

Abstract
A “longevity ” gene, sirtuin 1 (SIRT1), can attenuate age-dependent induction of left ventricular dysfunction. This study aimed to characterize the role of SIRT1 in the tolerance of aged heart to ischemic insults. Male C57BL/6 young (4–6 mo) and aged (24–26 mo) mice were used to determine the role of SIRT1 in myocardial ischemia/reperfusion (I/R) tolerance. SIRT1 localization was assessed by confocal microscopy. Immunoblotting was used to evaluate SIRT1 expression and translocation. The results demonstrated that SIRT1 is expressed predominantly as a sumoylated form in cardiomyocyte nuclei. Moreover, cardiac overexpression of desumoylase, sentrin-specific protease 2 (SENP2), significantly reduces nuclear sumoylated SIRT1 levels (P<0.05). Interestingly, I/R stress leads to desumoylation and translocation of nuclear SIRT1 into the cytoplasm in aged but not in young hearts. SIRT1 activity in ischemic young hearts was 3.2-fold higher than that seen in ischemic aged hearts, which suggests that aging causes impaired nucleocytoplasmic shuttling and activation of SIRT1 during ischemic stress. The infarct size in aged and Sirt1+/− knockout hearts was higher than that observed in young and Sirt1+/+ WT littermate hearts, respectively (all P<0.05). SIRT1 agonist, SRT1720, reduced myocardial infarction in both aged and Sirt1+/− hearts. Therefore, impaired cardiac SIRT1 activity plays a critical role in the observed increase in susceptibility of the aged heart to I/R injury. SIRT1 agonist can restore this aging-related loss of cardioprotection.—Tong, C., Morrison, A., Mattison, S., Qian, S., Bryniarski, M., Rankin, B., Wang, J., Thomas, D.P., Li, J. Impaired SIRT1 nucleocytoplasmic shuttling in the senescent heart during ischemic stress.
Keywords: Sirtuin 1, sumoylation, myocardial infarction
Sirtuin 1 (Sirt1), one of 7 mammalian homologs of Sir2 (1), an NAD+-dependent deacetylase, has long been assumed to be an antiaging protein due to the finding that Sir2 prolongs the life span of S. cerevisiae (2), C. elegans (3), and D. melanogaster (4). The studies on mammals have demonstrated that an absence of SIRT1 leads to chromosome abnormality (5), tumor genesis (6), and cell cycle arrest (7), and causes early neonatal death (8). In contrast, SIRT1 activation increases insulin sensitivity, decreases blood glucose level (3), and attenuates neuronal damage (9).
The nucleocytoplasmic shuttling of SIRT1 is involved in regulating the activity of SIRT1 (10, 11). Nuclear, but not cytoplasmic, SIRT1 is responsible for the resistance to oxidative stress (10). SIRT1's substrate NAD+ and product nicotinamide (NAM) are important for SIRT1 function (10). NAM phosphoribosyltransferase (NAMPT), the rate-limiting enzyme of the NAD+ biosynthesis pathway, has been shown to modulate SIRT1 activity (12). In the heart, SIRT1 plays a role in suppressing apoptosis (13) and resisting oxidative stress (14). The prevalence of ischemic heart disease, characterized by insufficient oxygen and blood supply to the heart, is significantly greater in the elderly population (15). This finding demonstrates that the aged heart is more susceptible to ischemia/reperfusion (I/R) injury (16). Within this context, as an antiaging protein with cardioprotective characteristics, SIRT1 is a potential interventional target for I/R injury management in the elderly.
In the present study, we report for the first time that cardiac SIRT1 is predominantly expressed in cardiomyocyte nuclei as a sumoylated form that is integral to the heart's tolerance for ischemic stress. The aged heart possesses lower SIRT1 protein levels and demonstrates an impaired SIRT1 activation in response to I/R. However, this depressed activation response can be revived pharmacologically and thus provides a potential mechanism whereby cardioprotection against I/R injury in senescent populations might be restored.
MATERIALS AND METHODS
Animal preparation
Male C57BL/6 mice (4–6 and 24–26 mo) were purchased from Charles River, Inc. (Wilmington, MA, USA). Sirt1 heterozygote Sirt1+/− mice were a generous gift from Dr. Jianping Ye (Louisiana State University, Baton Rouge, LA, USA; refs. 8, 17); sentrin-specific protease 2 (SENP2) transgenic mice (Tg-SENP2) were generated with the use of a cardiac-myosin heavy-chain (α-MHC) promoter (18). Mice were maintained on a 12-h light-dark cycle in a controlled environment with water ad libitum. All animal protocols were approved by the Institutional Animal Care and Use Committee of the State University of New York (SUNY) at Buffalo.
In vivo regional ischemia and myocardial infarct size measurement
Mice were anesthetized with sodium pentobarbital, then placed on a ventilator (Harvard Apparatus, Holliston, MA, USA; refs. 19–21). Body temperature was maintained at 37°C with a heating pad. After left lateral thoracotomy, the left anterior descending (LAD) coronary artery was occluded with an 8-0 nylon suture, and a small piece of gauze was placed between the suture and the heart to prevent arterial injury. ECG traces confirmed the ischemic hallmark of ST-segment elevation during coronary occlusion (ADInstruments, Colorado Springs, CO, USA). At different time points, the left ventricle (LV) was isolated before freeze-clamping in liquid nitrogen. Freeze-clamped LV was stored at −80°C for further immunoblotting analysis. For infarct size measurement experiments, the hearts were subjected to 20 min in vivo regional ischemia followed by 4 h reperfusion, after which they were processed with dual staining (20, 22). Non-necrotic tissue in the ischemic region was stained red with 2,3,5-triphenyltetrazolium chloride (TTC; 1%, w/v), and the nonischemic region was stained blue with Evan's blue (1%, w/v). Hearts were fixed by 4% formalin overnight at 4°C, then sectioned into 1-mm slices, photographed using a Leica microscope (Leica Microsystems, Wetzlar, Germany), and analyzed using ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA). The myocardial infarct size was calculated as the extent of myocardial necrosis to the ischemic area at risk (AAR; refs 20–22).
SIRT1 activity measurement
SIRT1 deacetylase activity assays were performed by incubation with nuclear or cytoplasmic extracts from heart samples. Trichostain A (0.2 mM; Sigma-Aldrich, St. Louis, MO, USA; ref. 23), components of Fluor de Lys SIRT1 Fluorescent Activity Assay/Drug Discovery Kit (Enzo Life Sciences, Farmingdale, NY, USA), including 100 μM fluorogenic peptide encompassing residues 379 to 382 of p53 with lysine 382 being acetylated, and 170 μM NAD+ at 37°C for 1 h, followed by incubation in developer for 15 min at room temperature according to the manufacturer's instructions. Fluorescent intensity was measured using a Bio-Tek Multifunction Plate Reader (BioTek Instruments, Winooski, VT, USA). No-enzyme and time 0 negative controls were generated by incubating developer II solution with 2 mM NAM before mixing the substrates with or without samples. SIRT1 activity was calculated with the corrected arbitrary fluorescence units of the tested samples to no-enzyme control and expressed as fluorescent units relative to the control.
Immunohistochemistry
Isolated hearts were perfused and incubated with 4% paraformaldehyde for 1 h at room temperature (24, 25). Tissues were then transferred to 25% sucrose overnight at 4°C, mounted in Tissue-Tek Optimal Cutting Temperature (OCT) compound (Sakura Finetek, Torrance, CA, USA), snap-frozen in liquid nitrogen, and stored at −80°C overnight. The samples were sliced into 30-μm-thick sections on a cryostat, mounted onto clean glass slides that had been charged by a chromium, potassium, and gelatin solution, thoroughly air-dried, and stored at −80°C until processing. For staining, slides were incubated with 0.2% Triton X-100 in PBS for 5 min at room temperature, washed with PBS, blocked with 5% BSA and 0.1% Triton-X in PBS for 2 h, and then incubated overnight with anti-SIRT1 antibody in a 5% BSA TBS-T solution (Cell Signaling, Danvers, MA, USA). The bound antibodies were labeled using Alexa Fluor 488-conjugated goat anti-rabbit secondary antibody (Cell Signaling). Sections were then covered using Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA, USA). Images were captured using a Zeiss Axioimager Motorized fluorescence microscope or Zeiss LSM 510 Meta confocal microscope (Carl Zeiss, Oberkochen, Germany) and analyzed by MATLAB software (MathWorks, Natick, MA, USA).
Nuclear-cytoplasmic fraction
The nuclear-cytoplasmic fraction of heart tissue or cells was conducted using the NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (Thermo Fisher Scientific, Rockford, IL, USA) according to the manufacturer's protocol.
Immunoblotting analysis
Immunoblottings were performed as described previously (22, 25–27). The heart tissue samples were homogenized in ice-cold lysis buffer. Bradford method (Bio-Rad Laboratories, Hercules, CA, USA) was used for measuring the lysate protein concentration. Heart homogenate proteins were then resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride (PVDF) membranes. For reprobing, membranes were stripped with Restore Western Blot Stripping Buffer (Thermo Fisher Scientific). Rabbit polyclonal antibodies against SIRT1, p53, and peroxisome proliferator-activated receptor γ coactivator 1-α (PGC1-α) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against acetyl-p53 (Lys 379), acetyl-lysine, GAPDH, TATA-binding protein (TBP), and horseradish peroxidase linked secondary antibodies were purchased from Cell Signaling.
Ex vivo Langendorff-perfused heart system
Mice were given an injection of heparin (100 U i.p.) 10 min prior to pentobarbital sodium (60 mg/kg i.p.)-induced euthanasia. The hearts were quickly excised and retrogradely perfused (4 ml/min) on a Heart Perfusion System (Radnoti Glass Technology, Monrovia, CA, USA) with 95% O2 and 5% CO2 equilibrated Krebs-Henseleit buffer containing 118 mM NaCl, 4.75 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 25 mM NaHCO3, 1.4 mM CaCl2, 10 μU/ml insulin, 7 mM glucose, 1% BSA, and 0.4 mM oleate. For generating the ex vivo ischemic model, buffer flow was cut off for 20 min after 30 min of preperfusion of buffer containing SRT1720 (1 μM in 0.01% DMSO) or vehicle (0.01% DMSO). The hearts were then reperfused with the same rate of buffer flow during reperfusion. The LabChart6 software (ADInstruments) was used to monitor the heart rate and left ventricular developed pressure.
NAD+ assay
NAD+ and NADH content in mouse hearts was measured using Amplite Fluorimetric NAD+/NADH Assay Kit (AAT Bioquest, Sunnyvale, CA, USA), according to the manufacturer's instructions. Heart lysates were prepared based on total protein concentration. To detect NADH only, NAD+ in the lysate was decomposed in advance by heating samples at 65°C for 30 min. Absorbance was monitored at 430 nm by xMark microplate spectrophotometer (Bio-Rad).
Apoptosis measurement in cardiac tissue sections
Heart longitudinal frozen sections were stained for apoptosis with the CardioTACS In Situ Apoptosis Dectection Kit (Trevigen, Gaithersburg, MD, USA). Cardiomyocytes that incorporated the labeled biotinylated nucleotides were considered terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) positive and were counted under a Meiji TC5300 inverted phase contrast microscope (Meiji Techno Co., Ltd., Saitama, Japan) with a ×40 objective. Apoptosis percentage was determined by the number of TUNEL-positive nuclei divided by the number of nuclei in 10 randomly chosen separate fields.
Fluorescent annexin V staining
Annexin V staining to determine cell apoptosis were performed as described previously (28). HL-1 cardiomyocytes were grown for 48 h in a 6-well cell culture plate in which an autoclaved glass coverslip was placed in the bottom of each well. Fresh Claycomb medium was applied to cells before treating with vehicle, SRT1720 (1 μM), and SRT1720 plus anacardiac acid (10 μM). The hypoxia groups were placed in 95% nitrogen and 5% carbon dioxide in a mixed-gas perfusion hypoxia chamber for 1 h, followed by 1 h normoxia at 37°C; the control groups were placed in nomoxia conditions at 37°C in a cell culture incubator. After incubation, coverslips were washed sequentially with cold PBS and annexin V binding buffer; then 100 μl of annexin V alex488 conjugate solution (1:20 dilution; Life Technologies, Grand Island, NY, USA) was applied for 15 min at room temperature, and coverslips were again washed twice with annexin-binding buffer. Mounting medium (containing DAPI; 20 μl) was then quickly applied, and the coverslips were flipped, mounted on autoclaved microscopy glass slides, and observed by Zeiss fluorescence microscope.
Statistical analysis
The sample size calculation was determined by defining the unpaired equal sample size test with 85% power, value of α = 0.05, and the estimated sd as one-third of the range of the random sample value. Data are expressed as means ± se. Significance was tested by Student's unpaired 2-tailed t test or 2-way repeated measures ANOVA with Bonferroni correction, as appropriate. A value of P < 0.05 was considered significant.
RESULTS
SIRT1 levels are impaired in aged heart
Cryostat sections of hearts from young and aged C57BL/6 mice were stained with anti-SIRT1 antibody. The results demonstrated that SIRT1 is localized predominantly in the nuclear portion of cardiomyocytes (Fig. 1A). The confirmatory results were obtained by analyzing SIRT1 protein expression levels in the nuclei, cytoplasm, and whole-heart lysates from young and aged hearts (Fig. 1B). Interestingly, nuclear SIRT1 demonstrates a 20-kDa increase in molecular mass as compared to cytoplasmic SIRT1 (Fig. 1B). To determine whether aging causes alterations in SIRT1 levels and localization, nuclear and cytoplasmic fractions from both young and aged hearts were separated and analyzed by immunoblotting. The results demonstrated that both nuclear (140 kDa) and cytoplasmic (120 kDa) SIRT1 protein levels are decreased in aged vs. young hearts (both P<0.05; Fig. 1B). Moreover, the whole heart lysates confirmed the lower levels of SIRT1 proteins in aged vs. young hearts (P<0.05; Fig. 1B). These results indicate that cardiac SIRT1 protein expression levels are significantly decreased with aging.
Figure 1.

Cardiac SIRT1 is predominantly localized in the nucleus. A) Immunofluorescent staining of SIRT1 in myocardial cryostat tissue sections from young and aged C57BL/6 mice. SIRT1 is shown in green, DAPI staining in blue. B) Immunoblotting shows SIRT1 protein expression levels in nuclei, cytoplasm and whole homogenates from young and aged hearts. Values are means ± se; n = 5–7. *P < 0.05 vs. young.
SIRT1 is sumoylated in the nucleus
Sumoylation, a common post-translational modification for nuclear proteins (29), may be responsible for the observed increase in molecular mass of nuclear compared to cytoplasmic SIRT1. Immunoblotting with anti-small ubiquitin-related modifier 1 (SUMO-1) antibody showed that the increase in molecular mass of cardiac nuclear SIRT1 is due to sumoylation by SUMO-1 proteins (Fig. 2A, top panel). The molecular mass of SUMO-1 is ∼11 kDa (30), but it appears larger on SDS-PAGE and adds ∼20 kDa to the apparent molecular mass of most substrates (30), which is consistent with our results of SUMO-SIRT1in the heart (Fig. 2A). We also performed SIRT1 immunoprecipitation and immunoblotting with anti-SUMO-1 antibody in both young and aged hearts to confirm that only 140-kDa SIRT1, and not 120-kDa SIRT1, is sumoylated (Supplemental Fig. S1). Furthermore, cardiac overexpression of a desumoylase, SENP2 (31), significantly decreased nuclear SIRT1 protein levels (Fig. 2B). Tg-SENP2 hearts also demonstrated decreased SIRT1 activity, as indicated by a higher level of acetylation of PGC-1α, a well-known downstream target of SIRT1 (32), in Tg-SENP2 hearts than that seen in WT hearts (Supplemental Fig. S2).
Figure 2.
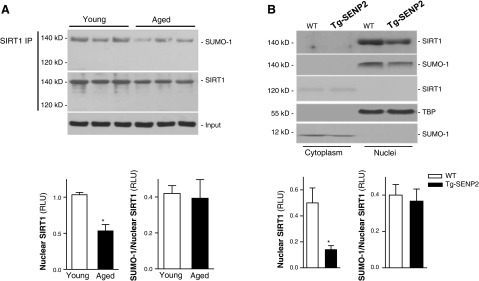
Sumoylation of cardiac SIRT1. A) Nuclear extracts from young and aged hearts were subjected to immunoprecipitation (IP) with SIRT1 antibody. The IP products were further analyzed by immunoblotting; anti-SIRT1 and anti-SUMO-1 antibodies were reciprocally used to confirm sumoylation of SIRT1. Anti-TBP was used as a input (to panel); bar graphs show relative levels of sumoylated SIRT1 in young vs. aged hearts (bottom panel). Values are means ± se; n = 6–7. *P < 0.05 vs. young. B) Fractionated heart homogenates from Tg-SENP2 mice and wild-type littermates were examined by immunoblotting. Anti-SIRT1 and anti-SUMO-1 antibodies were reciprocally used to confirm sumoylation of nuclear SIRT1 (top panel); bar graphs show relative levels of nuclear sumoylated SIRT1in WT and Tg-SENP2 hearts, respectively (bottom panel). Values are means ± se; n = 6. *P < 0.05 vs. WT.
Activation and nucleocytoplasmic shuttling of SIRT1 are blunted in aged heart during I/R
Although SIRT1 has been reported as being protective against oxidative stress in the heart (14), the mechanism underlying SIRT1 modulation of I/R injury remains unknown. To determine the role of SIRT1 in I/R, both young and aged mouse hearts were subjected to in vivo regional ischemia with 10 min of LAD coronary artery occlusion, followed by 20 min reperfusion. Nuclear SIRT1 activity was elevated 107.7% in response to ischemic stress in young hearts (Fig. 3A). However, the SIRT activity of ischemic aged hearts was blunted as compared to ischemic young hearts (Fig. 3A). Intriguingly, neither young nor aged heart cytoplasmic SIRT1 activity showed any change in response to ischemic stress (data not shown). The impairment of SIRT1 activity in aged hearts was confirmed by intracellular levels of NAD+, which drives SIRT1 activity (33). NAD+ levels were markedly elevated from 12.17 ± 3.71 to 30.82 ± 4.84 pmol/mg by 10 min of ischemia in young hearts (P<0.05), whereas the values were no different between sham treatment and ischemic conditions in aged hearts (6.42±2.16 and 6.80±0.91pmol/mg, respectively; Fig. 3B).
Figure 3.

Activation and nucleocytoplasmic shuttling of SIRT1 during I/R. A) LVs from young and aged hearts were harvested following a sham operation (sham), after 10 min in vivo regional ischemia (isch), and after 10 min of in vivo regional ischemia followed by 20 min reperfusion (I/R). Nuclear fractions of young and aged LVs were subjected to the SIRT1 activity assay. B) Intracellular NAD+ content measurement of LVs from young and aged heart samples were obtained from the same three aforementioned conditions. Values are means ± se; n = 6. *P < 0.05 vs. young sham; †P < 0.05 vs. young ischemia. C) Nuclear (left panel) and cytoplasmic (right panel) extracts from young and aged hearts were analyzed by immunoblotting; bar graphs show relative levels of nuclear SIRT1 (left panel) and cytoplasmic SIRT1 (right panel) from young and aged hearts. Values are means ± se; n = 6–8. *P < 0.05 vs. corresponding young group; †P < 0.05 vs. aged sham. D) Hearts from young and aged C57BL/6 mice were subjected to ex vivo global I/R with a Langendorff heart perfusion system. Hearts from control, ischemia (20 min), and I/R (20 min of ischemia followed by 30 min of reperfusion) treatment groups were collected for SIRT1 immunostaining (top panel); bar graph shows percentage of nuclear SIRT1 as a fraction of the total SIRT1 (bottom panel). Values are means ± se for 5 independent experiments. *P < 0.05 vs. aged control.
SIRT1 nucleocytoplasmic shuttling plays a critical role in regulating its activity (10). To determine whether I/R stress alters the rate of SIRT1 nuclear-to-cytosolic shuttling in the heart, young and aged hearts were subjected to in vivo regional ischemia with 10 min of LAD occlusion, followed by 20 min reperfusion. Nuclear SIRT1 in aged hearts was significantly decreased during ischemia or I/R vs. sham operation (P<0.05; Fig. 3C, left panel), while no significant changes occur in the nuclear SIRT1 of young hearts in response to ischemic stress (Fig. 3C, left panel). Intriguingly, cytoplasmic SIRT1 in aged hearts was significantly increased in response to ischemic stress as compared to sham operation (P<0.05, Fig. 3C, right panel), whereas no significant changes were observed in young hearts during ischemia or I/R (Fig. 3C, right panel). These findings were confirmed by immunofluorescent SIRT1 staining that showed a 30% drop in nuclear SIRT1 content in aged hearts following ischemia and/or reperfusion (both P<0.05 vs. control; Fig. 3D). In contrast, nuclear SIRT1 content did not alter following ischemia and/or reperfusion in young hearts (Fig. 3D). These results suggest that the nucleocytoplasmic shuttling of SIRT1 is impaired in the aged heart.
SIRT1 agonist, SRT1720, activates SIRT1 in the heart
The synthesized small molecule SRT1720 has been reported to activate SIRT1 more potently than a known SIRT1 activator, resveratrol (34). SRT1720 (20 μg/g) was administered to C57BL/6 mice via tail vein injection; hearts were harvested 30 min following SRT1720 injection. The nuclear fraction or whole lysate of heart tissue homogenates was used for immunoprecipitating of PGC-1α (Fig. 4A), a well-known downstream target of SIRT1 (32). The results demonstrated that SRT1720 treatment significantly decreased acetylation of PGC-1α, and enhanced the interaction between nuclear SIRT1 (140 kDa) and PGC-1α (Fig. 4A, bottom panel). Interestingly, no interaction occurred between cytoplasmic SIRT1 (120 kDa) and PGC-1α (Fig. 4A, bottom panel). In addition, to determine whether SRT1720 induces SIRT1 activation by increasing nuclear localization and sumoylation of SIRT1, the HL-1 cardiomyocyte (35) was treated with SRT1720 (1 μM). Notably, SRT1720 treatment resulted in an increase in the 140-kDa SIRT1, regarded as the sumoylated nuclear SIRT1, and a decrease in the 120-kDa SIRT1, the nonsumoylated cytoplasmic form (Fig. 4B, top panel). Moreover, nuclear and cytoplasmic fraction SIRT1 blots confirmed that SRT1720 induces SIRT1 translocation from cytoplasm to nuclei (Fig. 4B, bottom panels). SUMO-1 blots demonstrated that 140- but not 120-kDa SIRT1 is sumoylated (Fig. 4B, second blot panel). These results indicate that SRT1720 activates SIRT1 in the heart by promoting its sumoylation, leading to its nuclear localization.
Figure 4.

SRT1720 triggers cardiac SIRT1 activation. A) SRT1720 was administered to C57BL/6 mice through tail vein injection; the hearts were harvested under control, vehicle, or SRT1720 treatment conditions. Nuclear extracts or heart homogenates were used for immunoprecipitation by employing an anti-PGC-1α antibody, and then the immunoprecipitates were immunoblotted with anti-acetyl lysine and anti-PGC-1α (nuclear extracts) and anti-SIRT1 (heart homogenates), respectively (top panel); bar graphs indicate the relative amount of acetyl lysine (bottom left panel) and related amount of associated SIRT1 (bottom right panel) in the PGC-1α immunoprecipitates. Values are means ± se; n = 5–6. *P < 0.05 vs. control. B) HL-1 cardiomyocytes were treated with vehicle or SRT1720 (1 μM) for 1 h, then cell lysates were immunoblotted with anti-SIRT1 and anti-SUMO-1, respectively (first and second blot panels); the nuclear and cytoplasmic fractions were examined by immunoblotting with anti-SIRT1 antibody to confirm translocation of SIRT1 by SRT1720 treatment; bar graphs demonstrate the relative levels of sumoylation of SIRT1 in HL-1 cardiomyocytes in response to SRT1720 treatment. Values are means ± se; n = 3–4. *P < 0.05 vs. vehicle.
SRT1720 protects heart against I/R injury
To characterize the importance of SIRT1 activation in cardioprotection against I/R injury, the extent of myocardial infarction was evaluated after 20 min in vivo regional ischemia followed by 4 h reperfusion. The infarct size of aged C57BL/6 mice was 2.8-fold larger than that of young mice (P<0.05; Fig. 5A, C). However, SRT1720 (20 μg/g i.v.) given 30 min prior to ischemia significantly reduced infarct size by 82% in aged mice (P<0.01). To confirm that SIRT1 deficiency is a major factor involved in the vulnerability of aged hearts to I/R stress, SIRT1 deficient heterozygous (Sirt1+/−) mice (comparable cardiac SIRT1 protein level with aged mice) and wild type littermates (comparable cardiac SIRT1 protein level with young mice) were subjected to the identical I/R conditions. Sirt1+/− showed a comparable infarct size with aged hearts (Fig. 5A, C), while wild-type littermates had no apparent discrepancy in myocardial infarction compared to young hearts (Fig. 5A, C). SRT1720 treatment significantly decreased infarct size in Sirt1+/− mice (P<0.01; Fig. 5A, C). Moreover, ex vivo heart perfusion data showed that postischemic contractile function was impaired in aged vs. young hearts (P<0.05; Fig. 5D). Intriguingly, SRT1720 treatment significantly improved postischemic contractile function recovery (Fig. 5D) and reduced infarct size in aged hearts (Fig. 5E). These results indicate that pharmacological activation of SIRT1 protects against I/R injury in aging.
Figure 5.
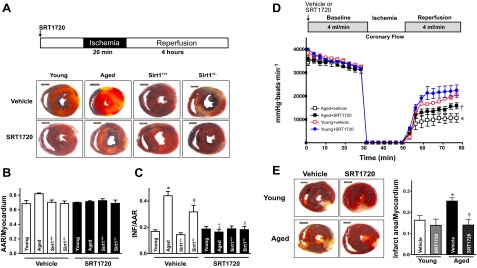
SRT1720 reduces I/R injury. Young and aged C57BL/6 mice, Sirt1+/− heterozygous knockout mice, and Sirt1+/+ wild-type littermate mice (C57BL/6 background) were subjected to 20 min of in vivo regional ischemia followed by 4 h reperfusion, and SRT1720 or vehicle was administered via tail vein 30 min before ischemia, as shown in the illustration. A) Representative tissue cross sections. B, C) Ratio of AAR to myocardium (B) and ratio of infarct area (INF) to AAR (C) in mouse hearts with vehicle or SRT1720 treatment. Values are means ± se for 6 independent experiments. *P < 0.01 vs. young vehicle; †P < 0.01 vs. Sirt1+/+ vehicle; ‡P < 0.01 vs. aged vehicle; §P < 0.01 vs. Sirt1+/− vehicle. D) Isolated hearts from young and aged C57BL/6 mice were subjected to ex vivo heart perfusion (Langendorff). Heart rate × left ventricular developed pressure was determined during baseline perfusion, global ischemia, and postischemic reperfusion with vehicle or SRT1720 administration; n = 4/group. *P < 0.05 vs. young vehicle during reperfusion; †P < 0.05 vs. aged vehicle during reperfusion. E) Isolated hearts from young and aged C57BL/6 mice were subjected to 20 min global ischemia and 2 h reperfusion with vehicle or SRT1720 administration. Left panel: representative sections. Right panel: ratio of infarct size to total myocardium. Values are means ± se; n = 5–6/group. *P < 0.05 vs. young vehicle; †P < 0.05 vs. aged vehicle.
Activation and sumoylation of SIRT1 are critical for its cardioprotective function during I/R
p53 is a biomarker of heart senescence and heart failure (36), with its transcriptional activity and stability known to be regulated by SIRT1 (37). To further study mechanisms underlying the increased vulnerability to I/R in the aged heart and whether pharmacological SIRT1 activation can diminish this vulnerability, acetylation of p53 that is related its activity and stability (37) was assessed in both young and aged ischemic hearts. Acetyl-p53 (Lys 379) was significantly higher in aged hearts under sham and I/R conditions compared to young hearts (Fig. 6A), indicating compromised SIRT1 activity in the aged heart. TUNEL staining assay was conducted on apical sections of isolated hearts exposed to ex vivo global 20 min of ischemia followed by 2 h reperfusion, the results demonstrated more positive apoptotic nuclei in the aged heart (Fig. 6B). However, SRT1720 treatment significantly reduced apoptosis caused by I/R in the aged heart (P<0.05; Fig. 6B).
Figure 6.
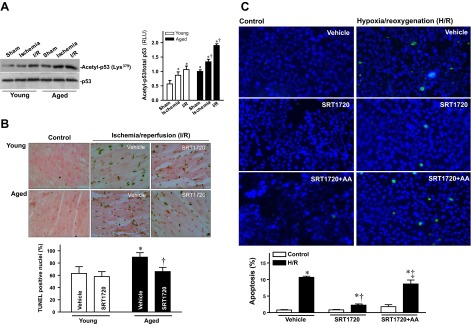
SRT1720 inhibits apoptosis and stimulates SIRT1 sumoylation. A) LVs from young and aged hearts were harvested following a sham operation (sham), 10 min in vivo regional ischemia, and 10 min of in vivo regional ischemia followed by 20 min reperfusion (I/R). Homogenates were analyzed by immunoblotting with anti-acetyl-p53 or anti-p53 antibody (left panel); bar graph demonstrates relative levels of acetyl-p53 in young and aged hearts during sham, ischemia, or I/R treatment conditions (right panel). Values are means ± se; n = 5–7/group. *P < 0.05 vs. young sham; †P < 0.05 vs. corresponding young group. B) TUNEL staining of young and aged hearts exposed to ex vivo 20 min of ischemia followed by 2 h of reperfusion with vehicle or SRT1720 administration (top panel), bar graph shows apoptosis percentage, calculated from number of TUNEL-positive nuclei divided by total number of nuclei (∼1000 nuclei were counted in 10 random microscopic fields). Values are means ± se; n = 5–7/group; *P < 0.05 vs. young vehicle; †P < 0.05 vs. aged vehicle. C) HL-1 cardiomyocytes were pretreated 1 h with vehicle, SRT1720 (1 μM) or AA (10 μM) plus SRT1720, and then treated with or without 1 h of hypoxia and 1 h reoxygenation (H/R). Top panel: representative high-power fields of fluorescent-annexin stained HL-1 cells. Bottom panel: percentage of apoptosis. HL-1 cells were examined in multiple high-power fields; positively stained cells (intense, punctate fluorescence) were enumerated (∼1000 cells were counted in 10 random microscopic fields). *P < 0.01 vs. vehicle control; †P < 0.05 vs. vehicle H/R; ‡P < 0.01 vs. SRT1720 H/R.
To verify the cardioprotection of SIRT1 sumoylation against I/R injury, anacardic acid (AA), a specific inhibitor of sumoylation (38), was used to characterize the role of SIRT1 sumoylation in preventing I/R-induced injury. HL-1 cardiomyocytes were pretreated for 1 h with vehicle or AA (10 μM) prior to hypoxia/reoxygenation (H/R) treatment with or without SRT1720 (1 μM). The results demonstrated that H/R markedly induced apoptosis of HL-1 cells (Fig. 6C); SRT1720 treatment inhibited H/R-induced apoptosis of HL-1 cardiomyocytes (Fig. 6C), while pretreated with a sumoylation inhibitor, anacardic acid, blocked the protective effect of SRT1720 on H/R-induced apoptosis (Fig. 6C). Taken together, these findings indicate that SIRT1 sumoylation is critical for its myocardial protection against I/R-induced injury.
DISCUSSION
SIRT1 has long been assumed to be an antiaging protein due to its ortholog Sir2 that prolongs life span in lower organisms (2–4). However, its functions in mammals are incompletely understood. In this study, we have demonstrated that SIRT1 is predominantly localized in the cardiomyocyte nuclei; this finding seems to conflict with previous reports (10) that paid little attention to modifications in SIRT1 caused by sumoylation. We report for the first time that nuclear SIRT1 in cardiomyocytes is predominantly sumoylated. Unlike ubiquitination, which is associated with protein degradation, sumoylation, a covalent modification of proteins with SUMO proteins, is known to regulate substrate translocation (39) stabilization (40) and transcriptional activity (41). Therefore, sumoylation may not only contribute to the nuclear localization of cardiac SIRT1 but also be involved in the regulation of SIRT1's activity. Moreover, the results demonstrated that cardiac overexpression of SENP2 can significantly reduce nuclear sumoylated SIRT1 in the heart. Recently, transgenic SENP2-overexpressing mice have been shown to demonstrate cardiac developmental defects, heart failure, and early death (31).
One of SIRT1's downstream targets, p53, is known to be activated and accumulates with senescence (42), heart failure (42), and myocardial I/R injury (36). Our data showed a significant decline in SIRT1 protein levels in the aged heart, in contrast to findings from earlier studies that reported cardiac SIRT1 protein levels to be elevated in senescence (14). The discrepancy could be due to the fact that investigators up to this point have not distinguished between cytoplasmic (nonsumoylated) and nuclear (sumoylated) forms. In this study, nuclear SIRT1 levels in aged heart are lower than those seen in young heart, and SIRT1 activation during I/R in aging was also blunted, indicating that, in addition to reduced SIRT1 expression, the aged heart cannot mount as robust a SIRT1 response to ischemic stress. Furthermore, we found that during acute ischemia, unlike the young heart, the aged heart failed to sequester SIRT1 into the nucleus, which resulted in an increase in nonsumoylated cytoplasmic SIRT1. These findings revealed that nuclear localization is important for sumoylation and activation of cardiac SIRT1. Aging related increase levels of reactive oxygen species (ROS) may impair the activities of SUMO-1 and desumoylase in the heart (43), which could lead to degradation and impaired nucleocytoplasmic shuttling of SIRT1 in the aged heart. Hsu et al. (44) reported that the cardiac SIRT1 expression levels decline in hearts exposed to I/R. However, we could not confirm this decrease in SIRT1 protein levels during acute I/R. One reason to explain this difference in findings is that our I/R protocol is different from that of Hsu et al., who performed ischemia for 20–30 min followed by 24 h of reperfusion, which is a long-duration I/R protocol that usually results in myocardial necrosis. In contrast our protocol consisted of a 10-min ischemic period followed by 20 min of reperfusion to investigate signaling mechanisms or 20 min ischemia followed by 4 h reperfusion to measure acute myocardial infarction.
As a known mechanism that drives SIRT1 deacetylase activity (45), intracellular NAD+ levels were monitored using an in vivo myocardial I/R model. Differences in NAD+ content between young and aged hearts during sham treatment, ischemia, and I/R conditions mimicked differences seen for SIRT1 activity between the two age groups. These data not only confirmed the SIRT1 findings with respect to aging but also strongly indicate that enzymes regulating intracellular NAD+ levels, such as NAMPT, a rate-limiting enzyme of the NAD+ salvage pathway (12), could be a potential upstream modulator of cardiac SIRT1 activity in the heart. The aged hearts did demonstrate lower protein levels of NAMPT than young hearts (Supplemental Fig. S3), but I/R stress did not change NAMPT levels in eithr young or aged hearts (data not shown); another possibility is that the NAMPT activity might be modulated by I/R stress.
Genetic overexpression of SIRT1 in the heart has been shown to be beneficial for oxidative stress-induced myocardial injury (14, 44). We also showed that a known SIRT1 agonist, resveratrol, significantly reduced infarct size in the aged heart, as well as preserving postischemic cardiac contractility (22). However, it is recognized that resveratrol can activate AMPK besides SIRT1 (46). Therefore, several more specific SIRT1 activators have been synthesized, among all of which SRT1720 demonstrates the highest potency for SIRT1 activation (34), which can increase insulin sensitivity and decrease blood glucose levels in diabetic mice (34). In this study, we discovered that SRT1720 enhanced the interaction between SIRT1 and its downstream target PGC-1α in the heart and consequently decreased acetyl- PGC-1α, which strongly supports the tenet that SRT1720 is a SIRT1 activator in the heart. Furthermore, HL-1 cardiomyocytes were utilized to elucidate the mechanism whereby SRT1720 induces SIRT1 activation, the results indicate that SRT1720 triggers transport of SIRT1 from the cytoplasm into the nucleus, and this translocation is coincident with sumoylation of SIRT1. Based on computational structural analysis of the interaction between SIRT1 and SRT1720 (Supplemental Fig. S4), SRT1720 docking to SIRT1 could lead to exposure of the SIRT1 nuclear localization signal (NLS) domain to the nuclear pore complex (NPC) containing SUMO (47), by which SIRT1 becomes sumoylated and allows nucleocytoplasmic transport in the nuclear pore (47).
Although SRT1720 has been shown to mimic the beneficial effects of caloric restriction in a diabetes animal model (34, 48), the potential role that it plays in limiting myocardial I/R-induced injury is unknown. Here we report that SRT1720 acting as a SIRT1 agonist confers resistance against acute myocardial injury, especially in senescent and genetically engineered SIRT1-deficient hearts. Intriguingly, SRT1720 markedly inhibits apoptosis caused by H/R in HL-1 cardiomyocytes, and the sumoylation inhibitor AA blocks the sumoylation and translocation of SIRT1 triggered by SRT1720 in HL-1 cells, consequently attenuating the antiapoptotic effect of SRT1720 in HL-1cells. These data indicate that SIRT1 sumoylated in the nuclei is critical for its cardioprotection against I/R injury. Therefore, cardiac SIRT1 sumoylation plays an important role in cardioprotection against I/R injury, and pharmacological SIRT1 activation could be a novel therapeutic strategy for ischemia heart disease, particularly in older adults.
Supplementary Material
Acknowledgments
This work was supported by grants awarded by the National Institute on Aging of the U.S. National Institutes of Health (AG 028163; J.L.), the American Heart Association (SDG0835169N and 12GRNT11620029; J.L.), and the American Diabetes Association (Basic Science grant 11-BS-92; J.L.).
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- AA
- anacardic acid
- AAR
- area at risk
- DAPI
- 4′,6-diamidino-2-phenylindole
- H/R
- hypoxia/reoxygenation
- I/R
- ischemia/reperfusion
- LAD
- left anterior descending
- LV
- left ventricle
- MHC
- myosin heavy chain
- NAM
- nicotinamide
- NAMPT
- nicotinamide phosphoribosyltransferase
- PGC-1α
- peroxisome proliferator-activated receptor γ coactivator 1-α
- SDS-PAGE
- sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- SENP2
- sentrin-specific protease 2
- SIRT1
- Sirtuin 1
- SUMO-1
- small ubiquitin-related modifier 1
- TBP
- TATA-binding protein
- TTC
- 2,3,5-triphenyltetrazolium chloride
- TUNEL
- terminal deoxynucleotidyl transferase dUTP nick end labeling
REFERENCES
- 1. Cohen H. Y., Miller C., Bitterman K. J., Wall N. R., Hekking B., Kessler B., Howitz K. T., Gorospe M., de Cabo R., Sinclair D. A. (2004) Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305, 390–392 [DOI] [PubMed] [Google Scholar]
- 2. Kaeberlein M., McVey M., Guarente L. (1999) The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 13, 2570–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tissenbaum H. A., Guarente L. (2001) Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 410, 227–230 [DOI] [PubMed] [Google Scholar]
- 4. Rogina B., Helfand S. L. (2004) Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc. Natl. Acad. Sci. U. S. A. 101, 15998–16003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang R. H., Sengupta K., Li C., Kim H. S., Cao L., Xiao C., Kim S., Xu X., Zheng Y., Chilton B., Jia R., Zheng Z. M., Appella E., Wang X. W., Ried T., Deng C. X. (2008) Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 14, 312–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boily G., He X. H., Pearce B., Jardine K., McBurney M. W. (2009) SirT1-null mice develop tumors at normal rates but are poorly protected by resveratrol. Oncogene 28, 2882–2893 [DOI] [PubMed] [Google Scholar]
- 7. Ota H., Tokunaga E., Chang K., Hikasa M., Iijima K., Eto M., Kozaki K., Akishita M., Ouchi Y., Kaneki M. (2006) Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene 25, 176–185 [DOI] [PubMed] [Google Scholar]
- 8. Cheng H. L., Mostoslavsky R., Saito S., Manis J. P., Gu Y., Patel P., Bronson R., Appella E., Alt F. W., Chua K. F. (2003) Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. U. S. A. 100, 10794–10799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shindler K. S., Ventura E., Rex T. S., Elliott P., Rostami A. (2007) SIRT1 activation confers neuroprotection in experimental optic neuritis. Invest. Ophthalmol. Vis. Sci. 48, 3602–3609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tanno M., Sakamoto J., Miura T., Shimamoto K., Horio Y. (2007) Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 282, 6823–6832 [DOI] [PubMed] [Google Scholar]
- 11. Yang Y., Fu W., Chen J., Olashaw N., Zhang X., Nicosia S. V., Bhalla K., Bai W. (2007) SIRT1 sumoylation regulates its deacetylase activity and cellular response to genotoxic stress. Nat. Cell Biol. 9, 1253–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fulco M., Cen Y., Zhao P., Hoffman E. P., McBurney M. W., Sauve A. A., Sartorelli V. (2008) Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell. 14, 661–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alcendor R. R., Kirshenbaum L. A., Imai S., Vatner S. F., Sadoshima J. (2004) Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ. Res. 95, 971–980 [DOI] [PubMed] [Google Scholar]
- 14. Alcendor R. R., Gao S., Zhai P., Zablocki D., Holle E., Yu X., Tian B., Wagner T., Vatner S. F., Sadoshima J. (2007) Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 100, 1512–1521 [DOI] [PubMed] [Google Scholar]
- 15. Mathers C., Fat D. M., Boerma J. T., eds. (2008) The global burden of disease: 2004 update. World Health Organization, Geneva, Switzerland/Ebrary, Inc., Palo Alto, CA, USA [Google Scholar]
- 16. Azhar G., Gao W., Liu L. X., Wei J. Y. (1999) Ischemia-reperfusion in the adult mouse heart influence of age. Exp. Gerontol. 34, 699–714 [DOI] [PubMed] [Google Scholar]
- 17. Xu F., Gao Z., Zhang J., Rivera C. A., Yin J., Weng J., Ye J. (2010) Lack of SIRT1 (mammalian Sirtuin 1) activity leads to liver steatosis in the SIRT1+/− mice: a role of lipid mobilization and inflammation. Endocrinology 151, 2504–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang J., Schwartz R. J. (2010) Sumoylation and regulation of cardiac gene expression. Circ. Res. 107, 19–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miller E. J., Li J., Leng L., McDonald C., Atsumi T., Bucala R., Young L. H. (2008) Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature 451, 578–582 [DOI] [PubMed] [Google Scholar]
- 20. Wang J., Yang L., Rezaie A. R., Li J. (2011) Activated protein C protects against myocardial ischemic/reperfusion injury through AMP-activated protein kinase signaling. J. Thromb. Haemost. 9, 1308–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morrison A., Yan X., Tong C., Li J. (2011) Acute rosiglitazone treatment is cardioprotective against ischemia-reperfusion injury by modulating AMPK, Akt, and JNK signaling in nondiabetic mice. Am. J. Physiol. Heart Circ. Physiol. 301, H895–H902 [DOI] [PubMed] [Google Scholar]
- 22. Ma H., Wang J., Thomas D. P., Tong C., Leng L., Wang W., Merk M., Zierow S., Bernhagen J., Ren J., Bucala R., Li J. (2010) Impaired macrophage migration inhibitory factor-AMP-activated protein kinase activation and ischemic recovery in the senescent heart. Circulation 122, 282–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin J. F., Lin S. M., Chih C. L., Nien M. W., Su H. H., Hu B. R., Huang S. S., Tsai S. K. (2008) Resveratrol reduces infarct size and improves ventricular function after myocardial ischemia in rats. Life Sci. 83, 313–317 [DOI] [PubMed] [Google Scholar]
- 24. Russell R. R., 3rd, Li J., Coven D. L., Pypaert M., Zechner C., Palmeri M., Giordano F. J., Mu J., Birnbaum M. J., Young L. H. (2004) AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J. Clin. Invest. 114, 495–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang J., Ma H., Tong C., Zhang H., Lawlis G. B., Li Y., Zang M., Ren J., Nijland M. J., Ford S. P., Nathanielsz P. W., Li J. (2010) Overnutrition and maternal obesity in sheep pregnancy alter the JNK-IRS-1 signaling cascades and cardiac function in the fetal heart. FASEB J. 24, 2066–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tong C., Morrison A., Yan X., Zhao P., Yeung E. D., Wang J., Xie J., Li J. (2010) Macrophage migration inhibitory factor deficiency augments cardiac dysfunction in type 1 diabetic murine cardiomyocytes. J. Diabetes. 2, 267–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yeung E. D., Morrison A., Plumeri D., Wang J., Tong C., Yan X., Li J. (2012) Alternol exerts prostate-selective antitumor effects through modulations of the AMPK signaling pathway. Prostate 72, 165–172 [DOI] [PubMed] [Google Scholar]
- 28. Shi X., Leng L., Wang T., Wang W., Du X., Li J., McDonald C., Chen Z., Murphy J. W., Lolis E., Noble P., Knudson W., Bucala R. (2006) CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity 25, 595–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hay R. T. (2005) SUMO: a history of modification. Mol. Cell 18, 1–12 [DOI] [PubMed] [Google Scholar]
- 30. Bayer P., Arndt A., Metzger S., Mahajan R., Melchior F., Jaenicke R., Becker J. (1998) Structure determination of the small ubiquitin-related modifier SUMO-1. J. Mol. Biol. 280, 275–286 [DOI] [PubMed] [Google Scholar]
- 31. Kim E. Y., Chen L., Ma Y., Yu W., Chang J., Moskowitz I. P., Wang J. (2012) Enhanced desumoylation in murine hearts by overexpressed SENP2 leads to congenital heart defects and cardiac dysfunction. J. Mol. Cell. Cardiol. 52, 638–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rodgers J. T., Lerin C., Haas W., Gygi S. P., Spiegelman B. M., Puigserver P. (2005) Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434, 113–118 [DOI] [PubMed] [Google Scholar]
- 33. Canto C., Gerhart-Hines Z., Feige J. N., Lagouge M., Noriega L., Milne J. C., Elliott P. J., Puigserver P., Auwerx J. (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Milne J. C., Lambert P. D., Schenk S., Carney D. P., Smith J. J., Gagne D. J., Jin L., Boss O., Perni R. B., Vu C. B., Bemis J. E., Xie R., Disch J. S., Ng P. Y., Nunes J. J., Lynch A. V., Yang H., Galonek H., Israelian K., Choy W., Iffland A., Lavu S., Medvedik O., Sinclair D. A., Olefsky J. M., Jirousek M. R., Elliott P. J., Westphal C. H. (2007) Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450, 712–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Claycomb W. C., Lanson N. A., Jr., Stallworth B. S., Egeland D. B., Delcarpio J. B., Bahinski A., Izzo N. J., Jr. (1998) HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. U. S. A. 95, 2979–2984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xie Z. L., Koyama T., Abe K., Fujii Y., Sawa H., Nagashima K. (2000) Upregulation of p53 protein in rat heart subjected to a transient occlusion of the coronary artery followed by reperfusion. Jpn. J. Physiol. 50, 159–162 [DOI] [PubMed] [Google Scholar]
- 37. Vaziri H., Dessain S. K., Eagon E. N., Imai S. I., Frye R. A., Pandita T. K., Guarente L., Weinberg R. A. (2001) hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107, 149–159 [DOI] [PubMed] [Google Scholar]
- 38. Fukuda I., Ito A., Hirai G., Nishimura S., Kawasaki H., Saitoh H., Kimura K., Sodeoka M., Yoshida M. (2009) Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem. Biol. 16, 133–140 [DOI] [PubMed] [Google Scholar]
- 39. Stade K., Vogel F., Schwienhorst I., Meusser B., Volkwein C., Nentwig B., Dohmen R. J., Sommer T. (2002) A lack of SUMO conjugation affects cNLS-dependent nuclear protein import in yeast. J. Biol. Chem. 277, 49554–49561 [DOI] [PubMed] [Google Scholar]
- 40. Cai Q., Robertson E. S. (2010) Ubiquitin/SUMO modification regulates VHL protein stability and nucleocytoplasmic localization. PLoS One 5, e12636. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41. Wei F., Scholer H. R., Atchison M. L. (2007) Sumoylation of Oct4 enhances its stability, DNA binding, and transactivation. J. Biol. Chem. 282, 21551–21560 [DOI] [PubMed] [Google Scholar]
- 42. Tyner S. D., Venkatachalam S., Choi J., Jones S., Ghebranious N., Igelmann H., Lu X. B., Soron G., Cooper B., Brayton C., Park S. H., Thompson T., Karsenty G., Bradley A., Donehower L. A. (2002) p53 mutant mice that display early ageing-associated phenotypes. Nature 415, 45–53 [DOI] [PubMed] [Google Scholar]
- 43. Li F., Zhang L., Craddock J., Bruce-Keller A. J., Dasuri K., Nguyen A., Keller J. N. (2008) Aging and dietary restriction effects on ubiquitination, sumoylation, and the proteasome in the heart. Mech. Ageing Dev. 129, 515–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hsu C. P., Zhai P., Yamamoto T., Maejima Y., Matsushima S., Hariharan N., Shao D., Takagi H., Oka S., Sadoshima J. (2010) Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation 122, 2170–2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang T. L., Sauve A. A. (2006) NAD metabolism and sirtuins: Metabolic regulation of protein deacetylation in stress and toxicity. AAPS J. 8, E632–E643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dasgupta B., Milbrandt J. (2007) Resveratrol stimulates AMP kinase activity in neurons. Proc. Natl. Acad. Sci. U. S. A. 104, 7217–7222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang H., Saitoh H., Matunis M. J. (2002) Enzymes of the SUMO modification pathway localize to filaments of the nuclear pore complex. Mol. Cell. Biol. 22, 6498–6508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Smith J. J., Kenney R. D., Gagne D. J., Frushour B. P., Ladd W., Galonek H. L., Israelian K., Song J., Razvadauskaite G., Lynch A. V., Carney D. P., Johnson R. J., Lavu S., Iffland A., Elliott P. J., Lambert P. D., Elliston K. O., Jirousek M. R., Milne J. C., Boss O. (2009) Small molecule activators of SIRT1 replicate signaling pathways triggered by calorie restriction in vivo. BMC Syst. Biol. 3, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.