

Abstract
Because little is known about the actions of botanical estrogens (BEs), widely consumed by menopausal women, we investigated the mechanistic and cellular activities of some major BEs. We examined the interactions of genistein, daidzein, equol, and liquiritigenin with estrogen receptors ERα and ERβ, with key coregulators (SRC3 and RIP140) and chromatin binding sites, and the regulation of gene expression and proliferation in MCF-7 breast cancer cells containing ERα and/or ERβ. Unlike the endogenous estrogen, estradiol (E2), BEs preferentially bind to ERβ, but their ERβ-potency selectivity in gene stimulation (340- to 830-fold vs. E2) is enhanced at several levels (coregulator recruitment, chromatin binding); nevertheless, at high (0.1 or 1 μM) concentrations, BEs also fully activate ERα. Because ERα drives breast cancer cell proliferation and ERβ dampens this, the relative levels of these two ERs in target cells and the BE dose greatly affect gene expression and proliferative response and will be crucial determinants of the potential benefits vs. risks of BEs. Our findings reveal key and novel mechanistic differences in the estrogenic activities of BEs vs. E2, with BEs displaying patterns of activity distinctly different from those seen with E2 and provide valuable information to inform future studies.—Jiang, Y., Gong, P., Madak-Erdogan, Z., Martin, T., Jeyakumar, M., Carlson, K., Khan, I., Smillie, T. J., Chittiboyina, A. G., Rotte, S. C. K., Helferich, W. G., Katzenellenbogen, J. A., Katzenellenbogen, B. S. Mechanisms enforcing the estrogen receptor β selectivity of botanical estrogens.
Keywords: chromatin binding, gene regulation, proliferation, breast cancer cells
As the average age of the U.S. population rises, an increasing number of women are postmenopausal for many years, and due to the dramatic reduction in levels of estrogens, they often experience hot flashes, night sweats, and mood changes, and they suffer from urogenital atrophy and loss of bone density. The traditional treatment for menopausal symptoms is to restore estrogen levels by hormone replacement therapy (HRT). Despite the well-known advantages of HRT, studies from the Women's Health Initiative revealed several adverse effects of estrogen alone or estrogen plus progestin, such as increased risk of heart disease (1), stroke (2), breast cancer (3), and dementia (4, 5). Consequently, there is great interest from researchers, clinicians, and the public in the development of new treatment strategies to avoid the adverse effects of HRT.
Botanicals containing estrogenic compounds are widely available and are consumed by women, in particular by older women seeking relief from menopausal symptoms, with the expectation that these botanical estrogens (BEs) may provide a safe, natural source of estrogens to replace the loss of endogenous estrogens in menopause. Soy-based products have drawn increasing attention lately as alternative treatments for relief of menopausal symptoms because their major isoflavone components, genistein and daidzein, are known to have estrogenic effects (6). However, preclinical studies (7, 8) and studies in humans have given inconclusive results regarding the efficacy of soy for this purpose (9–11). Because estrogens have stimulatory effects on many tissues, including increasing the growth of some breast cancers, the unregulated consumption of BEs might not be contributing uniformly to healthy aging in women (12–16).
Estrogens exert effects on diverse target tissues and cells, and they act through two different estrogen receptors (ERs), ERα and ERβ (17–20). ERα and ERβ are encoded by different genes and have different tissue distributions and different ligand binding specificities. ERα is generally the more potent regulator of gene expression and appears responsible for mediating the proliferative drive of estrogens in some target tissues, such as the uterus and some breast cancers, whereas ERβ when present together with ERα has a generally restraining effect on ERα activities (21–25). Because we and others have shown that many estrogens isolated from botanicals (e.g., genistein, daidzein, equol, liquiritigenin) are preferential ligands for binding to ERβ (6, 17, 26–28), they differ in this respect from endogenous estrogens and most estrogen pharmaceutical agents, which are ERα-binding preferential. Thus, one might expect the ERβ-preferential botanicals to have different biological activities than estradiol (E2).
To gain mechanistic information and to examine dose-dependent effects of BEs that might provide a new conceptual framework for understanding whether BEs have similar or unique activities compared to those of other estrogens, such as E2, we have studied in detail 4 BEs, genistein, daidzein, S-equol, and liquiritigenin. We measured their binding affinities to ERα and ERβ, and the affinity with which their complexes with ER recruit the key steroid receptor coactivator (SRC), SRC3; we examined the chromatin binding of these ligand-receptor complexes along with that of the coregulators SRC3 and receptor interacting protein 140 (RIP140) at estrogen-regulated genes by chromatin immunoprecipitation (ChIP) assays, and we assessed their potency and efficacy in regulating gene expression and their effects on proliferation of human breast cancer cells (MCF-7) containing only ERα, only ERβ, or both ERα and ERβ, thereby mimicking the different ratios of these two ERs present in different ER target tissues and human breast tumors. Our studies highlight that BEs bind, induce coactivator recruitment, and stimulate chromatin binding preferentially of ERβ to enhance expression of ERβ-regulated genes. In addition, our findings provide new insight that, compared to E2, the ERβ vs. ERα potency selectivity of BEs is enhanced at distinct levels (e.g., coregulator recruitment, chromatin binding) beyond their binding affinities for the two ERs. However, at high concentrations, rather than dampen cell proliferation through ERβ, they stimulate proliferation through ERα, highlighting that BE dose and the presence of ERα and/or ERβ in cells markedly affect the biological effects of BEs.
MATERIALS AND METHODS
Ligands, cell culture, and construction of MCF-7 breast cancer cells containing ERα, ERα, and ERβ, or ERβ only
MCF-7 cells [American Type Culture Collection (ATCC), Manassas, VA, USA] were cultured in DMEM (Gibco/Life Technologies, Grand Island, NY, USA), supplemented with 5% calf serum (HyClone, Logan, UT, USA) and 100 μg/ml penicillin/streptomycin (Invitrogen, Carlsbad, CA). For estrogen-free experiments, the cells were seeded in phenol red-free DMEM (Gibco/Life Technologies) plus 5%charcoal-dextran-treated calf serum at a density of 2.5 × 105 cells/well of 6-well plate (for mRNA studies) or 1 × 106 cells/10-cm tissue culture dish (for ChIP assays) for 3 d before siRNA transfection and adenovirus infection. Recombinant adenoviruses were constructed and prepared as described (22, 24). Cells were infected with either control adenovirus (Ad) expressing β-galactosidase or adenovirus expressing ERβ (AdERβ) for 72 h. Conditions used were those described previously (24, 29) to generate MCF-7 cells expressing levels of ERβ equal to that of the endogenously expressed ERα. The siRNA experiments for knockdown of the endogenous ERα in MCF-7 cells were performed as described previously and resulted in knockdown of ERα mRNA and protein by greater than 95% (24). siERα sequences (Dharmacon, Lafayette, CO, USA) were forward, 5′-UCAUCGCAUUCCUUGCAAAdTdT-3′, and reverse, 5′-UUUGCAAGGAAUGCGAUGAdTdT-3′ (24). Because ERα knockdown did not affect ERβ levels, the level of ERβ obtained in the ERβ-only cells (24) was similar to that of ERα in the original MCF-7 cells. E2, genistein, and formononetin were from Sigma-Aldrich (St. Louis, MO, USA), liquiritigenin from Tocris Bioscience (Bristol, UK), daidzein from Indofine Chemical Co. (Hillsborough, NJ, USA), R-equol from Cayman Chemical Co. (Ann Arbor, MI, USA). Racemic and S-equol were prepared as described (26, 30). All compounds were checked for identity and purity by mass spectrometry and NMR.
Relative binding affinity assay
Relative binding affinities were determined by a competitive radiometric binding assay as described previously (31, 32) using 2 nM [3H]-E2 as tracer ([2,4,6,7-3H]-estra-1,3,5(10)-triene-3,17β-diol, 70–115 Ci/mmol; Perkin Elmer, Waltham, MA, USA), and purified, full-length, human ERα and ERβ purchased from PanVera/Invitrogen. Incubations were for 18–24 h at 0°C. Hydroxyapatite (Bio-Rad, Hercules, CA, USA) was used to absorb the receptor-ligand complexes, and free ligand was washed away. The binding affinities are expressed as relative binding affinity (RBA) values with the RBA of E2 set to 100%. The values given are the average ± range or sd of 2 to 3 independent determinations. E2 binds to ERα with a Kd of 0.2 nM and to ERβ with a Kd of 0.5 nM (31).
Coactivator titration time-resolved fluorescence resonance energy transfer (FRET) assay to determine relative coactivator-binding affinity (RCA)
The assay was conducted exactly as described previously and utilized SRC3-fluorescein and biotinylated-ERα or ERβ labeled with a streptavidin-terbium conjugate (33–35).
RNA isolation and real-time PCR
Total RNA was isolated from cells using TRIzol (Invitrogen), RNA samples were reverse transcribed by MMTV reverse transcriptase (New England Biolabs, Ipswich, MA, USA), and real-time PCR was carried out on the ABI Prism 7900HT (Applied Biosystems, Foster City, CA, USA) using SYBR Green PCR Master Mix (Roche, Basel, Switzerland) as described previously (22).
Western blot analysis
Whole-cell extracts were prepared using 1× ChIP lysis buffer supplemented with 1× complete protease inhibitor (Roche). Proteins were separated on 10% SDS-PAGE gels and transferred to nitrocellulose membranes. Western blotting used antibodies against ERα (Santa Cruz Biotechnology, Santa Cruz, CA, USA), ERβ (CWK-F12 produced by our laboratory; ref. 36), and β-actin (Sigma-Aldrich).
ChIP assays
ChIP for ERα or ERβ was carried out as described previously (37) and used the ERα antibody HC-20 (Santa Cruz Biotechnology). ERβ antibodies were a combination with equal parts of CWK-F12 produced by our laboratory (36), GTX70182 (GeneTex, SanAntonio, TX, USA), GR40 (Calbiochem, La Jolla, CA, USA), and PA1-311 (Thermo Fisher Scientific, Rockford, IL, USA), RIP140 (H-300; Santa Cruz Biotechnology), and SRC3 (M397; Santa Cruz Biotechnology). The ChIP DNA was used for quantitative real-time PCR.
Cell proliferation assays
WST-1 assay (Roche) was used to quantify cell viability, as described previously (38). Absorbance was measured at 450 nm using a Bio-Rad 680 Microplate Reader, and all assays were performed in triplicate.
Statistics
Statistically significant differences of gene expression changes on different ligand treatments were analyzed by 1-way ANOVA followed by Bonferroni post hoc test in GraphPad (GraphPad, San Diego, CA, USA). Results are the average ± sem of ≥3 independent experiments. Values of P < 0.05 were considered significant.
RESULTS
BEs show preference in binding and coactivator recruitment to ERβ vs. ERα
The BEs studied include genistein, daidzein, racemic equol, R-equol, S-equol, liquiritigenin, biochanin A (the monomethyl ether of genistein), and formononetin (the monomethyl ether of daidzein). These compounds are displayed in Fig. 1 in the orientation that best shows their relationship to the structure of 17β-E2. Binding affinities for human full-length ERα and ERβ were determined by a radiometric competitive binding assay using tritiated 17β-E2 as tracer and E2 as standard (31, 32). Affinities are expressed as RBA values where the affinity of E2 for ERα or ERβ was set at 100% (Table 1). E2 has an absolute binding affinity (Kd) of 0.2 nM for ERα and 0.5 nM for ERβ (31). All of the BEs have ≥10-fold lower affinity for ERβ and ≥50-fold lower affinity for ERα, compared to E2, but all of the naturally occurring BEs are quite ERβ preferential in their binding affinities.
Figure 1.
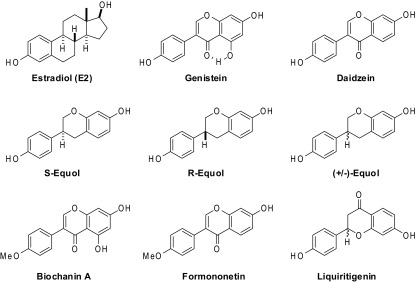
Structures of the BEs studied.
Table 1.
RBA of BEs for ERα and ERβ and comparison with E2
| BE | RBAERα (%) | RBAERβ (%) | β/α |
|---|---|---|---|
| E2 | 100 | 100 | 1 |
| Genistein | 0.021 ± 0.009 | 6.80 ± 1.1 | 324 |
| Daidzein | 0.003 ± 0.001 | 0.051 ± 0.02 | 17 |
| S-equol | 0.144 ± 0.02 | 3.50 ± 0.51 | 24 |
| R-equol | 0.374 ± 0.05 | 0.327 ± 0.02 | 0.9 |
| (±)-Equol | 0.201 ± 0.02 | 1.52 ± 0.41 | 8 |
| Biochanin A | ∼0.001 ± 0 | 0.024 ± 0.006 | 24 |
| Formononetin | 0.003 ± 0.001 | 0.003 ± 0.001 | 1 |
| Liquritigenin | <0.001 | 0.013 ± 0.003 | >13 |
RBA is calculated from the competitive radiometric binding curves: (IC50 E2/IC50 BE) × 100. β/α = RBAERβ/RBAERα.
As seen in Table 1, the highest-affinity ERβ ligand is genistein; its RBA of 6.8% corresponds to a Kd of 7.4 nM, and it also has by far the greatest ERβ selectivity (β/α=324). Daidzein, the most abundant isoflavone in soy, binds much less well to both ERs than genistein and with a reduced ERβ preference. The reduced affinity of daidzein compared to genistein has been attributed to the absence of the phenolic hydroxy group that in genistein forms an intramolecular hydrogen bond with the ketone; this leaves this polar functional group in daidzein isolated in the ER ligand-binding pocket, which lowers its affinity because the ER cannot supply a suitable polar residue to partner with this ketone (39).
Daidzein is metabolized in the gut by some microflora to S-equol (40), a conversion that increases its affinity for both ERs by 50- to 70-fold while still preserving substantial ERβ preference; thus, the ERβ affinity of S-equol rivals that of genistein. The unnatural enantiomer, R-equol, also binds much better than daidzein, but its ERα binding affinity is more markedly increased, so it is very slightly ERα preferential. The binding of (±)-equol is, as expected, the average of that of R- and S-equol on both ERs. Not surprisingly, the methyl ethers, formononetin, and biochanin A, also have very low binding affinities (<0.03%) for both receptors, reflecting the well-known importance of having a free phenol that can mimic the A-ring phenol of E2 for high affinity (41). Liquiritigenin also has a very low affinity for both ERs but is distinctly ERβ-binding preferential. And as opposed to the isoflavonoid structures of the other BEs, liquiritigenin has a flavonoid structure that is a poorer structural mimic of E2 (see Fig. 1).
We have developed a time-resolved fluorescence energy transfer (TR-FRET) assay to quantify the interaction of coactivators and corepressors with nuclear hormone receptors (33, 42, 43), and we used this method in a coactivator-titration mode (33) to compare the affinity with which SRC3, a major coregulator found at high levels in breast cancer (30, 44, 45), binds to ERα and ERβ complexes of these BEs. The titration curves are shown in Fig. 2, and affinities are summarized in Table 2. SRC3 binds to the ERα-E2 complex very well, with an EC50 of 0.13 nM (which for comparison with BEs is considered to be an RCA of 100). The BEs, by contrast, form complexes with ERα, with which SRC3 has lower affinity (E2 > genistein > liquiritigen > S-equol > R-equol). All of the BEs, however, form ERβ complexes with which SRC3 has very good affinity, similar to that of the ERβ-E2 complex. If one proposes that the transcriptional activity of ER requires that it bind both ligand and coactivator, then transcriptional potency must, in some way, reflect both ligand-binding affinity and coactivator-binding affinity (33). Thus, with the BEs, the selectivity of their transcriptional activity through ERβ (see below) should be enhanced by both ligand-binding preference and coactivator-binding preference (Table 3), an aspect that is discussed further in Discussion.
Figure 2.

Coactivator binding affinity to ERα or ERβ complexes with the BEs. In a coactivator titration protocol, increasing concentrations of the coactivator SRC3 (labeled with fluorescein) are added to a fixed concentration of the ER-BE complex (ER labeled through biotin with streptavidin-terbium). The SRC3-ER interaction is monitored by time-resolved FRET and expressed as receptor/donor emission intensities × 1000 (A/D*1000), as described previously (33, 42, 43). RCA, relative coactivator affinity with E2 set at 100%.
Table 2.
Binding affinity of the coregulator SRC3 to ERα and ERβ complexes with E2 and BEs expressed as RCA values
| BE | ERα |
ERβ |
β/α | ||
|---|---|---|---|---|---|
| EC50 (nM) | RCA | EC50 (nM) | RCA | ||
| E2 | 0.13 ± 0.004 | [100] | 0.12 ± 0.002 | [100] | [1.0] |
| Genistein | 1.1 ± 0.1 | 11.30 ± 0.7 | 0.20 ± 0.01 | 61 ± 0.9 | 5.4 |
| Daidzein | 14.2 ± 1.1 | 0.88 ± 0.05 | 0.22 ± 0.02 | 54.4 ± 2.1 | 61.8 |
| S-equol | 16.7 ± 0.6 | 0.73 ± 0.01 | 0.18 ± 0.01 | 78.5 ± 4.2 | 108.0 |
| R-equol | 33.9 ± 1.8 | 0.36 ± 0.008 | 0.33 ± 0.1 | 43 ± 2.1 | 119.0 |
| (±)-Equol | 25.9 ± 4.2 | 0.47 ± 0.02 | 0.25 ± 0.11 | 56 ± 4.2 | 119.0 |
| Biochanin A | 88.8 ± 6.4 | 0.14 ± 0.02 | 0.26 ± 0.01 | 46.7 ± 1.5 | 334.0 |
| Formononetin | ND | ND | 0.53 ± 0.02 | 22.9 ± 1.7 | — |
| Liquiritigenin | 2.2 ± 0.4 | 4.60 ± 0.5 | 0.13 ± 0.01 | 88.9 ± 6.7 | 19.3 |
RCA = (EC50 E2/EC50 BE) × 100. β/α = RCAERβ/RCAERα. ND, not detectable.
Table 3.
Potency of the BEs and their preferential activities through ERβ vs. ERα in cellular (gene stimulation) and cell-free (ligand binding and coactivator binding) assays
| Ligand | Potency from cellular activities |
Potency from cell-free activities |
||||
|---|---|---|---|---|---|---|
| Gene stimulation, EC50 (nM) |
β/α potency selectivity [relative to E2] | β/α selectivity RBA × RCA [relative to E2] | β/α RCA selectivity | β/α RBA selectivity | ||
| ERβ only, OTUB2 | ERα only, PgR | |||||
| E2 | 1.67 | 0.02 | 0.012 [1.0] | [1.0] | [1.00] | [1.00] |
| Genistein | 4 | 24 | 6 [500] | 1750 | 5.4 | 324 |
| S-equol | 1.4 | 14 | 10 [830] | 2600 | 108 | 24 |
| Daidzein | 50 | 200 | 4 [340] | 1024 | 62 | 17 |
| Liquiritigenin | 60 | 300 | 5 [420] | 400 | 19 | 21 |
OTUB2, otubain 2; PgR, progesterone receptor.
Relative levels of ERα and ERβ in cells affect response to different BEs
The potency of the BEs, acting through either ERα or ERβ, in regulating the expression of estrogen-responsive genes was evaluated in MCF-7 cells containing 3 different complements of ER: cells that contain ERα only, cells with ERα + ERβ at equal levels, or cells with ERβ only. These cells were constructed using ERα siRNA for ERα knockdown and/or adenovirus gene delivery for expression of ERβ, as we described previously (24, 46, 47). The expression of ERα and ERβ protein in the 3 cell types is shown in Fig. 3. ERα-only MCF-7 cells contain endogenous ERα, ERα + ERβ MCF-7 cells contain both ERα and ERβ, and ERβ-only MCF-7 cells contain ERβ and very little ERα.
Figure 3.

Western blots show ERα and ERβ levels in cells with the 3 complements of ERs. A) MCF-7 cells were infected with control adenovirus or ERβ-expressing adenovirus to generate cells containing ERα only and ERα + ERβ, respectively. B) Cells designated as ERβ only were generated by knockdown of ERα using ERα siRNA and then infection with ERβ adenovirus.
For our studies of endogenous gene expression regulation, we selected the 4 most significant BEs: genistein, daidzein, S-equol, and liquiritigenin. R-equol is not found naturally but was studied for comparison. We investigated the effect of the 4 BEs on the expression of the progesterone receptor (PgR), an ERα-responsive gene (48), and otubain 2 (OTUB2), an ERβ-responsive gene (24), in the ERα-only, ERα + ERβ, and ERβ-only cells over a dose range, comparing them with the response to E2. E2 showed a marked ERα potency preference in gene stimulation: In ERα-only cells, E2 had an EC50 of 0.02 nM for stimulating PgR gene expression, whereas in ERα + ERβ or ERβ-only cells, it had considerably higher EC50 values for stimulating OTUB2 gene expression (Fig. 4A). Thus, although E2 binds to ERα only 2–3-fold better than to ERβ, its ERα potency preference for gene regulation was much greater than this (∼50-fold; red curve is to the left of green curves), and it was especially poor in stimulating gene expression in ERβ-only cells.
Figure 4.
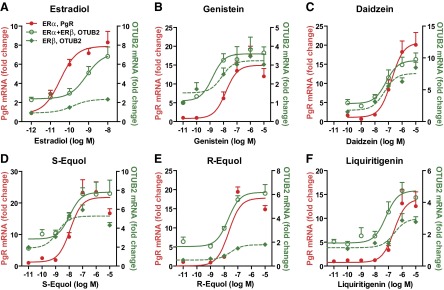
Effects of E2 and BEs on gene regulation in MCF-7 cells containing ERα only, ERα + ERβ, or ERβ only. Effects of E2 (A), genistein (B), daidzein (C), S-equol (D), R-equol (E), and liquiritigenin (F) on ERα-preferential progesterone receptor (PgR) gene expression (red) or ERβ-preferential OTUB2 gene expression (green) monitored after 4 h treatment.
Notably, by contrast, the 4 naturally occurring BEs showed a very different (more ERβ preferential) pattern of potency in gene regulation (green curves to left of red curve). As seen in Fig. 4B, low concentrations of genistein stimulated gene expression more effectively when ERβ was present in cells. Daidzein, S-equol, and liquiritigenin (Fig. 4C, D, F) also showed ERβ potency-preferential gene regulation. However, with all compounds at high concentrations, gene stimulation was also effectively elicited in cells containing ERα only (Fig. 4). In addition, gene stimulation by BEs and E2 in the 3 receptor cell backgrounds was completely suppressed by an excess of the antiestrogen ICI 182,780 (fulvestrant) implying ER mediation (data not shown).
The 4 BEs displayed a 5- to 10-fold ERβ potency preference for gene regulation. Considering that E2 has a gene regulation potency preference ∼50-fold in favor of ERα, the BEs thus have a 250- to 500-fold greater ERβ potency preference relative to E2 in terms of their regulation of gene expression (Table 3). This likely reflects both the preference of ERβ for binding the BEs and of coactivators for binding to the ERβ-BE complexes. The unnatural R-equol enantiomer showed little if any ERβ potency selectivity in gene regulation (Fig. 4E), consonant with its nearly equal binding affinity for ERα and ERβ (Table 1).
Potency and efficacy of liquiritigenin through ERβ are greater than expected from its ERβ binding affinity: gene stimulation and chromatin binding of receptors and coregulators
In terms of ligand binding, the RBA of liquiritigenin for ERα is only ∼0.001% and for ERβ is only 0.021% that of E2 (Table 1). Thus, the RBA of liquiritigenin is 105-fold lower than that of E2 for ERα and 5000-fold lower for ERβ. However, the EC50 values for liquiritigenin stimulation of OTUB2 gene expression in ERα + ERβ or ERβ-only cells are ∼100 nM (Fig. 4F), while that of E2 is ∼1 nM. This means that liquiritigenin only needs ∼100-fold higher concentrations than E2 to achieve the same effects through ERβ in ERα+ERβ or ERβ-only cells. Thus, liquiritigenin appears to have greater potency and efficacy through ERβ in gene regulation than expected from its ERβ-binding affinity, which could be a reflection of the very good SRC3 binding to the ERβ-liquiritigenin complex (Fig. 2 and Table 2).
Because liquiritigenin has been a BE of considerable interest (27, 44), we sought to further understand the mechanism for its increased potency and efficacy in stimulating gene expression by examining the recruitment of ERα, ERβ, and key coregulators to their binding sites on target genes. Based on the dose-response studies for liquiritigenin shown in Fig. 4, 2 concentrations were selected. The low concentration (50 nM) elicited little stimulation of PgR gene expression in ERα MCF-7 cells but did stimulate some OTUB2 gene expression when ERβ was present; by contrast, at the high concentration (1 μM), liquiritigenin was able to stimulate both PgR and OTUB2 gene expression in cells containing ERα or ERβ, respectively (Fig. 5B, E).
Figure 5.

Recruitment of ERα, ERβ, and coregulators SRC3 and RIP140 to chromatin by E2 or liquiritigenin to mediate PgR and OTUB2 gene expression. A, D) Binding sites identified by ChIP-seq for ERα, ERβ, and cofactors SRC3 and RIP140 at the PgR (A) and OTUB2 genes (D). B, E) Effects of 10 nM E2 and 50 nM or 1 μM liquiritigenin on PgR (B) or OTUB2 (E) gene expression monitored at 4 h. mRNA level in control vehicle MCF-7 ERα cells is set at 1. C, F) Recruitment of ERα or ERβ, SRC3, or RIP140 to chromatin binding sites at the PgR gene (C) or OTUB2 gene (F) in MCF-7 cells containing ERα only, ERα + ERβ, or ERβ only that were treated for 45 min with 0.1% control ethanol vehicle, 10 nM E2, or 50 nM or 1 μM liquiritigenin prior to ChIP.
These two concentrations of liquiritigenin (50 nM and 1 μM), and 10 nM E2 for comparison, were then used in ChIP assays in cells with these three different complements of ERs to examine the recruitment of ERα, ERβ, SRC3, and another coregulator, RIP140, to binding sites on the PgR and OTUB2 genes (Fig. 5). Using ChIP-DNA sequencing (ChIP-seq; ref. 47), we have identified binding sites of ERα, ERβ, and the coregulators SRC3 and RIP140 on the PgR and OTUB2 genes after treatment of cells with E2 (Fig. 5A, D). As expected, 10 nM E2 recruited ERα to binding sites on the PgR gene in ERα-only or ERα + ERβ MCF-7 cells, and it recruited ERβ to binding sites on PgR in ERα+ERβ or ERβ-only cells (Fig. 5C, solid bars). E2 also recruited SRC3 and RIP140 to the same binding sites on the PgR gene, with highest recruitment observed in ERα cells, lowest in ERβ cells, and intermediate in ERα + ERβ cells. The high concentration of liquiritigenin (Fig. 5C, shaded bars) recruited ERα, ERβ, SRC3, and RIP140 to binding sites on PgR, in a manner similar to that of 10 nM E2. The low concentration of liquiritigenin (Fig. 5C, hatched bars), however, showed a different pattern. Even though 50 nM liquiritigenin was able to recruit ERβ in ERα + ERβ or ERβ-only cells, it was not effective in recruiting ERα, SRC3, and RIP140 to the same binding sites on PgR (Fig. 5C). Since PgR is an ERα-responsive gene and needs ERα and cofactor recruitment to initiate gene expression, this explains the observation that the low concentration of liquritigenin did not stimulate PgR gene expression in the 3 types of MCF-7 cells (Fig. 5B).
Both low and high concentrations of liquiritigenin, as well as 10 nM E2, up-regulated OTUB2 gene expression in ERα + ERβ or ERβ-only MCF-7 cells (Fig. 5E). Both concentrations of liquiritigenin recruited ERβ, SRC3 and RIP140 to binding sites on OTUB2, but liquiritigenin was less good in recruitment of ERα, showing recruitment only at the high 1μM concentration (Fig. 5F). Since OTUB2 is an ERβ-responsive gene, its gene expression was highest in ERα + ERβ or ERβ-only cells, consistent with the strong recruitment of ERβ and the two coregulators in these cells (Fig. 5F). By contrast, E2 recruited ERα to binding sites on the OTUB2 gene in ERα-only or ERα + ERβ cells, and E2 recruited ERβ to the binding site on the OTUB2 gene in ERα + ERβ or ERβ-only cells. E2 also recruited SRC3 and RIP140 to the same binding sites on the OTUB2 gene, with highest recruitment in ERα + ERβ cells. These results indicate that despite its very low binding affinity for the ERs, relatively low concentrations of liquiritigenin can recruit ER and coactivator proteins and activate gene transcription in a markedly ERβ-preferential manner.
BEs vary in their effectiveness in promoting the ERβ-dampening effect on the proproliferative activity of ERα
We examined the effects of the BEs on the proliferation of cells containing ERα-only or ERα + ERβ (Fig. 6). E2 stimulated proliferation of MCF-7 cells containing ERα only at ligand concentrations as low as 10−12 M and elicited maximal stimulation at 10−11 M. Expression of ERβ along with ERα, however, greatly reduced the magnitude of E2-stimulated cell proliferation (Fig. 6A).
Figure 6.
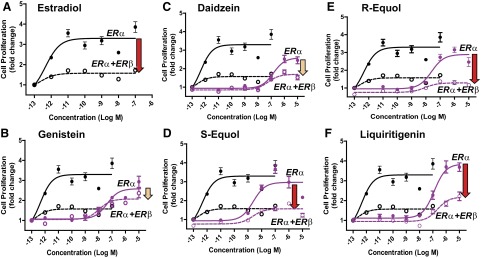
Effects of E2 (A), genistein (B), daidzein (C), S-equol (D), R-equol (E), and liquiritigenin (F) on the proliferation of ERα-only or ERα + ERβ cells. Proliferation was monitored after 5 d. Arrows indicate magnitude of suppression of proliferation by the specified ligand in cells containing ERα + ERβ vs. cells with ERα only. Red arrows indicate greater suppression; beige arrows indicate lesser suppression.
Consistent with the results of the dose-response gene expression studies, low concentrations (<10 nM) of genistein, daidzein, S-equol, R-equol, and liquiritigenin did not stimulate proliferation of ERα-only MCF-7 cells, but they were able to effectively promote proliferation of ERα-only cells at high concentrations (Fig. 6). S-equol was the most effective in stimulating proliferation through ERα, with good stimulation being observed at 10 nM. Notably, the copresence of ERβ dampened the stimulation of cell proliferation through ERα by the high concentrations of the BEs. The effect of ERβ in reducing proliferation was most marked with E2, liquiritigenin, and S-equol (Fig. 6, red arrows), and least with daidzein and genistein (Fig. 6, beige arrows).
In cells containing ERβ only, the basal, control rate of cell proliferation monitored by Ki67 gene expression was reduced compared to that of ERα-only cells, and 10 nM E2 enhanced expression of Ki67 mRNA in both ERα-only or ERβ-only cells (Fig. 7A). Consistent with the greater efficacy of BEs through ERβ vs. ERα, the low concentration of genistein and daidzein elevated Ki67 in cells containing ERβ but not ERα (Fig. 7B, C). At a high concentration (1 μM), all BEs except S-equol elicited increased Ki67 expression in both ERα-only and ERβ-only cells(Fig. 7B–E). S-equol did not elevate Ki67 in ERβ-only cells, but did at both the lower and higher concentration in ERα-only cells, perhaps reflecting the fact that S-equol had the highest binding affinity of the natural BEs for ERα (Table 1).
Figure 7.

Effects of E2 (A), genistein (B), daidzein (C), S-equol (D), and liquiritigenin (E) on expression of the proliferation-related gene Ki67 in ERα-only or ERβ-only cells. RNA was isolated from cells after 24 h, and gene expression was assessed by qPCR.
DISCUSSION
ERβ selectivity of BEs becomes progressively amplified along the ER signaling axis
We undertook these studies to examine whether the estrogenic activity of BEs was similar to or different from that of E2, and if different, through what mechanistic components did these differences arise. While it was well known that BEs have preferential binding affinity for ERβ, our findings are novel in revealing that, relative to E2, the ERβ-preferential potency of the BEs becomes progressively reinforced as their activity proceeds down the ER axis and involves key coregulator and chromatin binding aspects. Although the 4 major naturally occurring BEs we studied in detail bind to ERβ with a 20- to 300-fold preference over ERα, using our TR-FRET assay, we found that the BEs recruit SRC3 to ERβ as well as does E2, whereas their potency in recruiting SRC3 to ERα was only 1–10% that of E2, thereby reinforcing their ERβ preference at a second (coregulator) level. Further evidence for the amplification of the ERβ potency preference of the BEs came from dose-response studies of ERα- vs. ERβ-responsive gene expression in breast cancer cells containing different complements of ERα and ERβ. Here we found that the relative potency of BEs for activating ERβ- vs. ERα-regulated genes compared to E2 was as high as 500. Consequently, at low concentrations, the BEs regulated gene expression that was preferentially through ERβ; however, this preferential regulation of estrogen-responsive gene expression by ERβ was no longer observed in cells treated with higher concentrations of BEs, when ERα and ERβ were equally well recruited to gene regulatory sites on chromatin and both functioned as effective ligand-regulated transcription factors and stimulators of proliferation through ERα. We previously noted this dose-dependent ERβ vs. ERα selectivity for genistein in gene stimulation (24).
Table 3 contains a summary of our dataset, arranged in terms of the preference of the 4 principal BEs for activity through ERβ vs. ERα from both cell-free assays and cell-based assays. When the cellular potency selectivity values are referenced to the selectivity of E2, the ERβ potency preference of the 4 BEs in cells ranged from 340 to 830. These values are greater than their ERβ-binding selectivities and are notably similar to that predicted from the product of their ERβ-binding selectivity (RBA) and their SRC3 coactivator recruitment potency selectivity (RCA) (400–2600). Thus, it appears that both their ERβ ligand-binding preference and their coactivator ERβ-binding preference (33) make significant contributions to the very high ERβ potency selectivity of the BEs in a cellular context.
S-equol and liquiritigenin as unusual BEs: mechanism and potency
Two other aspects of altered potency relate to S-equol and liquiritigenin. S-equol is an intestinal metabolite of daidzein, the most abundant isoflavone in soy, and this metabolic conversion results in a 50- and 70-fold increase in binding affinity for ERα and ERβ, respectively. Thus, unlike its low-affinity metabolic precursor, daidzein, S-equol had a potency similar to that of genistein in terms of stimulation of gene expression and regulation of proliferation through ERα or ERβ. The enteric production of S-equol from daidzein, however, depends on the constituent microflora, with individuals being “low” and “high” equol producers (40). Thus, the exposure of individuals to S-equol depends not only on their intake of the precursor, daidzein, but the degree to which daidzein undergoes metabolic conversion to S-equol.
There is much current interest in understanding the effects of liquiritigenin, the most active estrogenic compound from licorice root (Glycyrrhizae uralensis). Liquiritigenin is known to be a highly selective ERβ agonist (27) and has been shown to significantly reduce tumor growth in a glioma xenograft model that contains only ERβ (49). Even though liquiritigenin has a very low affinity for both ERα and ERβ, its ERβ preferential binding and activity were evident in our studies. In the 3 types of MCF-7 cells, liquiritigenin proved to be more potent in stimulating gene expression than expected based on its binding affinity. Our results suggest that this increment in potency might be due to the competence of the ER-liquiritigenin complex for the recruitment of coactivators and for binding to regulatory chromatin sites of estrogen-responsive genes. As observed with the other 3 BEs, low doses of liquiritigenin did not stimulate the proliferation of ERα-only cells, while high concentrations stimulated ERα-only MCF-7 cell proliferation to an extent as great as E2. A recent report on the dynamic racemization of the liquiritigenin enantiomers and the equilibration of these flavonoids with the ring-opened isoliquiritigenin chalcone isomer suggests that the net biological activity of this compound may actually be the combined result of the activity from an equilibrating mixture of these 3 different chemical species, regardless of which component has been administered (44); this is most likely true for most, cell-free, cellular, and in vivo studies with this compound.
Metabolism of other BEs may also be occurring in MCF-7 cells, since there is evidence for phase II metabolism and its up-regulation by botanicals, e.g., UGT 2B15 up-regulation by E2 and genistein (50) and sulfation of genistein and biochanin A (51). However, the extent of metabolism and its relationship with oral first-pass effects in vivo are not known.
BE proliferative effects on breast cancer cells: ligand dose and ERα and ERβ cell context
It is now well established that ERα is the major driver of the proliferative effects of estrogens in both breast cancer and normal reproductive tissues, with ERβ serving largely as a brake on ERα-driven proliferation (22, 23, 48, 52). Notably, while normal breast tissue contains both ERα and ERβ, ERβ levels decline with the development and progression of breast cancer to a more malignant state, so that ERα becomes the major player in hormone-responsive advanced breast cancer (53–55). The MCF-7 breast cancer cells we studied having ERα only, ERα + ERβ, and ERβ only provide a convenient model system to examine the proliferative and gene regulatory effects of various estrogens through the two ERs in breast cancer (22, 24). Previously we and others reported that E2 markedly stimulated cell proliferation in ERα-only containing breast cancer cells, while proliferation in ERα + ERβ cells was more limited (23–25, 29, 47).
All 4 BEs had little or no effect on ERα-only MCF-7 cell proliferation at low concentrations, whereas at high concentrations they stimulated proliferation of these cells quite well. And, as with E2, expression of ERβ along with ERα reduced the proliferative response of cells to the BEs at all concentrations. Thus, stimulation of proliferation appears to track with the potency of a BE for ERα, with ERβ always having a growth-suppressive effect on ERα, which was observed with all BEs and with E2. Of note, however, the copresence of ERβ along with ERα reduced proliferation with E2, liquiritigenin, and S-equol more so than with daidzein and genistein. Also, we found that proliferation was slower in ERβ-only vs. ERα-only cells. In this regard, it is of interest that patients with human breast tumors that express ERβ in the absence of ERα are reported to have a generally good prognosis and clinical outcome (56).
Potential health effects of BEs
BEs are being widely consumed by women seeking relief from menopausal symptoms, and while their efficacy for this therapeutic purpose is uncertain, much still remains to be understood about potential benefits and risks of BEs. To provide a better basis for further understanding of these aspects, we investigated the estrogenic character of 4 major BEs in a broader context; this involved examination of their binding to ERα and ERβ, interaction of the ligand-receptor complexes with coregulators, receptor and coregulator recruitment to chromatin binding sites of estrogen-responsive genes, and effects on the regulation of ER-subtype-selective gene expression and breast cancer cell proliferation. The BEs are clearly different from E2 in affinity and potency, but most notably in their selectivity for driving the function of ERβ over ERα. One can imagine that the consequences of this altered potency and selectivity might therefore depend greatly on dose and target tissue context, and the relative cellular levels of ERα and ERβ.
When ERβ is present, estrogen action through this ER subtype appears to have important and generally beneficial effects. This is the case in brain, where ERβ mediates antidepressive and neuroprotective effects and controls aspects of cognitive function, as well as mediating antiinflammatory and protective effects on bone and the cardiovascular system (18, 45, 57, 58). As we observed in breast cancer cells here, in target tissues such as the prostate, which contain both ERα and ERβ, estrogen action through ERβ appears to mute the hyperproliferative effect of ERα (59, 60). Thus, BEs might be expected to have net beneficial effects when present at concentrations at which they exert ERβ-preferential activities. In women consuming soy foods and soy supplements, blood levels of isoflavones are usually in the 1–2 μM range for total isoflavones (i.e., free plus conjugated). Since the free is presumed to be the only active form and ∼98% is conjugated, the blood levels of free isoflavones would be ∼40 nM, which should be sufficient to have some ERβ activity. Other BEs may have similar blood levels of conjugates, but may have a different profile of the aglycone form.
Our findings have highlighted that the ERβ-selective activity of the BEs is dose-dependent and is lost at high concentrations, when ERα becomes activated to the same extent as elicited by E2. Thus, in targets in which ERα may be driving proliferation without or with only minimal restraint by ERβ, such as in certain hormone-responsive advanced breast cancers, high doses of BEs could have the same potential detrimental effects as the endogenously produced E2, whereas lower doses of BEs could be beneficial. Thus, our findings underscore that the dose and the target cell, with its relative levels of these two ERs, are crucial determinants of the potential benefits vs. risks of BEs. Our studies reveal novel mechanistic differences underlying the estrogenic activities of the BEs vs. E2, which provide valuable information to inform future clinical studies.
Acknowledgments
This project was made possible by U.S. National Institutes of Health (NIH) grant P50AT006268 (to W.G.H., I.K., B.S.K., and J.A.K.) from the National Center for Complementary and Alternative Medicines (NCCAM), the Office of Dietary Supplements (ODS), and the National Cancer Institute (NCI).
Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NCCAM, ODS, NCI, or NIH. Partial support to Z.M.E. and T.M. was from NIH T32ES007326.
Footnotes
- BE
- botanical estrogen
- ChIP
- chromatin immunoprecipitation
- ChIP-seq
- chromatin immunoprecipitation-DNA sequencing
- E2
- estradiol
- ER
- estrogen receptor
- FRET
- fluorescence resonance energy transfer
- HRT
- hormone replacement therapy
- OTUB2
- otubain 2
- PgR
- progesterone receptor
- RBA
- relative binding affinity
- RCA
- relative coactivator binding affinity
- RIP140
- receptor interacting protein 140
- SRC
- steroid receptor coactivator
- TR-FRET
- time-resolved fluorescence resonance energy transfer
REFERENCES
- 1. Manson J. E., Hsia J., Johnson K. C., Rossouw J. E., Assaf A. R., Lasser N. L., Trevisan M., Black H. R., Heckbert S. R., Detrano R., Strickland O. L., Wong N. D., Crouse J. R., Stein E., Cushman M. (2003) Estrogen plus progestin and the risk of coronary heart disease. N. Engl. J. Med. 349, 523–534 [DOI] [PubMed] [Google Scholar]
- 2. Wassertheil-Smoller S., Hendrix S. L., Limacher M., Heiss G., Kooperberg C., Baird A., Kotchen T., Curb J. D., Black H., Rossouw J. E., Aragaki A., Safford M., Stein E., Laowattana S., Mysiw W. J. (2003) Effect of estrogen plus progestin on stroke in postmenopausal women: the Women's Health Initiative: a randomized trial. JAMA 289, 2673–2684 [DOI] [PubMed] [Google Scholar]
- 3. Chlebowski R. T., Hendrix S. L., Langer R. D., Stefanick M. L., Gass M., Lane D., Rodabough R. J., Gilligan M. A., Cyr M. G., Thomson C. A., Khandekar J., Petrovitch H., McTiernan A. (2003) Influence of estrogen plus progestin on breast cancer and mammography in healthy postmenopausal women: the Women's Health Initiative Randomized Trial. JAMA 289, 3243–3253 [DOI] [PubMed] [Google Scholar]
- 4. Shumaker S. A., Legault C., Rapp S. R., Thal L., Wallace R. B., Ockene J. K., Hendrix S. L., Jones B. N., 3rd, Assaf A. R., Jackson R. D., Kotchen J. M., Wassertheil-Smoller S., Wactawski-Wende J. (2003) Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women's Health Initiative Memory Study: a randomized controlled trial. JAMA 289, 2651–2662 [DOI] [PubMed] [Google Scholar]
- 5. Shumaker S. A., Legault C., Kuller L., Rapp S. R., Thal L., Lane D. S., Fillit H., Stefanick M. L., Hendrix S. L., Lewis C. E., Masaki K., Coker L. H. (2004) Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women's Health Initiative Memory Study. JAMA 291, 2947–2958 [DOI] [PubMed] [Google Scholar]
- 6. Kuiper G. G., Lemmen J. G., Carlsson B., Corton J. C., Safe S. H., van der Saag P. T., van der Burg B., Gustafsson J. A. (1998) Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 139, 4252–4263 [DOI] [PubMed] [Google Scholar]
- 7. Allred C. D., Allred K. F., Ju Y. H., Virant S. M., Helferich W. G. (2001) Soy diets containing varying amounts of genistein stimulate growth of estrogen-dependent (MCF-7) tumors in a dose-dependent manner. Cancer Res. 61, 5045–5050 [PubMed] [Google Scholar]
- 8. Hsieh C. Y., Santell R. C., Haslam S. Z., Helferich W. G. (1998) Estrogenic effects of genistein on the growth of estrogen receptor-positive human breast cancer (MCF-7) cells in vitro and in vivo. Cancer Res. 58, 3833–3838 [PubMed] [Google Scholar]
- 9. Huntley A. L., Ernst E. (2004) Soy for the treatment of perimenopausal symptoms: a systematic review. Maturitas 47, 1–9 [DOI] [PubMed] [Google Scholar]
- 10. Taku K., Melby M. K., Kronenberg F., Kurzer M. S., Messina M. (2012) Extracted or synthesized soybean isoflavones reduce menopausal hot flash frequency and severity: systematic review and meta-analysis of randomized controlled trials. Menopause 19, 776–790 [DOI] [PubMed] [Google Scholar]
- 11. Siow R. C., Li F. Y., Rowlands D. J., de Winter P., Mann G. E. (2007) Cardiovascular targets for estrogens and phytoestrogens: transcriptional regulation of nitric oxide synthase and antioxidant defense genes. Free Radic. Biol. Med. 42, 909–925 [DOI] [PubMed] [Google Scholar]
- 12. Butler L. M., Wu A. H., Wang R., Koh W. P., Yuan J. M., Yu M. C. (2010) A vegetable-fruit-soy dietary pattern protects against breast cancer among postmenopausal Singapore Chinese women. Am. J. Clin. Nutr. 91, 1013–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Levis S., Strickman-Stein N., Ganjei-Azar P., Xu P., Doerge D. R., Krischer J. (2011) Soy isoflavones in the prevention of menopausal bone loss and menopausal symptoms: a randomized, double-blind trial. Arch. Intern. Med. 171, 1363–1369 [DOI] [PubMed] [Google Scholar]
- 14. Pitkin J. (2012) Alternative and complementary therapies for the menopause. Menopause Int. 18, 20–27 [DOI] [PubMed] [Google Scholar]
- 15. Sacks F. M., Lichtenstein A., Van Horn L., Harris W., Kris-Etherton P., Winston M. (2006) Soy protein, isoflavones, and cardiovascular health: an American Heart Association Science Advisory for professionals from the Nutrition Committee. Circulation 113, 1034–1044 [DOI] [PubMed] [Google Scholar]
- 16. Hilakivi-Clarke L., Andrade J. E., Helferich W. (2010) Is soy consumption good or bad for the breast? J. Nutr. 140, 2326S–2334S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kuiper G. G., Carlsson B., Grandien K., Enmark E., Haggblad J., Nilsson S., Gustafsson J. A. (1997) Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 138, 863–870 [DOI] [PubMed] [Google Scholar]
- 18. Harris H. A. (2007) Estrogen receptor-beta: recent lessons from in vivo studies. Mol. Endocrinol. 21, 1–13 [DOI] [PubMed] [Google Scholar]
- 19. Pettersson K., Gustafsson J. A. (2001) Role of estrogen receptor beta in estrogen action. Annu. Rev. Physiol. 63, 165–192 [DOI] [PubMed] [Google Scholar]
- 20. Katzenellenbogen B. S., Korach K. S. (1997) A new actor in the estrogen receptor drama: enter ER-beta. Endocrinology 138, 861–862 [DOI] [PubMed] [Google Scholar]
- 21. Sun J., Meyers M. J., Fink B. E., Rajendran R., Katzenellenbogen J. A., Katzenellenbogen B. S. (1999) Novel ligands that function as selective estrogens or antiestrogens for estrogen receptor-alpha or estrogen receptor-beta. Endocrinology 140, 800–804 [DOI] [PubMed] [Google Scholar]
- 22. Frasor J., Chang E. C., Komm B., Lin C. Y., Vega V. B., Liu E. T., Miller L. D., Smeds J., Bergh J., Katzenellenbogen B. S. (2006) Gene expression preferentially regulated by tamoxifen in breast cancer cells and correlations with clinical outcome. Cancer Res. 66, 7334–7340 [DOI] [PubMed] [Google Scholar]
- 23. Paruthiyil S., Parmar H., Kerekatte V., Cunha G. R., Firestone G. L., Leitman D. C. (2004) Estrogen receptor beta inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell cycle arrest. Cancer Res. 64, 423–428 [DOI] [PubMed] [Google Scholar]
- 24. Chang E. C., Charn T. H., Park S. H., Helferich W. G., Komm B., Katzenellenbogen J. A., Katzenellenbogen B. S. (2008) Estrogen receptors alpha and beta as determinants of gene expression: influence of ligand, dose, and chromatin binding. Mol. Endocrinol. 22, 1032–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Strom A., Hartman J., Foster J. S., Kietz S., Wimalasena J., Gustafsson J. A. (2004) Estrogen receptor beta inhibits 17beta-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proc. Natl. Acad. Sci. U. S. A. 101, 1566–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Muthyala R. S., Ju Y. H., Sheng S., Williams L. D., Doerge D. R., Katzenellenbogen B. S., Helferich W. G., Katzenellenbogen J. A. (2004) Equol, a natural estrogenic metabolite from soy isoflavones: convenient preparation and resolution of R- and S-equols and their differing binding and biological activity through estrogen receptors alpha and beta. Bioorg. Med. Chem. 12, 1559–1567 [DOI] [PubMed] [Google Scholar]
- 27. Mersereau J. E., Levy N., Staub R. E., Baggett S., Zogovic T., Chow S., Ricke W. A., Tagliaferri M., Cohen I., Bjeldanes L. F., Leitman D. C. (2008) Liquiritigenin is a plant-derived highly selective estrogen receptor beta agonist. Mol. Cell. Endocrinol. 283, 49–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Paruthiyil S., Cvoro A., Zhao X., Wu Z., Sui Y., Staub R. E., Baggett S., Herber C. B., Griffin C., Tagliaferri M., Harris H. A., Cohen I., Bjeldanes L. F., Speed T. P., Schaufele F., Leitman D. C. (2009) Drug and cell type-specific regulation of genes with different classes of estrogen receptor beta-selective agonists. PLoS ONE 4, e6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chang E. C., Frasor J., Komm B., Katzenellenbogen B. S. (2006) Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology 147, 4831–4842 [DOI] [PubMed] [Google Scholar]
- 30. Heemstra J. M., Kerrigan S. A., Doerge D. R., Helferich W. G., Boulanger W. A. (2006) Total synthesis of (S)-equol. Org. Lett. 8, 5441–5443 [DOI] [PubMed] [Google Scholar]
- 31. Carlson K. E., Choi I., Gee A., Katzenellenbogen B. S., Katzenellenbogen J. A. (1997) Altered ligand binding properties and enhanced stability of a constitutively active estrogen receptor: evidence that an open pocket conformation is required for ligand interaction. Biochemistry 36, 14897–14905 [DOI] [PubMed] [Google Scholar]
- 32. Katzenellenbogen J. A., Johnson H. J., Jr., Carlson K. E. (1973) Studies on the uterine, cytoplasmic estrogen binding protein: thermal stability and ligand dissociation rate: an assay of empty and filled sites by exchange. Biochemistry 12, 4092–4099 [DOI] [PubMed] [Google Scholar]
- 33. Jeyakumar M., Carlson K. E., Gunther J. R., Katzenellenbogen J. A. (2011) Exploration of dimensions of estrogen potency: parsing ligand binding and coactivator binding affinities. J. Biol. Chem. 286, 12971–12982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tamrazi A., Carlson K. E., Daniels J. R., Hurth K. M., Katzenellenbogen J. A. (2002) Estrogen receptor dimerization: ligand binding regulates dimer affinity and dimer dissociation rate. Mol. Endocrinol. 16, 2706–2719 [DOI] [PubMed] [Google Scholar]
- 35. Gunther J. R., Du Y., Rhoden E., Lewis I., Revennaugh B., Moore T. W., Kim S. H., Dingledine R., Fu H., Katzenellenbogen J. A. (2009) A set of time-resolved fluorescence resonance energy transfer assays for the discovery of inhibitors of estrogen receptor-coactivator binding. J. Biomol. Screen. 14, 181–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Choi I., Ko C., Park-Sarge O. K., Nie R., Hess R. A., Graves C., Katzenellenbogen B. S. (2001) Human estrogen receptor beta-specific monoclonal antibodies: characterization and use in studies of estrogen receptor beta protein expression in reproductive tissues. Mol. Cell. Endocrinol. 181, 139–150 [DOI] [PubMed] [Google Scholar]
- 37. Barnett D. H., Sheng S., Charn T. H., Waheed A., Sly W. S., Lin C. Y., Liu E. T., Katzenellenbogen B. S. (2008) Estrogen receptor regulation of carbonic anhydrase XII through a distal enhancer in breast cancer. Cancer Res. 68, 3505–3515 [DOI] [PubMed] [Google Scholar]
- 38. Bergamaschi A., Katzenellenbogen B. S. (2012) Tamoxifen downregulation of miR-451 increases 14-3-3zeta and promotes breast cancer cell survival and endocrine resistance. Oncogene 31, 39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pike A. C., Brzozowski A. M., Hubbard R. E., Bonn T., Thorsell A. G., Engstrom O., Ljunggren J., Gustafsson J. A., Carlquist M. (1999) Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. EMBO J. 18, 4608–4618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wong J. M., Kendall C. W., Marchie A., Liu Z., Vidgen E., Holmes C., Jackson C. J., Josse R. G., Pencharz P. B., Rao A. V., Vuksan V., Singer W., Jenkins D. J. (2012) Equol status and blood lipid profile in hyperlipidemia after consumption of diets containing soy foods. Am. J. Clin. Nutr. 95, 564–571 [DOI] [PubMed] [Google Scholar]
- 41. Anstead G. M., Carlson K. E., Katzenellenbogen J. A. (1997) The estradiol pharmacophore: ligand structure-estrogen receptor binding affinity relationships and a model for the receptor binding site. Steroids 62, 268–303 [DOI] [PubMed] [Google Scholar]
- 42. Jeyakumar M., Katzenellenbogen J. A. (2009) A dual-acceptor time-resolved Foster resonance energy transfer assay for simultaneous determination of thyroid hormone regulation of corepressor and coactivator binding to the thyroid hormone receptor: mimicking the cellular context of thyroid hormone action. Anal. Biochem. 386, 73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jeyakumar M., Webb P., Baxter J. D., Scanlan T. S., Katzenellenbogen J. A. (2008) Quantification of ligand-regulated nuclear receptor corepressor and coactivator binding, key interactions determining ligand potency and efficacy for the thyroid hormone receptor. Biochemistry 47, 7465–7476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Simmler C., Hajirahimkhan A., Lankin D. C., Bolton J. L., Jones T., Soejarto D. D., Chen S. N., Pauli G. F. (2013) Dynamic residual complexity of the isoliquiritigenin-liquiritigenin interconversion during bioassay. J. Agric. Food Chem. 61, 2146–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Saijo K., Collier J. G., Li A. C., Katzenellenbogen J. A., Glass C. K. (2011) An ADIOL-ERbeta-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell 145, 584–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Charn T. H., Liu E. T., Chang E. C., Lee Y. K., Katzenellenbogen J. A., Katzenellenbogen B. S. (2010) Genome-wide dynamics of chromatin binding of estrogen receptors alpha and beta: mutual restriction and competitive site selection. Mol. Endocrinol. 24, 47–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Madak-Erdogan Z., Charn T.-H., Jiang Y., Liu E. T., Katzenellenbogen J. A., Katzenellenbogen B. S. (2013) Integrative genomics of gene and metabolic regulation by estrogen receptors α and β and their coregulators. Mol. Syst. Biol. 9, 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Williams C., Edvardsson K., Lewandowski S. A., Strom A., Gustafsson J. A. (2008) A genome-wide study of the repressive effects of estrogen receptor beta on estrogen receptor alpha signaling in breast cancer cells. Oncogene 27, 1019–1032 [DOI] [PubMed] [Google Scholar]
- 49. Sareddy G. R., Nair B. C., Gonugunta V. K., Zhang Q. G., Brenner A., Brann D. W., Tekmal R. R., Vadlamudi R. K. (2012) Therapeutic significance of estrogen receptor beta agonists in gliomas. Mol. Cancer Ther. 11, 1174–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Harrington W. R., Sengupta S., Katzenellenbogen B. S. (2006) Estrogen regulation of the glucuronidation enzyme UGT2B15 in estrogen receptor-positive breast cancer cells. Endocrinology 147, 3843–3850 [DOI] [PubMed] [Google Scholar]
- 51. Peterson T. G., Ji G. P., Kirk M., Coward L., Falany C. N., Barnes S. (1998) Metabolism of the isoflavones genistein and biochanin A in human breast cancer cell lines. Am. J. Clin Nutr. 68, 1505S–1511S [DOI] [PubMed] [Google Scholar]
- 52. Lindberg M. K., Moverare S., Skrtic S., Gao H., Dahlman-Wright K., Gustafsson J. A., Ohlsson C. (2003) Estrogen receptor (ER)-beta reduces ERalpha-regulated gene transcription, supporting a “ying yang” relationship between ERalpha and ERbeta in mice. Mol. Endocrinol. 17, 203–208 [DOI] [PubMed] [Google Scholar]
- 53. Saji S., Hirose M., Toi M. (2005) Clinical significance of estrogen receptor beta in breast cancer. Cancer Chemother. Pharmacol. 56(Suppl. 1), 21–26 [DOI] [PubMed] [Google Scholar]
- 54. Shaaban A. M., O'Neill P. A., Davies M. P., Sibson R., West C. R., Smith P. H., Foster C. S. (2003) Declining estrogen receptor-beta expression defines malignant progression of human breast neoplasia. Am. J. Surg. Pathol. 27, 1502–1512 [DOI] [PubMed] [Google Scholar]
- 55. Speirs V., Carder P. J., Lane S., Dodwell D., Lansdown M. R., Hanby A. M. (2004) Oestrogen receptor beta: what it means for patients with breast cancer. Lancet Oncol. 5, 174–181 [DOI] [PubMed] [Google Scholar]
- 56. Gruvberger-Saal S. K., Bendahl P. O., Saal L. H., Laakso M., Hegardt C., Eden P., Peterson C., Malmstrom P., Isola J., Borg A., Ferno M. (2007) Estrogen receptor beta expression is associated with tamoxifen response in ERalpha-negative breast carcinoma. Clin. Cancer Res. 13, 1987–1994 [DOI] [PubMed] [Google Scholar]
- 57. Nilsson S., Koehler K. F., Gustafsson J. A. (2011) Development of subtype-selective oestrogen receptor-based therapeutics. Nat. Rev. Drug Discov. 10, 778–792 [DOI] [PubMed] [Google Scholar]
- 58. Suzuki S., Gerhold L. M., Bottner M., Rau S. W., Dela Cruz C., Yang E., Zhu H., Yu J., Cashion A. B., Kindy M. S., Merchenthaler I., Gage F. H., Wise P. M. (2007) Estradiol enhances neurogenesis following ischemic stroke through estrogen receptors alpha and beta. J. Comp. Neurol. 500, 1064–1075 [DOI] [PubMed] [Google Scholar]
- 59. Hartman J., Strom A., Gustafsson J. A. (2012) Current concepts and significance of estrogen receptor beta in prostate cancer. Steroids 77, 1262–1266 [DOI] [PubMed] [Google Scholar]
- 60. Prins G. S., Korach K. S. (2008) The role of estrogens and estrogen receptors in normal prostate growth and disease. Steroids 73, 233–244 [DOI] [PMC free article] [PubMed] [Google Scholar]