

Abstract
Transforming growth factor β (TGFβ) has significant profibrotic activity both in vitro and in vivo. This reflects its capacity to stimulate fibrogenic mediators and induce the expression of other profibrotic cytokines such as platelet-derived growth factor (PDGF) and epidermal growth factor (EGF/ErbB) ligands. Here we address both the mechanisms by which TGFβ induced ErbB ligands and the physiological significance of inhibiting multiple TGFβ-regulated processes. The data document that ErbB ligand induction requires PDGF receptor (PDGFR) mediation and engages a positive autocrine/paracrine feedback loop via ErbB receptors. Whereas PDGFRs are essential for TGFβ-stimulated ErbB ligand up-regulation, TGFβ-specific signals are also required for ErbB receptor activation. Subsequent profibrotic responses are shown to involve the cooperative action of PDGF and ErbB signaling. Moreover, using a murine treatment model of bleomycin-induced pulmonary fibrosis we found that inhibition of TGFβ/PDGF and ErbB pathways with imatinib plus lapatinib, respectively, not only prevented myofibroblast gene expression to a greater extent than either drug alone, but also essentially stabilized gas exchange (oxygen saturation) as an overall measure of lung function. These observations provide important mechanistic insights into profibrotic TGFβ signaling and indicate that targeting multiple cytokines represents a possible strategy to ameliorate organ fibrosis dependent on TGFβ.—Andrianifahanana, M., Wilkes, M. C., Gupta, S. K., Rahimi, R. R., Repellin, C. E., Edens, M., Wittenberger, J., Yin, X., Maidl, E., Becker, J., Leof, E. B. Profibrotic TGFβ responses require the cooperative action of PDGF and ErbB receptor tyrosine kinases.
Keywords: EGF, pulmonary fibrosis
Fibroproliferative disorders are a leading cause of morbidity and mortality and encompass a wide spectrum of pathological conditions featuring localized or systemic tissue and organ fibrosis. Management of these diseases remains an ongoing challenge due to their inherently complex and often undefined etiology (1–4). Significant advances in delineating the molecular events leading to fibrosis have been recorded over the past. However, attempts to devise efficient therapeutic treatments in clinical contexts have generally met with limited success (2, 4, 5), warranting renewed efforts to further characterize the mechanisms regulating fibrogenesis. In that regard, the role of transforming growth factor β (TGFβ) is clearly paramount, considering its implication in the pathogenesis of nearly all types of fibrotic diseases (6, 7).
Signaling by TGFβ begins at the cell surface via ligand binding to the type II TGFβ receptor, a constitutively active, membrane-bound serine/threonine kinase. This triggers recruitment and activating phosphorylation of the type I receptor, which in turn phosphorylates the receptor-regulated transcription coregulators (R-Smads) Smad2 and Smad3. Heteromerization of R-Smads with the common mediator, Smad4, and nuclear translocation subsequently ensue, culminating in modulation (induction or repression) of Smad-regulated genes (8, 9). Although a large number of TGFβ-regulated functions have been attributed to Smads (7, 10, 11), non-Smad signaling pathways are also equally important in mediating TGFβ's effects (7, 12). Moreover, instances of crosstalk between the two have been reported (13).
TGFβ exerts its profibrotic effects essentially by promoting fibroblast proliferation, myofibroblast differentiation, and extracellular matrix remodeling. Various studies have documented the mechanisms by which this pleiotropic cytokine carries out these functions (6, 11). Smad activation enables expression of fibrogenic intermediate effectors, such as platelet-derived growth factor (PDGF; refs. 14–16) or connective tissue growth factor (CTGF/CCN2; refs. 17, 18), via transcriptional regulation. Non-Smad pathways also exhibit potent profibrotic activities; examples include the p21-activated kinase 2 (PAK2)/cAbl (19, 20) and Akt/mTOR (21) modules, both of which operate downstream of the focal adhesion kinase (22), and phosphoinositide 3-kinase (23). Similar roles have been described for different members of the mitogen-activated protein kinase (MAPK) family (24, 25). Of note, we have recently reported that the epidermal growth factor receptor (EGFR/ErbB) pathway serves as a critical mediator of profibrotic TGFβ signaling (26). Activation of this mitogenic pathway was shown to require Smad mediation (26), although the detailed regulatory mechanisms remained undefined.
Our initial interest in the role of ErbB in fibrogenesis took its origins from previously reported links between ErbB and the pathogenesis of fibrosis (27, 28). Having identified ErbB as an important downstream mediator of TGFβ (26), we deemed it critical to address the processes involved. Here we investigate the molecular mechanisms and physiological implications of ErbB activation by TGFβ in the context of fibrogenesis. We describe a hitherto unknown mechanism whereby TGFβ-induced ErbB activation requires the integration of autocrine signals from the PDGF receptor (PDGFR) and additional signals from the TGFβ receptor complex. Moreover, we show that up-regulation of ErbB ligands by TGFβ is mediated by the PDGFR via MEK [MAPK and extracellular signal-regulated kinase (ERK) kinase] and engages a positive feedback loop through ErbB. Finally, using a TGFβ-driven murine model of lung fibrosis we demonstrate the physiological relevance of PDGFR-ErbB cooperation, where disease progression is prevented by a dual inhibition of these receptor tyrosine kinases. These findings uncover novel aspects of profibrotic TGFβ signaling and present an opportunity for improving current therapeutic strategies.
MATERIALS AND METHODS
Cell culture
Murine fibroblasts (AKR-2B) and derived cell lines expressing shRNA specific for different target genes were used in the experiments. Cells were routinely maintained in high-glucose DMEM (Invitrogen, Gaithersburg, MD, USA) supplemented with 10% FBS (Hyclone, Logan, UT, USA). In all experiments, cells were grown under reduced serum conditions for 24 h before subjecting to specific test reagents.
Antibodies and other reagents
Anti-phospho-AKT (Ser473; cat. 9271), anti-AKT (cat. 9272), anti-PDGFRβ (cat. 3175), anti-phospho-Smad3 (cat. 9520), and anti-phospho-ERK (cat. 9106) were purchased from Cell Signaling Technology (Beverly, MA, USA), whereas anti-ErbB1 (cat. sc-373746), anti-PDGFRα (cat. sc-338), anti-PDGFRβ (cat. sc-1627), and anti-ERK1/2 (cat. sc-94) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-SMAD3 (cat. ab28379) was purchased from Abcam (Cambridge, MA, USA), and anti-GAPDH (cat. MAB374), anti-phosphotyrosine (clone 4G10; cat. 05-321), anti-ErbB1 (cat. 06-847), and anti-ErbB2 (cat. 06-562) were acquired from Millipore (Charlottesville, MA, USA). A phospho-Smad3-specific antibody (immunogen: COOH-GSPSIRCSpSVpS) was generated in our laboratory (20). AG1295 was obtained from Millipore, lapatinib from LC Laboratories (Woburn, MA, USA), and U0126 from Merck (Whitehouse Station, NJ, USA). Imatinib was prepared as described previously (29). Human TGFβ1 was obtained from R&D Systems (Minneapolis, MN, USA), whereas recombinant human PDGF-AA, -AB, and -BB were purchased from Sigma-Aldrich (St. Louis, MO, USA). All cytokines were used at a final concentration of 10 ng/ml throughout the experiments.
Protein extraction, immunoprecipitation (IP), and Western blotting
Total proteins were extracted in modified RIPA buffer and processed for IP and Western blotting as described previously (26) with minor modifications. PDGFRβ was immunoprecipitated in the same extraction buffer, whereas detergent concentrations were adjusted to 1% Na-deoxycholate and 0.1% sodium dodecyl sulfate for ErbB1 IP.
RT-PCR analysis
Experimental conditions were as described previously (26). Briefly, complementary DNAs were subjected to amplification by PCR using previously published parameters (26). The characteristics of additional primers used are as follows: Pdgf-a (forward: 5′-GCCTGTGCCCATTCGCAGGA-3′; reverse: 5′-AGACCGCACGCACATTGGCA-3′; amplicon size: 458 bp; accession no. NM_008808.3), Pdgf-b (forward: 5′-TGAAATGCTGAGCGACCAC-3′; reverse: 5′-GGGTCACTACTGTCTCACA-3′; amplicon size: 467 bp; accession no. NM_011057), Pdgf-c (forward: 5′-AGTCCTTCGGTGTTGCCCCCT-3′; reverse: 5′-TCCTCGTGGTGTTCCAGAGCCA-3′; amplicon size: 488 bp; accession no. NM_019971).
RNA interference
Plasmids (pLKO.1-puro) encoding shRNA-targeting ErbB1, ErbB2, PDGFRα, and PDGFRβ were purchased from the Mayo Clinic Jacksonville RNA Interference Shared Resource. The production of lentivirus and transduction of AKR-2B cells were as described previously (21). Stable clones were generated in the presence of 1.5 μg/ml puromycin.
Plasmid and transfection
The construct encoding a FLAG-tagged, constitutively active version of MEK1 (FLAG-MEK1-S218-222D) was generously provided by Dr. Scott Eblen (Medical University of South Carolina, Charleston, SC, USA). Transfection procedures using the TransIT-2020 reagent (Mirus Bio, Madison, WI, USA) were per the manufacturer's recommendations.
Enzyme-linked immunosorbent assay (ELISA)
AKR-2B fibroblasts were seeded in 6-well plates at 4 × 105 cells/well and allowed to adhere overnight. The next day, cells were serum starved in DMEM supplemented with 0.1% FBS for 24 h, and then the medium was replaced with 1 ml OPTI-MEM I medium (Invitrogen). Following 1 h incubation the medium was replaced with 1 ml of fresh OPTI-MEM I, to which growth factors (TGFβ1, PDGF-AB, or PDGF-BB) were added. Treatments were applied for various time periods, after which conditioned media were collected and centrifuged at 16,060 g for 15 min to remove debris. Aliquots (100 μl) of the supernatants were subjected to ELISA for the detection of secreted murine epiregulin (Ereg) using the DuoSet ELISA kit (R&D Systems). Assays were performed as per manufacturer's recommendations. Following removal of the conditioned media, cells were washed twice in PBS (pH 7.4) and harvested in 500 μl PBS containing 1% Triton X-100. Total proteins were extracted on ice, and lysates were cleared by centrifugation at 16,060 g for 10 min. Aliquots (50 μl) of the cell lysates were used in Western blot analyses of β-tubulin expression to confirm equal amounts of starting material.
Soft agar assay
The experimental procedures for soft agar assay were essentially as described previously (21). The ability of cells to undergo anchorage-independent growth following TGFβ treatment was evaluated by counting colonies >50 μm in diameter using a Gelcount apparatus (Oxford Optronics, Milton, Abingdon, UK).
Cell migration assay
Transwells with 8-μm-pore membrane (Corning, Tewksbury, MA, USA) were coated with 10 μg/ml plasma fibronectin for 1 h at 37°C, and then blocked with 1% bovine serum albumin for 30 min (22). Serum-starved AKR-2B cultures were grown in DMEM supplemented with TGFβ1 and in the presence or absence of lapatinib (5 μM), AG1295 (20 μM), or U0126 (10 μM) for 24 h. Cells were trypsinized, washed in PBS, and then suspended in DMEM containing 0.5% FBS. Cell suspension (100 μl; ∼104 cells) was added to the top chamber, whereas the bottom chamber was filled with DMEM containing 0.5% FBS to serve as a chemoattractant. Cell migration was allowed to proceed for 4–6 h at 37°C. Nonmigrated cells were removed from the upper surface of the membrane, and cells that migrated to the underside were washed with PBS, fixed, and stained using the Hema 3 kit (Fisher Scientific, Pittsburgh, PA, USA). Migrated cells were counted in 4 randomly selected high-power fields (×100), and photomicrographs were captured. The relative percentage of cell migration was calculated using cell counts from untreated cells as a baseline (100%).
Bleomycin (BLM)-induced lung fibrosis
Female C57 black mice (Jackson Laboratories, Bar Harbor, ME, USA) were used for all experiments and handled according to the standard care and methodologies approved by the Mayo Clinic Institutional Animal Care and Use Committee. Eight-week-old (∼20 g) mice were administered BLM (0.1 U diluted in 75 μl of 0.9% normal saline) or 75 μl of 0.9% normal saline alone by tracheal instillation using an intratracheal aerosolizer (Penn-Century, Wyndmoor, PA, USA) on d 0, while under ketamine/xylazine anesthesia. At this time, animals were shaved around the collar region to allow monitoring of dissolved oxygen levels on room air using a MouseOx collar clip monitoring system (Starr Life Sciences Corp., Holliston, MA, USA) and ear punched for identification. Accuracy of the readings was determined by cross-referencing MouseOx live readings with blood gasses obtained from cardiac bleed immediately postsacrifice in a subset of animals. Imatinib and lapatinib were prepared by thoroughly blending with Methocel (Dow Chemical Company, Midland, MI, USA) at a ratio of 1:7 before solubilization in 0.9% normal saline at 4°C with agitation for 60 min.
Prior to drug treatment, BLM-challenged mice were categorized into 5 groups ranging in degree of physical symptoms. Following removal of those animals suffering from severe respiratory distress, each treatment group was populated with a minimum of 6 animals normalized for the severity of symptoms and treated once daily (0.2 ml) by intraperitoneal injection commencing 15 d after BLM insult. Mouse weights, heart rate, respiratory rate, and dissolved oxygen, as well as overall well-being, were monitored daily. On d 28, mice were euthanized by overdose of pentobarbital (100 mg/kg), lungs were dissected, and samples were collected for immunohistochemistry and collagen synthesis analyses as described previously (29, 30).
Collagen synthesis
A portion of each lung was used for hydroxyproline assay as described previously (29) with minor modifications. Briefly, explanted lung was washed in PBS, weighed, and homogenized using TissueLyser LT (Qiagen, Germantown, MD, USA) and diluted in 10 ml PBS per milligram lung weight. Samples (100 μl) were then boiled overnight in 12M HCl, and duplicate samples were analyzed by hydroxyproline assay.
Histological analyses
Histological analyses were performed as described previously (30).
Normalization and quantitation of lung fibrosis
To control for differences in the severity of BLM-induced fibrosis, negative (saline insult:saline treatment) and positive (BLM insult:saline treatment) controls were performed in parallel. Comparisons within experimental batches were made possible by converting raw values to a percentage of fibrosis induced relative to the mean value of the batch positive control. To allow comparisons across experimental batches, correction values to accommodate the differing degrees of fibrosis induced by BLM in the positive controls of each batch were determined. Once variation between positive controls was normalized, the same correction factors were applied to experimental treatment groups. Data comparisons between experimental and control groups were analyzed by unpaired 2-tailed Student's t tests, and values of P < 0.05 were considered significant.
RESULTS
TGFβ induction of ErbB ligands requires both PDGF and ErbB receptor signaling by way of MEK
We previously determined that profibrotic TGFβ signaling is mediated through cell type-specific induction of ErbB ligands (26). Furthermore, while this was primarily contingent on Smad3-dependent signaling, the kinetics of ligand expression and ErbB receptor activation (i.e., occurring >12 h after TGFβ treatment) indicated the need for additional autocrine/paracrine factors. In that regard, one group of profibrotic cytokines showing both a TGFβ and Smad3 tropism is the PDGF family (16, 31–33). To investigate whether PDGF expression was consistent with that hypothesis, PDGF message and receptor phosphorylation was shown to occur within 1–6 h following TGFβ stimulation. In contrast, analogous effects on the ErbB pathway were delayed an approximate 9 additional hours (Supplemental Fig. S1A). As numerous events can occur over such times, both pharmacologic and genetic approaches were used to document causality (Fig. 1A and Supplemental Fig. S1B, respectively). Specifically, use of either the PDGF receptor inhibitors AG1295/imatinib or shRNA to α and/or β PDGF receptor subunits significantly diminished ErbB ligand mRNA or receptor activation. Of note, as the greatest inhibition required knockdown of the PDGFRβ subunit and/or β plus α subunits (Fig. 1B), this would indicate that PDGF BB or AB is the primary PDGF isoform mediating TGFβ actions. Next, we determined whether ErbB ligand mRNA was similarly induced by various PDGF isoforms. As shown in Fig. 1C, while PDGF-AB and -BB stimulated amphiregulin (Areg), epiregulin (Ereg), and heparin binding EGF-like growth factor (Hbegf) mRNA to a comparable extent, PDGF-AA was significantly less active. The inability of PDGF-AA to generate a robust ErbB response suggests a role for PDGF-B ligand as the critical mediator. This is in agreement with our observation that when added at equimolar concentrations, only PDGF-AB and PDGF-BB (but not PDGF-AA) can provide a sustained activation of PDGFR's downstream effectors (data not shown). Furthermore, the kinetics of ErbB expression was significantly faster than that observed for TGFβ (compare Fig. 1C and Supplemental Fig. S1A; see ref. 26), consistent with a direct action of PDGF on ErbB message rather than the indirect response (i.e., via PDGF) of TGFβ.
Figure 1.

TGFβ induces ErbB signaling via autocrine activation of the PDGFR pathway. A) RT-PCR analyses of ErbB ligands (12 h post-treatment) and Western blot analysis of ErbB1 phosphorylation (15 h) following cell stimulation with TGFβ1 in the presence of the PDGFR inhibitor AG1295. The profile of PDGFR phosphorylation (12 h) confirms the efficacy of AG1295, whereas that of Smad3 (3 h) proves its specificity. B) RT-PCR analyses of ErbB ligands (12 h post-treatment) upon TGFβ1 stimulation of cells transduced with shRNA targeting PDGFRα, PDGFRβ, or both (shRα+shRβ). NT denotes nontargeting control. Western blot analyses show consistent Smad3 phosphorylation and efficient knockdown of PDGFRα and PDGFRβ (3 h). The data presented further corroborate the role of PDGFR in mediating up-regulation of ErbB ligands by TGFβ. C) Time course of ErbB ligand induction by PDGF in AKR-2B cells. RT-PCR analyses of ErbB ligands show a fairly rapid up-regulation of ErbB ligands by PDGF isoforms, with isoforms containing the B chain being the most potent.
ErbB ligands are known to autoregulate (Fig. 2A and refs. 34–36). Considering the extended timeframe in which ErbB mRNA is expressed following TGFβ treatment (Supplemental Fig. S1A and ref. 26), we next examined whether this might also reflect some component of ErbB autoinduction and/or paracrine activity. To initially address this question, cultures were stimulated with TGFβ in the presence of the ErbB1/ErbB2 kinase inhibitor lapatinib. As shown in Fig. 2B, there was a dose-dependent decrease in Areg, Ereg, and Hbegf mRNA expression as well as ErbB1 tyrosine phosphorylation. This occurred downstream of canonical TGFβ signaling as lapatinib had no effect on Smad3 phosphorylation. These findings were independently confirmed by generating stable cell lines expressing shRNA to ErbB1, ErbB2, or ErbB1 plus ErbB2 (Fig. 2C). While knockdown of ErbB1 was only partially effective in reducing TGFβ-stimulated ErbB ligand mRNA, consistent with previous publications documenting that the preferred receptor signaling complex is a heterodimer of ErbB1 and ErbB2 (26, 37), shRNA to ErbB2, or ErbB1 plus ErbB2 reduced expression to nearly basal levels.
Figure 2.
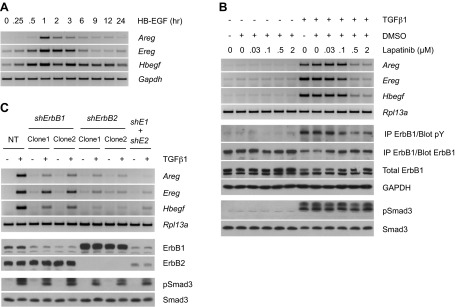
TGFβ stimulates ErbB ligand expression via ErbB autoinduction. A) RT-PCR analyses of ErbB ligand expression in HB-EGF-treated AKR-2B cells. Activation of ErbB induces expression of its own ligands. B) RT-PCR analyses of ErbB ligands and Western blot analysis of ErbB1 phosphorylation (15 h post-treatment) following cell stimulation with TGFβ1 in the presence of the ErbB1/ErbB2 inhibitor, lapatinib. The lack of effect on Smad3 phosphorylation (3 h) confirms the specificity of lapatinib. Stimulation of ErbB ligand expression by TGFβ requires ErbB autoinduction. C) RT-PCR analyses of ErbB ligands (12 h post-treatment) in response to TGFβ1 stimulation of cells stably expressing shRNA targeting ErbB1, ErbB2, or both (shE1+shE2). Western blot analyses (1 h) show efficient knockdown of ErbB1 and ErbB2 and uniform activation of Smad3. Results further support the role of ErbB autoinduction in ErbB ligand up-regulation by TGFβ.
The preceding data document that ErbB receptor activation following addition of TGFβ requires the concerted action of multiple autocrine/paracrine acting cytokines. While Smad3 is essential (Figs. 1A, B and 2B and ref. 26) for PDGF expression and subsequent ErbB autoinduction, additional signaling downstream of PDGF and ErbB is clearly necessary. One such mechanism common to both PDGF and ErbB ligands is the MEK/ERK pathway. As results from previous studies also suggested a role for Ras and its downstream effector MEK in Ereg induction (38, 39), we sought to determine whether MEK/ERK activation was critical for the cooperative action of these growth factors. To that end, the effect of the MEK inhibitor U0126 on TGFβ-stimulated Areg, Ereg, and Hbegf expression was examined. As shown in Fig. 3A, while Smad3 phosphorylation was unaffected, a dose-dependent loss in ErbB ligand message was observed coincident with the inhibition of ERK1/2 phosphorylation. Of note, experiments involving pharmacological inhibition of receptor activity demonstrated that activation of MEK/ERK by TGFβ occurred downstream of the PDGFR pathway (Supplemental Fig. S2A, B). In that both PDGF and ErbB isoforms similarly activate ERK signaling (40) and are required for TGFβ-dependent as well as independent induction of ErbB ligands (Figs. 1 and 2), the requirement for MEK activity in PDGF-AB and HB-EGF responses was examined. As would be expected, U0126 similarly inhibited Areg, Ereg, and Hbegf expression induced by either PDGF-AB or HB-EGF (Fig. 3B, C, respectively). Given that MEK emerged as the central mediator of ErbB ligand induction, we determined whether activation of this component alone would be sufficient for the process. Consistent with that hypothesis, exogenous expression of a constitutively active version of MEK1 functioned downstream of TGFβ and dose-dependently stimulated up-regulation of ErbB ligands (Fig. 3D).
Figure 3.

Up-regulation of ErbB ligands by TGFβ requires MEK activation. A) RT-PCR analyses of ErbB ligands (12 h post-treatment) on TGFβ1 stimulation of cells in the presence of the MEK inhibitor, U0126. Inhibition of TGFβ1-stimulated ERK1/2 phosphorylation (9 h) demonstrates the efficacy of U0126, and the lack of effect on Smad3 phosphorylation (3 h) verifies its specificity. Induction of ErbB ligands by TGFβ is mediated by MEK. B, C) RT-PCR analyses of ErbB ligands following stimulation of cells with PDGF-AB (2 h post-treatment) or HB-EGF (1 h post-treatment) in the presence of U0126. Western blot analyses of ERK1/2 and AKT phosphorylation (15 min) confirm drug efficacy and specificity, respectively. PDGFR- and ErbB-driven ErbB ligand up-regulation is MEK-dependent. D) RT-PCR analyses of ErbB ligands in cells transfected with control vector or FLAG-tagged constitutively active MEK1 (FLAG-MEK1-S218-222D) for 48 h. TGFβ1-stimulated cells (12 h) were used as a positive control for ErbB ligand induction. Phosphorylation of the downstream targets ERK1/2 indicates MEK activation. The results support the notion that activation of MEK is sufficient for ErbB ligand induction.
Differential effects of TGFβ and PDGF on ErbB receptor activation
The preceding data (Figs. 1 and 3) document the critical role of PDGF in profibrotic TGFβ signaling. As PDGFR ligands were shown to induce ErbB ligand expression, we determined whether ErbB1 was accordingly phosphorylated. While PDGF-AB and -BB were as active as TGFβ in stimulating ErbB ligand mRNA (Supplemental Fig. S1A and Figs. 1C and 3B), neither PDGF isoform was capable of inducing ErbB1 tyrosine phosphorylation (Fig. 4A). This was extremely surprising in that both (as expected) stimulated ERK1/2 phosphorylation (Fig. 4B). To address this further, an ELISA for epiregulin was performed on the conditioned media from AKR-2B cells stimulated with TGFβ, PDGF-AB, or PDGF-BB. Although maximal Ereg message was induced by both PDGF isoforms between 1 and 3 h stimulation (Fig. 1C), we were unable to detect secreted epiregulin protein at either 3 or 12 h following PDGF addition (Fig. 4C). This is in contrast to that observed for TGFβ as significant epiregulin secretion was observed consistent with the time course of ErbB1 receptor activation (Supplemental Fig. S1A, C). A model that incorporates the cooperative actions of PDGF and ErbB ligands in profibrotic TGFβ signaling is described in Fig. 4D.
Figure 4.
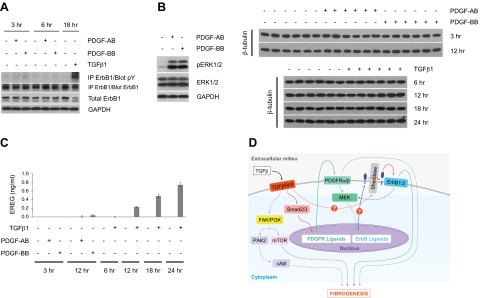
TGFβ stimulates the ErbB pathway through integration of autocrine PDGFR signals and additional TGFβ-specific input. A) Western blot analysis of ErbB1 phosphorylation following stimulation of cells with TGFβ1 or PDGF isoforms. Whereas both TGFβ and PDGF are able to induce ErbB ligand expression, only TGFβ can trigger ErbB phosphorylation. B) Western blot analyses showing phosphorylation of ERK1/2 by both PDGF isoforms (15 min). C) ELISA assay of conditioned media from cells stimulated with TGFβ1 or PDGF isoforms. Equivalent aliquots of cell lysates from 6 replicates used in ELISA were analyzed by Western blotting for β-tubulin expression. Data shown are from one representative experiment, repeated twice. Error bars = sd. Only TGFβ, but not PDGF, is capable of inducing EREG shedding. Results presented in A–C demonstrate that PDGFR activation is necessary and sufficient for TGFβ-induced ErbB ligand up-regulation, but not ErbB phosphorylation. D) Proposed model for profibrotic TGFβ signaling. Engagement of the TGFβ receptor (TGFβR) complex activates the canonical Smad pathway, leading to up-regulation of PDGFR ligands. Subsequent PDGFR phosphorylation promotes activation of the MEK pathway and stimulates ErbB ligand up-regulation. Additional TGFβR signals facilitate shedding of ErbB ligands and activation of cognate receptors, which further generate a positive feedback loop to induce its own ligands via MEK. Question marks denote potential mechanisms whereby TGFβ may independently regulate sheddase activity or, alternatively, control translation of ErbB ligand message. The PDGFR and ErbB pathways, in concert with the noncanonical TGFβ-activated signaling modules PAK2/cAbl and PI3K/AKT/mTOR (19–23, 25, 26, 29), all contribute to the fibrogenic program directed by TGFβ.
TGFβ-stimulated migration, soft agar colony formation, and lung fibrosis is dependent on multiple growth factors
In that the previous figures provide evidence for the synergistic action of multiple growth factors mediating the cellular response to TGFβ, we next investigated whether the proposed mechanisms were biologically relevant to TGFβ-stimulated colony formation in soft agar (Fig. 5A) as well as migration (Fig. 5B, C). Two questions were initially addressed. First, were these phenotypes dependent on the pathways discussed in Fig. 4D (i.e., cAbl/non-Smad as well as PDGF and ErbB receptors); and, second, would the response provide support to extend the analyses to in vivo models? As shown in Fig. 5, inhibition of PDGF and/or ErbB receptor signaling significantly attenuated the proliferative response to TGFβ. Of particular note, suboptimal imatinib plus lapatinib was equally as effective as 3- to 10-fold higher concentrations of either drug alone in preventing soft agar growth (Fig. 5A). Analogous effects were also observed vis-à-vis TGFβ-induced cell migration in that pharmacological inhibition of PDGFR, ErbB, or their common downstream effector, MEK, virtually abrogated TGFβ's effects (Fig. 5B, C).
Figure 5.
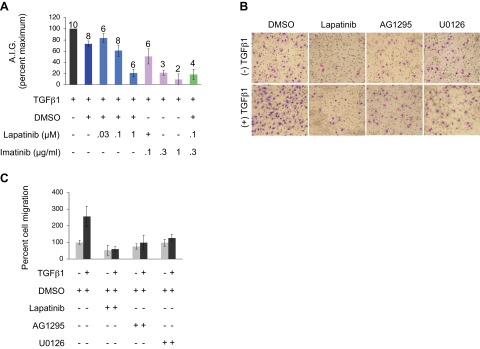
TGFβ-induced anchorage-independent growth (AIG) and migration require PDGFR and ErbB mediation. A) Analyses of TGFβ1-stimulated AIG in the presence of lapatinib and/or imatinib. Treatments were done in triplicate; number of experiments included for each particular treatment is indicated. Error bars = sem. Significant inhibition of ErbB and PDGFR can be attained using higher concentrations of lapatinib and imatinib, respectively, whereas a combination of both drugs at suboptimal doses yields a synergistic effect. B) Analyses of TGFβ1-induced cell migration in the presence of pharmacological inhibitors of ErbB (lapatinib; 5 μM), PDGFR (AG1295; 20 μM), and MEK (U0126; 10 μM). Representative photomicrographs are shown. C) Quantitative analysis of B, where values are from 2 independent experiments, with 4 replicates/treatment. Error bars = sd. Results in B and C show that all 3 pathways are required for TGFβ-induced cell migration.
One of the best characterized in vivo models of organ fibrosis dependent on TGFβ and PDGF is intratracheal administration of BLM (41–43). Consistent with a TGFβ/PDGF directed mechanism, we and others have previously shown that BLM-induced lung fibrosis can be ameliorated by intraperitoneal imatinib administered in either a prevention (i.e., starting 1 d following BLM; 29, 44) or treatment (i.e., initiated ∼halfway in the protocol; 45) regimen. While each approach has strengths as well as weaknesses, a treatment protocol more rigorously reflects the relevant human situation. Therefore, we next investigated whether combination therapy of imatinib plus lapatinib might show greater efficacy in ameliorating the biochemical and/or physiological effects of BLM on murine lung function. As shown in Fig. 6, although each agent alone, as expected, improved lung histology and showed a dose-dependent diminution in hydroxyproline content as well as profibrotic gene expression (Fig. 6A, B, D and Supplemental Tables S1 and S3; see Supplemental Fig. S3C for corresponding in vitro data), greater effectiveness was observed in all parameters when animals were treated with imatinib plus lapatinib (Fig. 6A, C, E and Supplemental Tables S2 and S4). This finding is most clearly documented by the oxygen saturation (SpO2) curves depicted in Fig. 7 and Supplemental Table S5. While untreated animals' SpO2 decreased 24% from d 14 to d 28, and single-agent imatinib or lapatinib showed an approximate 50% improvement (i.e., 14.2 and 12.5% drop from d 14, respectively), treatment with imatinib plus lapatinib essentially stabilized gas exchange (i.e., only a 4.5% decrease from d 14). Thus, by inhibiting the direct as well as indirect actions of multiple cytokines with imatinib plus lapatinib, one of the most critical reflections of normal lung physiology is maintained.
Figure 6.
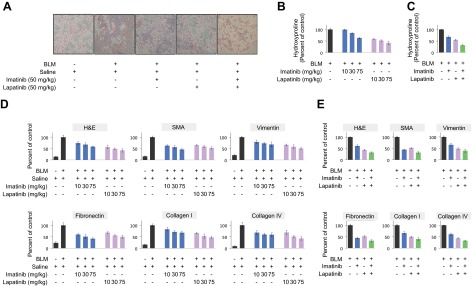
Imatinib and lapatinib cooperate to attenuate BLM-induced lung remodeling. A) Masson's Trichrome staining of paraffin-embedded lung sections from mice challenged with BLM (or saline) for 28 d and treated daily with imatinib and/or lapatinib beginning 15 d following initial BLM insult. Representative photomicrographs are shown (original view ×10). B, C) Quantitative analysis of hydroxyproline contents of lung sections generated from mice treated as in A. D, E) Pixellation scores (expressed as percentage of positive BLM control; ref. 30) quantifying H&E or immunohistochemical staining of lung sections prepared as in A. Data show expression or deposition of smooth muscle actin (SMA), vimentin, fibronectin, collagen I or collagen IV. H&E staining facilitates a direct visualization of collagen. Errors bars = sd. For C and E, both imatinib and lapatinib were used at 50 mg/kg/d. Results support a cooperative therapeutic role of imatinib and lapatinib in attenuating lung remodeling, as evidenced by reduced extracellular matrix production/deposition.
Figure 7.

Dual therapy with imatinib and lapatinib stabilizes lung gas exchange in BLM-challenged mice. Time-dependent fluctuation of oxygen saturation (SpO2) levels (determined on room air) in mice challenged with BLM (or saline) for 28 d and treated with imatinib and/or lapatinib (both at 50 mg/kg/d) beginning 15 d after initial BLM insult. Errors bars = sem (n=6). While administration of individual drugs resulted in partial recovery, a dual treatment yielded a cooperative effect on lung physiology, as indicated by the respective increases in dissolved oxygen levels.
DISCUSSION
Fibroproliferative disorders have been proposed as causal to upwards of 45% of all deaths in the Western world (3). This likely reflects (a minimum of) two realities: first, no organ or organ system is immune to fibrotic changes and destruction; and, second, effective intervention strategies are limited or unavailable (2, 4, 5). Since TGFβ directly and/or indirectly controls a number of profibrotic mediators, including PDGF, endothelin 1, angiotensin II, connective tissue growth factor, Wnt signaling, and micro-RNA expression, to name a few (46–49), characterizing the mechanisms through which its profibrotic actions are mediated is critical. In that regard, a number of Smad and non-Smad pathways have been implicated in the mesenchymal cell response to TGFβ (7, 20, 21, 26, 29, 48). For instance, while Smad3 has been documented to be profibrotic (50, 51), Smad2 is antifibrotic (52). Furthermore, in that inhibiting non-Smad signaling via Pak2/cAbl (as well as PDGF receptor) with imatinib has been shown to be efficacious in rodent models of lung and kidney fibrosis (29, 30), it has been examined in clinical trials of idiopathic pulmonary fibrosis (53), chronic graft-versus-host disease (54, 55), and systemic sclerosis (56–59). While imatinib was ineffective for idiopathic pulmonary fibrosis and in one of the systemic sclerosis trials, patient improvement was observed in the other 5 trials. Although these findings are very encouraging, they need to be interpreted cautiously as the studies were small, necessitating the need for larger randomized controlled trials (60). An additional caveat is that while imatinib would affect both cAbl as well as PDGF receptor signaling, as discussed above, a number of pathways/cytokines have been shown to be profibrotic and would be unaffected by imatinib. Thus, as clearly documented in the oncologic literature, the time may have passed for using single agent therapies in the treatment of various fibroproliferative disorders.
One of the complexities in deciphering TGFβ's mechanisms of action relates to the finding that it has both direct as well as indirect actions mediated through the induction of other growth factors such as PDGF (14–16) and CTGF (17, 18). Recently we have also added ErbB ligands, primarily Areg, Ereg, and Hbegf, to that list (26). As ErbB inhibitors are most often used following receptor amplification, our work not only introduced a novel conceptual framework for understanding the mechanisms underlying fibrogenesis, but suggested that targeting ErbB family members may also be effective in tumors with normal receptor expression whose proliferation and/or invasion is dependent on factors released by an activated stroma. We extend those findings in the current study in 2 important ways. First, a molecular mechanism by which TGFβ regulates ErbB signaling dependent on the integrated action of receptor tyrosine and serine/threonine kinases is defined (Fig. 4); and, second, we provide preclinical evidence supporting the aforementioned contention that greater antifibrotic efficacy in vitro as well as in vivo can be attained using multiple agent therapies (Figs. 5–7).
The results identify and characterize a novel mechanism whereby TGFβ activates the ErbB pathway through the concerted action of receptor tyrosine and serine/threonine kinase signaling. Specifically, we found that while the platelet-derived growth factor receptor is required for activation of the ErbB pathway by TGFβ, and PDGF induces ErbB ligand mRNA, surprisingly, PDGF is unable to induce ErbB receptor phosphorylation/activation. These data clearly show the complex and interdependent actions of multiple growth factors in fibrosis. Current projects are focused on defining the operative mechanisms behind this difference with our preliminary evidence, indicating that PDGF signaling (in contrast to TGFβ), per se, is unable to cleave the transmembrane ErbB ligand precursor.
Last, while our previous work (19, 20, 29) has directly led to promising clinical trial results for various fibroproliferative disorders (54–58), we have broadened our approach and tested whether an optimal antifibrotic response in vivo can be obtained by simultaneously targeting multiple TGFβ-regulated pathways. Utilizing a treatment model of lung fibrosis (i.e., in contrast to a prevention regimen to more closely mimic the human condition) where drugs were initiated 2 wk following BLM insult, we found that imatinib plus lapatinib [inhibits TGFβ (as well as other cytokine)-activated PDGF, cAbl, and ErbB pathways] was not only more effective than either treatment alone, but also stabilized a critical physiological parameter (i.e., dissolved blood oxygen) of pulmonary function (Fig. 7). This finding is extremely exciting as first, stabilization, in contrast to complete lung remodeling, is the best one could realistically expect/hope when treatment is only given for 2 wk; and, second, if analogous findings were observed in subsequent clinical trials, this would provide a major improvement in quality of life.
Supplementary Material
Acknowledgments
This work was supported by U.S. Public Health Service grants GM-55816 and GM-54200 from the National Institute of General Medical Sciences, the Scleroderma Foundation, and the Mayo Foundation (to E.B.L.). M.A. received a Fraternal Order of Eagles Cancer Research Fund (Rochester, MN, USA) fellowship grant.
The authors declare no conflicts of interest.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- BLM
- bleomycin
- CTGF
- connective tissue growth factor
- EGFR
- epidermal growth factor receptor
- ELISA
- enzyme-linked immunosorbent assay
- Ereg
- epiregulin
- ERK
- extracellular signal-regulated kinase
- IP
- immunoprecipitation
- MAPK
- mitogen-activated protein kinase
- MEK
- MAPK and ERK kinase
- PAK2
- p21-activated kinase 2
- PDGF
- platelet-derived growth factor
- PDGFR
- platelet-derived growth factor receptor
- TGFβ
- transforming growth factor β
REFERENCES
- 1. Steele M. P., Schwartz D. A. (2012) Molecular mechanisms in progressive idiopathic pulmonary fibrosis. Annu. Rev. Med. 64, 265–276 [DOI] [PubMed] [Google Scholar]
- 2. Todd N. W., Luzina I. G., Atamas S. P. (2012) Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogenesis Tissue Repair 5, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wynn T. A. (2007) Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Invest. 117, 524–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wynn T. A., Ramalingam T. R. (2012) Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat. Med. 18, 1028–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sivakumar P., Ntolios P., Jenkins G., Laurent G. (2012) Into the matrix: targeting fibroblasts in pulmonary fibrosis. Curr. Opin. Pulm. Med. 18, 462–469 [DOI] [PubMed] [Google Scholar]
- 6. Nakerakanti S., Trojanowska M. (2012) The role of TGF-beta receptors in fibrosis. Open Rheumatol. J. 6, 156–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rahimi R. A., Leof E. B. (2007) TGF-beta signaling: a tale of two responses. J. Cell. Biochem. 102, 593–608 [DOI] [PubMed] [Google Scholar]
- 8. De Caestecker M. (2004) The transforming growth factor-beta superfamily of receptors. Cytokine Growth Factor Rev. 15, 1–11 [DOI] [PubMed] [Google Scholar]
- 9. Ross S., Hill C. S. (2008) How the Smads regulate transcription. Int. J. Biochem. Cell Biol. 40, 383–408 [DOI] [PubMed] [Google Scholar]
- 10. Feng X.-H., Derynck R. (2005) Specificity and versatility in TGF-b signaling through Smads. Annu. Rev. Cell Dev. Biol. 21, 659–693 [DOI] [PubMed] [Google Scholar]
- 11. Prud'homme G. J. (2007) Pathobiology of transforming growth factor beta in cancer, fibrosis and immunologic disease, and therapeutic considerations. Lab. Invest. 87, 1077–1091 [DOI] [PubMed] [Google Scholar]
- 12. Zhang Y. E. (2009) Non-Smad pathways in TGF-beta signaling. Cell Res. 19, 128–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mu Y., Gudey S. K., Landstrom M. (2012) Non-Smad signaling pathways. Cell Tissue Res. 347, 11–20 [DOI] [PubMed] [Google Scholar]
- 14. Leof E. B., Proper J. A., Goustin A. S., Shipley G. D., DiCorleto P. E., Moses H. L. (1986) Induction of c-sis mRNA and activity similar to platelet-derived growth factor by transforming growth factor beta: a proposed model for indirect mitogenesis involving autocrine activity. Proc. Natl. Acad. Sci. U. S. A. 83, 2453–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Taylor L. M., Khachigian L. M. (2000) Induction of platelet-derived growth factor B-chain expression by transforming growth factor-beta involves transactivation by Smads. J. Biol. Chem. 275, 16709–16716 [DOI] [PubMed] [Google Scholar]
- 16. Daniel T. O., Gibbs V. C., Milfay D. F., Williams L. T. (1987) Agents that increase cAMP accumulation block endothelial c-sis induction by thrombin and transforming growth factor-β. J. Biol. Chem. 262, 11893–11896 [PubMed] [Google Scholar]
- 17. Chen Y., Blom I. E., Sa S., Goldschmeding R., Abraham D. J., Leask A. (2002) CTGF expression in mesangial cells: involvement of SMADs, MAP kinase, and PKC. Kidney Int. 62, 1149–1159 [DOI] [PubMed] [Google Scholar]
- 18. Phanish M. K., Wahab N. A., Colville-Nash P., Hendry B. M., Dockrell M. E. (2006) The differential role of Smad2 and Smad3 in the regulation of pro-fibrotic TGFbeta1 responses in human proximal-tubule epithelial cells. Biochem. J. 393, 601–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wilkes M. C., Leof E. B. (2006) TGF-beta activation of c-Abl is independent of receptor internalization and regulated by PI3K and PAK2 in mesenchymal cultures. J. Biol. Chem. 281, 27846–27854 [DOI] [PubMed] [Google Scholar]
- 20. Wilkes M. C., Murphy S. J., Garamszegi N., Leof E. B. (2003) Cell-type-specific activation of PAK2 by transforming growth factor β independent of Smad2 and Smad3. Mol. Cell. Biol. 23, 8878–8889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rahimi R., Andrianifahanana M., Wilkes M. C., Edens M. E., Kottom T. J., Blenis J., Leof E. B. (2009) Distinct roles for mammalian target of rapamycin complexes in the fibroblast response to transforming growth factor-β. Cancer Res. 69, 84–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hong M., Wilkes M. C., Penheiter S. G., Gupta S. K., Edens M., Leof E. B. (2011) Non-Smad transforming growth factor-beta signaling regulated by focal adhesion kinase binding the p85 subunit of phosphatidylinositol 3-kinase. J. Biol. Chem. 286, 17841–17850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wilkes M. C., Mitchell H., Gulati-Penheiter S., Doré J. J., Suzuki K., Edens M., Sharma D. K., Pagano R. E., Leof E. B. (2005) Transforming growth factor-b activation of phosphatidylinositol 3-kinase is independent of Smad2 and Smad3 and regulates fibroblast responses via p21-activated kinase-2. Cancer Res. 65, 10431–10440 [DOI] [PubMed] [Google Scholar]
- 24. Kolosova I., Nethery D., Kern J. A. (2011) Role of Smad2/3 and p38 MAP kinase in TGF-beta1-induced epithelial-mesenchymal transition of pulmonary epithelial cells. J. Cell. Physiol. 226, 1248–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Suzuki K., Wilkes M. C., Garamszegi N., Edens M., Leof E. B. (2007) Transforming growth factor beta signaling via Ras in mesenchymal cells requires p21-activated kinase 2 for extracellular signal-regulated kinase-dependent transcriptional responses. Cancer Res. 67, 3673–3682 [DOI] [PubMed] [Google Scholar]
- 26. Andrianifahanana M., Wilkes M. C., Repellin C. E., Edens M., Kottom T. J., Rahimi R. A., Leof E. B. (2010) ERBB receptor activation is required for profibrotic responses to transforming growth factor beta. Cancer Res. 70, 7421–7430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ishii Y., Fujimoto S., Fukuda T. (2006) Gefitinib prevents bleomycin-induced lung fibrosis in mice. Am. J. Respir. Crit. Care Med. 174, 550–556 [DOI] [PubMed] [Google Scholar]
- 28. Rice A. B., Moomaw C. R., Morgan D. L., Bonner J. C. (1999) Specific inhibitors of platelet-derived growth factor or epidermal growth factor receptor tyrosine kinase reduce pulmonary fibrosis in rats. Am. J. Pathol. 155, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Daniels C. E., Wilkes M. C., Edens M., Kottom T. J., Murphy S. J., Limper A. H., Leof E. B. (2004) Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J. Clin. Invest. 114, 1308–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang S., Wilkes M. C., Leof E. B., Hirschberg R. (2005) Imatinib mesylate blocks a non-Smad TGFb pathway and reduces fibrogenesis in experimental obstructive nephropathy. FASEB J. 19, 1–11 [DOI] [PubMed] [Google Scholar]
- 31. Bruna A., Darken R. S., Rojo F., Ocana A., Penuelas S., Arias A., Paris R., Tortosa A., Mora J., Baselga J., Seoane J. (2007) High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell. 11, 147–160 [DOI] [PubMed] [Google Scholar]
- 32. Goustin A. S., Leof E. B., Shipley G. D., Moses H. L. (1986) Growth factors and cancer [Review]. Cancer Res. 46, 1015–1029 [PubMed] [Google Scholar]
- 33. Leask A., Abraham D. J. (2004) TGF-beta signaling and the fibrotic response. FASEB J. 18, 816–827 [DOI] [PubMed] [Google Scholar]
- 34. Barrientos S., Stojadinovic O., Golinko M. S., Brem H., Tomic-Canic M. (2008) Growth factors and cytokines in wound healing. Wound Repair Regen. 16, 585–601 [DOI] [PubMed] [Google Scholar]
- 35. Coffey R. J., Jr., Derynck R., Wilcox J. N., Bringman T. S., Goustin A. S., Moses H. L., Pittelkow M. R. (1987) Production and auto-induction of transforming growth factor-alpha in human keratinocytes. Nature 328, 817–820 [DOI] [PubMed] [Google Scholar]
- 36. Harris R. C., Chung E., Coffey R. J. (2003) EGF receptor ligands. Exp. Cell Res. 284, 2–13 [DOI] [PubMed] [Google Scholar]
- 37. Bublil E. M., Yarden Y. (2007) The EGF receptor family: spearheading a merger of signaling and therapeutics. Curr. Opin. Cell Biol. 19, 124–134 [DOI] [PubMed] [Google Scholar]
- 38. Cho M. C., Choi H. S., Lee S., Kim B. Y., Jung M., Park S. N., Yoon D. Y. (2008) Epiregulin expression by Ets-1 and ERK signaling pathway in Ki-ras-transformed cells. Biochem. Biophys. Res. Commun. 377, 832–837 [DOI] [PubMed] [Google Scholar]
- 39. Takahashi M., Hayashi K., Yoshida K., Ohkawa Y., Komurasaki T., Kitabatake A., Ogawa A., Nishida W., Yano M., Monden M., Sobue K. (2003) Epiregulin as a major autocrine/paracrine factor released from ERK- and p38MAPK-activated vascular smooth muscle cells. Circulation 108, 2524–2529 [DOI] [PubMed] [Google Scholar]
- 40. Schlessinger J. (2000) Cell signaling by receptor tyrosine kinases. Cell 103, 211–225 [DOI] [PubMed] [Google Scholar]
- 41. Adamson I. Y., Bowden D. J. (1974) The pathogensis of bleomycin-induced pulmonary fibrosis in mice. Am. J. Pathol. 77, 185–197 [PMC free article] [PubMed] [Google Scholar]
- 42. Moseley P. L., Hemken C., Hunninghake G. W. (1986) Augmentationn of fibroblast proliferation by bleomycin. J. Clin. Invest. 78, 1150–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sime P. J., O'Reilly K. M. (2001) Fibrosis of the lung and other tissues: new concepts in pathogenesis and treatment. Clin. Immunol. 99, 308–319 [DOI] [PubMed] [Google Scholar]
- 44. Aono Y., Nishioka Y., Inayama M., Ugai M., Kishi J., Uehara H., Izumi K., Sone S. (2005) Imatinib as a novel antifibrotic agent in bleomycin-induced pulmonary fibrosis in mice. Am. J. Respir. Crit. Care Med. 171, 1279–1285 [DOI] [PubMed] [Google Scholar]
- 45. Chaudhary N. I., Schnapp A., Park J. E. (2006) Pharmacologic differentiation of inflammation and fibrosis in the rat bleomycin model. Am. J. Respir. Crit. Care Med. 173, 769–776 [DOI] [PubMed] [Google Scholar]
- 46. Akhmetshina A., Palumbo K., Dees C., Bergmann C., Venalis P., Zerr P., Horn A., Kireva T., Beyer C., Zwerina J., Schneider H., Sadowski A., Riener M. O., MacDougald O. A., Distler O., Schett G., Distler J. H. (2012) Activation of canonical Wnt signalling is required for TGF-beta-mediated fibrosis. Nat. Commun. 3, 735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Leask A. (2010) Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ. Res. 106, 1675–1680 [DOI] [PubMed] [Google Scholar]
- 48. Verrecchia F., Mauviel A. (2007) Transforming growth factor-beta and fibrosis. World J. Gastroenterol. 13, 3056–3062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xie T., Liang J., Guo R., Liu N., Noble P. W., Jiang D. (2011) Comprehensive microRNA analysis in bleomycin-induced pulmonary fibrosis identifies multiple sites of molecular regulation. Physiol. Genomics 43, 479–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sato M., Muragaki Y., Saika S., Roberts A. B., Ooshima A. (2003) Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Invest. 112, 1486–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhao J., Shi W., Wang Y. L., Chen H., Bringas P., Jr., Datto M. B., Frederick J. P., Wang X. F., Warburton D. (2002) Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 282, L585–L593 [DOI] [PubMed] [Google Scholar]
- 52. Meng X. M., Huang X. R., Chung A. C., Qin W., Shao X., Igarashi P., Ju W., Bottinger E. P., Lan H. Y. (2010) Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J. Am. Soc. Nephrol. 21, 1477–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Daniels C. E., Lasky J. A., Limper A. H., Mieras K., Gabor E., Schroeder D. R. (2010) Imatinib treatment for idiopathic pulmonary fibrosis: randomized placebo-controlled trial results. Am. J. Respir. Crit. Care Med. 181, 604–610 [DOI] [PubMed] [Google Scholar]
- 54. Magro L., Catteau B., Coiteux V., Bruno B., Jouet J. P., Yakoub-Agha I. (2008) Efficacy of imatinib mesylate in the treatment of refractory sclerodermatous chronic GVHD. Bone Marrow Transplant. 42, 757–760 [DOI] [PubMed] [Google Scholar]
- 55. Olivieri A., Locatelli F., Zecca M., Sanna A., Cimminiello M., Raimondi R., Gini G., Mordini N., Balduzzi A., Leoni P., Gabrielli A., Bacigalupo A. (2009) Imatinib for refractory chronic graft-versus-host disease with fibrotic features. Blood 114, 709–718 [DOI] [PubMed] [Google Scholar]
- 56. Khanna D., Saggar R., Mayes M. D., Abtin F., Clements P. J., Maranian P., Assassi S., Singh R. R., Furst D. E. (2011) A one-year, phase I/IIa, open-label pilot trial of imatinib mesylate in the treatment of systemic sclerosis-associated active interstitial lung disease. Arthritis Rheum. 63, 3540–3546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pope J., McBain D., Petrlich L., Watson S., Vanderhoek L., de Leon F., Seney S., Summers K. (2011) Imatinib in active diffuse cutaneous systemic sclerosis: Results of a six-month, randomized, double-blind, placebo-controlled, proof-of-concept pilot study at a single center. Arthritis Rheum. 63, 3547–3551 [DOI] [PubMed] [Google Scholar]
- 58. Spiera R. F., Gordon J. K., Mersten J. N., Magro C. M., Mehta M., Wildman H. F., Kloiber S., Kirou K. A., Lyman S., Crow M. K. (2011) Imatinib mesylate (Gleevec) in the treatment of diffuse cutaneous systemic sclerosis: results of a 1-year, phase IIa, single-arm, open-label clinical trial. Ann. Rheum. Dis. 70, 1003–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Distler O., Distler J. H., Varga J., Denton C. P., Lafyatis R. A., Wigley F. M., Schett G. (2010) A multi-center, open-label, proof of concept study of imatinib mesylate demonstrates no benefit for the treatment of fibrosis in patients with early, diffuse systemic sclerosis. Arthritis Rheum. 62(Suppl. 10), 560 [Google Scholar]
- 60. Beyer C., Distler O., Distler J. H. (2012) Innovative antifibrotic therapies in systemic sclerosis. Curr. Opin. Rheumatol. 24, 274–280 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.