

Abstract
Cyclopropenimine 1 is shown to catalyze Mannich reactions between glycine imines and N-Boc-aldimines with high levels of enantio- and diastereocontrol. The reactivity of 1 is shown to be substantially greater than a widely used thiourea cinchona alkaloid derived catalyst. A variety of aryl and aliphatic N-Boc-aldimines are effective substrates for this transformation. A preparative scale reaction to deliver >90 mmol of product is shown using 1 mol% catalyst. The product of this transformation is converted into several useful derivatives.
Vicinal diamino stereoarrays represent a prominent chemical motif found in bioactive natural products, synthetic building blocks, and metal ligands.1 The preparation of α,β-diaminoacid derivatives via enantioselective direct Mannich reactions of glycinate Schiff bases offers a powerful means to access such stereoarrays (Figure 1).2 Although a number of enantioselective catalyst systems have been reported that deliver α-amino Mannich products efficiently and with high stereoselectivity, these methods suffer from important limitations of substrate scope, catalyst reactivity, or stereocontrol, and thus a more broadly effective solution to the challenge of enantioselective Mannich reactions remains to be developed. We recently introduced chiral cyclopropenimines as a potent new class of enantioselective Brønsted base catalyst.3 Here we report that the readily available4,5 chiral cyclopropenimine 1 is a highly efficient and selective catalyst for direct Mannich reactions that operates even in the context of the challenging coupling of t-butyl glycinate donors and N-Boc-imine acceptors.
Figure 1.
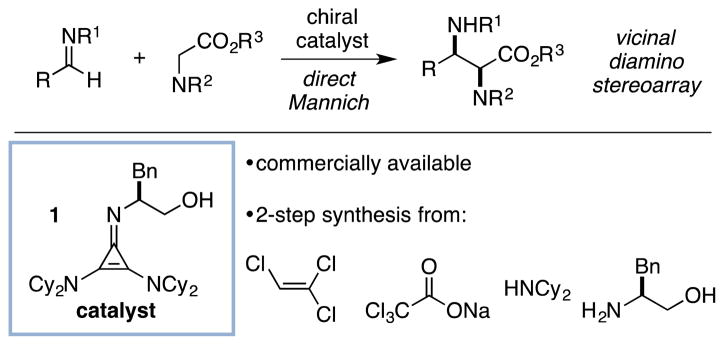
Vincinal diamino stereocenters via direct glycinate Mannich reaction.
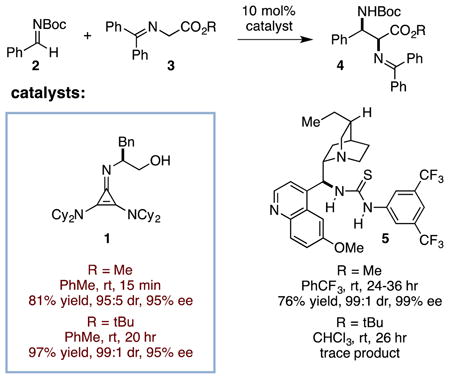
Enantio- and diastereoselective Mannich reactions of glycinate Schiff bases6 have been accomplished by a number of groups using copper-based catalysis; however these methods have employed N-sulfonyl imine electrophiles, which result in products that are challenging to deprotect.7,8 More synthetically versatile products have been obtained with chiral phase-transfer catalysis,9 but typically with only moderate levels of stereoselectivity.10 Arguably the most attractive approach to this class of reactions is via direct chiral Brønsted-base catalysis,11 which has been shown to be compatible with the use of N-carbamoyl imine substrates. Indeed, the state-of-the-art catalyst for this transformation is the widely employed bifunctional thiourea 5,12 which relies on strong H-bond activation coupled with a relatively weak tertiary amine Brønsted base (eq 1). Barbas has reported that 5 catalyzes the stereoselective Mannich reaction of methyl glycinate substrates in 24–36 h but is ineffective for the more sterically demanding t-butyl esters, which are desirable because of their amenability to acidic deprotection.13 Notably, the more strongly basic catalyst tetramethylguanidine (TMG) is also ineffective at catalyzing the reaction shown in equation 1 (R = Me).14
 |
(1) |
Given the remarkably high reactivity that we had previously observed for Michael additions using cyclopropenimine 1,3 we wondered whether this catalyst could also offer a more broadly effective approach to glycinate Mannich reactions and thereby help to alleviate the substrate limitations noted above. In fact, we found that 10 mol% 1 catalyzed the reaction of glycinate 3 (R = Me) with N-Boc-benzaldimine 2 to produce the diamine adduct 4 in 81% yield with 95:5 dr (syn:anti) and 95% ee in only 15 min at room temperature, a remarkable enhancement of reactivity over the thiourea catalyst 5. Moreover, 1 was also found to catalyze the reaction of t-butyl glycinate 3 in 20 hr to deliver the Mannich product in 97% yield with 99:1 dr and 94% ee.15 These results clearly demonstrate the substantially greater reactivity of 1 versus less basic catalyst frameworks.16
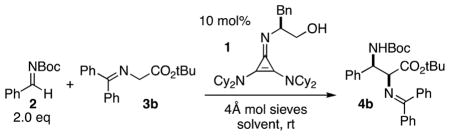
A portion of the optimization studies leading to the reaction shown in equation 1 are presented in Table 1. In all cases, molecular sieves were added to inhibit hydrolysis of the imine 2, which 1 slowly catalyzes in the presence of water. Although the use of solvents such as ethyl acetate and diethyl ether resulted in promising selectivities (entries 1–2), we found that enantioselectivity was increased in aromatic solvents (entries 3–5), with toluene providing optimal results. Dilution of the reaction mixture increased selectivity, albeit at a cost of reaction time (entries 5–9). Additionally, we found that using ground molecular sieves improved the efficiency and selectivity of the reaction (entry 10). Notably, the stoichiometry of the electrophile could be reduced to 1.5 equiv with no loss in efficiency or selectivity (entry 11).
Table 1.
Optimization of enantioselective cyclopropenimine catalyzed Mannich reaction.a
| |||||
|---|---|---|---|---|---|
| entry | solvent | cone (M) | time (h) | syn/anti | ee (%) |
| 1 | EtOAc | 0.35 | 12 | 98:2 | 83 |
| 2 | Et2O | 0.35 | 8 | 98:2 | 85 |
| 3 | PhH | 0.35 | 20 | 99:1 | 89 |
| 4 | m-xylene | 0.35 | 20 | 99:1 | 91 |
| 5 | PhMe | 0.35 | 16 | 99:1 | 91 |
| 6 | PhMe | 0.45 | 6 | 99:1 | 90 |
| 7 | PhMe | 0.25 | 24 | 99:1 | 92 |
| 8 | PhMe | 0.15 | 36 | 99:1 | 94 |
| 9 | PhMe | 0.08 | 48 | 98:2 | 93 |
| 10b | PhMe | 0.25 | 20 | 98:2 | 95 |
| 11b,c | PhMe | 0.25 | 26 | 98:2 | 95 |
All reactions proceeded to ≥95% conversion of glycine imine.
Ground 4Å mol sieves were used.
1.5 eq of 2 were used.
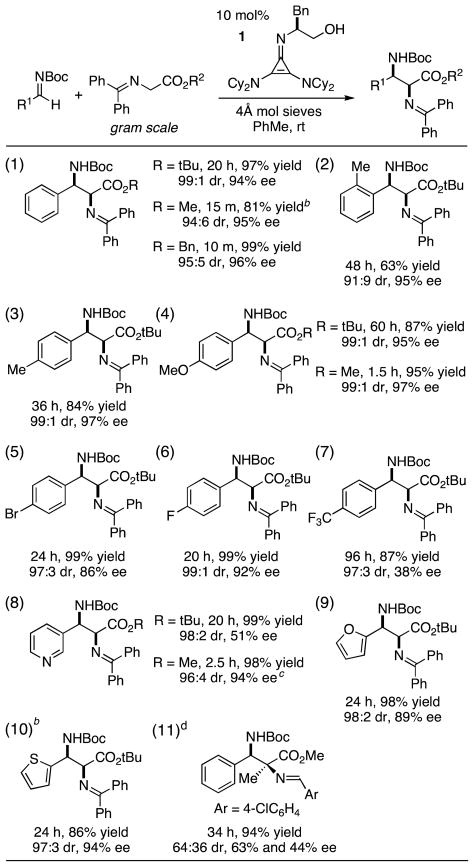
The substrate scope of the cyclopropenimine-catalyzed Mannich reaction was next explored. To emphasize the practicability and simplicity of this chemistry, each of the reactions shown was performed using one gram of glycinate substrate under conditions that included exposure to the atmosphere. Methyl, benzyl and t-butyl glycinates all showed good reactivity and selectivity at this scale with N-Boc-benzaldimine (entry 1). Given the incompatibility of t-butyl glycinates with many other Brønsted base catalysts, we chose to explore the reaction of this substrate with other N-Boc-aldimines.
Both o- and p-tolualdimines yielded diamine products with high diastereo- and enantioselectivity (entries 2 and 3). The reaction with the more electron-rich anisaldimine was also efficient and stereoselective (entry 4), although the reaction time was significantly longer (60 h) than with electron-neutral substrates. In this case, use of the methyl glycinate substrate led to product in only 1.5 h with comparably good stereoselectivity. Halogenated substrates were found to perform well (entries 5 and 6); however the highly electron-deficient p-trifluoromethylbenzaldimine was relatively unreactive, and yielded diamine with severely diminished ee (entry 7). Notably, heterocyclic imines, including those with pyridine, furan, and thiophene ring systems, were all excellent substrates for this transformation (entries 8–10). Interestingly, although the enantioselectivity with the pyridyl substrate was subpar using the t-butyl glycinate donor, high selectivity was restored with the use of the methyl glycinate (entry 8). An alanine derived imine substrate gave rise to a product bearing a tetrasubstituted stereocenter (entry 11), however the diastero- and enantioselectivity of this substrate type were suboptimal with the current catalyst system.
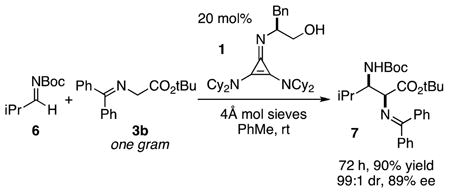
Of particular note is the fact that an aliphatic aldimine 6 was also found to be a suitable substrate for reaction with the t-butyl glycinate donor 3b (eq 2). Although this reaction required 20 mol% catalyst loading to achieve a reasonable rate, the procedure was readily performed on a gram scale to produce adduct 7 with high yield and stereoselectivity. To the best of our knowledge, this represents the first use of aliphatic N-Boc-aldimines for a highly enantioselective direct Mannich reaction with glycine imines.
 |
(2) |
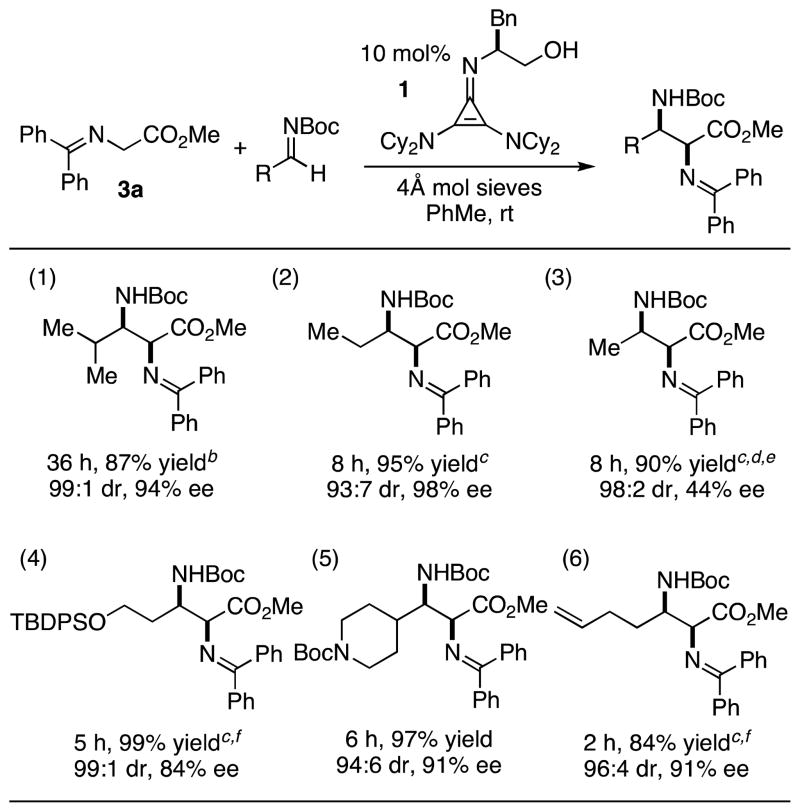
We further explored the substrate scope of the aliphatic N-Boc-aldimine Mannich reaction. A major concern with aliphatic N-Boc-aldimines is the potential for base-promoted enamine formation, leading to undesired side products. We were able to avoid competitive N-Boc-aldimine decomposition by the judicious selection of several parameters. Thus, methyl glycinate 3a was selected as the enolate for this screen in order to decrease reaction times and allow for lower catalyst loadings. Additionally, some reactions were carried out with excess N-Boc-aldimine and at lowered temperatures. In this way, an array of aliphatic N-Boc-aldimines with a variety of substitution patterns and functional groups were found to be suitable substrates for this reaction (Table 3).
Table 3.
Aliphatic substrate scope of Mannich reaction.a
|
Yields based on purified products. Diastereomeric ratios (dr) and enantiomeric excesses (ee) were determined by HPLC.
One gram scale of glycine imine.
4 equiv of N-Boc-Imine used.
0 ºC.
N-Boc-Imine made in situ with Cs2CO3; 20 mol% catalyst used.
−25 ºC.
Substrates bearing isopropyl and ethyl groups resulted in high diastereo- and enantioselectivities (entries 1 and 2). Entry 3 shows the production of protected 2,3-diaminobutanoic acid, an α,β-diaminoacid widely found as a key motif in a number of naturally occurring peptides.1 For this substrate, the N-Boc-aldimine was formed in situ due to its instability and difficulty of isolation. At 0 ºC, the reaction proceeded with good conversion, albeit with only 44% ee. The selectivity could be increased to 83% ee upon cooling to −78 ºC, however at this temperature the reaction did not reach completion.17 We also found silyl protected alcohol (entry 4) and N-Boc protected amine (entry 5) functional groups to be well accommodated. Additionally, a substrate bearing a terminal olefin worked well with high selectivities (entry 6). The tolerance of diverse functional groups in this reaction should enable an enhanced synthetic utilization of these vicinal diamino stereoarrays.
Our mechanistic rationale for this reaction is shown in Figure 2. Deprotonation of glycine imine 2 and H-bond engagement of N-Boc imine 3 by the catalyst 1 leads to pre-transition state complex 8a or 8b, with subsequent C–C bond formation as the rate- and enantiodetermining step. This latter hypothesis is supported by (1) the slow reaction rate of the less electrophilic p-methoxybenzaldimine (entry 3, Table 2), and (2) the significantly slower rate and diminished enantioselectivity of p-trifluoromethylbenzaldimine (entry 5), which can be rationalized as a consequence of the decreased H-bond acceptor capacity of this substrate. The precise organization of the transition state, including the question of whether the enolate is OH-bound (8a) or NH-bound (8b), is a subject currently under investigation.
Figure 2.

Proposed catalytic cycle for cyclopropenimine-catalyzed Mannich reaction. The red double-headed arrow indicates conformational locking of the stereocenter due to steric conflict with a cyclohexyl ring.
Table 2.
Gram-scale substrate scope of Mannich reaction.a
|
Yields based on purified products. Diastereomeric ratios (dr) and enantiomeric excesses (ee) were determined by HPLC.
Yield determined by 1H NMR versus Bn2O as a standard; product characterized after hydrolysis of the benzophenone imine.
Reaction performed at a concentration of 0.07 M.
20 mol% catalyst was used; 0.9 mmol scale.
Finally, we have investigated the performance of catalyst 1 for the production of preparative amounts of chiral diamino acid derivatives (Scheme 1, eq 3). Using 1 mol% of catalyst 1, 26.7 g of the diamino ester 9 was prepared (73% yield, 96:4 dr, 93% ee) in 8 h following imine hydrolysis with citric acid. Adduct 9 was subsequently converted to several useful derivatives, including diketopiperazine 10, β-lactam 11, amidine 12, and tripeptide 13, in preparative quantities in three or less operations (Scheme 1). The efficient performance of catalyst 1 coupled with its ready availability should make this chemistry a practical tool for the preparation of 1,2-diamino stereocenters.
Scheme 1.

Preparative scale synthesis and derivatization of diamine 8. See supporting information for full experimental details. (a) TFA, CH2Cl2, rt; (b) dimethyloxylate, MeOH, reflux; (c) CBzCl, Na2CO3 (aq), PhMe, rt; (d) TMSCl, CH2Cl2, 0 ºC then tBuMgCl; (e) 4-bromobenzaldehyde, TEA, CH2Cl2, rt then NBS, 0 ºC; (f) Z-Ala-OH, EDC, HOBt, TEA, CH2Cl2, 0 ºC to rt; (g) Boc-Phe-OH, EDC, HOBt, TEA, CH2Cl2, 0 ºC to rt.
Supplementary Material
Acknowledgments
Financial support was provided by NIHGMS (RO1 GM102611). THL is grateful for an Ely Lilly Grantee Award. J.S.B. is grateful for NDSEG and NSF graduate fellowships. We thank the Leighton group (Columbia University) for use of their instrumentation.
Footnotes
Supporting Information Available: Experimental procedures and product characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Viso A, de la Pradilla RF, Garcia A, Flores A. Chem Rev. 2005;105:3167. doi: 10.1021/cr0406561. [DOI] [PubMed] [Google Scholar]; (b) Viso A, de la Pradilla RF, Tortosa M, García A, Flores A. Chem Rev. 2011;111:PR1. doi: 10.1021/cr100127y. [DOI] [PubMed] [Google Scholar]; (c) Lucet D, Le Galle T, Mioskowski C. Angew Chem Int Ed. 1998;37:2580. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2580::AID-ANIE2580>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 2.For a review, see: Gómez Arrayás R, Carretero JC. Chem Soc Rev. 2009;38:1940. doi: 10.1039/b820303b.
- 3.Bandar JS, Lambert TH. J Am Chem Soc. 2012;134:5552. doi: 10.1021/ja3015764. [DOI] [PubMed] [Google Scholar]
- 4.See supporting information for an improved synthesis of cyclopropenimine 1 directly from pentachlorocyclopropane.
- 5.(S)-2-(2,3-bis(dicyclohexylamino)cyclopropenimine)-3-phenylpropan-1-ol hydrochloride (1•HCl) can be purchased from Sigma-Aldrich (product #L511188).
- 6.For selected examples of enantioselective Mannich reactions using other donors, see: Zhang WQ, Cheng LF, Gong LZ. Angew Chem Int Ed. 2012;51:4085. doi: 10.1002/anie.201107741.Liu X, Deng L, Jiang X, Yan W, Liu C, Wang R. Org Lett. 2010;12:876. doi: 10.1021/ol902916s.Uragachi D, Ueki Y, Ooi T. J Am Chem Soc. 2008;130:14088. doi: 10.1021/ja806311e.Shi SH, Huang FP, Zhu P, Dong ZW, Hui XP. Org Lett. 2012;14:2010. doi: 10.1021/ol300510b.Cutting GA, Stainforth NE, John MP, Kociok-Köhn G, Willis MC. J Am Chem Soc. 2007;129:10632. doi: 10.1021/ja073473f.Li L, Ganesh M, Seidel D. J Am Chem Soc. 2009;131:11648. doi: 10.1021/ja9034494.Jiang J, Xu HD, Xi JB, Ren BY, Lv FP, Guo X, Jiang LQ, Zhang ZY, Hu WH. J Am Chem Soc. 2011;133:8428. doi: 10.1021/ja201589k.ChenXDong S, Qiao Z, Zhu Y, Xie M, Lin L, Liu X, Feng X. Chem Eur J. 2011;17:2583. doi: 10.1002/chem.201002571.Lu G, Yoshino T, Morimoto H, Matsunaga S, Shibasaki M. Angew Chem Int Ed. 2011;50:4382. doi: 10.1002/anie.201101034.Nakamura S, Maeno Y, Ohara M, Yamamura A, Funahashi Y, Shibata N. Org Lett. 2012;14:2960. doi: 10.1021/ol301256q.Melhado AD, Amarante GW, Wang ZJ, Luparia M, Toste FD. J Am Chem Soc. 2011;133:3517. doi: 10.1021/ja1095045.Chowdari NS, Ahmad M, Albertshofer K, Tanaka F, Barbas CF., III Org Lett. 2006;8:2839. doi: 10.1021/ol060980d.
- 7.Bernardi L, Gothelf AS, Hazell RG, Jørgensen KA. J Org Chem. 2003;68:2583. doi: 10.1021/jo026766u. [DOI] [PubMed] [Google Scholar]
- 8.For metal catalyzed Mannich reactions of glycine imines, see: Hernández-Toribio J, Gómez Arrayás R, Carretero JC. Chem Eur J. 2010;16:1153. doi: 10.1002/chem.200902258.Hernández-Toribio J, Gómez Arrayás R, Carretero JC. J Am Chem Soc. 2008;130:16150. doi: 10.1021/ja807583n.Liang G, Tong MC, Tao H, Wang CJ. Adv Synth Catal. 2010;352:1851.Shang D, Liu Y, Zhou X, Liu X, Feng X. Chem Eur J. 2009;15:3678. doi: 10.1002/chem.200900118.Yan XX, Peng Q, Li Q, Zhang K, Yao J, Hou XL, Wu YD. J Am Chem Soc. 2008;130:14362. doi: 10.1021/ja804527r.Hernando E, Gómez Arrayás R, Carretero JC. Chem Commun. 2012;48:9622. doi: 10.1039/c2cc35160a.Salter MM, Kobayashi J, Shimizu Y, Kobayashi S. Org Lett. 2006;8:3533. doi: 10.1021/ol0613012.
- 9.For recent reviews see: Ooi T, Maruoka K. Angew Chem Int Ed. 2007;46:4222. doi: 10.1002/anie.200601737.Hashimoto T, Maruoka K. Chem Rev. 2007;107:5656. doi: 10.1021/cr068368n.O’Donnell MJ. Acc Chem Res. 2004;37:506. doi: 10.1021/ar0300625.
- 10.(a) Ooi T, Kameda M, Fujii J, Maruoka K. Org Lett. 2004;6:2397. doi: 10.1021/ol049215u. [DOI] [PubMed] [Google Scholar]; (b) Okada A, Shibuguchi T, Ohshima T, Masu H, Yamaguchi K, Shibasaki M. Angew Chem Int Ed. 2005;44:4564. doi: 10.1002/anie.200500927. [DOI] [PubMed] [Google Scholar]; (c) Shibuguchi T, Mihara H, Kuramochi A, Ohshima T, Shibasaki M. Chem Asian J. 2007;2:794. doi: 10.1002/asia.200700070. [DOI] [PubMed] [Google Scholar]
- 11.(a) Ishikawa T, editor. Superbases for Organic Synthesis: Guanidines, Amidines, and Phosphazenesand Related Organocatalysts. John Wiley and Sons; 2009. [Google Scholar]; (b) Palomo C, Oiarbide M, Lopez R. Chem Soc Rev. 2009;38:632. doi: 10.1039/b708453f. [DOI] [PubMed] [Google Scholar]; (c) Marcelli T, Hiemstra H. Synthesis. 2010:1229. [Google Scholar]; (d) Leow D, Tan CH. Synlett. 2010:1589. [Google Scholar]; (e) Fu X, Tan CH. Chem Commun. 2011;47:8210. doi: 10.1039/c0cc03691a. [DOI] [PubMed] [Google Scholar]
- 12.For a review, see: Connon SJ. Chem Commun. 2008:2499. doi: 10.1039/b719249e.
- 13.Zhang H, Syed S, Barbas CF., III Org Lett. 2010;12:708. doi: 10.1021/ol902722y. [DOI] [PubMed] [Google Scholar]
- 14.Notably, a chiral guanidine has been used for enantioselective Mannich reactions with significantly more acidic fluorenone-derived glycinate Schiff bases. Kobayashi S, Yazaki R, Seki K, Yamashita Y. Angew Chem Int Ed. 2008;47:5613. doi: 10.1002/anie.200801322.
- 15.Resubjection of product 4b to the reaction conditions does not lead to any racemization or change in diastereomeric ratio.
- 16.Using dimethyl malonate as the enolate in place of 3 resulted in 94% yield of Mannich product in 2 hours, although no enantioselectivity was observed.
- 17.At −78 ºC, the reaction stalled at 15% conversion and yielded a product with 83% ee and 99:1 dr.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.