

Abstract
Matrix metalloproteinase (MMP)-13 is one of the mammalian collagenases that play key roles in tissue remodelling and repair and in progression of diseases such as cancer, arthritis, atherosclerosis, and aneurysm. For collagenase to cleave triple helical collagens, the triple helical structure has to be locally unwound before hydrolysis, but this process is not well understood. We report crystal structures of catalytically inactive full-length human MMP-13(E223A) in complex with peptides of 14–26 aa derived from the cleaved prodomain during activation. Peptides are bound to the active site of the enzyme by forming an extended β-strand with Glu40 or Tyr46 inserted into the S1′ specificity pocket. The structure of the N-terminal part of the peptides is variable and interacts with different parts of the catalytic domain. Those areas are designated substrate-dependent exosites, in that they accommodate different peptide structures, whereas the precise positioning of the substrate backbone is maintained in the active site. These modes of peptide-MMP-13 interactions have led us to propose how triple helical collagen strands fit into the active site cleft of the collagenase.—Stura, E. A., Visse, R., Cuniasse, P., Dive, V., Nagase, H. Crystal structure of full-length human collagenase 3 (MMP-13) with peptides in the active site defines exosites in the catalytic domain.
Keywords: matrix metalloproteinases, collagen/substrate binding, X-ray crystallography, extracellular matrix
Matrix metalloproteinases (MMPs) play key roles in the turnover of numerous extracellular matrix molecules in embryonic development, morphogenesis, tissue remodelling, and tissue repair (1). Aberrant extracellular matrix turnover is associated with many diseases, such as arthritis, cancer, aneurysm, nephritis, tissue ulcers, and fibrosis (2, 3). Twenty-three MMPs are expressed in humans, and they share the metzincin signature sequence HEXGHXXGXXH and the conserved Met turn (4). MMPs are further classified according to their structural and functional properties into collagenases, gelatinases, stromelysins, membrane-bound MMPs, and others (5). MMP-1 (collagenase 1), MMP-8 (neutrophil collagenase), and MMP-13 (collagenase 3) belong to the collagenase subgroup. They cleave fibrillar collagen types I, II, and III at a specific locus and generate ¾ and ¼ fragments. The archetypal collagenase consists of a catalytic (Cat) domain, a linker region, and a hemopexin (Hpx) domain (6). It has long been established that to efficiently cleave triple helical collagen but not other substrates, collagenases must possess both the Cat and Hpx domains (7). The two domains together form a collagen-binding site that is unique to collagenases (8), and it has been shown that MMP-1 locally alters the structure of collagen on binding (9), whereas Stultz and coworkers (10, 11) concluded that collagenases bind a cleavable vulnerable state in collagen. The recent structural studies of MMP-1 in complex with a triple helical collagen peptide showed insight into the role of the Hpx domain in collagenolysis (12, 13). They showed extensive interaction of the Cat and Hpx domains with different strands of the triple helical collagen, but the structure did not reveal the details as to how a single peptide of the triple helix inserts into the active site cleft (12).
MMP-13 is not expressed in most adult tissues (14); it is induced in stromal cells in cancer and is associated with invasive and metastatic potential (15). MMP-13 is involved in cartilage collagen destruction in osteoarthritis and rheumatoid arthritis (16, 17), making MMP-13 a target for selective inhibition, but only limited information is available on the substrate binding; Bode et al. (18) showed the binding of a 3-aa peptide-based inhibitor to the nonprimed side of the MMP-8 active site cleft, and Bertini et al. (19) solved the structure of MMP-12 with 3-aa cleaved peptide products. We set out to crystallize full-length MMP-13 with single-stranded peptides containing the collagenase cleavage sequence and a phosphinic moiety (PO2 CH2) to mimic the transition state structure (20). So far, we have been unsuccessful in obtaining cocrystals of these peptides with the full-length enzyme, but we serendipitously found that the catalytically inactive MMP-13(E223A) mutant (numbering after preproMMP-13 sequence) spontaneously cocrystallized with peptides of 14–26 aa generated from the prodomain during the activation process. The structure revealed an extended peptide substrate in the active site. This study revealed exosites where the N-terminal section of the peptides interacts with the Cat domain. The nature of the interactions in this region seems adaptable, as the bound peptides can make different interactions, depending on their local conformation and environment. The mode of binding of the peptide in the active site cleft has provided a basis to postulate how triple helical collagen fits into the active site cleft of collagenases.
CH2) to mimic the transition state structure (20). So far, we have been unsuccessful in obtaining cocrystals of these peptides with the full-length enzyme, but we serendipitously found that the catalytically inactive MMP-13(E223A) mutant (numbering after preproMMP-13 sequence) spontaneously cocrystallized with peptides of 14–26 aa generated from the prodomain during the activation process. The structure revealed an extended peptide substrate in the active site. This study revealed exosites where the N-terminal section of the peptides interacts with the Cat domain. The nature of the interactions in this region seems adaptable, as the bound peptides can make different interactions, depending on their local conformation and environment. The mode of binding of the peptide in the active site cleft has provided a basis to postulate how triple helical collagen fits into the active site cleft of collagenases.
MATERIALS AND METHODS
MMP-13(E223A) expression and purification
ProMMP-13(E223A) was expressed, refolded, and purified as described for proMMP-1 (21). ProMMP13(E223A) was activated with 4-aminophenylmercuric acetate and the MMP-3 Cat domain, as described elsewhere (9), and MMP-13(E223A) was purified on an S200 Sephadex column (GE Healthcare, Little Chalfont, UK) in 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10 mM CaCl2, and 0.02% NaN3 (TNC buffer). After concentration to ∼1 mg/ml, the samples were stored at 4°C.
Crystal preparation
Crystals of various morphologies formed in the protein solution. Stability screening of the crystals by sitting-drop vapor diffusion at 20°C showed good tolerance for high-polyethylene glycol (PEG) concentrations. A syringe connected to a capillary was used to transfer several crystals to a sitting-drop plate, at room temperature (22).
Cryoprotectant vs. diffraction screening
The crystals were directly transferred to a cryoprotectant solution or preequilibrated in PEG solutions. For initial cryoprotectant screening, the crystals were soaked for 20 s in multicomponent cryosolutions (23): solution A [20% monomethyl PEG 2000 (MPEG-2K), 5% MPEG 550, 15% glycerol, and 200 mM imidazole malate, pH 6.5) or solution B (20% MPEG-2K, 10% MPEG 550, 10% ethylene glycol, and 90 mM imidazole malate, pH 6.0). The crystal was picked up with a LithoLoop (Molecular Dimensions, Newmarket, UK), mounted on Structural Proteomics in Europe–compatible standard caps, and plunged into liquid nitrogen. Only 1 crystal transferred directly from the Eppendorf tube to cryoprotectant solution B gave useful diffraction at the European Synchrotron Radiation Facility (ESRF) Beamline ID14-4 (Grenoble, France) at 100 K. For further screening, the crystals were transferred directly to the cryoprotectant solutions. Four solutions were tested: solution C [10% PEG-10K, 200 mM NaCl, 10% diethylene glycol, 10% 1,2-propanediol, 10% glycerol, and 10% 8:2 PCTP (propionic acid, cacodylate, bis-tris propane) buffer]; solution D [same contents as solution C, but with 9:1 AAB (sodium acetate, ADA [N-(2-acetamido)iminodiacetic acid], and bicine) instead of PCTP]; solution E (35% MPEG-5K, 15% PEG 400, and 15% 9:1 AAB); and solution F (12% PEG-10K, 4.2% MPEG-2K, 25% glycerol, and 25 mM NaMES, pH 5.5). AAB and PCTP are part of the Really Useful Buffer Kit (Molecular Dimensions Ltd, Newmarket, UK; ref. 24). 8:2 PCTP consists of 80% sodium propionate, sodium cacodylate, and bis-tris propane at pH 4 and 20% of the same mixture at pH 9.5; and 9:1 AAB consists of 90% sodium acetate, ADA, and bicine at pH 4 and 10% of the same mixture at pH 9.0. Cryoprotectant C gave the best results, followed by cryoprotectant D. Two more crystals were tested later, in which the concentration of 1,2-propanediol was increased with respect to the other components: 10% PEG-10K, 200 mM NaCl, 5% diethylene glycol, 20% 1,2-propanediol, 5% glycerol, and 10% 8:2 PCTP. Both crystals diffracted well.
Data collection, structure determination, and refinement
Complete data sets were collected to a 2.7-Å resolution, but were useful to only 2.85 Å for the first crystal and to 2.44 Å for the second; the third crystal diffracted to 2.6 Å. Intensity integration was done with MOSFLM (25) and XDS (26). Data reduction with SCALA (CCP4) showed that the first two crystals belong to the orthorhombic space group C2221, with two molecules in the asymmetric unit, whereas the third merged with acceptable statistics only in P21, although the packing is similar to that of the other two. To solve the first structure, separate Cat (2OW9; ref. 27) and Hpx (1PEX; ref. 28) domains were used for molecular replacement with MOLREP (29); rebuilding was carried out with XtalView/Xfit (30) and REFMAC for refinement (31). The second structure was solved by rigid body refinement with phenix. refine (32) and rebuilt with COOT (33). Phaser_MR (34) was used to solve the P21 molecular replacement problem with REFMAC for refinement (31) and rebuilding with COOT (33). The refinement statistics are given in Table 1 for data reprocessed with more recent versions of XDS, REFMAC, and phenix. refine. The figures were made using PyMOL (35).
Table 1.
Crystallization conditions and data collection statistics
| Statistic | PDB code |
||
|---|---|---|---|
| 4FU4 | 4FVL | 4G0D | |
| Data collection | |||
| Beamline | ESRF-ID14-4 | Soleil-Proxima 1 | ESRF-ID23-2 |
| Cryoprotectant | 20% MPEG-2K, 10% MPEG 550, 10% ethylene glycol, and 90 mM imidazole malate (pH 6.0) | 10% PEG-10K, 10% diethylene glycol, 10% 1.2-propanediol, 10% glycerol, 200 mM NaCl, and 10% PCTP (8:2 ratio) | 10% PEG-10K, 5% diethylene glycol, 20% 1.2-propanediol, 5% glycerol, 200 mM NaCl, and 10% PCTP (8:2 ratio) |
| Space group | C2221 | C2221 | P21 |
| Cell parameters (Å) | 127.1, 156.6, 106.1 | 125.8, 157.3, 105.6 | 101.3, 105.9, 101.2; β = 102.1 |
| Wavelength (Å) | 0.9395 | 0.9395 | 0.9395 |
| Mol/asym | 2 | 2 | 4 |
| VM (Å3/Da) | 2.91 | 2.91 | 2.92 |
| Resolution range (Å) | 50.−2.85 (3.02–2.85) | 50.−2.44 (2.58–2.44) | 50.−2.54 (2.69–2.54) |
| Reflections observed | 190,162 | 220,219 | 278,883 |
| Unique reflections | 23,888 | 37,803 | 68,882 |
| Completeness (%) | 99.8 (99.0) | 95.7 (96.6) | 99.7 (99.4) |
| I/〈σ(I)〉 | 8.81 | 11.2 | 8.65 |
| Rfactor/Rpim | 0.17 | 0.15 | 0.22 |
| Redundancy | 7.78 (8.23) | 5.82 (5.69) | 4.05 (4.01) |
| Structure | |||
| Resolution shells (Å) | 49.34–2.85 | 49.11–2.44 | 49.5–2.54 |
| Average B factor (Å2) | 39.3 | 33.1 | 28.9 |
| Rcryst/Rfree (%) | 17.8/24.7 | 17.1/22.4 | 16.9/24.1 |
| Favored regions (%) | 93.5 | 96.6 | 96.2 |
| Allowed (%) | 5.9 | 3.1 | 3.6 |
Crystallization was induced by prodomain peptide fragments after concentration of MMP-13(E223A) to 1 mg/ml and stored in 150 mM NaCl, 10 mM CaCl2, 0.02% sodium azide, and 50 mM Tris-HCl (pH 7.5) at 4°C. Data were collected with synchrotron radiation at 100 K. Structure was solved by molecular replacement. Rfactor = ∑hkl (|Ihkl ∑i)) − Ihkl]/(∑(Ihkl ∑i)) (ref. 26). Rpim = (1/N − 1)1/2 ∑hkl ∑I|Ii ∑ 〈I〉| ∑hkl ∑i Ii (ref. 25). Rcryst = ∑hkl ∥Fobs| − k|Fcalc∥/∑hkl |Fobs|. Rfree was calculated from 5% of the data excluded from refinement.
Analytical gel filtration and analytical ultracentrifugation
MMP-13(E223A) (200 μl of 1 μM) in TNC buffer was run on a Superdex 75 HR 10/30 column (GE Healthcare) at 0.8 ml/min. The column was calibrated with albumin, ovalbumin, chymotrypsinogen, and ribonuclease, using Stokes radii as published (36). For the analytical ultracentrifugation (AUC), MMP-13(E223A) was dialyzed against TNC buffer and run on a Proteome lab XL-I analytical ultracentrifuge with an AnTi50 rotor (Beckman Coulter, Brea, CA, USA) at 50,000 rpm at the AUC facility of University College London (London, UK). A280 absorption spectra were processed with SEDFIT (37) to calculate the sedimentation coefficient and molecular weight. The theoretical Stokes radius and the sedimentation coefficient were calculated by HYDROPRO (38).
Protein Data Bank accession numbers
The coordinates and structure factors for the MMP-13(E223A)-peptide complexes have been deposited in the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank with ID codes 4FU4 (C2221), 4FVL (C2221), and 4G0D (P21).
RESULTS
Overall structure
Three datasets describe the structure of full-length human MMP-13(E223A). Unaccounted for electron density stretching across the catalytic site was identified as a prodomain-derived peptide. (Table 1 and Figs. 1 and 3, numbering based on the preproMMP-13 sequence). MMP-13(E223A) consists of a Cat domain, a linker region, and an Hpx domain. In the crystal packing, MMP-13(E223A) is assembled as a dimer, mainly through Hpx blade 4 interactions; two dimers form a tetrameric unit. In the orthorhombic space group C2221, with two molecules in the asymmetric unit, the interface between two dimers is a perfect 2-fold. This symmetry is broken in the P21 form, hence the 4 molecules in the asymmetric unit. The molecules in the dimeric unit were designated A and B type on account of differences in the peptide conformation and the crystal-packing environment. The peptides bound in the active site cleft contribute to crystal-packing interactions and are the likely explanation for the spontaneous crystallization.
Figure 1.

The asymmetric unit of MMP-13 is a dimer, but it is a monomer in solution. A) The asymmetric unit of 4FU4 (C2221), with 2 molecules in the asymmetric unit designated A type (blue) and B type (green). Bound peptides, purple; Zn2+, pink sphere; Ca2+, green spheres; and Cl−, orange spheres. B) The asymmetric unit of 4G0D (P21) with 2 dimers. Molecules A (dark blue) and C (light blue) are A type; B (dark green) and D (light green) are B-type. C) Superdex 75 tracing of MMP-13(E223A) and MMP-1(E219A). Inset: calibration of the gel filtration column. D) Sedimentation velocity of 0.6 mg/ml MMP-13(E223A) at 20°C.
Figure 3.

Peptides bound to MMP-13 are prodomain derived. A) Cartoon representation of the peptides in A- and B-type molecules. MMP-13(E223A) is shown as a gray cartoon with Zn2+ as purple spheres. A-type molecules: peptide a (red), peptide c (green), peptide w (orange), and peptide y (blue). B-type molecules: peptide b (red), peptide d (green), peptide x (orange); peptide z (blue). Dashed lines: parts of the peptides that contribute to crystal packing. B) Schematic of the active site peptides. Sequence shows the MMP-13 prodomain with predicted helix 1. The 8 peptides are shown schematically, with the S1′-bound residue in bold and as a solid circle. C) Superimposed prodomains of MMP-2 (dark gray) showing helices 1–3 (H1–H3) in walleye stereo. The zymogen-maintaining cysteine (Cys) is indicated. Representative A- and B-type peptides are superimposed on H1. Active site cleft binding β-strand section with the P1′ Tyr residue is indicated; arrows indicate cleavage sites.
MMP-13 is monomeric in solution
The interface of the MMP-13(E223A) dimers in the crystal is located between the two Hpx domains, similar to that observed in MMP-9 (39) and the crystallographic interface seen in MMP-2 (40). When the MMP-13 Hpx domain was crystallized alone, this interface was not observed (28). Analytical gel filtration indicated that MMP-13(E223A) has a Stokes radius of RS = 2.93 nm, eluting slightly earlier than MMP-1(E219A) with a Stokes radius of RS = 2.66 nm (Fig. 1C). Analytical ultracentrifugation showed that MMP-13(E223A) was monodisperse, with a molecular mass of 43.1 kDa (Fig. 1D). The theoretical Stokes radii calculated by HYDROPRO (38) for MMP-13(E223A) were, as a monomer with a peptide in the active site cleft, RS = 3.14 nm; without a peptide, RS = 2.99 nm; and as a dimer with peptides, RS = 4.157 nm, or without peptides, RS = 4.02 nm. These data indicate that MMP-13 is a monomer in solution.
Cat, linker, and Hpx domains
The Cat domain of full-length MMP-13(E223A) superimposes well on available MMP-13Cat structures (see ref. 41 for a review). In the absence of Glu223, a water and a Cl− ion are found coordinating the active site zinc. A difference from most MMP-13Cat structures is that the N-terminal Tyr104 makes a salt bridge with Asp257 of helix 3, like those found for MMP-1 (6, 42) and MMP-8 (43). This salt bridge is considered essential for full collagenase activity (44). The Cat domain accommodates long peptides without major conformational changes.
The MMP-13 linker region, Gly267-Lys283, is 16 residues connecting the Cat and Hpx domains that share a small interface (313 Å2). The Hpx domain structure forms a flat disc consisting of 4 blades and is similar to the structure of the MMP-13 Hpx crystallized alone (28). The S10′ pocket (Schechter and Berger subsite nomenclature; ref. 45), which binds the P10′ Leu residue of a triple helical collagen peptide identified in MMP-1 (12), is conserved in the MMP-13 Hpx domain and could be involved in collagen binding. Superimposing the 8 MMP-13(E223A) structures on their Cat domain shows a very good fit (Fig. 2A). Because of crystal packing, the Hpx domain in B-type molecules is slightly rotated compared with the A type.
Figure 2.
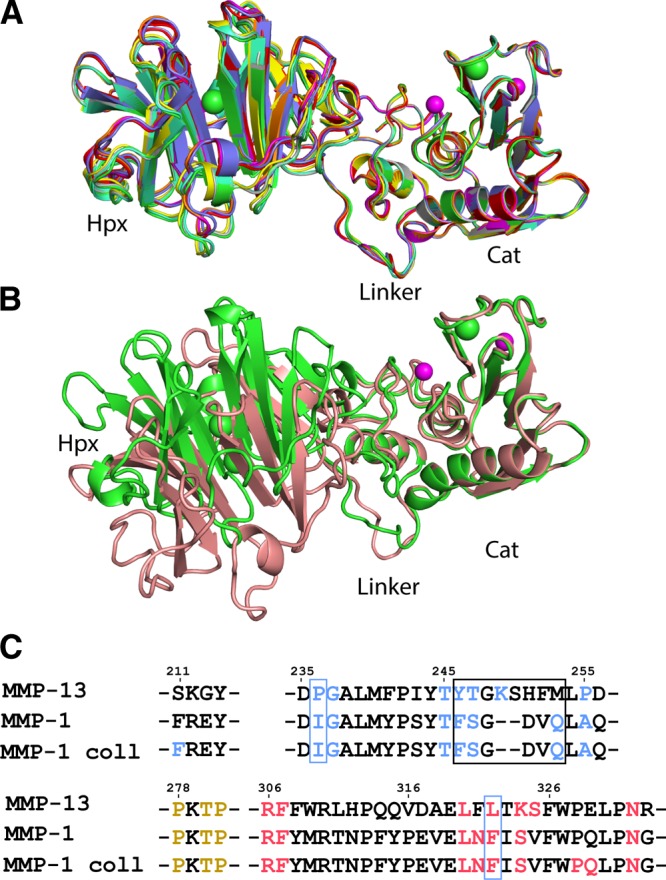
Superimposition of MMP-13(E223A) and MMP-1 structures. A) Superimposition of the 8 MMP-13 structures on their Cat domains: 4FU4-A, red; 4FU4-B, green; 4FVL-A, blue; 4FVL-B, yellow; 4G0D-A, purple; 4G0D-B, cyan; 4G0D-C, orange; and 4G0D-D, gray. Zn2+, pink sphere; Ca2+, green spheres. B) MMP-1(E219A) (2CLT; pink) superimposed on the Cat domain of MMP-13(E223A) (green). Root mean square deviation (RMSD) = 0.57 Å2 on all atoms. Zn2+, purple spheres; Ca2+, green spheres. C) Sequence alignment of MMP-13 and MMP-1 and MMP-1–collagen complex (4AUO), highlighting residues of the Cat-Hpx interface. Cat domain residues, blue; linker, orange; and Hpx domain residues, red. Black box, residues not conserved between MMP-1 and MMP-13; blue box, residues that contribute to the different Cat-Hpx conformation between MMP-1 and MMP-13.
Structural comparison with other full-length MMPs
Superimposing human full-length MMP-1(E219A) (42) and MMP-13(E223A) on their Cat domains shows a different Cat-Hpx conformation (Fig. 2B); MMP-13 Hpx is rotated by ∼30° and translated. Most distal, in blade 3, the relative displacement is 11.7 Å. The N-terminal section of the MMP-13 linker is in a position different from that in MMP-1 (Fig. 2B), but the C-terminal 7 linker residues superimpose well; therefore, the linker region is not causal of the conformational difference. Rather, it is due to the Cat-Hpx interface. Conserved hydrophobic residues form the core of the interface with Arg306 as a pivot point (Fig. 2C). The Tyr246-Met253 loop of MMP-13 is not conserved in MMP-1. The residues considered responsible for the different Cat-Hpx conformation of MMP-13 are Pro236/Leu322, which are less bulky than the corresponding Ile and Phe in MMP-1. Because of this, the MMP-13 Cat and Hpx domains move closer together than in MMP-1, changing their relative orientation. In collagen-bound MMP-1 (12), additional Cat and Hpx contacts are made (Fig. 2C), indicating that the binding of collagen has slightly changed the Cat-Hpx domain configuration. The recently discovered S10′ pocket, important in MMP-1 collagen binding (12), is therefore in a different position in MMP-13, with one edge of the pocket Arg297 moved 7.3 Å relative to MMP-1. The Pro/Leu pair is also found in the Cat-Hpx interface of MMP-2, and its Cat-Hpx configuration is almost identical to MMP-13.
Peptides bound to MMP-13
The bound peptides were derived from the N-terminal part of the propeptide, but with different lengths ending with Thr50. These are assigned a, b, c, d, w, x, y, and z and bind in an extended conformation (A type) or partly at an angle to the active site cleft (B type) (Fig. 3A, B). These differences most likely result from crystal packing. The longest resolved peptides (a, b, and x) are 26 aa long (G25GDEDDLSEEDLQFAERYLRSYYHPT50). The electron density in the N-terminal region of peptides is less clear, suggesting that a single peptide species is present. These peptides were generated during the activation of proMMP-13(E223A) by MMP-3Cat in the presence of 4-aminophenylmercuric acetate and must have been copurified. Six peptides have an N-terminal α-helix (Fig. 3A, B; b, d, w, x, y, and z), and two have a random coil with an extended structure (a and c). The propensity of this sequence to form an α helix is most likely due to its prodomain origin. All MMP prodomain structures solved so far consist of 3 α helices connected through loops (40, 46–48), and MMP-13 is likely to have a similar prodomain structure. The peptides include the predicted first helix (D35LQFAERYLRSYY47) of the MMP-13 prodomain and show alignment with the N-terminal section of helix 1 of the MMP-1 and MMP-2 prodomains (Fig. 3C). However, only a part of this sequence forms an α helix when bound to MMP-13; in the active site cleft, it forms a β strand (Fig. 3B, C).
The MMP-13 active site cleft
The β strand of the peptide in the active site cleft is a continuation of the Cat domain β sheet, forming hydrogen bonds with antiparallel β-strand IV and Pro242 and Tyr244 of the S1′ pocket wall (Fig. 4A, B). These backbone contacts are identical in all 8 peptides and delineate the interaction with primed and nonprimed substrate-binding subsites S4 to S3′. The S1′ pocket is considered to be an important component in substrate specificity. With the exception of peptide a, which has Glu40, all peptides have Tyr46 in the S1′ pocket (Fig. 4C, D). Glu40 is not usually found as a P1′ residue, but it coordinates a water and makes indirect hydrogen bonds with the carbonyl groups of Leu239, Phe241, and Ile243, the latter two are part of the S1′ pocket entrance (Fig. 4C). In the S1′ pocket, the phenyl ring of Tyr46 is stacked against the His222 imidazole ring, its hydroxyl group coordinates two waters. One of these makes indirect hydrogen bonds with the carbonyls of Leu239 and Phe241, the other with the carbonyl of Leu218 (Fig. 4D).
Figure 4.

Peptide binding the MMP-13 active site cleft subsites. A) Peptide d as a representative example (purple sticks and labels), Cat domain residues with at least 1 atom within 4 Å (gray sticks and labels), and hydrogen bonds between the peptide substrate and MMP-13(E223A) Cat domain (broken orange sticks). B) Schematic representation of the interactions shown in A. C) Stereo close-up of the S1′ pocket with E40 of peptide a. Water (small green spheres) and MMP-13 surface within 4 Å of the peptide (green shading). D) S1′ pocket with Y46 bound in stereo (peptide d).
The S2′ site is defined by the P2′ Tyr side chain, adopting two alternative positions, suggesting that the pocket can accommodate even larger residues. It makes a hydrophobic crystallographic contact with Trp470. The P3′ His side chain points down into the active site cleft. The S3′ pocket is formed by Leu185, contacting the peptide backbone and the Tyr214 and Tyr244 side chains. The P3′ side chain's most distal part is solvent exposed. The C-terminal backbone conformation beyond P3′ may be influenced by crystal packing.
On the nonprimed side, His226 and Phe107 contribute to the S2 subsite. Asp231, also part of this subsite, makes a salt bridge with the P2 arginine side chain (in all but peptide a). The S3 subsite is mainly hydrophobic, formed by Tyr176, His187, and Phe189. MMP-13 has a strong preference for Pro in P3 (49), which correlates with the collagen sequence cleaved. The structures reveal that other amino acids can be accommodated in the S3 pocket without affecting the active site cleft H-bonding network. The side chains of P4 residues are in slightly different positions, but they are close enough to designate S4, with hydrophobic interactions provided by Phe107 and Pro108, Leu111 and Pro190.
The P1 carbonyl coordinates to the catalytic site zinc (Fig. 4A, B). In some of the structures, a water molecule is present (Fig. 4D, top left), but MMP-13(E223A) lacks Glu223 to polarize the water molecule needed to attack the P1-P1′ peptide bond.
Exosites in the Cat domain
While the structure of the P4-P3′ regions of substrate is constrained by its interaction with the active site cleft, outside the active cleft, the substrate binding subsites of the enzyme vary with the peptide conformations. Six peptides have an N-terminal α helix; the remaining two are random coil. The A-type peptides have an extended conformation, whereas in the B-type peptides, the α helix is angled out from the active site cleft. Hydrophobic residues are positioned on 1 face of the α helix (−L36QFAERYLRS∼Y; hydrophobic residues in bold, helix underscored, ∼ indicates, potential cleavage site), and those make the main contacts with MMP-13 and stabilize the helix (Fig. 5A, B). The lack of Leu36 may explain why peptide c does not form a helix. Peptide a has a shifted binding mode and does not have similarly spaced hydrophobic residues (−D30LSEEDLQFA∼E).
Figure 5.
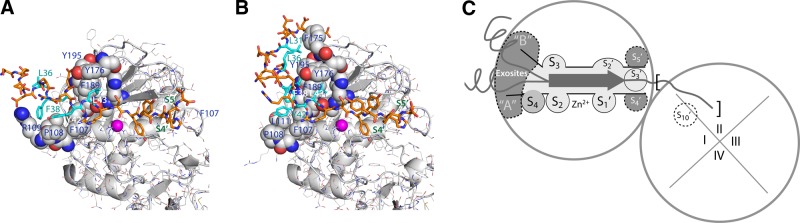
Exosites in the MMP-13 Cat domain defined by peptide binding. A) N-terminal region of peptide w, a representative A type (orange sticks with hydrophobic residues in cyan). Interacting residues on the Cat domain are represented by van der Waals spheres. B) Peptide b, a representative B-type peptide. Color scheme as in A. C) Exosites in MMP-13. The main exosites (dark gray) are the extended entrance to the active site. In the active site, the S4–S3′ subsites are each labeled.
The A- and B-type peptides share contacts with several MMP-13(E223A) residues (Leu111, Phe189, Pro190, Phe175, Tyr176, and Tyr195; Fig. 5), which define the exosites. S4 is directly adjacent to them and could be functionally part of them. The A- and B-type peptides make additional contacts unique to each type (Fig. 5A, B), but whether they are peptide specific or crystal-packing dependent cannot be distinguished. C-terminal minor interactions are observed with sites equivalent to S4′ (Ile234) and S5′ (Ser182). Nevertheless, all these interactions contribute to the overall free energy of peptide binding. Such interactions between the bound peptide and the exosites seem to be rather adaptable and adjust depending on the local structure and amino acid composition of the substrate. There is another interesting example of an interaction in this area, the AB-loop of tissue inhibitor of metalloproteinases 2 (TIMP-2) binds the same part of the MMP-13 exosite, as does the B-type peptide α helix (ref. 50 and Fig. 6A). The F175YP exosite loop, which is not conserved in MMP-1 or MMP-8 (the loop has the sequence NSP instead), seems structurally adaptable to its environment, with Phe175 solvent exposed and found as different rotamers, depending on the bound peptides. This finding suggests that the mode of substrate interaction can be influenced by the type of substrate, and it could be important, especially for larger substrates, such as collagen at the entrance to the active site cleft of MMP-13.
Figure 6.
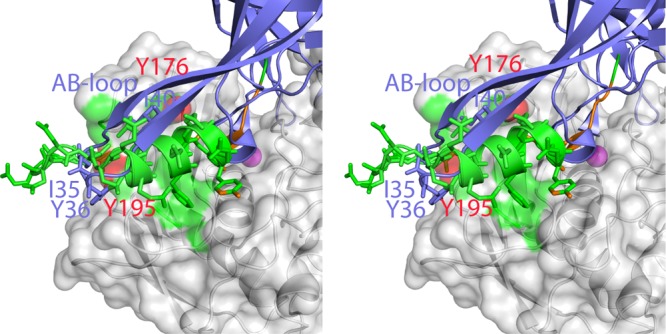
Exosite of MMP13 interacts with TIMP-2. B-type peptides interact with the same area as the TIMP-2 AB loop in MMP-13 (2E2D). B-type peptide contacts (green) and contacts shared with TIMP-2 (red). (Phe175 of MMP-13 is in a different conformation in MMP-13-TIMP-2 and is not highlighted.) TIMP-2 structure (blue).
Implication for collagenolysis
The recently reported structure of MMP-1 with a collagen peptide revealed the binding mode of collagen to MMP-1 (12). We predicted that collagen would interact with the Cat domain of MMP-13 in a similar manner. To compare the putative collagen interaction with the peptides bound in the active site and exosites of MMP-13, we superimposed the MMP-1–collagen productive model (12) on the MMP-13 Cat domain. In this configuration the “trailing” strand in the productive complex model is positioned closest to the active site of MMP-13 and the bound peptide (Fig. 7A, B; red strand). The middle strand also makes major contacts with the Cat domain and binds to the top part of the substrate-binding cleft (Fig. 7A, B; green strand), illustrating that the N-terminal section of these strands occupy the same area as the A- and B-type peptide exosites (Fig. 7A, B; orange strand) and that several residues are involved in both single peptide binding and collagen peptide binding (Fig. 7C). This observation suggests that triple helical collagen behaves as a single substrate in a broader context near the active site (Fig. 7C).
Figure 7.
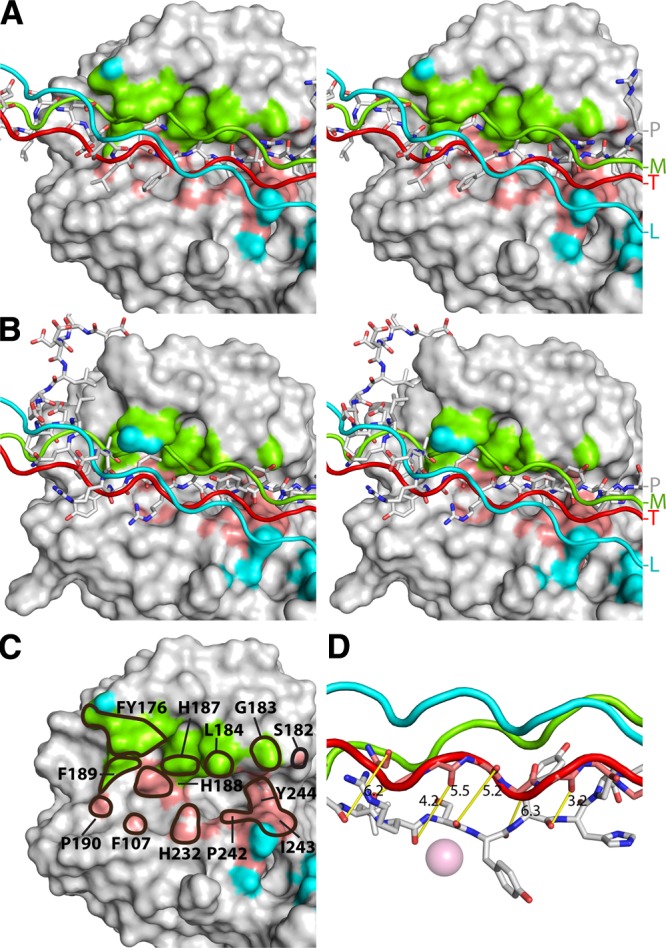
Comparison of MMP-13 peptide contacts and potential collagen contacts. A) The productive model of the MMP-1–collagen complex (12) is superimposed on the MMP-13 Cat domain. MMP-13 is shown as a Connolly surface and the peptides as sticks. Collagen leading strand contacts (cyan), middle strand contacts (green), and trailing strand contacts (red). B) MMP-13 with peptide b and superimposed collagen as in A) C) Cat domain showing the collagen contacts (color scheme as in A). Residues also involved in peptide contacts (solid outline), with the residue numbers. D) The distance between equivalent collagen Cα and peptide Cα (yellow rods). Representative peptide as bound in the MMP-13(E223A) active site, with Y46 in the S1′ pocket. The superimposed collagen structure, with the protein backbone of the strand that will be positioned in the active site (gray sticks).
The 8 peptides show very good superimposition within the active site cleft, from S4 to S3′, but much less outside the active site cleft. Thus, most substrates, including collagen, are likely to adopt a similar backbone conformation between S4 and S3′, just before the P1-P1′ peptide bond is cleaved. This implies that “looping out” of at least 6–7 residues of the trailing strand of the collagen triple helix is needed for the correct placement of the strand to be cleaved. In our superimposition model of peptides, the backbone of the collagen strand to be cleaved has to move from 3 to 6 Å into the active site cleft with relatively moderate changes in the backbone angles (Fig. 7D). If the central 6–7 residues fit the active site, then the predicted interaction of collagen with the exosites is likely to be with an unfolded triple helix. The funnel shape of the entrance to the active site cleft would be wide enough to accommodate such interaction.
DISCUSSION
MMPs require extended residues on both N- and C-terminal sides of the scissile bond for efficient hydrolysis (51), but only limited structural information is available on how MMPs interact with peptide substrates at their active sites. Structurally characterized peptides that bind to MMP active sites are the cysteine switch regions of the MMP propeptides (40, 46–48) and the MMP-2-selective inhibitory peptide derived from β-amyloid precursor protein; the latter defines the S5′–S3′ subsites of MMP-2 (52). However, the orientation of these peptides in the active site is opposite to that of a peptide substrate, indicating their function as an inhibitor. Grams et al. (53) modeled a peptide substrate in MMP-8 (collagenase 2), based on thiol- and hydroxamate-containing peptidic inhibitors that bind to nonprimed and primed binding sites, respectively. Later, Bertini et al. (19) solved the structure of MMP-8 and MMP-12 with 3-aa cleaved peptide products. Those have shown indirectly how MMPs can accommodate an uncleaved peptide substrate within the S3 to S3′ subsites, along with backbone hydrogen bond interactions. The 14- to 26-residue peptides bound to MMP-13(E223A), described herein, clearly show the subsites S4 to S3′ and the backbone hydrogen contacts, which are identical to those modeled with the PLG∼FAG peptide in MMP-8 (53) (Fig. 4A, B). Of note, the N terminus of the “cleaved” peptide MMP-12 structure is pulled more into the active site cleft, because of a direct interaction with the active site glutamic acid, suggesting a conformational rearrangement upon cleavage (19). The present study revealed extended exosites on the nonprimed side in the metalloproteinase, which shows its versatility in accepting variable structures.
MMP-13 has a preference for Gly in P4, a strong preference for Pro in P3, and not a clear preference in P2, but P1 favors Gly. The S1′ pocket prefers hydrophobic residues including Leu, Ile, Tyr, and Phe; the P2′ position Arg; and the P3′ Gly and Ala (54) (Peptidase Database, Wellcome Trust Sanger Institute, Cambridge UK; http://merops.sanger.ac.uk). The peptides in the active site of MMP-13(E223A) have Tyr or Glu in the S1′ pocket. The YLRS∼YYH sequence may not be an optimal sequence, but the exosites likely contribute to the binding energy of the substrate and residues, which would be considered unfavorable in a short peptide substrate may thus be brought to the active site in extended substrates. An interesting finding is that P3′ His makes a hydrogen-bond to Tyr214, a residue shown to be important in collagenase activity (21). Studies with MMP-12 demonstrated distal exosites for elastin binding that affect kcat and Km (54). Exosites are particularly relevant for larger extracellular matrix substrates, like collagen and elastin, which are shown to interact with the enzyme at sites removed from the active site (12, 13, 55, 56). Our studies showed that both an A-type peptide with an extended conformation and a B-type in a bent conformation interact with extended exosites, which together form the “entrance” to the active site cleft and accommodate different structures: the B-type peptides as an α helix, the A-type as an α helix or as extended peptides, and the AB loop of TIMP-2 (50). In the case of the B-type peptides, the orientation of the helix appears to be dictated by crystallographic contacts, but it is not difficult to envisage how a large protein substrate could impose a structural conformation on a section N-terminal of the cleavage site. For example, the MMP-13 cleavage site in the fibrinogen β-chain is located in an α-helix (57, 58), suggesting that interaction with the exosite is essential for cleavage. Thus, MMP-13 appears to be adaptable to such structures, and the N-terminal structure of the substrate may have an impact on the efficiency of substrate cleavage, as it may affect the binding affinity and thus could contribute directly to the substrate specificity of the enzyme. This area of MMP-13Cat can be considered a collection of substrate-dependent exosites (i.e., versatile exosites that adjust to the substrate). The exosites including S4 are not conserved in two other human collagenases, MMP-1 and MMP-8, suggesting a structural basis for broader substrate specificity of MMP-13.
The crystal structure of full-length MMP-1 bound to a triple helical collagen peptide (12) showed that collagen contacts the enzyme with all three strands and identified essential exosites both on the Cat and Hpx domains. The latter includes an S10′ site that is key for collagen interaction. MMP-13 has a conserved S10′ subsite, but the orientation of the Cat and Hpx domains is shifted, and docking of collagen to MMP-13 causes some clashes, suggesting that the MMP-13 ground state may be less favorable for collagen binding. Binding to the long triple helical collagen may result in an MMP-13 conformation similar to that of MMP-1, which reiterates that interdomain flexibility is an important accompaniment to the mechanism of collagenolysis (12, 13, 48, 59). The interdomain mobility may be higher than suggested by the crystal structures, since solution studies of MMP-1 and triple helical collagen with nuclear magnetic resonance (NMR) and small-angle X-ray scattering indicate a dissociation between Cat and Hpx (13, 59). Regardless of how collagen binds MMP-13, the collagenase-cleavable bond in the triple helix cannot fit in the active site on the Cat domain without structural changes in the local collagen structure (12). The result of this should at least in part correspond to the cocrystallized peptides in MMP-13. In earlier work, we have shown that the collagen structure is locally unwound by the cooperative action of the Cat and Hpx domains, leading to the correct positioning of the cleavable bond (9, 12). In fact, this process is blocked by a synthetic, active site–directed inhibitor, suggesting that looping out of a strand into the active site is essential. This process is assisted by the temperature-dependent propensity of the imino acid–poor section near the cleavage site, rendering it a less tight triple helix (12, 60, 61). In a recent free-energy simulation study, Lu and Stultz (62) indicated that the MMP cleavage site could be sufficiently distorted to fit 1 strand in the active site in the presence of the catalytic domain of MMP-8, but not without the enzyme, supporting that the enzyme influences the local structure of collagen.
What do the MMP-13 structures with extended peptides tell us about collagenases' ability to cleave triple helical collagens? Our superimposition of the peptides shown in this study and the triple helix in a productive complex (12) indicate that the collagen strand located close to the active site overlaps reasonably well with both A- and B-type peptides, but few contacts are made at the distal substrate-dependent exosites that were defined by A- and B-type peptides. We speculate that the substrate-dependent exosites in the Cat domain are involved in collagen contacts in the unwinding stage of the process, specifically the FYP(175–177) loop, due to its structural adaptability. On binding to collagenase, the collagen strand closest to the active site would move 3–6 Å with the help of Cat-Hpx domain flexing and would be placed into the substrate binding site, which is essential for the initial cleavage of the triple helix. This would also induce the two other strands to unfold locally; such unwound structures may be accommodated in the exosites with sufficient space. Once the first strand is cleaved, it would trigger further structural unraveling in the second and third strands, rendering them susceptible to rapid hydrolysis by the enzyme. Thus, these exosites likely play an important role in collagenolysis and its specificity. We therefore propose that the substrate-dependent exosites are important regions to target for the design of new MMP-13 substrate-specific inhibitors.
Acknowledgments
The authors thank Noriko Itoh for construction of the MMP-13(E223A) expression vector; Alan Lyons and Toyoko Nakamura for help in expressing and purifying the protein; Laura Vera for efforts with cryoprotection to improve data quality; the European Synchrotron Radiation Facility (ESRF; Grenoble, France) and Soleil for beam time; and Drs. Andrew McCarthy on line ID14-4, Max Nanao on line ID23-2; and Andrew Thomson on Proxima 1 for assistance with the synchrotron experiments.
This work was supported by the Wellcome Trust (grant 075473), Arthritis Research UK, and the U.S. National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Disease (NIH/NIAMS; grant AR40994; R.V. and H.N.).
Footnotes
- AAB
- sodium acetate, ADA, and bicine
- ADA
- N-(2-acetamido)iminodiacetic acid
- Cat
- catalytic
- Hpx
- hemopexin
- MPEG-2K
- monomethyl polyethylene glycol 2000
- MMP
- matrix metalloproteinase
- PCTP
- propionic acid, cacodylate, bis-tris propane
- PEG
- polyethylene glycol
- TIMP
- tissue inhibitor of matrix metalloproteinase
REFERENCES
- 1. Page-McCaw A., Ewald A. J., Werb Z. (2007) Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 8, 221–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Woessner J. F. (1998) The matrixmetalloproteinase family. In Matrix Metalloproteinases (Parks W. C., Mecham R. P., eds) pp. 1–14, Academic Press, San Diego, CA, USA [Google Scholar]
- 3. Murphy G., Nagase H. (2008) Progress in matrix metalloproteinase research. Mol. Aspects Med. 29, 290–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gomis-Rüth F. X. (2008) Structure and mechanism of metallocarboxypeptidases. Crit. Rev. Biochem. Mol. Biol. 43, 319–345 [DOI] [PubMed] [Google Scholar]
- 5. Visse R., Nagase H. (2003) Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ. Res. 92, 827–839 [DOI] [PubMed] [Google Scholar]
- 6. Li J., Brick P., O'Hare M. C., Skarzynski T., Lloyd L. F., Curry V. A., Clark I. M., Bigg H. F., Hazleman B. L., Cawston T. E. (1995) Structure of full-length porcine synovial collagenase reveals a C-terminal domain containing a calcium-linked, four-bladed beta-propeller. Structure 3, 541–549 [DOI] [PubMed] [Google Scholar]
- 7. Nagase H., Visse R. (2011) Triple helicase activity and the structural basis of collagenolysis. In Extracellular Matrix Degradation (Parks W., Mecham R. P., eds) pp. 95–122, Springerverlag, Berlin/Heidelberg [Google Scholar]
- 8. Murphy G., Allan J. A., Willenbrock F., Cockett M. I., O'Connell J. P., Docherty A. J. (1992) The role of the C-terminal domain in collagenase and stromelysin specificity. J. Biol. Chem. 267, 9612–9618 [PubMed] [Google Scholar]
- 9. Chung L., Dinakarpandian D., Yoshida N., Lauer-Fields J. L., Fields G. B., Visse R., Nagase H. (2004) Collagenase unwinds triple-helical collagen prior to peptide bond hydrolysis. EMBO J. 23, 3020–3030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Salsas-Escat R., Nerenberg P. S., Stultz C. M. (2010) Cleavage site specificity and conformational selection in type I collagen degradation. Biochemistry 49, 4147–4158 [DOI] [PubMed] [Google Scholar]
- 11. Nerenberg P. S., Stultz C. M. (2008) Differential unfolding of alpha1 and alpha2 chains in type I collagen and collagenolysis. J. Mol. Biol. 382, 246–256 [DOI] [PubMed] [Google Scholar]
- 12. Manka S. W., Carafoli F., Visse R., Bihan D., Raynal N., Farndale R. W., Murphy G., Enghild J. J., Hohenester E., Nagase H. (2012) Structural insights into triple-helical collagen cleavage by matrix metalloproteinase 1. Proc. Natl. Acad. Sci. U. S. A. 109, 12461–12466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bertini I., Fragai M., Luchinat C., Melikian M., Toccafondi M., Lauer J. L., Fields G. B. (2012) Structural basis for matrix metalloproteinase 1-catalyzed collagenolysis. J. Am. Chem. Soc. 134, 2100–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ala-aho R., Kähäri V.-M. (2005) Collagenases in cancer. Biochimie (Paris) 87, 273–286 [DOI] [PubMed] [Google Scholar]
- 15. Pendás A. M., Uría J. A., Jiménez M. G., Balbín M., Freije J. P., López-Otín C. (2000) An overview of collagenase-3 expression in malignant tumors and analysis of its potential value as a target in antitumor therapies. Clin. Chim. Acta 291, 137–155 [DOI] [PubMed] [Google Scholar]
- 16. Takaishi H., Kimura T., Dalal S., Okada Y., D'Armiento J. (2008) Joint diseases and matrix metalloproteinases: a role for MMP-13. Curr. Pharmaceut. Biotechnol. 9, 47–54 [DOI] [PubMed] [Google Scholar]
- 17. Goldring M. B. (2012) Chondrogenesis, chondrocyte differentiation, and articular cartilage metabolism in health and osteoarthritis. Ther. Adv. Musculoskelet. Dis. 4, 269–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bode W., Reinemer P., Huber R., Kleine T., Schnierer S., Tschesche H. (1994) The X-ray crystal structure of the catalytic domain of human neutrophil collagenase inhibited by a substrate analogue reveals the essentials for catalysis and specificity. EMBO J. 13, 1263–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bertini I., Calderone V., Fragai M., Luchinat C., Maletta M., Yeo K. J. (2006) Snapshots of the reaction mechanism of matrix metalloproteinases. Angew. Chem. Int. Ed. Engl. 45, 7952–7955 [DOI] [PubMed] [Google Scholar]
- 20. Gall A. L., Ruff M., Kannan R., Cuniasse P., Yiotakis A., Dive V., Rio M. C., Basset P., Moras D. (2001) Crystal structure of the stromelysin-3 (MMP-11) catalytic domain complexed with a phosphinic inhibitor mimicking the transition-state. J. Mol. Biol. 307, 577–586 [DOI] [PubMed] [Google Scholar]
- 21. Chung L., Shimokawa K., Dinakarpandian D., Grams F., Fields G. B., Nagase H. (2000) Identification of the (183)RWTNNFREY(191) region as a critical segment of matrix metalloproteinase 1 for the expression of collagenolytic activity. J. Biol. Chem. 275, 29610–29617 [DOI] [PubMed] [Google Scholar]
- 22. Stura E. A. (1999) Seeding techniques. In Crystallization of Nucleic Acids and Proteins: a Practical Approach (Ducruix A., Giegé G., eds) pp. 177–208, Oxford University Press, Oxford, UK [Google Scholar]
- 23. Vera L., Stura E. A. (2013) Strategies for protein cryocrystallography [E-pub ahead of print]. Cryst. Growth Des. 10.1021/cg301531f [DOI] [Google Scholar]
- 24. Newman J. J. (2004) Novel buffer systems for macromolecular crystallization. Acta Crystallogr. D Biol. Crystallogr. 60, 610–612 [DOI] [PubMed] [Google Scholar]
- 25. Leslie A. G. W. (2006) The integration of macromolecular diffraction data. Acta Crystallogr. D Biol. Crystallogr. 62, 48–57 [DOI] [PubMed] [Google Scholar]
- 26. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Johnson A. R., Pavlovsky A. G., Ortwine D. F., Prior F., Man C.-F., Bornemeier D. A., Banotai C. A., Mueller W. T., McConnell P., Yan C., Baragi V., Lesch C., Roark W. H., Wilson M., Datta K., Guzman R., Han H.-K., Dyer R. D. (2007) Discovery and characterization of a novel inhibitor of matrix metalloprotease-13 that reduces cartilage damage in vivo without joint fibroplasia side effects. J. Biol. Chem. 282, 27781–27791 [DOI] [PubMed] [Google Scholar]
- 28. Gomis-Rüth F. X., Gohlke U., Betz M., Knäuper V., Murphy G., López-Otín C., Bode W. (1996) The helping hand of collagenase-3 (MMP-13): 2.7 a crystal structure of its C-terminal haemopexin-like domain. J. Mol. Biol. 264, 556–566 [DOI] [PubMed] [Google Scholar]
- 29. Vagin A., Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 30. McRee D. E. (1999) XtalView/Xfit: a versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 125, 156–165 [DOI] [PubMed] [Google Scholar]
- 31. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 32. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. DeLano W. L. (2002) Unraveling hot spots in binding interfaces: progress and challenges. Curr. Opin. Struct. Biol. 12, 14–20 [DOI] [PubMed] [Google Scholar]
- 36. Erickson H. P. (2009) Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol. Proced. Online 11, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. García De La Torre J., Huertas M. L., Carrasco B. (2000) Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys. J. 78, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cha H., Kopetzki E., Huber R., Lanzendörfer M., Brandstetter H. (2002) Structural basis of the adaptive molecular recognition by MMP9. J. Mol. Biol. 320, 1065–1079 [DOI] [PubMed] [Google Scholar]
- 40. Morgunova E., Tuuttila A., Bergmann U., Isupov M., Lindqvist Y., Schneider G., Tryggvason K. (1999) Structure of human pro-matrix metalloproteinase-2: activation mechanism revealed. Science 284, 1667–1670 [DOI] [PubMed] [Google Scholar]
- 41. Tallant C., Marrero A., Gomis-Rüth F. X. (2010) Matrix metalloproteinases: fold and function of their catalytic domains. Biochim. Biophys. Acta 1803, 20–28 [DOI] [PubMed] [Google Scholar]
- 42. Iyer S., Visse R., Nagase H., Acharya K. R. (2006) Crystal structure of an active form of human MMP-1. J. Mol. Biol. 362, 78–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reinemer P., Grams F., Huber R., Kleine T., Schnierer S., Piper M., Tschesche H., Bode W. (1994) Structural implications for the role of the N terminus in the ‘superactivation’ of collagenases: a crystallographic study. FEBS Lett. 338, 227–233 [DOI] [PubMed] [Google Scholar]
- 44. Nagase H. (1997) Activation mechanisms of matrix metalloproteinases. Biol. Chem. 378, 151–160 [PubMed] [Google Scholar]
- 45. Schechter I., Berger A. (1967) On the size of the active site in proteases: I, papain. Biochem. Biophys. Res. Commun. 27, 157–162 [DOI] [PubMed] [Google Scholar]
- 46. Becker J. W., Marcy A. I., Rokosz L. L., Axel M. G., Burbaum J. J., Fitzgerald P. M., Cameron P. M., Esser C. K., Hagmann W. K., Hermes J. D. (1995) Stromelysin-1: three-dimensional structure of the inhibited catalytic domain and of the C-truncated proenzyme. Protein Sci. 4, 1966–1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Elkins P. A., Ho Y. S., Smith W. W., Janson C. A., D'Alessio K. J., McQueney M. S., Cummings M. D., Romanic A. M. (2002) Structure of the C-terminally truncated human ProMMP9, a gelatin-binding matrix metalloproteinase. Acta Crystallogr. D Biol. Crystallogr. 58, 1182–1192 [DOI] [PubMed] [Google Scholar]
- 48. Jozic D., Bourenkov G., Lim N. H., Visse R., Nagase H., Bode W., Maskos K. (2005) X-ray structure of human proMMP-1: new insights into procollagenase activation and collagen binding. J. Biol. Chem. 280, 9578–9585 [DOI] [PubMed] [Google Scholar]
- 49. Deng S. J., Bickett D. M., Mitchell J. L., Lambert M. H., Blackburn R. K., Carter H. L., Neugebauer J., Pahel G., Weiner M. P., Moss M. L. (2000) Substrate specificity of human collagenase 3 assessed using a phage-displayed peptide library. J. Biol. Chem. 275, 31422–31427 [DOI] [PubMed] [Google Scholar]
- 50. Maskos K., Lang R., Tschesche H., Bode W. (2007) Flexibility and variability of TIMP binding: X-ray structure of the complex between collagenase-3/MMP-13 and TIMP-2. J. Mol. Biol. 366, 1222–1231 [DOI] [PubMed] [Google Scholar]
- 51. Nagase H., Fields G. B. (1996) Human matrix metalloproteinase specificity studies using collagen sequence-based synthetic peptides. Biopolymers. 40, 399–416 [DOI] [PubMed] [Google Scholar]
- 52. Hashimoto H., Takeuchi T., Komatsu K., Miyazaki K., Sato M., Higashi S. (2011) Structural basis for matrix metalloproteinase-2 (MMP-2)-selective inhibitory action of β-amyloid precursor protein-derived inhibitor. J. Biol. Chem. 286, 33236–33243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Grams F., Reinemer P., Powers J. C., Kleine T., Pieper M., Tschesche H., Huber R., Bode W. (1995) X-ray structures of human neutrophil collagenase complexed with peptide hydroxamate and peptide thiol inhibitors. Implications for substrate binding and rational drug design. Eur. J. Biochem. 228, 830–841 [DOI] [PubMed] [Google Scholar]
- 54. Igarashi Y., Eroshkin A., Gramatikova S., Gramatikoff K., Zhang Y., Smith J. W., Osterman A. L., Godzik A. (2007) CutDB: a proteolytic event database. Nucleic Acids Res. 35, D546–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fulcher Y. G., Van Doren S. R. (2011) Remote exosites of the catalytic domain of matrix metalloproteinase-12 enhance elastin degradation. Biochemistry 50, 9488–9499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Robichaud T. K., Steffensen B., Fields G. B. (2011) Exosite interactions impact matrix metalloproteinase collagen specificities. J. Biol. Chem. 286, 37535–37542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hiller O., Lichte A., Oberpichler A., Kocourek A., Tschesche H. (2000) Matrix metalloproteinases collagenase-2, macrophage elastase, collagenase-3, and membrane type 1-matrix metalloproteinase impair clotting by degradation of fibrinogen and factor XII. J. Biol. Chem. 275, 33008–33013 [DOI] [PubMed] [Google Scholar]
- 58. Kollman J. M., Pandi L., Sawaya M. R., Riley M., Doolittle R. F. (2009) Crystal structure of human fibrinogen. Biochemistry 48, 3877–3886 [DOI] [PubMed] [Google Scholar]
- 59. Bertini I., Fragai M., Luchinat C., Melikian M., Mylonas E., Sarti N., Svergun D. I. (2009) Interdomain flexibility in full-length matrix metalloproteinase-1 (MMP-1). J. Biol. Chem. 284, 12821–12828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fields G. B. (1991) A model for interstitial collagen catabolism by mammalian collagenases. J. Theor. Biol. 153, 585–602 [DOI] [PubMed] [Google Scholar]
- 61. Stultz C. M. (2002) Localized unfolding of collagen explains collagenase cleavage near imino-poor sites. J. Mol. Biol. 319, 997–1003 [DOI] [PubMed] [Google Scholar]
- 62. Lu K. G., Stultz C. M. (2013) Insight into the degradation of type-I collagen fibrils by MMP-8. J. Mol. Biol. [DOI] [PubMed] [Google Scholar]