

Abstract
We investigated the novel role of HCA2 (GPR109A) and its ligand nicotinic acid in regulating macrophage function. Hca2 expression in the RAW264.7 murine macrophage cell line is strongly induced by LPS treatment and correlates with the expression of TNF-α. Treatment with 300 μM nicotinic acid (reported EC50 3 μM, peak plasma concentration 50–300 μM), significantly inhibited TNF-α, IL-6, IL-12p40, and IL-1β production (P<0.05) in LPS (1 ng/ml)-stimulated wild-type murine bone marrow-derived macrophages (BMMs) but failed to do so in Hca2−/− BMMs. Treatment with nicotinic acid reduced nuclear factor κB (NF-κB) activation levels by 43% (P<0.03) in wild-type BMMs 6 h after LPS stimulation but not in Hca2−/− BMMs. Nicotinic acid significantly inhibited wild-type BMM chemotaxis (P<0.001), but had no effect on the chemotaxis of Hca2−/− BMMs. A significant increase in low-density lipoprotein uptake by both wild-type (P<0.006) and Hca2−/− BMMs (P<0.03) in response to LPS was observed, which was significantly suppressed by nicotinic acid in wild-type BMMs (P<0.04) but not in Hca2−/− BMMs. Our results suggest that the nicotinic acid-HCA2 axis is a novel negative regulator of macrophage activation.—Zandi-Nejad, K., Takakura, A., Jurewicz, M., Chandraker, A. K., Offermanns, S., Mount, D., Abdi, R. The role of HCA2 (GPR109A) in regulating macrophage function.
Keywords: nicotinic acid, inflammation, PUMA-G, HM74A
The role of inflammation in the pathogenesis of prevalent diseases such as atherosclerosis has increasingly been recognized. In parallel, macrophages have become the focus of intense research due to their pathogenic role in the development and process of inflammation. Macrophages produce proinflammatory cytokines, such as tumor necrosis factor α (TNF-α) and interleukin 1β (IL-1β) through activation of toll-like receptors (TLRs) and nuclear factor κB (NF-κB)/Rel family with subsequent regulation of inflammatory genes. These cytokines may then induce additional cytokines in a paracrine and/or an autocrine fashion (1). While the TLR system of macrophages has been well characterized, significant strides to identify novel key mechanisms regulating macrophage function have been made over the past several years (2).
The hydroxycarboxylic acid 2 (HCA2) receptor is a G-protein-coupled receptor (GPCR) also known as protein up-regulated in macrophages by IFN-γ (PUMA-G; murine ortholog), HM74A (human ortholog), and GPR109A. It was discovered as a GPCR primarily expressed in adipocytes and activated macrophages; hence, the original name PUMA-G (3). It has been reported that nicotinic acid (NA) binds to this receptor (4–6) and that the lipolytic effect of NA in adipocytes is mediated through this GPCR (5). Several endogenous and synthetic ligands have been identified since, all of which share a hydroxycarboxylic acid moiety (7); hence, the HCA2 receptor nomenclature (8), which will be used here.
Although IFN-γ induces a large number of genes and cellular responses in both macrophages and embryonic fibroblasts, a subset of genes, including Hca2, is induced only in macrophages. In addition, Hca2 induction in a variety of murine macrophage cell lines stimulated with cytosine-phosphate-guanine oligodeoxynucleotides (CpG-ODNs; synthetic oligonucleotides containing unmethylated CpG dinucleotides that are frequently present in bacterial DNA), or lipopolysaccharide (LPS) suggests that Hca2 may be induced not only by the IFN-γR but also directly through TLRs. Of note, neither granulocyte-macrophage colony-stimulating factor (GM-CSF) nor GpC-ODNs (the unstimulatory control for CpG-ODNs) induce Hca2 expression in macrophages (3). Taken together, these data suggest a role for HCA2 in the early innate immune response. However, the role of HCA2 function in macrophages remains to be fully explored.
Here, we show that LPS-induced proinflammatory cytokines in both the murine macrophage-like RAW264.7 cell line and primary murine bone marrow-derived macrophages (BMMs) are significantly reduced by treatment with NA and that the inhibitory effect of NA on cytokine production is mediated by HCA2 through NF-κB signaling. Furthermore, we demonstrate that NA inhibits low-density lipoprotein (LDL) uptake and chemotaxis via HCA2. Finally, we also show that cytokine production induced by other proinflammatory stimuli (IFN-γ and uric acid) is also suppressed by NA and other ligands of HCA2 receptor, namely lactate and β-hydroxybutyrate.
MATERIALS AND METHODS
Cell culture
RAW264.7 cells [American Type Culture Collection (ATCC), Manassas, VA, USA] were cultured in DMEM containing 10% FBS (ATCC) and were maintained at 37°C in a humidified chamber of 5% CO2. The culture medium was replaced with fresh medium every other day.
Real-time reverse transcriptase polymerase chain reaction (RT-PCR)
Total RNA was extracted from RAW264.7 cells by homogenization in TRIzol reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions. Complementary DNA (cDNA) was generated from 5 μg of total RNA using RT (Invitrogen). Real-time RT-PCR was performed using the following primers: β-actin F, β-actin R, TNF-α F, TNF-α R, Hca2 F, and Hca2 R.
Preparation of monosodium urate monohydrate (MSU) crystals
MSU crystals were prepared by crystallization of supersaturated uric acid solutions (4–5 mg/ml; Sigma-Aldrich, St. Louis, MO, USA) in 0.1 M borate acid (pH 8.5) at room temperature for >48 h, followed by washing with ethanol and acetone.
Mice
Mice were studied according to the animal experimental guidelines issued by the Animal Care and Use Committee at Harvard University. Hca2 (Puma-g) heterozygous mice (5) were backcrossed 7–9 times to the C57BL/6 strain (The Jackson Laboratory, Bar Harbor, ME, USA), and homozygous offspring were obtained by intercrossing Hca2+/− mice. Genotyping of the Hca2 allele was performed by PCR using the following primers (5): Hca2 knockout, 5′-CCTCTTCGCTATTACGCCAGC-3′, and Hca2 reverse, 5′-TCAGATCTGACTCGTCCACC-3′, for the deletion allele; and Hca2 wild type, 5′-CCATTGCCCAGGAGTCCGAAC-3′, and Hca2 reverse for the wild-type allele.
BMM cell culture
Eight-week-old Hca2−/− and littermate wild-type Hca2+/+ mice were euthanized, and the femoral bones were flushed with DMEM/F12 containing 100 U of penicillin and streptomycin and 10% FBS. Cells were seeded in bacterial culture Petri dishes in 80% DMEM/F12 containing 100 U of penicillin and streptomycin, 10% FBS, and 20% conditioned medium from L929 fibrosarcoma cells (kindly provided by Dr. Joseph Bonventre, Brigham and Women's Hospital, Boston, MA, USA). Medium was changed every other day. On d 6, the cells were trypsinized and seeded at a density of 1 × 104 cells/ml for enzyme-linked immunosorbent assay (ELISA) and Luminex analyses.
Luminex
The Luminex assay (Millipore, Billerica, MA, USA) was performed according to the manufacturer's instructions. Briefly, BMMs from Hca2+/+ and Hca2−/− mice were plated at 1 × 106 cells/10 cm2 dish in 80% DMEM/F12 containing 10% FBS and 20% conditioned medium from L929 fibrosarcoma cells. At 24 h after plating, cells were cultured in fresh DMEM with 1 ng/ml of LPS and/or 300 μM NA for 48 h. Culture supernatants harvested from macrophage cultures were assessed for production of GM-CSF, IL-10, IL-12p40, IL-12p70, IL-1α, IL-1β, IL-2, IL-6, interferon-inducible protein 10 (IP-10), monocyte chemoattractant protein 1 (MCP-1), monokine induced by γ-interferon (MIG), and tumor necrosis factor α (TNF-α). Beads conjugated to cytokines were incubated with supernatants, followed by incubation with biotinylated beads and streptavidin.
ELISA
ELISA (R&D Systems, Minneapolis, MN, USA) was performed according to the manufacturer's instructions. Briefly, the culture supernatants harvested from RAW264.7 cells or BMM cultures were assessed for production of TNF-α and IL-6.
Flow cytometric analysis
Cells were run on a FACSCalibur (Becton Dickinson, Franklin Lakes, NJ, USA) following 30 min staining with anti-mouse F4/80, CD11c, or DEC-205 (1:100; BD Biosciences, San Jose, CA, USA). Data were analyzed using FlowJo 6.3.2 software (Treestar, Ashland, OR, USA).
Western blot analysis
Cells were lysed in RIPA buffer (Thermo Scientific, Rockford, IL, USA) containing protease inhibitors (Roche, South San Francisco, CA, USA), and cell lysates were cleared by centrifugation. Equal amounts of lysates (100 μg) were separated on 10% SDS-PAGE. Proteins were transferred onto nitrocellulose membranes (Amersham, Woburn, MA, USA), and the membranes were then blocked with 5% BSA in PBS for 30 min and incubated overnight at 4°C with antibody to phospho NF-κB (pNF-κB), NF-κB, inhibitor κB (IκB) (1:1000; Cell Signaling, Danvers, MA, USA), or α-tubulin (1:5000; Abcam, Cambridge, MA, USA). After 3 washings with PBS, the membranes were incubated with peroxidase-conjugated goat anti-mouse IgG or anti-rabbit IgG (1:5000; Amersham) for 1 h. Finally, blots were developed using the enhanced chemiluminescence (ECL) method. Membranes were then stripped of antibodies using Restore Western blot Stripping Buffer (Thermo Scientific, Billerica, MA, USA) for 30 min at 37°C. Membranes were then reprobed with primary antibody and secondary antibodies.
LDL uptake in macrophages
BMMs from Hca2+/+ and Hca2−/− mice were plated at 1 × 106 cells/well in 24-well plates with 80% DMEM/F12 containing 10% FBS and 20% conditioned medium from L929 fibrosarcoma cells. At 24 h after plating, cells were cultured in fresh DMEM with 1 ng/ml of LPS and/or 300 μM NA for 48 h. LDL conjugated to DyLight 549 (Cayman Chemical Co., Ann Arbor, MI, USA) was added per the manufacturer's instructions, and cells were incubated for another 24 h at 37°C (internalization). After washing with ice-cold PBS 3 times, cells were lysed in RIPA buffer, transferred to 96-well plates, and analyzed for fluorescence using a scanning fluorometer (Flexstation; Molecular Probes, Carlsbad, CA, USA), with excitation and emission wavelengths set at 544 and 590 nm, respectively.
Chemotaxis assay
BMMs from Hca2+/+ and Hca2−/− mice were plated at 1 × 106 cells per upper chamber of a 24-well Transwell system (5 μm pore size; Corning, Tewksbury, MA, USA) in 80% DMEM/F12 containing 10% FBS and 20% conditioned medium from L929 fibrosarcoma cells. At 24 h after plating, the culture medium was changed to DMEM containing either PBS or 1 ng/ml LPS and/or 300 μM NA, and fresh DMEM containing either PBS or 100 ng/ml MCP-1 was replaced in the lower chamber. Cells were allowed to migrate for 2 h, and the cell content of the lower chambers was then collected and counted. The chemotaxis index was calculated by dividing the number of migrated cells in the presence of MCP-1 by the number of cells migrated in its absence.
Statistical analysis
Statistical significance was performed using Student's t test. Values of P < 0.05 were considered statistically significant.
RESULTS
Hca2 expression in response to LPS is associated with production of TNF-α in RAW264.7 cells
The Hca2 cDNA was initially cloned from the murine macrophage cell line ANA-1 stimulated with IFN-γ and TNF-α (3). To investigate whether a correlation exists between Hca2 expression and the degree of macrophage activation, we performed real-time RT-PCR for Hca2 and TNF-α at several time points following LPS stimulation. RAW264.7 cells, a murine macrophage-like cell line, were thus cultured in the presence of different concentrations of LPS for 6, 24, or 48 h. Hca2 mRNA expression level was detected as early as 6 h after LPS stimulation and significantly increased both dose and time dependently (Fig. 1A). As expected, TNF-α mRNA expression in response to LPS paralleled the expression of Hca2 (Fig. 1B). Consistent with this finding, IFN-γ also induced Hca2 and TNF-α mRNA expression in a dose-dependent manner (Supplemental Fig. S1A, B). To measure TNF-α production, culture supernatants obtained from LPS stimulation of RAW264.7 cells were subjected to ELISA assays. TNF-α protein levels were also significantly increased dose and time dependently (Fig. 1C), as were IL-6 protein levels (Fig. 1D). Given that TNF-α production increases as early as 1 h after stimulation with LPS, we examined LPS-induced Hca2 expression in both RAW264.7 and primary bone marrow-derived macrophages. This revealed that Hca2 mRNA is expressed at low levels in unstimulated conditions and is induced in a time-dependent fashion in response to LPS similar to TNF-α production, suggestive of a role for the HCA2 receptor in the early stages of macrophage activation (Supplemental Fig. S1C, D).
Figure 1.
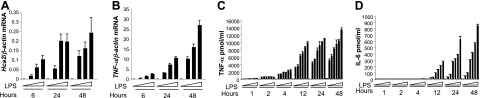
Hca2 expression correlates with murine macrophage activation. A, B) RAW264.7 cells were treated with 0, 0.5, 1, or 10 ng/ml LPS for the indicated times. Hca2 (A) and TNF-α (B) mRNA expression was quantified by quantitative real-time RT-PCR and normalized to β-actin mRNA expression. Data are shown as means ± se of 3 independent experiments. C, D) RAW267.4 cells were treated with 0, 0.5, 1, 5, 10, 20, or 50 ng/ml of LPS for the indicated times. Culture supernatants were subjected to ELISA assays for TNF-α (C) and IL-6 (D) production.
NA suppresses macrophage activation
We first tested and confirmed that NA in the concentration range (100–300 μM) used in previous studies (5, 7, 9) reduces TNF-α production in LPS-treated RAW264.7 cells (Supplemental Fig. S1E); of note, this concentration range corresponds to peak plasma concentration (50–300 μM) of NA after administration of a standard pharmacological dose (10, 11).
It has been shown that resident tissue macrophages, which have a highly differentiated phenotype, contribute to acute MSU crystal-induced inflammation (12). To investigate whether NA inhibits MSU-induced macrophage activation, RAW274.7 cells were cultured with MSU crystals, IFN-γ, and ΜSU crystals plus IFN-γ, in the presence or absence of NA. While the morphology of cells in the presence of MSU crystals alone was similar to unstimulated cells, IFN-γ-treated cells displayed short spike-like structures and small vacuoles (Fig. 2Aa–c). In stark contrast, treatment with a combination of MSU crystals and IFN-γ lead to numerous small and large vacuoles in RAW264.7 cells (Fig. 2Ad). More notably, NA significantly suppressed these morphological changes in IFN-γ-treated cells and in cells treated with both IFN-γ and MSU crystals (P<0.001; Fig. 2Ag, h; B). However, NA did not affect the morphology of unstimulated cells and MSU crystal-treated cells (Fig. 2Ae, f). Consistent with previous reports that alteration in cell morphology is a common feature of macrophage activation, TNF-α mRNA levels were greatly up-regulated after stimulation with IFN-γ and MSU crystals (Fig. 2C). To confirm that these changes are due specifically to MSU crystals and not simply to any crystalline stimulus, we repeated this experiment with allopurinol crystals, which are structurally similar to uric acid; indeed, the addition of these crystals to IFN-γ did not increase TNF-α, confirming the specific role of MSU crystals in induction of the observed changes (Supplemental Fig. S2). Of note, in addition to NA (P<0.01), other hydroxycarboxylates, such as β-hydroxybutyrate (P<0.002) and lactate (P<0.002), reduced TNF-α mRNA expression as well (Fig. 2C).
Figure 2.

NA suppresses macrophage activation. A) RAW264.7 cells were treated with either PBS (a, e), 0.05 mg/ml MSU crystals (b, f), 1 ng/ml IFN-γ (c, g), or MSU crystals and IFN-γ (d, h) in the absence (a–d) or presence (e–h) of 100 μM NA. Of note, MSU and IFN-γ in combination have a synergistic effect on vacuolization, which was diminished by NA. Arrows indicate vacuoles. Images are representative of 5 independent experiments. B) Cells were cultured in the conditions mentioned in A. A total of 500 cells per condition and experiment were randomly counted and scored for the vacuolization phenotype. Data are shown as means ± sd of 3 independent experiments. *P < 0.001 vs. MSU/IFN-γ. C) RAW264.7 cells were treated with MSU crystals and IFN-γ in the presence of either 100 μM NA, 2 mM β-hydroxybutyrate (βΗ), or 10 mM lactate (Lac). Data are shown as means ± sd of 3 independent experiments. *P < 0.01, **P < 0.002 vs. MSU/IFN-γ.
Effect of NA on macrophage activation is mediated by HCA2
We utilized primary BMMs from Hca2−/− mice or their wild-type littermates to test whether the anti-inflammatory effect of NA is mediated by HCA2. Bone marrow cells were isolated from Hca2−/− or wild-type littermates and were cultured with L929-conditioned medium to proliferate and differentiate into a homogenous population of mature BMMs. The efficiency of differentiation into macrophages was assessed by surface staining for F4/80, CD11c, and DEC205. Approximately 98% cells were positive for the macrophage marker F4/80 and were negative for the dendritic cell markers CD11c and DEC205; the cell population was also shown to be very homogeneous 6 d after seeding (Fig. 3A and Supplemental Fig. S3). Mature BMMs were seeded at a density of 1 × 104 cells/ml and were stimulated with LPS ± NA, and culture supernatants were subjected to ELISA analysis for TNF-α production. Consistent with data obtained using the RAW264.7 cell line, TNF-α levels were increased in supernatants from LPS-treated wild-type BMM cultures, compared to those of the untreated wild-type BMM cultures (Fig. 3B). Furthermore, NA significantly decreased TNF-α production in LPS-treated wild-type BMM cultures (n=6 mice, P<0.02). In contrast, we found that NA had no effect on LPS-induced increased TNF-α production in Hca2−/− BMM cultures (n=5 mice; P>0.05). Taken together, these findings suggest that the anti-inflammatory effect of NA is mediated by HCA2. To examine the production of other cytokines in response to LPS in the presence or absence of NA, we performed a Luminex assay for GM-CSF, IL-10, IL-12p40, IL-12p70, IL-1α, IL-1β, IL-2, IL-6, IP-10, MCP-1, MIG, and TNF-α. Luminex assay analyses revealed that all cytokines except MIG, which is known to be induced by IFN-γ but not by LPS (13, 14), were increased in response to LPS (Fig. 3C and Supplemental Fig. S4), indicating a successful experiment. Excitingly, NA significantly reduced levels of the LPS-induced proinflammatory cytokines TNF-α, IL-6, IL-12p40, and IL-1β in wild-type BMM cultures (n=3 mice, P<0.05) but failed to do so in Hca2−/− BMMs (n=3 mice; Fig. 3C and Supplemental Fig. S4). These data suggest that NA inhibits LPS-induced production of proinflammatory cytokines IL-6, IL-12p40, IL-1β, and TNF-α through HCA2.
Figure 3.

Inhibitory effect of NA on macrophage activation is mediated by HCA2. A) Highly pure (∼98%) BMMs from HCA2-knockout mice and their wild-type littermates were obtained. B) BMMs were treated with 1 ng/ml LPS in the presence or absence of 300 μM NA for 48 h. Culture supernatants were then subjected to a TNF-α ELISA assay. While up-regulation of TNF-α production in LPS-treated wild-type BMMs was decreased after NA treatment (n=6 mice; P<0.02), NA did not alter TNF-α production in LPS-treated Hca2−/− BMMs (n=5 mice). Data are shown as means ± se. C) Proinflammatory cytokines in BMM culture supernatants were measured using a Luminex assay. TNF-α, IL-6, IL-12p40, and IL-1β levels were dramatically increased in both LPS-treated wild-type and Hca2−/− BMMs, compared to vehicle-treated BMMs (n=3 mice/genotype). NA significantly inhibited proinflammatory cytokine production in wild-type (P=0.033 for TNF-α; P=0.0265 for IL-6; P=0.0335 for IL-12p40; and P=0.046 for IL-1β) but not in Hca2−/− BMM cultures. Data are shown as means ± se. n.s., not significant. *P < 0.05.
NA down-regulates NF-κB activation via HCA2
NF-κB is a well-characterized transcription factor that regulates the expression of inflammatory genes (15), and binding of LPS to TLR4 is known to activate NF-κB followed by degradation of IκB. To elucidate the inhibitory mechanism of NA on proinflammatory cytokine production by macrophages, we examined NF-κB signaling in response to LPS in BMMs isolated from Hca2−/− mice or their littermate wild-type controls. Cells were seeded at a density of 1 × 105 cells/ml, and protein was harvested at 0.5, 2, 6, and 12 h after LPS stimulation in the presence or absence of NA. Cell lysates were subjected to Western blot for pNF-κB, NF-κB, and IκB. In both Hca2−/− and wild-type BMMs, active NF-κB (pNF-κB) peaked at 0.5 h after LPS stimulation and was accompanied by IκB degradation (Fig. 4A, B). Levels of pNF-κB were maintained for 6 h; however, a striking reduction in the level of pNF-κB was seen 12 h after LPS stimulation. As expected, NA significantly reduced pNF-κB levels by 43% (P<0.03; n=4) in wild-type BMMs 6 h after LPS stimulation (Fig. 4A, B). On the other hand, NA did not alter the expression levels of pNF-κB in Hca2−/− BMMs at any time points after LPS stimulation (Fig. 4C, D). These findings suggest that the inhibition of LPS-induced NF-κB activation by NA is mediated by HCA2.
Figure 4.

NA down-regulates NF-κB activation via HCA2. BMMs were treated with 0 or 1 ng/ml LPS for the indicated times in the presence or absence of 300 μM NA. Western blot was performed with the indicated antibodies. At 6 h after LPS stimulation, a significant reduction in pNF-κB levels was observed in NA-treated wild-type BMM lysates (A, B). In contrast, no difference was seen in pNF-κB levels between vehicle- and NA-treated Hca2−/− BMMs (C, D). α-Tubulin was used as a loading control. Quantification of band intensity is shown in graphical form (B, D). Data are shown as means ± se of 4 independent experiments (4 mice/genotype). *P < 0.03.
Effect of NA on chemotaxis and LDL uptake
Given that chemokines, including MCP-1, recruit monocytes and macrophages to sites of inflammation, such as atherosclerotic lesions (16, 17), we performed a chemotaxis assay using the transwell system. BMMs were cultured for 2 h in the presence or absence of MCP-1 and/or NA. The migration (chemotaxis) of wild-type BMMs cultured in the upper chamber toward the lower chamber containing MCP-1 was significantly inhibited by the addition of NA (P<0.01; n=3; Fig. 5A). In contrast, NA did not affect MCP-1-induced chemotaxis in Hca2−/− BMMs (Fig. 5B). Whereas LPS treatment alone did not affect chemotaxis in either wild-type or Hca2−/− BMMs, the combination of LPS and MCP-1 enhanced chemotactic behavior of BMMs (P<0.03; n=3) but only to the same extent as MCP-1 alone (Fig. 5). These data suggest that MCP-1 induces BMM migration and that NA inhibits MCP-1-induced chemotaxis through HCA2.
Figure 5.

Effect of NA on chemotaxis. Chemotaxis indices of BMM were determined in the absence or presence of MCP-1 (100 ng/ml) and/or LPS (1 ng/ml) and/or NA (300 μM). A) Inhibition of MCP-1-induced chemotaxis of wild-type BMMs by NA was observed. B) No effect of NA on MCP-1-induced chemotaxis of Hca2−/− BMMs was seen. Data are shown as means ± se of 3 independent experiments. ns, not significant. *P < 0.009, **P < 0.02.
Recent studies have shown that both native LDL and oxidized LDL are taken up by macrophages (18–21) and that accumulation of LDL in macrophages causes foam cell formation and development of atherosclerotic plaques. To investigate whether NA inhibits LDL uptake by macrophages, we incubated LPS-stimulated BMMs with native LDL in the presence or absence of NA. BMMs were cultured for 48 h in the presence or absence of LPS and/or NA. Cells were then cultured for another 24 h in the presence of LDL conjugated to DyLightTM 549. A significant increase in LDL uptake was observed in both LPS-stimulated wild-type and Hca2−/− BMMs, compared to unstimulated BMMs (Fig. 6) (P<0.006 for wild-type, P<0.03 for Hca2−/−, n=3 mice/genotype). Notably, NA significantly reduced LDL uptake by LPS-stimulated wild-type BMMs (P<0.04; n=3 mice). In contrast, no significant reduction in LDL uptake by LPS-stimulated Hca2−/− BMMs was seen in the presence of NA (P>0.4; n=3 mice). Furthermore, we did not observe any effect of NA on LDL uptake by unstimulated BMMs. Taken together, these results indicate that the inhibitory effect of NA on chemotaxis and LDL uptake in macrophages is mediated by HCA2.
Figure 6.

Effect of NA on LDL uptake. BMMs were treated with 1 ng/ml LPS in the presence or absence of NA (300 μM) for 48 h, followed by incubation with DyLightTM-nLDL for 24 h. LDL uptake was assessed by a scanning fluorometer. Data are shown as means ± se of 3 independent experiments (3 mice/genotype). *P < 0.0052, **P < 0.035, ***P < 0.027.
DISCUSSION
Macrophages not only constitute a primary line of defense against pathogens, but they are also the dominant cells present in inflamed tissues, exerting a central role in the process of inflammation. Understanding the regulatory pathways that control their function will not only be a major step to better understand their role in inflammation but also will serve as a basis for developing novel targeted therapies.
In this study, we found that the level of Hca2 expression correlates with the degree of macrophage activation, as measured by proinflammatory cytokine production. Therefore, we hypothesized that activated macrophages may be subjected to negative feedback mechanisms via HCA2 signaling. Indeed, NA, a ligand of HCA2, significantly suppressed LPS-induced proinflammatory cytokine production, including TNF-α, IL-6, IL-12p40, and IL-1β in wild-type BMMs. These cytokines have been found to play a critical role in a multitude of inflammatory diseases, including rheumatoid arthritis (22, 23), diabetes (24), and cancer (25). Notably, these cytokines have also been found in macrophages of atherosclerotic lesions and appear to play an important role in the pathogenesis of atherosclerosis, where they promote smooth muscle cell proliferation and the recruitment of monocytes and T cells into lesions, as well as in the subsequent differentiation and activation of these cells (26–30). Interestingly, NA did not significantly suppress levels of LPS-induced GM-CSF, IL-10, IL-12p70, IL-1α, IL-2, IP-10, and MCP-1, suggesting a selective role for HCA2.
We also investigated the role of HCA2 in MSU crystal-induced inflammation, which is known to lead to gout (12). In the present study, we found that the combination of MSU and IFN-γ significantly induces TNF-α production, and that changes in macrophage morphology are more pronounced by adding MSU crystals to IFN-γ treatment. Consistent with our observations, it has been reported that peritoneal resident cells (primarily macrophages) treated with MSU ex vivo produce proinflammatory cytokines (31). MSU crystals also are reported to up-regulate HCA2 mRNA in cultured mouse peritoneal macrophages (32). We further showed that treatment with NA abolishes the combination of MSU and IFN-γ-induced morphological changes and proinflammatory cytokine production. Of note, NA is known to cause hyperuricemia by increasing renal uric acid reabsorption in the proximal tubule, precipitating gout (33); presumably, the local protective effect of NA is overwhelmed under these circumstances. It is noteworthy that β-hydroxybutyrate, the proposed endogenous ligand for HCA2 (7), also reduced MSU/IFN-γ-induced TNF-α production, suggesting a potential anti-inflammatory role.
Given that NF-κB is a key transcription factor that regulates expression of proinflammatory genes (15), we examined whether HCA2-mediated signaling pathways modulate NF-κB signaling. Consistent with recent in vivo studies in which high doses of NA were shown to suppress LPS-induced NF-κB activation and IL-6 and TNF-α expression in whole lung tissue (34) and cultured human monocytes (35), we found that NA inhibits proinflammatory cytokines via NF-κB inactivation in macrophages. Furthermore, we demonstrated that the inhibitory effect of NA is mediated by HCA2. However, how HCA2 regulates NF-κB activation in macrophages remains unknown.
The role of cyclic adenosine monophosphate (cAMP) in this process remains to be explored, as previous studies have yielded conflicting results. It has been shown that NA lowers lipid levels by inhibition of cAMP production (36) through Gαi-mediated inhibition of adenylyl cyclase in adipose tissue (37, 38). On the other hand, cAMP levels are also a key element for macrophage activation. Increased cAMP levels by forskolin or prostaglandin E2 are known to inhibit LPS-induced proinflammatory cytokine production in fully activated macrophages (39, 40); however, increased bacteria-derived cAMP within macrophages results in cAMP response element-binding protein (CREB) phosphorylation and TNF-α production (41). Furthermore, LPS stimulation has been demonstrated to increase cAMP in macrophages, and a subset of cAMP-induced gene transcription could be a normal inflammatory response (42, 43). Thus, further investigation is necessary to determine whether the mechanism of reduction of NF-κB activation by NA is cAMP-dependent.
Another feature of macrophage activation is chemotaxis, which is crucial for the trafficking of leukocytes during the early stages of inflammation and atherosclerosis. We found that NA diminishes MCP-1-induced chemotaxis in wild-type BMMs but not in Hca2−/− BMMs. Lukasova et al. (44) also demonstrated that NA reduces peritoneal macrophage recruitment to atherosclerotic plaques, as well as into the peritoneal cavity in mice. It has been shown that different macrophage populations (peritoneal vs. bone marrow) do not respond identically to microenvironments (45, 46); however, we have found similar effects of MCP-1 and NA on BMMs. Surprisingly, the MCP-1-induced chemotaxis index in LPS-stimulated BMMs was lower than that of unstimulated BMMs. It is therefore tempting to speculate that activated macrophages may tend to remain at the site of inflammation as suggested by our data. The inhibitory effect of NA on chemotaxis was observed within 2 h and at relatively low Hca2 expression levels, suggestive of NA's inhibitory effect on the early stages of macrophage activation. It has been demonstrated that whereas cAMP acts as a positive regulator of a number of proinflammatory chemokine genes during differentiation from monocytes to macrophages (42), it is the increase in the guanosine 3′, 5′-cyclic monophosphate (cGMP) level and the activation of cGMP-dependent protein kinase (PKA) in macrophages that are involved in α-elastin-induced macrophage chemotaxis (47).
In contrast to the results of chemotaxis of LPS-stimulated macrophages, we found that compared to untreated BMMs, LPS-treated BMMs significantly increased their uptake of LDL. Furthermore, NA suppressed LDL uptake by LPS-treated wild-type BMMs but not by Hca2−/− BMMs. Accumulation of cholesterol in macrophages and the formation of foam cells are the initial steps in the formation and progression of atherosclerosis. These data indicate and confirm the notion that macrophages activated by means other than cholesterol show increased cholesterol uptake, a process that can be inhibited by NA through HCA2. Interestingly, increased cAMP levels are known to inhibit macrophage phagocytosis of apoptotic cells (48).
NA has shown to exert beneficial effects on cardiovascular outcomes, attributed to its lipid-modifying effects seen in humans (49). However, a recent study using a humanized genetic mouse model for atherosclerosis, as well as data from human clinical trials using synthetic HCA2 receptor agonists, show that the lipid-modifying effects of NA are independent of the HCA2 receptor (50). Furthermore, recent evidence indicates that lipid-independent HCA2-mediated beneficial effects on the progression of atherosclerosis in mice were accompanied by reduced recruitment of macrophages to atherosclerotic plaques (44). Our studies further complement these findings by showing that NA reduces LDL uptake by LPS-stimulated macrophages in an HCA2 receptor-dependent manner. Taken together, it appears that the beneficial effects of NA on atherosclerotic cardiovascular outcomes are based on both its lipid-modifying (HCA2 receptor-independent) and its anti-inflammatory (HCA2 receptor-dependent) functions.
In summary, our data suggest that HCA2 suppresses macrophage proinflammatory function by inhibiting proinflammatory cytokine production, LDL uptake, and chemotaxis. Thus, HCA2-mediated signaling pathways may offer potential targets for therapeutic intervention to prevent or slow the progression of chronic inflammatory diseases.
Supplementary Material
Acknowledgments
This study was supported, in part, by a Genzyme Renal Innovation Program (GRIP) grant from Genzyme (Cambridge, MA, USA) (K.Z.-N.), a Scientist Development grant from the American Heart Association (A.T.), and U.S. National Institutes of Health grants DK070756 and DK57708 (D.M.), and RO1-AI091930-01A 1 (R.A.).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, American Heart Association, or Genzyme.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- BMM
- bone marrow-derived macrophage
- CpG-ODN
- cytosine-phosphate-guanine oligodeoxynucleotide
- cAMP
- cyclic adenosine monophosphate
- cDNA
- complementary DNA
- ELISA
- enzyme-linked immunosorbent assay
- GM-CSF
- granulocyte-macrophage colony-stimulating factor
- GpC-ODN
- guanine-phosphate-cytosine oligodeoxynucleotide
- GPCR
- G-protein-coupled receptor
- HCA2
- hydroxycarboxylic acid 2
- IκB
- inhibitor κB
- IL
- interleukin
- IP-10
- interferon-inducible protein 10
- LDL
- low-density lipoprotein
- LPS
- lipopolysaccharide
- MCP-1
- monocyte chemoattractant protein 1
- MIG
- monokine induced by γ-interferon
- MSU
- monosodium urate monohydrate
- NA
- nicotinic acid
- NF-κB
- nuclear factor κB
- pNF-κB
- phospho nuclear factor κB
- PUMA-G
- protein upregulated in macrophages by IFN-γ
- RT-PCR
- reverse transcriptase polymerase chain reaction
- TLR
- toll-like receptor
- TNF-α
- tumor necrosis factor α
REFERENCES
- 1. Seymour R. M., Henderson B. (2001) Pro-inflammatory–anti-inflammatory cytokine dynamics mediated by cytokine-receptor dynamics in monocytes. IMA J. Math. Appl. Med. Biol. 18, 159–192 [PubMed] [Google Scholar]
- 2. O'Neill L. A., Bowie A. G. (2007) The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 7, 353–364 [DOI] [PubMed] [Google Scholar]
- 3. Schaub A., Futterer A., Pfeffer K. (2001) PUMA-G, an IFN-γ-inducible gene in macrophages is a novel member of the seven transmembrane spanning receptor superfamily. Eur. J. Immunol. 31, 3714–3725 [DOI] [PubMed] [Google Scholar]
- 4. Soga T., Kamohara M., Takasaki J., Matsumoto S., Saito T., Ohishi T., Hiyama H., Matsuo A., Matsushime H., Furuichi K. (2003) Molecular identification of nicotinic acid receptor. Biochem. Biophys. Res. Commun. 303, 364–369 [DOI] [PubMed] [Google Scholar]
- 5. Tunaru S., Kero J., Schaub A., Wufka C., Blaukat A., Pfeffer K., Offermanns S. (2003) PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat. Med. 9, 352–355 [DOI] [PubMed] [Google Scholar]
- 6. Wise A., Foord S. M., Fraser N. J., Barnes A. A., Elshourbagy N., Eilert M., Ignar D. M., Murdock P. R., Steplewski K., Green A., Brown A. J., Dowell S. J., Szekeres P. G., Hassall D. G., Marshall F. H., Wilson S., Pike N. B. (2003) Molecular identification of high and low affinity receptors for nicotinic acid. J. Biol. Chem. 278, 9869–9874 [DOI] [PubMed] [Google Scholar]
- 7. Taggart A. K., Kero J., Gan X., Cai T. Q., Cheng K., Ippolito M., Ren N., Kaplan R., Wu K., Wu T. J., Jin L., Liaw C., Chen R., Richman J., Connolly D., Offermanns S., Wright S. D., Waters M. G. (2005) (D)-beta-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J. Biol. Chem. 280, 26649–26652 [DOI] [PubMed] [Google Scholar]
- 8. Offermanns S., Colletti S. L., Lovenberg T. W., Semple G., Wise A., AP I. J. (2011) International Union of Basic and Clinical Pharmacology. LXXXII: Nomenclature and Classification of hydroxy-carboxylic acid receptors (GPR81, GPR109A, and GPR109B). Pharmacol Rev. 63, 269–290 [DOI] [PubMed] [Google Scholar]
- 9. Benyo Z., Gille A., Kero J., Csiky M., Suchankova M. C., Nusing R. M., Moers A., Pfeffer K., Offermanns S. (2005) GPR109A (PUMA-G/HM74A) mediates nicotinic acid-induced flushing. J. Clin. Invest. 115, 3634–3640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hotz W. (1983) Nicotinic acid and its derivatives: a short survey. Adv. Lipid Res. 20, 195–217 [PubMed] [Google Scholar]
- 11. Carlson L. A., Oro L., Ostman J. (1968) Effect of a single dose of nicotinic acid on plasma lipids in patients with hyperlipoproteinemia. Acta Med. Scand. 183, 457–465 [DOI] [PubMed] [Google Scholar]
- 12. Martin W. J., Walton M., Harper J. (2009) Resident macrophages initiating and driving inflammation in a monosodium urate monohydrate crystal-induced murine peritoneal model of acute gout. Arthritis Rheum. 60, 281–289 [DOI] [PubMed] [Google Scholar]
- 13. Farber J. M. (1990) A macrophage mRNA selectively induced by γ-interferon encodes a member of the platelet factor 4 family of cytokines. Proc. Natl. Acad. Sci. U. S. A. 87, 5238–5242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Amichay D., Gazzinelli R. T., Karupiah G., Moench T. R., Sher A., Farber J. M. (1996) Genes for chemokines MuMig and Crg-2 are induced in protozoan and viral infections in response to IFN-γ with patterns of tissue expression that suggest nonredundant roles in vivo. J. Immunol. 157, 4511–4520 [PubMed] [Google Scholar]
- 15. Rothwarf D. M., Karin M. (1999) The NF-κB activation pathway: a paradigm in information transfer from membrane to nucleus. Sci. STKE 1999, RE1. [DOI] [PubMed] [Google Scholar]
- 16. Matsushima K., Larsen C. G., DuBois G. C., Oppenheim J. J. (1989) Purification and characterization of a novel monocyte chemotactic and activating factor produced by a human myelomonocytic cell line. J. Exp. Med. 169, 1485–1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Charo I. F., Myers S. J., Herman A., Franci C., Connolly A. J., Coughlin S. R. (1994) Molecular cloning and functional expression of two monocyte chemoattractant protein 1 receptors reveals alternative splicing of the carboxyl-terminal tails. Proc. Natl. Acad. Sci. U. S. A. 91, 2752–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Funk J. L., Feingold K. R., Moser A. H., Grunfeld C. (1993) Lipopolysaccharide stimulation of RAW 264.7 macrophages induces lipid accumulation and foam cell formation. Atherosclerosis 98, 67–82 [DOI] [PubMed] [Google Scholar]
- 19. Hendriks W. L., van der Boom H., van Vark L. C., Havekes L. M. (1996) Lipoprotein lipase stimulates the binding and uptake of moderately oxidized low-density lipoprotein by J774 macrophages. Biochem. J. 314, 563–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kruth H. S., Huang W., Ishii I., Zhang W. Y. (2002) Macrophage foam cell formation with native low density lipoprotein. J. Biol. Chem. 277, 34573–34580 [DOI] [PubMed] [Google Scholar]
- 21. Yasuda T., Hirata K., Ishida T., Kojima Y., Tanaka H., Okada T., Quertermous T., Yokoyama M. (2007) Endothelial lipase is increased by inflammation and promotes LDL uptake in macrophages. J. Atheroscler. Thromb. 14, 192–201 [DOI] [PubMed] [Google Scholar]
- 22. Deane K. D., O'Donnell C. I., Hueber W., Majka D. S., Lazar A. A., Derber L. A., Gilliland W. R., Edison J. D., Norris J. M., Robinson W. H., Holers V. M. (2010) The number of elevated cytokines and chemokines in preclinical seropositive rheumatoid arthritis predicts time to diagnosis in an age-dependent manner. Arthritis Rheum. 62, 3161–3172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kobayashi T., Murasawa A., Komatsu Y., Yokoyama T., Ishida K., Abe A., Yamamoto K., Yoshie H. (2010) Serum cytokine and periodontal profiles in relation to disease activity of rheumatoid arthritis in Japanese adults. J. Periodontol. 81, 650–657 [DOI] [PubMed] [Google Scholar]
- 24. Mohammad M. K., Morran M., Slotterbeck B., Leaman D. W., Sun Y., Grafenstein H., Hong S. C., McInerney M. F. (2006) Dysregulated Toll-like receptor expression and signaling in bone marrow-derived macrophages at the onset of diabetes in the non-obese diabetic mouse. Int. Immunol. 18, 1101–1113 [DOI] [PubMed] [Google Scholar]
- 25. Yurkovetsky Z. R., Kirkwood J. M., Edington H. D., Marrangoni A. M., Velikokhatnaya L., Winans M. T., Gorelik E., Lokshin A. E. (2007) Multiplex analysis of serum cytokines in melanoma patients treated with interferon-α2b. Clin. Cancer Res. 13, 2422–2428 [DOI] [PubMed] [Google Scholar]
- 26. Barath P., Fishbein M. C., Cao J., Berenson J., Helfant R. H., Forrester J. S. (1990) Detection and localization of tumor necrosis factor in human atheroma. Am. J. Cardiol. 65, 297–302 [DOI] [PubMed] [Google Scholar]
- 27. Tipping P. G., Hancock W. W. (1993) Production of tumor necrosis factor and interleukin-1 by macrophages from human atheromatous plaques. Am. J. Pathol. 142, 1721–1728 [PMC free article] [PubMed] [Google Scholar]
- 28. Seino Y., Ikeda U., Ikeda M., Yamamoto K., Misawa Y., Hasegawa T., Kano S., Shimada K. (1994) Interleukin 6 gene transcripts are expressed in human atherosclerotic lesions. Cytokine 6, 87–91 [DOI] [PubMed] [Google Scholar]
- 29. Kirii H., Niwa T., Yamada Y., Wada H., Saito K., Iwakura Y., Asano M., Moriwaki H., Seishima M. (2003) Lack of interleukin-1β decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 23, 656–660 [DOI] [PubMed] [Google Scholar]
- 30. Lee T. S., Yen H. C., Pan C. C., Chau L. Y. (1999) The role of interleukin 12 in the development of atherosclerosis in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 19, 734–742 [DOI] [PubMed] [Google Scholar]
- 31. Chen C. J., Shi Y., Hearn A., Fitzgerald K., Golenbock D., Reed G., Akira S., Rock K. L. (2006) MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J. Clin. Invest. 116, 2262–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pessler F., Mayer C. T., Jung S. M., Behrens E. M., Dai L., Menetski J. P., Schumacher H. R. (2008) Identification of novel monosodium urate crystal regulated mRNAs by transcript profiling of dissected murine air pouch membranes. Arthritis Res. Ther. 10, R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mount D. B., Kwon C. Y., Zandi-Nejad K. (2006) Renal urate transport. Rheum. Dis. Clin. North Am. 32, 313–331, vi [DOI] [PubMed] [Google Scholar]
- 34. Kwon W. Y., Suh G. J., Kim K. S., Kwak Y. H. (2011) Niacin attenuates lung inflammation and improves survival during sepsis by downregulating the nuclear factor-κB pathway. Crit. Care Med. 39, 328–334 [DOI] [PubMed] [Google Scholar]
- 35. Digby J. E., Martinez F., Jefferson A., Ruparelia N., Chai J., Wamil M., Greaves D. R., Choudhury R. P. (2012) Anti-inflammatory effects of nicotinic acid in human monocytes are mediated by GPR109A dependent mechanisms. Arterioscler. Thromb. Vasc. Biol. 32, 669–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Butcher R. W., Baird C. E., Sutherland E. W. (1968) Effects of lipolytic and antilipolytic substances on adenosine 3′,5′-monophosphate levels in isolated fat cells. J. Biol. Chem. 243, 1705–1712 [PubMed] [Google Scholar]
- 37. Aktories K., Schultz G., Jakobs K. H. (1980) Regulation of adenylate cyclase activity in hamster adipocytes. Inhibition by prostaglandins, alpha-adrenergic agonists and nicotinic acid. Naunyn Schmiedebergs Arch. Pharmacol. 312, 167–173 [DOI] [PubMed] [Google Scholar]
- 38. Aktories K., Jakobs K. H., Schultz G. (1980) Nicotinic acid inhibits adipocyte adenylate cyclase in a hormone-like manner. FEBS Lett. 115, 11–14 [DOI] [PubMed] [Google Scholar]
- 39. Serezani C. H., Ballinger M. N., Aronoff D. M., Peters-Golden M. (2008) Cyclic AMP: master regulator of innate immune cell function. Am. J. Respir. Cell Mol. Biol. 39, 127–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wall E. A., Zavzavadjian J. R., Chang M. S., Randhawa B., Zhu X., Hsueh R. C., Liu J., Driver A., Bao X. R., Sternweis P. C., Simon M. I., Fraser I. D. (2009) Suppression of LPS-induced TNF-α production in macrophages by cAMP is mediated by PKA-AKAP95-p105. Sci. Signal. 2, ra28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Agarwal N., Lamichhane G., Gupta R., Nolan S., Bishai W. R. (2009) Cyclic AMP intoxication of macrophages by a Mycobacterium tuberculosis adenylate cyclase. Nature 460, 98–102 [DOI] [PubMed] [Google Scholar]
- 42. Hertz A. L., Bender A. T., Smith K. C., Gilchrist M., Amieux P. S., Aderem A., Beavo J. A. (2009) Elevated cyclic AMP and PDE4 inhibition induce chemokine expression in human monocyte-derived macrophages. Proc. Natl. Acad. Sci. U. S. A. 106, 21978–21983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jin S. L., Lan L., Zoudilova M., Conti M. (2005) Specific role of phosphodiesterase 4B in lipopolysaccharide-induced signaling in mouse macrophages. J. Immunol. 175, 1523–1531 [DOI] [PubMed] [Google Scholar]
- 44. Lukasova M., Malaval C., Gille A., Kero J., Offermanns S. (2011) Nicotinic acid inhibits progression of atherosclerosis in mice through its receptor GPR109A expressed by immune cells. J. Clin. Invest. 121, 1163–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stout R. D., Suttles J. (2004) Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J. Leukoc. Biol. 76, 509–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stout R. D., Jiang C., Matta B., Tietzel I., Watkins S. K., Suttles J. (2005) Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J. Immunol. 175, 342–349 [DOI] [PubMed] [Google Scholar]
- 47. Kamisato S., Uemura Y., Takami N., Okamoto K. (1997) Involvement of intracellular cyclic GMP and cyclic GMP-dependent protein kinase in α-elastin-induced macrophage chemotaxis. J. Biochem. 121, 862–867 [DOI] [PubMed] [Google Scholar]
- 48. Rossi A. G., McCutcheon J. C., Roy N., Chilvers E. R., Haslett C., Dransfield I. (1998) Regulation of macrophage phagocytosis of apoptotic cells by cAMP. J. Immunol. 160, 3562–3568 [PubMed] [Google Scholar]
- 49. Offermanns S. (2012) It ain't over 'til the fat lady sings. Sci. Transl. Med. 4, 148fs130 [DOI] [PubMed] [Google Scholar]
- 50. Lauring B., Taggart A. K., Tata J. R., Dunbar R., Caro L., Cheng K., Chin J., Colletti S. L., Cote J., Khalilieh S., Liu J., Luo W. L., Maclean A. A., Peterson L. B., Polis A. B., Sirah W., Wu T. J., Liu X., Jin L., Wu K., Boatman P. D., Semple G., Behan D. P., Connolly D. T., Lai E., Wagner J. A., Wright S. D., Cuffie C., Mitchel Y. B., Rader D. J., Paolini J. F., Waters M. G., Plump A. (2012) Niacin lipid efficacy is independent of both the niacin receptor GPR109A and free fatty acid suppression. Sci. Transl. Med. 4, 148ra115. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.