there was a one case report of successful PEG-IFNα, RBV and DCV combination therapy for recurrent cholestatic HCV after liver retransplantation.42 Three months after the second transplant, DCV (20 mg/day), PEG-IFNα (180 μg/week), and RBV (800 mg/day) were prescribed for an early recurrent cholestatic HCV patient. Serum HCV RNA became undetectable at week 3 of treatment and remained undetectable during 24 weeks of triple therapy and during the posttreatment follow up. DCV was well tolerated and the minimum drug concentrations were within the targeted range throughout the treatment.
IFN-free regimens
Phase IIa clinical study by Lok et al43
This was a preliminary study of two antiviral agents, including the NS5 inhibitor DCV and the NS3 protease inhibitor asunaprevir (ASV), in HCV GT 1 patients.43 This open-label, Phase IIa study recruited 21 patients with chronic HCV GT 1 infection who had not had a response to previous treatment with PEG-IFNα and RBV (null responders). Eleven patients (group A) were treated with the NS5A inhibitor DCV (60 mg once daily) and the NS3 protease inhibitor ASV (600 mg twice daily) alone. The other ten patients (group B) were treated with DCV and ASV in combination with PEG-IFNα and RBV for 24 weeks. A sustained virologic response (SVR) 12 weeks after the end of the treatment period was achieved in four patients in group A (36%; two of nine with HCV GT 1a and two of two with GT 1b). Six patients (all with HCV GT 1a) had VBT while receiving therapy, and resistance mutations to both antiviral agents were found in all cases. One patient had a viral response at the end of treatment but had a relapse after the treatment period. On the other hand, all ten patients in group B had an SVR at 12 weeks after treatment; nine patients in group B had an SVR at 24 weeks after treatment. Diarrhea was the most common adverse event in both groups. Six patients had transient elevations of alanine aminotransferase levels to more than three times the upper limit of the normal range. This clinical study further supported a proof of concept that SVR can be achieved by two DAAs without PEG-IFNα and RBV therapy. Nonoverlapping resistance profiles were also found in DCV- and ASV-treated patients. This clinical trial presented proof that the use of DAAs could provide a clinical cure for HCV in patients who are refractory to the current standard of care.
Phase IIa clinical study by Sulkowski et al44,45
This was a randomized, open-label, two-stage, parallel-group, Phase IIa study aiming to evaluate the efficacy and safety of all-oral and once-daily combination of DCV and sofosbuvir (SOF), a nucleotide analog NS5B polymerase inhibitor, with or without RBV, for 24 weeks in treatment-naïve patients infected with HCV GT 1, 2, and 3.44 A total of 126 GT 1 and 44 GT 2/3 patients were randomly assigned to receive a once-daily dose of 60 mg of DCV plus 400 mg of SOF, with or without a prior once-daily dose of 400 mg of SOF for 7 days, or with or without RBV. The primary efficacy end point was undetectable HCV RNA at 24 weeks after the end of treatment (SVR24). It was seen that cotreatment of DCV and SOF with or without RBV achieved SVR24 in more than 93% of patients with HCV GT 1, 2, or 3. The virologic response did not vary according to IL28B GT, viral subtype, or the administration of RBV. The DCV plus SOF regimen was well tolerated, and a low hemoglobin was observed only in patients taking RBV.
In order to extend the potential of the all-oral and once-daily combination of DCV plus SOF regimen in patients who previously failed prior treatment with telaprevir or boceprevir plus PEG IFNα/RBV, another similarly designed clinical study was performed. In this randomized, open-label, parallel-group, Phase IIa clinical study, those 41 difficult-to-treat GT 1 patients, including those with prior nonresponse, relapse, or viral breakthrough, were randomly assigned to receive a once-daily dose of 60 mg of DCV plus 400 mg of SOF with or without RBV.45 The primary efficacy end point was undetectable HCV RNA at 12 weeks after the end of treatment (SVR12). Surprisingly, the all-oral, once-daily combination of DCV and SOF with or without RBV achieved SVR12 in all 41 HCV GT1–infected patients who failed prior treatment. This clinical trial also provided evidence that the use of DCV with other DAA with a high genetic barrier, such as SOF, could result in a clinical elimination of HCV in patients who previously failed current standard of care.
Phase IIa clinical study by Chayama et al46
This was a dual open-label, Phase IIa study with the NS5A inhibitor, DCV, and the NS3 protease inhibitor, ASV, initiated in HCV virus GT 1b–infected null responders, including ten patients with chronic HCV GT 1b infection and previous null response to PEG-IFNα and RBV.46 The patients received dual DAA treatment for 24 weeks with DCV (60 mg once-daily) and ASV (initially 600 mg twice-daily, then subsequently reduced to 200 mg twice daily). The primary efficacy end point was SVR12. Nine patients completed the full course of treatment. Surprisingly, all nine patients achieved SVR12 and SVR24. The HCV RNA also remained undetectable posttreatment, even in the patient who discontinued after 2 weeks. No VBT was detected. Diarrhea and mild headache were the most common adverse events. Transaminase elevations were reported in three patients but did not result in discontinuation. This study clearly showed that a dual therapy with DCV and ASV, without PEG-IFNα and RBV, can achieve high SVR rates in difficult-to-treat patients with HCV GT 1b infection and previous null response to PEG-IFNα and RBV.
Phase IIa clinical study by Suzuki et al47
In this open-label, Phase IIa study, a dual oral therapy with DCV and ASV for 24 weeks was conducted for Japanese patients with HCV GT 1b infection and limited treatment options.47 Among the patients were 21 null responders (<2log10 HCV RNA reduction after 12 weeks of PEG-IFNα/RBV) and 22 patients who were intolerant to or medically ineligible for PEG-IFNα with or without RBV therapy. The patients were treated with a once-daily dose of 60 mg of DCV and a twice-daily dose of 200 mg of ASV for 24 weeks. The primary efficacy end point was SVR12. In all, 36 of the 43 enrolled patients completed 24 weeks of therapy. Serum HCV RNA levels declined rapidly, becoming undetectable in all patients on therapy by week 8, and 33 patients (76.7%) achieved SVR12 and SVR24, 19 (90.5%) of the null responders and 14 (63.6%) of the ineligible/intolerant patients. No virologic failures were observed among the null responders. Three ineligible/intolerant patients experienced VBT, and four relapsed posttreatment. Diarrhea, nasopharyngitis, headache, and mild increases of alanine/aspartate aminotransferases were the most common adverse events. This study indicated that a dual therapy with DCV and ASV, without PEG-IFNα/RBV, was well tolerated and achieved high SVR rates in two groups of difficult-to-treat patients with HCV virus GT 1b infection.
In vitro and in vivo correlation study by Karino et al48
This study was a characterization of virologic escape in HCV GT 1b patients treated with DAAs, DCV, and ASV, including previously described three patients with VBT and four patients with relapse.48 Baseline NS3 polymorphisms (T54S, Q80L, and V170M) at amino acid positions previously associated with low-level resistance (< ninefold) to select NS3 protease inhibitors were detected in four null responders and three ineligibles, but they were not associated with virologic failure. Baseline NS5A polymorphisms (L28M, L31M, and Y93H) associated with DCV resistance (<25-fold) were detected in five null responders and six ineligibles. All three of the patients with VBT and two of the four patients with relapse carried a baseline NS5A-Y93H polymorphism. NS3 and NS5A resistance–associated variants were detected together (NS3-D168A/V and NS5A-L31M/V-Y93H) after virologic failure. While the DCV-resistant substitutions persisted through 48 weeks posttreatment, the ASV-resistant substitutions were no longer detectable. In addition, five out of ten patients with a baseline NS5A-Y93H polymorphism experienced virologic failure, whereas five out of ten achieved SVR.
Polymorphism and HCV clearance
A baseline polymorphism in patients is an important indicator to determine the potential clinical outcome of HCV therapy with a specific DAA. When the impact of a baseline polymorphism on the emergence of resistance to DCV was examined, a Q30R variant was observed at VBT in one of the GT 1a–infected subjects.49 However, another baseline polymorphism (E62D) did not contribute resistance to DCV.49 The linked variant, Q30R-E62D, conferred high-level resistance in vitro and is likely responsible for VBT in vivo.49 A baseline polymorphism with a minimal effect on the anti-HCV effect of DCV can affect the emergence of resistance and significantly affect clinical outcome.49 NS5A sequence heterogeneity present in GT 1a and GT 1b baseline specimens was found to have a minimal effect on the potency of DCV.49 Low rates of naturally occurring resistant variants to DCV were also detected in HCV 1 null responders.50 Due to the low rates of naturally occurring resistant variants in the NS5A region found in HCV 1 null responders to PEG-IFNα plus RBV, routine direct sequencing of HCV before starting DCV was not recommended.50 The prevalence of HCV variants resistant to NS3 protease inhibitors or DCV was also examined in hepatitis patients with GT 1b.51 In this study, drug-resistant mutations were determined in 362 hepatitis patients infected with HCV GT-1b who had not received DAAs before.51 Amino acid substitutions resistant to NS3 inhibitors (V36A, T54S, Q80H, and D168E) were detected in 15 of the 307 (4.9%) patients who had been examined for NS3 substitutions, and in these patients, T54S substitutions (3.3%) predominated over V36A (0.3%), Q80R (0.7%), and D168E (0.7%).51 Amino acid substitutions resistant to DCV (L31M and/or Y93H) were also detected in 33 of the 294 (11.2%) patients who were examined for NS5 substitutions, and the Y93H substitutions (8.2%) were found to predominate over the L31M substitutions (2.7%).51 One of the 239 (0.4%) patients who had been examined for amino acid substitutions in both NS3 and NS5A regions was found to possess HCV GT 1b variants resistant to NS3 inhibitors (T54S) and DCV (L31M).51
The prevalence of natural polymorphisms at the HCV NS5A gene associated with resistance to DCC was also studied in all IFNα treatment-naïve as well as in HIV-HCV-coinfected patients.52 Specifically, changes reported to be associated with DCV resistance in the in vitro replication system for the HCV GT subtypes 1a and 1b (M28T, Q30H/R, L31F/M/V, P32L, and Y93C/H/N) were examined.52 When the samples from 78 HIV-HCV-coinfected individuals, deposited at Los Alamos HCV database, were analyzed, none of the NS5A sequences from HCV GT 1a or HCV GT 3 showed changes associated with DCV resistance, although all NS5A sequences from HCV GT 4 harbored L31M.52 The double mutant L31M+Y93H was found in 7% of HCV GT 1b and in 13% of HCV GT 4.52 Primary resistance mutations to DCV were not seen as natural polymorphisms in HCV GT 1a or in HCV GT 3.52 They could be recognized in most HCV GT 1b and HCV GT 4 strains, regardless of HIV coinfection.52
Potential role of DCV in the future HCV therapy
Since the introduction of IFNα- and RBV-based combination therapy, there have been several medically unmet needs in HCV treatment. These include the need for the improvement of antiviral efficacy in HCV GT 1 and 4, the reduction of viral resistance, minimization of side effects, the shortening of duration and frequency of treatment, an all-oral delivery, and greater affordability. In regards to the improvement of antiviral efficacy in GT 1 and 4, DCV displayed a superior pangenotypic potency in both in vitro and in vivo studies. Therefore, DCV could serve as a critical component of combination therapy, especially for GT 1 patients, who comprised the majority of HCV patients. In addition, the analysis of DCV-induced viral resistance profiles proved its nonoverlapping property with other DAAs. On top of this, ability of DCV to maintain subnanomolar potency against all DCV-resistant variants would be another of its merits. This strongly suggests DCV as the best candidate to achieve the most synergistic antiviral effect when combined with other mechanistically distinct DAAs, by suppressing the emergence of all possible multiple resistant variants. Since most of the serious side effects of standard HCV therapy come from the nonspecific actions of IFNα and RBV, the minimization of side effects also could be achieved by an IFNα-free regimen consisting of DCV and other DAAs, including NS3 protease inhibitors and NS5B polymerase inhibitors. The feasibility of these DCV-containing and IFNα-free DAA combination regimens was already proved by several clinical trials.43,46,47 In order to shorten the duration and frequency of current anti-HCV treatment, DCV also would be a valuable combination option, thanks to its extraordinary antiviral potency and desirable pharmacokinetic profile, which make a once-daily dosing possible. Finally, the all-oral delivery of anti-HCV therapeutics could be realized by implementing a DCV-containing, all-DAA regimen in practice, with no need for muscular injections of IFNα. This would provide a great convenience for HCV patients. In regards to affordability, elimination of the recombinant protein-based IFNα would greatly help to bring down overall cost of current (IFNα-based) combination therapy.
In spite of all the strengths of DCV mentioned above, the most important limitation of DCV turned out to be its low genetic barrier. In vitro and clinical studies demonstrated selection of HCV variants resistant to DCV, more easily for GT 1a isolates than for GT 1b isolates.27,38 Single substitutions in the N terminal region of NS5A were found to confer more than 1000-fold resistance in GT 1a isolates. In order to supplement this weakness, DCV needs to be combined with other DAAs with high genetic barriers and different mechanisms of action. In this regard, a recently developed, second generation NS3 protease inhibitor, asunaprevir, and a nucleotide analog NS5B polymerase inhibitor, sofosbuvir, enjoyed relatively high genetic resistances, and their combined administration with DCV achieved a great antiviral response without the development of viral resistance in HCV patients.44,45,53–55 Therefore, these would potentially serve as great partners for DCV in combination therapy, in the near future.
Conclusion
This review summarizes key preclinical and clinical data of an HCV NS5A inhibitor, DCV, describing its discovery, mechanism of action, resistance profile, in vitro and in vitro efficacy and toxicity, and polymorphism. It also discusses the potential role of DCV in future HCV combination therapy, by satisfying several unmet medical needs in the HCV therapy area, and its limitation due to its low barrier to resistance. Given its extraordinary antiviral potency and high safety profile, its novel mechanism of action, nonoverlapping resistance profile, desirable pharmacokinetic characteristic, and superior synergism with other DAAs, DCV would make an invaluable component of the most ideal, IFNα-free, all-oral anti-HCV combination therapy. A careful evaluation of the relevant Phase III clinical trial results will be required to for DCV to fulfill its critical role in HCV treatment in the near future.
Acknowledgments
This work was supported by the GRRC program of Gyeonggi province (Study of control of viral diseases [grant number GRRC-DONGGUK2011-A01]) and by the Bio and Medical Technology Development Program of the National Research Foundation (NRF), funded by the Korean government (MEST) (grant number 2012053532).
Footnotes
Disclosure
The author declares no conflict of interest in this work.
References
- 1.Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5(10):558–567. doi: 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- 2.Alter MJ. Epidemiology of hepatitis C virus infection. World J Gastroenterol. 2007;13(17):2436–2441. doi: 10.3748/wjg.v13.i17.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alter MJ, Kruszon-Moran D, Nainan OV, et al. The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N Engl J Med. 1999;341(8):556–562. doi: 10.1056/NEJM199908193410802. [DOI] [PubMed] [Google Scholar]
- 4.Di Bisceglie AM. Natural history of hepatitis C: its impact on clinical management. Hepatology. 2000;31(4):1014–1018. doi: 10.1053/he.2000.5762. [DOI] [PubMed] [Google Scholar]
- 5.Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med. 2006;144(10):705–714. doi: 10.7326/0003-4819-144-10-200605160-00004. [DOI] [PubMed] [Google Scholar]
- 6.Pawlotsky JM. The results of Phase III clinical trials with telaprevir and boceprevir presented at the Liver Meeting 2010: a new standard of care for hepatitis C virus genotype 1 infection, but with issues still pending. Gastroenterology. 2011;140(3):746–754. doi: 10.1053/j.gastro.2011.01.028. [DOI] [PubMed] [Google Scholar]
- 7.Gentile I, Viola C, Reynaud L, et al. Hemolytic anemia during pegylated IFN-alpha2b plus ribavirin treatment for chronic hepatitis C: ribavirin is not always the culprit. J Interferon Cytokine Res. 2005;25(5):283–285. doi: 10.1089/jir.2005.25.283. [DOI] [PubMed] [Google Scholar]
- 8.Hegade VS, Sood R, Saralaya D, Moreea S. Pulmonary complications of treatment with pegylated interferon for hepatitis C infection-two case reports. Ann Hepatol. 2013;12(4):629–633. [PubMed] [Google Scholar]
- 9.Slavenburg S, Heijdra YF, Drenth JP. Pneumonitis as a consequence of (peg)interferon-ribavirin combination therapy for hepatitis C: a review of the literature. Dig Dis Sci. 2010;55(3):579–585. doi: 10.1007/s10620-009-0797-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tosone G, Borgia G, Gentile I, et al. A case of pegylated interferon alpha-related diabetic ketoacidosis: can this complication be avoided? Acta Diabetol. 2007;44(3):167–169. doi: 10.1007/s00592-007-0259-1. [DOI] [PubMed] [Google Scholar]
- 11.Liang TJ, Rehermann B, Seeff LB, Hoofnagle JH. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Ann Intern Med. 2000;132(4):296–305. doi: 10.7326/0003-4819-132-4-200002150-00008. [DOI] [PubMed] [Google Scholar]
- 12.Zeuzem S, Feinman SV, Rasenack J, et al. Peginterferon alfa-2a in patients with chronic hepatitis C. N Engl J Med. 2000;343(23):1666–1672. doi: 10.1056/NEJM200012073432301. [DOI] [PubMed] [Google Scholar]
- 13.Grakoui A, McCourt DW, Wychowski C, Feinstone SM, Rice CM. A second hepatitis C virus-encoded proteinase. Proc Natl Acad Sci U S A. 1993;90(22):10583–10587. doi: 10.1073/pnas.90.22.10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grakoui A, Wychowski C, Lin C, Feinstone SM, Rice CM. Expression and identification of hepatitis C virus polyprotein cleavage products. J Virol. 1993;67(3):1385–1395. doi: 10.1128/jvi.67.3.1385-1395.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee C. Interaction of Hepatitis C Virus Core Protein with Janus Kinase Is Required for Efficient Production of Infectious Viruses. Biomol Ther (Seoul) 21:97–106. doi: 10.4062/biomolther.2013.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5(6):453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 17.Blight KJ, Kolykhalov AA, Rice CM. Efficient initiation of HCV RNA replication in cell culture. Science. 2000;290(5498):1972–1974. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- 18.Lohmann V, Körner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285(5424):110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 19.Lee C. Discovery of hepatitis C virus NS5A inhibitors as a new class of anti-HCV therapy. Arch Pharm Res. 2011;34(9):1403–1407. doi: 10.1007/s12272-011-0921-6. [DOI] [PubMed] [Google Scholar]
- 20.Fridell RA, Qiu D, Valera L, Wang C, Rose RE, Gao M. Distinct functions of NS5A in hepatitis C virus RNA replication uncovered by studies with the NS5A inhibitor BMS-790052. J Virol. 2011;85(14):7312–7320. doi: 10.1128/JVI.00253-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foster TL, Belyaeva T, Stonehouse NJ, Pearson AR, Harris M. All three domains of the hepatitis C virus nonstructural NS5A protein contribute to RNA binding. J Virol. 2010;84(18):9267–9277. doi: 10.1128/JVI.00616-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scheel TK, Rice CM. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med. 2013;19(7):837–849. doi: 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elazar M, Cheong KH, Liu P, Greenberg HB, Rice CM, Glenn JS. Amphipathic helix-dependent localization of NS5A mediates hepatitis C virus RNA replication. J Virol. 2003;77(10):6055–6061. doi: 10.1128/JVI.77.10.6055-6061.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tellinghuisen TL, Marcotrigiano J, Rice CM. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature. 2005;435(7040):374–379. doi: 10.1038/nature03580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lohmann V, Hoffmann S, Herian U, Penin F, Bartenschlager R. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J Virol. 2003;77(5):3007–3019. doi: 10.1128/JVI.77.5.3007-3019.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Appel N, Zayas M, Miller S, et al. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 2008;4(3):e1000035. doi: 10.1371/journal.ppat.1000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao M, Nettles RE, Belema M, et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature. 2010;465(7294):96–100. doi: 10.1038/nature08960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pol S, Ghalib RH, Rustgi VK, et al. Daclatasvir for previously untreated chronic hepatitis C genotype-1 infection: a randomised, parallel-group, double-blind, placebo-controlled, dose-finding, phase 2a trial. Lancet Infect Dis. 2012;12(9):671–677. doi: 10.1016/S1473-3099(12)70138-X. [DOI] [PubMed] [Google Scholar]
- 29.Lemm JA, O’Boyle D, Liu M, et al. Identification of hepatitis C virus NS5A inhibitors. J Virol. 2010;84(1):482–491. doi: 10.1128/JVI.01360-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang C, Valera L, Jia L, Kirk MJ, Gao M, Fridell RA. In vitro activity of daclatasvir on hepatitis C virus genotype 3 NS5A. Antimicrob Agents Chemother. 2013;57(1):611–613. doi: 10.1128/AAC.01874-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang C, Jia L, Huang H, et al. In vitro activity of BMS-790052 on hepatitis C virus genotype 4 NS5A. Antimicrob Agents Chemother. 2012;56(3):1588–1590. doi: 10.1128/AAC.06169-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pelosi LA, Voss S, Liu M, Gao M, Lemm JA. Effect on hepatitis C virus replication of combinations of direct-acting antivirals, including NS5A inhibitor daclatasvir. Antimicrob Agents Chemother. 2012;56(10):5230–5239. doi: 10.1128/AAC.01209-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang C, Huang H, Valera L, et al. Hepatitis C virus RNA elimination and development of resistance in replicon cells treated with BMS-790052. Antimicrob Agents Chemother. 2012;56(3):1350–1358. doi: 10.1128/AAC.05977-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qiu D, Lemm JA, O’Boyle DR, et al. The effects of NS5A inhibitors on NS5A phosphorylation, polyprotein processing and localization. J Gen Virol. 2011;92(Pt 11):2502–2511. doi: 10.1099/vir.0.034801-0. [DOI] [PubMed] [Google Scholar]
- 35.Lee C, Ma H, Hang JQ, et al. The hepatitis C virus NS5A inhibitor (BMS-790052) alters the subcellular localization of the NS5A nonstructural viral protein. Virology. 2011;414(1):10–18. doi: 10.1016/j.virol.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Targett-Adams P, Graham EJ, Middleton J, et al. Small molecules targeting hepatitis C virus-encoded NS5A cause subcellular redistribution of their target: insights into compound modes of action. J Virol. 2011;85(13):6353–6368. doi: 10.1128/JVI.00215-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guedj J, et al. Modeling shows that the NS5A inhibitor daclatasvir has two modes of action and yields a shorter estimate of the hepatitis C virus half-life. Proc Natl Acad Sci. USA. 2013;110:3991–3996. doi: 10.1073/pnas.1203110110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fridell RA, Qiu D, Wang C, Valera L, Gao M. Resistance analysis of the hepatitis C virus NS5A inhibitor BMS-790052 in an in vitro replicon system. Antimicrob Agents Chemother. 2010;54(9):3641–3650. doi: 10.1128/AAC.00556-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nettles RE, Gao M, Bifano M, et al. Multiple ascending dose study of BMS-790052, a nonstructural protein 5A replication complex inhibitor, in patients infected with hepatitis C virus genotype 1. Hepatology. 2011;54(6):1956–1965. doi: 10.1002/hep.24609. [DOI] [PubMed] [Google Scholar]
- 40.Fridell RA, Wang C, Sun JH, et al. Genotypic and phenotypic analysis of variants resistant to hepatitis C virus nonstructural protein 5A replication complex inhibitor BMS-790052 in humans: in vitro and in vivo correlations. Hepatology. 2011;54(6):1924–1935. doi: 10.1002/hep.24594. [DOI] [PubMed] [Google Scholar]
- 41.Bifano M, Sevinsky H, Persson A, et al. Single-dose pharmacokinetics of daclatasvir (DCV; BMS-790052) in subjects with hepatic impairment compared with healthy subjects. Presented at: 62nd Annual Meeting of the American Association for the Study of Liver Diseases; November 6–9, 2011; San Francisco, CA. [Google Scholar]
- 42.Fontana RJ, Hughes EA, Appelman H, Hindes R, Dimitrova D, Bifano M. Case report of successful peginterferon, ribavirin, and daclatasvir therapy for recurrent cholestatic hepatitis C after liver retransplantation. Liver Transpl. 2012;18(9):1053–1059. doi: 10.1002/lt.23482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med. 2012;366(3):216–224. doi: 10.1056/NEJMoa1104430. [DOI] [PubMed] [Google Scholar]
- 44.Sulkowski MS, Gardiner DF, Rodriguez-Torres M, et al. AI444-040 Study Group Sustained virologic response with daclatasvir plus sofosbuvir ± ribavirin (RBV) in chronic HCV genotype (GT) 1-infected patients who previously failed telaprevir (TVR) or boceprevir (BOC); Presented at: 48th Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, The Netherlands. [Google Scholar]
- 45.Sulkowski MS, Gardiner DF, Rodriguez-Torres M, et al. AI444040 Study Group High rate of sustained virologic response with the all-oral combination of daclatasvir (NS5A inhibitor) plus sofosbuvir (nucleotide NS5B inhibitor), with or without ribavirin, in treatment-naive patients chronically infected with HCV GT 1, 2, or 3. Hepatology. 2012;56:1516–1517. [Google Scholar]
- 46.Chayama K, Takahashi S, Toyota J, et al. Dual therapy with the nonstructural protein 5A inhibitor, daclatasvir, and the nonstructural protein 3 protease inhibitor, asunaprevir, in hepatitis C virus genotype 1b-infected null responders. Hepatology. 2012;55(3):742–748. doi: 10.1002/hep.24724. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki Y, Ikeda K, Suzuki F, et al. Dual oral therapy with daclatasvir and asunaprevir for patients with HCV genotype 1b infection and limited treatment options. J Hepatol. 2013;58(4):655–662. doi: 10.1016/j.jhep.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 48.Karino Y, Toyota J, Ikeda K, et al. Characterization of virologic escape in hepatitis C virus genotype 1b patients treated with the direct-acting antivirals daclatasvir and asunaprevir. J Hepatol. 2013;58(4):646–654. doi: 10.1016/j.jhep.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 49.Sun JH, O’Boyle Ii DR, Zhang Y, et al. Impact of a baseline polymorphism on the emergence of resistance to the hepatitis C virus nonstructural protein 5A replication complex inhibitor, BMS-790052. Hepatology. 2012;55(6):1692–1699. doi: 10.1002/hep.25581. [DOI] [PubMed] [Google Scholar]
- 50.Galmozzi E, Aghemo A, Colombo M. Low rates of naturally occurring resistant variants to the NS5A inhibitor daclatasvir in HCV-1 null responders. Hepatology. 2013;57(5):2087. doi: 10.1002/hep.25924. [DOI] [PubMed] [Google Scholar]
- 51.Suzuki F, Sezaki H, Akuta N, et al. Prevalence of hepatitis C virus variants resistant to NS3 protease inhibitors or the NS5A inhibitor (BMS-790052) in hepatitis patients with genotype 1b. J Clin Virol. 2012;54(4):352–354. doi: 10.1016/j.jcv.2012.04.024. [DOI] [PubMed] [Google Scholar]
- 52.Plaza Z, Soriano V, Vispo E, et al. Prevalence of natural polymorphisms at the HCV NS5A gene associated with resistance to daclatasvir, an NS5A inhibitor. Antivir Ther (Lond) 2012;17(5):921–926. doi: 10.3851/IMP2091. [DOI] [PubMed] [Google Scholar]
- 53.Asselah T. Sofosbuvir-based interferon-free therapy for patients with HCV infection. J Hepatol. July2013 doi: 10.1016/j.jhep.2013.07.023. Epub. [DOI] [PubMed] [Google Scholar]
- 54.Chae HB, Park SM, Youn SJ. Direct-acting antivirals for the treatment of chronic hepatitis C: open issues and future perspectives. Scientific World Journal. 2013;2013:704912. doi: 10.1155/2013/704912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gentile I, Borgia F, Buonomo AR, Castaldo G, Borgia G. A novel promising therapeutic option against hepatitis C virus: an oral nucleotide NS5B polymerase inhibitor sofosbuvir. Curr Med Chem. 2013;20(30):3733–3742. doi: 10.2174/09298673113209990178. [DOI] [PubMed] [Google Scholar]