

Abstract
Advances in the treatment of non-small cell lung cancer (NSCLC) over the last decade have predominantly involved the development of therapies directed at molecular targets such as mutations in the epidermal growth factor receptor (EGFR) or rearrangements in the anaplastic lymphoma kinase (ALK) gene. Other targets have been discovered at low frequency, with multiple agents approved or in development for treatment of these rare molecular subtypes. The tumour microenvironment has also provided opportunities for therapies targeting angiogenesis and the host immune response. This review will provide an overview of current targeted therapies in NSCLC and promising treatment approaches on the horizon.
KEY WORDS : Non-small-cell lung carcinoma (NSCLC), molecular targeted therapy, immunotherapy, epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase (ALK)
Introduction
Delivering a high chance of benefit and avoiding futile treatment is crucial in the management of advanced lung cancer where quality of life is constantly at risk from disease progression or treatment toxicity. This ideal is now achievable with the realisation of targeted therapy in non-small cell lung cancer (NSCLC). Targeted therapy refers to pharmaceutical agents that affect a known molecular target in the cancer cell or tumour microenvironment. In some cases, the presence of the target is determined prior to treatment by interrogating tumour samples with a variety of histological and molecular techniques. In other cases, the presence of the target is assumed to be present in the majority of patients on the basis of prior analyses on large numbers of samples. Detectable targets that indicate a high chance of treatment benefit with a given therapy are termed predictive biomarkers. This is in contrast to prognostic biomarkers, which merely indicate an influence on prognosis rather than treatment response. Testing for mutations in the epidermal growth factor receptor (EGFR) gene and rearrangements of the anaplastic lymphoma kinase (ALK) gene in adenocarcinoma of the lung are now in routine clinical use as predictive genomic biomarkers in the management of advanced lung cancer. The group of patients with lung adenocarcinomas that harbour either of these genomic alterations (15-50% depending on the population studied) are already benefiting from targeted therapy with oral kinase inhibitors such as erlotinib and crizotinib. Other potential predictive genomic biomarkers in known oncogenes such as BRAF, ROS1, MET and PIK3CA have been identified in a systematic fashion and efforts are underway to target them with novel drug compounds.
It is clear now that lung cancer represents a constellation of diseases with distinct molecular profiles and sensitivity to treatment. This re-imagining of the classification of lung cancer has been paralleled by the discovery that squamous cell carcinoma and adenocarcinoma of the lung have very different molecular architectures, and distinguishing the two on histological grounds remains a crucial first step to guide subsequent molecular analyses. Determining the molecular subtypes of lung cancer in the clinic requires an ongoing effort to develop reliable molecular diagnostics, as has occurred with testing for EGFR mutation and ALK rearrangement. Lung cancer therapy is also likely to benefit from the nascent field of cancer immunotherapy, with preliminary evidence that targeting the host immune response to lung cancer will be a successful and versatile treatment modality in the future. This review will summarise the current state of targeted therapy for lung cancer with a focus on NSCLC, and discuss promising agents in development.
Targeting oncogenic mutations and chromosomal aberrations in NSCLC
EGFR-mutant NSCLC
Mutations in the EGFR gene found in adenocarcinoma of the lung was the first biomarker predictive of benefit from a targeted therapy in NSCLC, and was exemplary of the impressive efficacy that could be expected from this paradigm. Small molecule inhibitors of EGFR were originally developed and tested in unselected lung cancer populations, where some patients were noted to have dramatic responses (1,2). Subsequent studies revealed that tumours with mutations in the intracellular tyrosine kinase domain that mediates downstream signalling of the EGFR gene product had substantial clinical responses to oral tyrosine kinase inhibitors (TKIs) such as gefitinib or erlotinib (3-5).
Before EGFR mutation was known to be a predictive biomarker, certain patient populations were seen to benefit more from EGFR TKIs, namely those with lung adenocarcinomas, Asian ethnicity, females and never-smokers. It is now known that the enhanced efficacy in these populations is explained by the greater likelihood that their tumours harbour EGFR mutations (5-8) and that such mutations are almost exclusively found in adenocarcinoma of the lung (7-9). There is however no clinical characteristic that can be used in lieu of EGFR mutation testing.
The efficacy of EGFR TKIs in advanced EGFR-mutant lung cancer has now been established in eight randomised phase III clinical trials. The first of these was the pivotal IPASS study which evaluated the efficacy of gefitinib versus first line chemotherapy with carboplatin and paclitaxel in an Asian population of light or never smokers with advanced lung cancer (10). As part of this study which involved over 1,200 patients, 437 patients had tumour samples assayed for EGFR mutations. In the overall population, the study showed a non-inferior progression free survival for gefitinib compared to chemotherapy. It was also found that EGFR mutation was a very strong predictor of improved progression free survival with gefitinib, and that gefitinib was inferior to chemotherapy in patients without EGFR mutations. These results were confirmed in the phase III First-SIGNAL study which also compared gefitinib to chemotherapy in never-smokers with advanced lung cancer (11).
In addition to IPASS and First-SIGNAL, there have been six randomised controlled phase III trials comparing the EGFR TKIs gefitinib, erlotinib or afatinib to chemotherapy in patients with exclusively EGFR-mutant lung cancer, both in Asian and Caucasian populations. These studies which are summarised in Table 1 (12-17), uniformly show superior response rates, progression free survival and quality of life with EGFR TKIs compared to cytotoxic chemotherapy. Despite mature follow up data (18-20), no trial of a first line EGFR TKI has shown an overall survival benefit, most likely explained by the large numbers of patients in the chemotherapy arms of these trials that crossed over to EGFR TKI treatment after progression. Although there has been no direct comparison, the second generation EGFR TKI afatinib appears to have more toxicity compared to gefitinib and erlotinib, with higher rates of severe diarrhoea and skin rash (16).
Table 1. Phase III trials of EGFR TKIs in exclusively EGFR-mutant advanced NSCLC.
| Trial | Patients | Targeted agent | Comparator arm | Primary endpoint |
|---|---|---|---|---|
| Western Japan Thoracic Oncology Group 3405 (12) | 172 | Gefitinib | Cisplatin + Docetaxel | Median PFS 9.2 versus 6.3 months (HR 0.49, 95% CI: 0.34-0.71, P<0.0001) |
| North East Japan Study Group 002 (13) | 230 | Gefitinib | Carboplatin + Paclitaxel | Median PFS 10.8 versus 5.4 months (HR 0.3, 95% CI: 0.22-0.41, P<0.001) |
| OPTIMAL (14) | 165 | Erlotinib | Carboplatin + Gemcitabine | Median PFS 13.1 versus 4.6 months (HR 0.16, 95% CI: 0.1-0.26, P<0.0001) |
| EURTAC (15) | 174 | Erlotinib | Cisplatin + Docetaxel or Gemcitabine | Median PFS 9.7 versus 5.2 months (HR 0.37, 95% CI: 0.25-0.54, P<0.0001) |
| LUX-Lung 3 (16) | 345 | Afatinib | Cisplatin + Pemetrexed | Median PFS 11.1 versus 6.9 months (HR 0.58, 95% CI: 0.43-0.78, P=0.001) |
| LUX-Lung 6 (17) | 364 | Afatinib | Cisplatin + Gemcitabine | Median PFS 11 versus 5.6 months (HR 0.28, P<0.0001) |
PFS, Pogression free survival; HR, Hazard ratio; CI, Confidence interval.
It is now recommended that all patients with advanced adenocarcinoma of the lung be tested for EGFR mutations (21), which is typically carried out using DNA sequencing of archival formalin fixed tumour tissue obtained at biopsy. The frequency of EGFR mutation in current or former smokers is approximately 10%, and in never smokers can be up to 40-50% (8,22). Due to the superior response rates and quality of life seen with erlotinib or gefitinib compared to chemotherapy, it is also recommended that all patients with EGFR-mutant NSCLC receive these treatments as first line therapy (23-25).
EGFR TKIs continue to have a role in NSCLC without EGFR mutations, where they may inhibit the overexpressed non-mutant protein, so-called wild-type EGFR. Erlotinib was found to improve overall survival in advanced NSCLC compared to placebo following progression on second or third line chemotherapy in the NCIC Clinical Trials Group BR.21 phase III study (26). This study was conducted before the link between EGFR mutation and EGFR TKI response was known, but subsequent subgroup analysis showed that the benefit was maintained in patients with wild-type EGFR and non-adenocarcinoma histology. A similar phase III study comparing gefitinib to placebo in a heavily pre-treated population failed to meet statistical significance, but there was a trend towards improved survival (27) with gefitinib.
Only one phase III study has compared EGFR TKIs to chemotherapy as second line therapy in a population that is specifically EGFR wild-type (28). Although this study suggested that docetaxel was a superior treatment in this group, final publication of results is awaited. A variety of studies have been conducted in unselected populations, showing that EGFR TKIs are non-inferior to second line chemotherapy (29), have a role as maintenance therapy after first line chemotherapy (30), and have similar efficacy to second line chemotherapy in patients that have failed to respond to first line treatment (31). There are no data to suggest the use of EGFR TKIs as first line therapy in EGFR wild-type disease, and this strategy appeared to be detrimental in IPASS (10) and also in the phase III TORCH study of erlotinib followed by chemotherapy versus chemotherapy followed by erlotinib (32).
Second generation EGFR TKIs are irreversible inhibitors of mutant EGFR, and also inhibit other receptors in the epidermal growth factor family. Afatinib, an ErbB receptor family blocker, is one such drug that has progressed furthest in development. In a phase IIb/III study of afatinib versus best supportive care in an unselected population of patients who had progressed on two chemotherapy regimens as well as either erlotinib or gefitinib, there was a modest prolongation of progression free survival by 2 months, but no overall survival benefit (33). Afatinib has also been tested in two phase III randomised trials as first line therapy in patients with EGFR-mutant NSCLC (Table 1) where it showed superior progression free survival compared to chemotherapy (16,17). It has been approved by the United States Food and Drug Administration (FDA) for this indication. Another second generation EGFR TKI dacomitinib has shown superior progression free survival compared to erlotinib when given after failure of prior chemotherapy in a phase II study of 188 patients (34), and is currently under investigation in two phase III studies compared to erlotinib (ARCHER) or placebo (BR26).
An alternative approach to targeting EGFR in NSCLC has been the use of monoclonal antibodies engineered to have strong affinity for the EGFR protein, such as cetuximab (35). Two randomised phase III trials have been conducted comparing chemotherapy to chemotherapy plus cetuximab in advanced NSCLC. The FLEX study of 1,125 patients with advanced NSCLC showed a modest improvement in overall survival of around 1 month with the addition of cetuximab to chemotherapy (36). A similar study failed to show benefit in the primary endpoint of progression free survival (37). Data about the role of EGFR protein expression in predicting benefit have been conflicting, although a retrospective subgroup analysis showed high EGFR expression was predictive of longer survival with cetuximab in the FLEX study (38,39). The lack of clear benefit and uncertainty over an appropriate biomarker has limited the use of cetuximab.
Acquired treatment resistance to EGFR TKIs
There is now little doubt about the effectiveness of EGFR TKIs in EGFR-mutant NSCLC. However, despite high initial response rates, drug resistance and clinical failure is inevitable with the use of these agents over the course of a patient’s treatment, so-called acquired resistance. In contrast to cytotoxic chemotherapy, the well defined mechanism of action of EGFR TKIs means that treatment resistance is a potentially tractable problem. Serial biopsies of tumours before and after treatment with EGFR TKIs have provided insight into the mechanisms of treatment failure (40-43), and have now been performed in sufficient numbers of patients to give an overview of the most common resistance mechanisms. In approximately 60% of cases, treatment failure is mediated by the presence of the secondary EGFR mutation T790M that is resistant to inhibition by current EGFR TKIs (40,43). This is presumed to develop from a resistant population of cells already present in low numbers before treatment with EGFR TKIs (44). In another 5-15% of cases, activation of alternative pathways within the cell that free it from dependence on EGFR signalling occurs, most commonly involving amplification of the MET gene (40-42,45) and mutations in PIK3CA (41). Mutations in BRAF have also been seen, and confirmed to confer resistance in cell line models (46), as has amplification of HER2 (47). Activation of the AXL kinase appears to be another mechanism of acquired resistance (48). Unexpectedly, transformation to small cell histology has been observed in approximately 5% of cases (41,42) and several of these patients responded to conventional chemotherapy regimens used for small cell lung cancer (41). It is of note that several mechanisms of resistance may co-exist in the same tumour (41-43), such as T790M mutation and MET amplification.
The great value in understanding the mechanism of acquired resistance is that it provides a pathway to developing improved therapeutic strategies. Given that T790M mutations are the most common mechanism of acquired resistance, developing EGFR TKIs that inhibit T790M mutant EGFR is a logical next step. There is in vitro evidence that second generation EGFR TKIs such as afatinib may have better efficacy against T790M mutations (49), although response rates in trials with populations expected to have significant numbers of T790M mutations have been poor (33). A phase II study of afatinib combined with cetuximab has however shown promising results, controlling disease in all 22 patients enrolled with 36% showing partial responses (50). Toxicity has been a problem with this combination however. Finally, third generation mutation-selective EGFR TKIs such as CO-1868 have been developed that specifically inhibit the T790M mutant EGFR protein. CO-1868 is currently being tested in a phase I trial in patients with advanced EGFR-mutant NSCLC that have progressed on other EGFR TKIs, where it has shown preliminary evidence of efficacy in resistant disease and a favourable toxicity profile (51). AP26113 is another third generation EGFR TKI with T790M activity that is in phase I/II testing (52).
Targeted therapies already exist or are in development for other molecular pathways that may mediate acquired resistance, such as those involving HER2, BRAF, PIK3CA and MET. Combining such therapies with EGFR TKIs may provide an avenue for preventing or delaying acquired resistance. This has been applied in vitro where EGFR TKI resistance was reversed by co-administration of a MET inhibitor (53,54). Challenges remain in designing trials of tailored drug combinations in this setting and managing the potential toxicities that arise.
ALK-positive NSCLC
ALK was first detected as a fusion oncogene in lung adenocarcinoma in 2007 (55,56), although it had previously been identified as a fusion oncogene arising from a translocation between chromosome 2p and 5q in a subset of anaplastic large cell lymphomas (57). In the context of NSCLC the most frequent ALK gene rearrangement arises due to a short inversion in chromosome 2p where the ALK gene is fused with the echinoderm microtubule-associated protein-like 4 gene (EML4). The aberrant fusion protein EML4-ALK promotes cell growth, and is sufficient to transform cells into a malignant phenotype in vitro (55). ALK-positive cells seem to rely almost exclusively on the fusion protein to drive cell growth and survival, a concept termed ‘oncogene addiction’ that also applies to EGFR-mutant NSCLC (58). In this context, inhibition of oncogene function in EML4-ALK addicted tumours should result in growth arrest and cell death, and this was observed in animal models using small molecule kinase inhibitors targeting ALK (59,60).
Although developed originally as a small molecule inhibitor of the oncogene c-MET, crizotinib was also found to inhibit the ALK kinase (61), and was already in phase I trials when ALK was discovered to play a role in lung cancer. A reliable diagnostic method was also developed to detect ALK fusions in archival lung tissue using fluorescence in situ hybridisation (FISH) with break-apart probes. This enabled patients with advanced ALK-positive lung cancer to be enrolled rapidly into a phase I trial of crizotinib, where an impressive response rate of 60% was demonstrated (62,63). Most of these patients had received prior chemotherapy. A subsequent report with more mature data compared the overall survival of patients who received crizotinib in the phase I study to ALK-positive patients that were not enrolled and also ALK negative patients. Although not a randomised comparison, use of crizotinib was associated with improved survival compared to historical cohorts (64). It was also noted that the presence of an ALK fusion was not prognostic for survival in the absence of crizotinib.
Of the 1,500 patients screened for ALK fusions in the phase I study, only 5% were positive (62). In a similar fashion to EGFR mutations, some clinicopathologic characteristics predict a higher likelihood of ALK positivity, including young age, lack of smoking history and adenocarcinoma with solid, acinar or signet-ring histologic patterns. In an unselected population with NSCLC the frequency of ALK positivity is approximately 4% (62,65-68). ALK fusions are only very rarely found in lung cancers that have mutations in other oncogenes such as EGFR or KRAS (67).
Crizotinib has since been compared to standard second line chemotherapy in a multi-centre phase III randomised controlled trial in 342 patients with advanced ALK-positive lung cancer that had progressed after first line chemotherapy (69). Almost all of the patients in the standard arm received pemetrexed or docetaxel. The study was clearly positive for the primary endpoint with a median progression free survival of 7.7 months in the crizotinib arm and 3.0 months in the chemotherapy arm, shown in Figure 1 (HR 0.49, 95% CI: 0.37-0.64, P<0.0001) (69). Crizotinib also improved baseline symptoms and delayed subsequent worsening to a greater degree than chemotherapy in quality of life analyses. There was no overall survival benefit seen, most likely because at least 64% of patients in the chemotherapy arm subsequently received crizotinib. A phase III trial of crizotinib as first line treatment for ALK-positive lung cancer has recently completed accrual. Crizotinib has received regulatory approval in Europe and the United States. It is recommended by international guidelines that testing for the presence of an ALK fusion be considered for all patients with adenocarcinoma of the lung (23,70).
Figure 1.
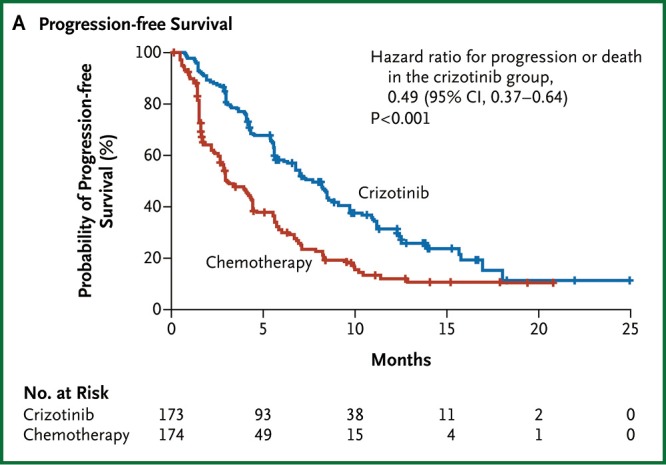
Progression free survival for second line crizotinib versus chemotherapy in ALK-positive NSCLC. From “Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013;368:2385-94. Copyright © 2013 Massachusetts Medical Society”. Reprinted with permission.
Crizotinib and ALK positive lung cancer is a unique example of the promise of targeted therapy. It has taken only 4 years from the original discovery of the EML4-ALK fusion in lung cancer to the FDA approval of crizotinib and its widespread clinical use for this indication.
Acquired resistance to crizotinib
With time, resistance to ALK inhibition with crizotinib is inevitable. The median progression free survival in the largest study of crizotinib was 7.7 months (69). In a similar fashion to EGFR TKIs, biopsy of progressing lesions in patients treated with crizotinib has provided insight into resistance mechanisms (71-74). Mutations in the ALK gene appear to mediate resistance in around one third of patients, although there is a much wider spectrum of mutations than that seen in EGFR-mutant lung cancer where T790M dominates as discussed previously. Activation of alternate signalling pathways involving EGFR and c-KIT (an oncogene targeted by imatinib) may also play a role in mediating resistance (71). In vitro studies suggest that targeting the alternative pathway with existing agents such as gefitinib in the case of EGFR or imatinib for c-KIT may reverse resistance to crizotinib (71). The mechanism of crizotinib resistance in ALK positive tumours currently remains unknown in around one third of cases (75). Of concern, multiple different resistance mechanisms may occur simultaneously in the same patient (71).
Next generation ALK inhibitors with different properties to crizotinib have been developed to have greater potency and potentially target resistance mutations. One agent CH5424802, has been tested in phase I and phase II trials in crizotinib naïve ALK-positive NSCLC, and is notable for the 93% overall response rate seen (76). Another agent LDK378 has shown efficacy in a phase I trial which included both crizotinib resistant and naïve ALK-positive NSCLC (77), with a response rate of 70%. LDK378 also appeared effective in the presence of resistant ALK mutations.
KRAS-mutant NSCLC
KRAS mutations occur in around 30% of NSCLC (73), making them the most common driver mutation seen in an unselected population. Adenocarcinomas make up the majority of NSCLC with KRAS mutations (78), and there is a positive association with smoking history (79). KRAS mutations may predict a lack of benefit from EGFR TKIs in patient with wild-type EGFR, but data have been conflicting (80-82). Despite much research, it has not proved possible to directly target KRAS, although recent progress has been made (83). Alternative strategies have involved targeting the down stream signalling pathway of KRAS (84), a role fulfilled by the MEK inhibitor selumetinib (85). In a randomised phase II trial of second line therapy in KRAS-mutant advanced NSCLC, selumetinib plus docetaxel was superior to docetaxel in response rate and progression free survival (86). Other approaches to targeting KRAS-mutant NSCLC in early phase trials include PIK3CA/mTOR/AKT pathway inhibitors in combination with MEK inhibitors to effectively block downstream KRAS signalling (87).
Other oncogenes in NSCLC
With the advent of next generation sequencing technology, driver oncogenes beyond EGFR, ALK and KRAS have been characterised in NSCLC, often at frequencies of less than 5% (88). As targeted therapies already exist for several of these altered genes and are in use in other cancer types, there is currently a focus on identifying lung cancer patients with these alterations and matching them to appropriate therapies within early phase trials (89). There are clear differences between squamous cell and adenocarcinoma histologies in terms of driver oncogenes (9,90), so these will be discussed separately. The pattern and frequency of alterations are summarised in Figure 2.
Figure 2.

Relative frequency of genomic alterations in adenocarcinoma and squamous cell carcinoma. Data adapted from multiple references (see text) and are estimates only.
Adenocarcinomas
ROS1 translocation
Fusion genes involving the receptor tyrosine kinase ROS1 have been found in 1-2% of NSCLC typically in never or light smokers with adenocarcinoma (91,92). This fusion is notable as it appears sensitive to inhibition with crizotinib (91,93), and defines a molecular subclass of lung cancers with clinical similarity to ALK-positive cancers.
MET amplification
MET is the gene for the hepatocyte growth factor receptor (HGFR). Activation of MET signalling is sufficient to transform cells to a malignant phenotype, and has effects on the cell cycle and survival. NSCLC cells commonly overexpress MET, and MET amplification is a defined pathway of resistance to EGFR TKIs (40-42,45). The monoclonal antibody onartuzumab (MetMAb) blocks binding of HGF to the MET receptor. It was combined with erlotinib in a randomised phase II trial in advanced NSCLC after failure of prior therapy. In patients with MET over-expression, combination therapy significantly prolonged overall survival from 4.6 to 12.6 months (HR 0.37, 95% CI: 0.2-0.71, P=0.002) compared to erlotinib alone. Tivantinib, a small molecule MET inhibitor was tested in a phase III trial in combination with erlotinib, but the study was closed early for futility (Press Release, ArQule Inc. and Daiichi Sankyo Co.).
BRAF mutations
BRAF is a well characterised driver mutation in metastatic melanoma, where it is treated with oral BRAF inhibitors such as vemurafenib or dabrafenib. A phase II trial of dabrafenib in BRAF mutant NSCLC is ongoing, with 7 out of the first 17 patients on trial demonstrating a partial response (94). The frequency of BRAF mutation in NSCLC is 1-5% (88,95,96), and appears to be at least equally as common in current or former smokers as non-smokers. The classic sensitising V600E mutation was only found in 50% of the BRAF mutant lung cancers, which may limit the use of currently available BRAF inhibitors (95).
HER2 amplification and mutations
HER2 amplification or mutation is known to exist in some lung cancers with a frequency of around 3% (97). Attempts at treating HER2 amplified NSCLC with the monoclonal anti-HER2 antibody trastuzumab were unsuccessful (98). HER2 mutation in exon 20 is a more promising molecular subgroup, and there exist several small molecule inhibitors of the HER2 tyrosine kinase such as afatinib or dacomitinib (99). There have been early reports of some responses to these drugs in patients with HER2 mutations (100), and trials are ongoing.
RET translocations
Fusions involving the receptor tyrosine kinase RET gene have recently been indentified in lung adenocarcinomas, and in vitro studies have confirmed the oncogenic potential of at least some of the identified fusions (101). The prevalence of RET rearrangements is estimated at between 1-2%, being higher in never or light smokers (92,101). The RET kinase inhibitor vandetanib (102) is a well established treatment for medullary thyroid carcinoma and may be a treatment option for RET positive adenocarcinoma of the lung.
PIK3CA mutation
PIK3CA is a known oncogene central to the phosphatidylinositide 3-kinase (PI3K) pathway that is deregulated in multiple cancer types (103). PIK3CA has been found altered in 1-2% of lung adenocarcinomas, and may co-exist with other mutant oncogenes (104-106). There is considerable effort to target this gene in other cancer types, and early phase trials are underway with PIK3CA targeted therapy for lung cancer both as monotherapy and in combination with other targeted agents and chemotherapy.
Squamous cell carcinomas
Recent progress has identified three potential therapeutic targets in squamous cell carcinoma of the lung. The fibroblast growth factor receptor 1 (FGFR1) is one such target, which is amplified in 21-22% of squamous cell carcinomas in recent studies (107,108). These studies also showed that FGFR1 amplified cells underwent apoptosis when treated with a small molecule FGFR1 inhibitor, and FGFR1 amplified tumours in mice shrank with inhibitor therapy, suggesting that FGFR1 is an important driver in some squamous cell carcinomas. Multiple small molecule inhibitors of FGFR1 are in development and entering early phase trials, with promising preliminary activity (109).
Mutations in the receptor tyrosine kinase DDR2 gene have been seen in 2% of squamous cell carcinomas of the lung (9,110). TKIs widely used in treating chronic myeloid leukaemia such as dasatinib also have activity against DDR2. Dasatinib has produced partial responses in some squamous NSCLC patients in phase I trials (111,112). In one of the patients with a response, sequencing of a tumour biopsy revealed a DDR2 mutation (110). Phase II trials of dasatinib specifically in squamous cell carcinoma of the lung are underway.
Alterations in genes playing a role in the PI3K pathway are present in 30-50% of squamous cell carcinomas, mostly comprising PIK3CA amplification and mutation, and deletion of the tumour suppressor gene PTEN (9,106). This pathway is important to maintaining cell survival and promoting growth (103), but the relationship between alterations in this pathway and response to inhibitors is complex. Phase I trials of PIK3CA inhibitors are underway in squamous NSCLC.
Targeting the tumour microenvironment
Angiogenesis in lung cancer
Angiogenesis has emerged as a broadly available target in multiple cancer types, as any sizeable tumour requires the ability to form a new blood supply to survive (113,114). The most well studied pathway mediating angiogenesis involves the vascular endothelial growth factor (VEGF) family of ligands and associated receptors which have intracellular tyrosine kinase domains that mediate downstream signalling (115). Targeting VEGF receptor tyrosine kinase signalling using small molecule inhibitors has generally proven unsuccessful, despite multiple agents having been tested in phase III trials (116-122). The VEGF and FGF receptor inhibitor nintedanib combined with chemotherapy has shown a marginal benefit of less than one month in progression free survival over chemotherapy alone, as second line treatment of advanced NSCLC in two phase III trials (123,124).
Bevacizumab is the most widely used anti-angiogenic agent in routine practice. It is a recombinant humanised monoclonal antibody that binds to VEGF, specifically the VEGF-A isoform, and prevents activation of the VEGF receptor (125). The Eastern Cooperative Oncology Group E4599 trial was performed in 878 patients with advanced NSCLC, and compared bevacizumab plus chemotherapy with carboplatin and paclitaxel to chemotherapy alone (126). Bevacizumab was continued as maintenance therapy until progression after 6 cycles of chemotherapy. Median overall survival was superior with bevacizumab at 12.3 versus 10.3 months (HR 0.79, 95% CI: 0.67-0.92; P=0.003). Progression free survival and response rate were also superior with bevacizumab in a second phase III trial AVAiL, although overall survival was no different (127). Toxicities of bevacizumab include arterial thromboembolism, hypertension, augmented chemotherapy-related haematological toxicity and bleeding (126). Due to the higher risk of significant haemoptysis, bevacizumab should not be used for squamous cell histology. Bevacizumab has not had widespread uptake as standard first line therapy outside of the United States due to concerns about toxicity, cost and the lack of a biomarker predictive of benefit.
Immunotherapy
Recent advances in tumour immunology have revealed that the immune system plays an important role in controlling malignant growth, and shapes the characteristics of the tumour that eventually manifests clinically (128). Harnessing the immune system as a therapeutic modality has already shown success in advanced melanoma (129) and prostate cancer (130). Although traditionally not considered to be an immunogenic tumour type, there is evidence that markers of a host immune response to lung cancer have a significant prognostic impact in both the adjuvant setting and advanced disease (131-134). Enhancing the immune response may therefore represent a rational therapeutic target. Immunotherapy in lung cancer consists primarily of two approaches: vaccines derived from lung cancer cell lines or tumour associated antigens, and immuno-stimulatory checkpoint antibodies.
Vaccines
Several vaccines have shown promising results in phase II trials, and are currently being evaluated in randomised phase III trials. The largest trials will be discussed here.
Belagenpumatucel-L is an irradiated whole cell product consisting of multiple lung cancer cell lines reflecting adenocarcinoma, large cell carcinoma and squamous cell carcinoma histologies together with an immuno-adjuvant (135). A small single arm phase II trial conducted in a mixed population of early stage and advanced lung cancer demonstrated radiological responses in 15% of patients with measurable disease and a positive correlation between prolonged overall survival and higher vaccine dose (135). Belagenpumatucel-L is being further evaluated in a phase III trial recruiting patients with stage III-IV disease that is stable or responding after first line therapy.
Other vaccines consist of antigens expressed exclusively or predominantly in lung cancer cells. Melanoma-associated antigen-A3 (MAGE-A3) is expressed in 35% of NSCLC (136), and has been prepared as a mono-antigenic vaccine. This was tested in a randomised placebo-controlled phase II trial following resection of stage I-II NSCLC showing cellular expression of MAGE-A3 (137). Following surgery, the disease free survival and overall survival were no different between vaccine and placebo groups, but there were numerically fewer recurrences in the vaccine group after a median of 44 months post surgery (35% versus 43% in placebo group). 2,270 patients have been recruited to a phase III trial of the MAGE-A3 vaccine, with results awaited.
MUC-1 is an epithelial cell protein that is differentially glycosylated in malignant cells (138) and overexpressed in NSCLC (139,140). The BLP25 vaccine contains the MUC-1 peptide and an immuno-adjuvant encased in a liposomal delivery system (141). In a phase III randomised trial comparing BLP25 to placebo after concurrent or sequential chemoradiotherapy for stage III NSCLC, patients who had received concurrent treatment showed a median overall survival of 30.8 months compared to 20.6 months with placebo (HR 0.78, 95% CI: 0.64-0.95; P=0.016) (142). BLP25 also prolonged survival in a phase II study in advanced NSCLC compared to best supportive care but this was not statistically significant (141). TG4010 is an alternative approach to MUC-1 vaccination, incorporating an attenuated but replication competent vaccinia virus that encodes for the MUC-1 protein and interleukin-2 (143). In a randomised phase II study, cisplatin and gemcitabine plus TG4010 was compared to cisplatin and gemcitabine alone in 148 patients with advanced NSCLC (144). Progression free survival at 6 months was 43% with the vaccine versus 35% without, but this difference was not statistically significant. Further studies with BLP25 and TG4010 are awaited.
Immune checkpoint blockade
Immune checkpoints refer to the molecular mechanisms that control T-cell responses to foreign antigens. Part of the immune checkpoint system encompasses stimulatory or suppressive co-receptors that modulate the interaction of the T-cell receptor (TCR) with human leukocyte antigen (HLA) expressed on the target cell. Two such receptors have emerged as important therapeutic targets in cancer. The cytotoxic T-lymphocyte antigen 4 (CTLA-4) receptor is expressed on T-cells following activation by antigen, and serves to dampen the T-cell response to promote self-tolerance and prevent autoimmune activation. Programmed cell death protein 1 (PD1) is also expressed on T-cells and similarly provides a mechanism for down-regulating the T-cell response if the ligand (programmed cell death 1 ligand 1 or PD-L1, also known as B7) is encountered. Preventing T-cell suppression at the tumour-immune interface by disrupting immunosuppressive signals forms a promising therapeutic strategy for advanced lung cancer that may also extend to adjuvant treatment.
The toxicities of the various immune checkpoint antibodies are similar and relate to autoimmune phenomena such as colitis, skin rash, pneumonitis and endocrinopathies. As these do not overlap with chemotherapy toxicity, combining these treatments with chemotherapy is a feasible approach. Ipilimumab is a humanised IgG1 anti-CTLA-4 receptor antibody, and is already an established therapy for advanced melanoma (129). A randomised placebo controlled trial was conducted comparing ipilimumab plus carboplatin and paclitaxel chemotherapy to placebo plus chemotherapy in 204 patients with advanced NSCLC (145). Ipilimumab was given in two schedules in the treatment arms: concurrent treatment starting from the first cycle of chemotherapy and phased treatment starting after two cycles of chemotherapy. In light of experience with melanoma that ipilimumab may cause an initial worsening in the radiological appearance of lesions used to assess progression free survival, modified immune-related radiological response criteria were used (146). The study was positive for the primary endpoint of immune-related progression free survival, which was 5.7 months in the phased treatment group compared to 4.6 months in the control group (HR 0.72, P=0.05). Efficacy was most pronounced in patients with squamous cell histology. A similar randomised phase II trial was carried out in 130 patients with extensive stage small cell lung cancer, and showed a trend towards improvement in immune-related progression free survival for the phased regimen in combination with chemotherapy compared to chemotherapy alone (6.4 versus 5.3 months; HR 0.64; 95% CI: 0.4-1.02; P=0.03) (147). Further trials for squamous cell lung cancer and small cell lung cancer are planned.
Multiple tumour types express the PD-L1 ligand on their cell surface, highlighting the role of the PD-1 receptor in suppressing anti-tumour T-cell responses (148). Monoclonal antibodies to both PD-1 and PD-L1 have been tested in several phase I trials that enrolled considerable numbers of patients with NSCLC (148,149). In one such trial the anti-PD-1 antibody nivolumab (formerly known as BMS-936558/MDX-1106) produced an unprecedented response rate of 18% amongst 129 NSCLC patients that were heavily pre-treated, with half of these patients having received three or more previous lines of therapy (148). In addition, the anti-PD-L1 antibody BMS-936559 produced response rates of 10% in a phase I trial that included 49 patients with NSCLC (149). The benefit was evident for both squamous cell carcinomas and adenocarcinomas. From these two trials there is early evidence that expression of the PD-L1 ligand in the tumour microenvironment, which can be evaluated with immunohistochemistry, may predict benefit from anti-PD-1/PD-L1 therapies. In addition to nivolumab, lambrolizumab is another anti-PD-1 antibody that has shown efficacy in melanoma and is being evaluated in lung cancer. Upcoming trials involving nivolumab and lambrolizumab are shown in Table 2.
Table 2. Upcoming trials of anti-PD-1 therapy in advanced NSCLC.
| Population | Treatment arms | Phase |
|---|---|---|
| Squamous cell carcinomas of the lung | Nivolumab versus Docetaxel | Phase III |
| Non-squamous carcinoma of the lung | Nivolumab versus Docetaxel | Phase III |
| All NSCLC, no previous therapy | Nivolumab monotherapy; Nivolumab + cisplatin/pemetrexed; Nivolumab + carboplatin/paclitaxel; Nivolumab + cisplatin/gemcitabine | Phase I |
| Standard first line chemotherapy followed by nivolumab and bevacizumab maintenance | Phase I | |
| Ipilimumab + nivolumab | Phase I | |
| EGFR-mutant NSCLC | Nivolumab + erlotinib | Phase I |
| All NSCLC | Lambrolizumab monotherapy; Lambrolizumab + standard chemotherapy; Lambrolizumab in NSCLC overexpressing PD-L1 | Phase I |
Conclusions
The last ten years have seen a revolution in the way that lung cancer is conceptualised and treated, born out by advances in genomics, cell biology and drug development technologies. The same advances that facilitated this revolution will continue to provide a roadmap for ongoing improvements by identifying new targets and defining the mechanisms of treatment failure and resistance. The transition of crizotinib from an investigational compound to an approved therapy in a mere 4 years also provides hope that there will be a rapid expansion in therapeutic options available to patients in the near feature. Similarly, immunotherapy represents an entirely new class of agents with a promising efficacy and toxicity profile. With the arrival of targeted therapy come multiple challenges however. The development of targeted therapies is often at odds with the traditional clinical trial structure required by regulatory authorities, where phase III trials illustrating an overall survival benefit are considered the gold standard. In addition, targeted therapies carry high costs to the patient or funding agency, and the long term economic viability of the current drug development cycle is uncertain. Finally, it is still the case that the majority of patients with advanced lung cancer have no targeted therapy available to them at the current time, either due to a lack of known targets in their tumour or poor access to novel agents. Addressing both these issues will remain a priority if the successes of the past decade are to be maintained.
Acknowledgements
Disclosure: Brett Hughes has served on Advisory Boards for Roche, Pfizer and Boehringer Inglheim. Benjamin Solomon has served on Advisory Boards for Roche, Pfizer, Novartis, Astra Zeneca, Eli Lilly, Clovis Oncology and Boehringer Ingelheim.
References
- 1.Fukuoka M, Yano S, Giaccone G, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected] J Clin Oncol 2003;21:2237-46 [DOI] [PubMed] [Google Scholar]
- 2.Kris MG, Natale RB, Herbst RS, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA 2003;290:2149-58 [DOI] [PubMed] [Google Scholar]
- 3.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129-39 [DOI] [PubMed] [Google Scholar]
- 4.Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004;304:1497-500 [DOI] [PubMed] [Google Scholar]
- 5.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 2004;101:13306-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller VA, Kris MG, Shah N, et al. Bronchioloalveolar pathologic subtype and smoking history predict sensitivity to gefitinib in advanced non-small-cell lung cancer. J Clin Oncol 2004;22:1103-9 [DOI] [PubMed] [Google Scholar]
- 7.Marchetti A, Martella C, Felicioni L, et al. EGFR mutations in non-small-cell lung cancer: analysis of a large series of cases and development of a rapid and sensitive method for diagnostic screening with potential implications on pharmacologic treatment. J Clin Oncol 2005;23:857-65 [DOI] [PubMed] [Google Scholar]
- 8.Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361:958-67 [DOI] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012;489:519-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947-57 [DOI] [PubMed] [Google Scholar]
- 11.Han JY, Park K, Kim SW, et al. First-SIGNAL: first-line single-agent iressa versus gemcitabine and cisplatin trial in never-smokers with adenocarcinoma of the lung. J Clin Oncol 2012;30:1122-8 [DOI] [PubMed] [Google Scholar]
- 12.Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010;11:121-8 [DOI] [PubMed] [Google Scholar]
- 13.Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 2010;362:2380-8 [DOI] [PubMed] [Google Scholar]
- 14.Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2011;12:735-42 [DOI] [PubMed] [Google Scholar]
- 15.Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239-46 [DOI] [PubMed] [Google Scholar]
- 16.Sequist LV, Yang JC, Yamamoto N, et al. Phase III Study of Afatinib or Cisplatin Plus Pemetrexed in Patients With Metastatic Lung Adenocarcinoma With EGFR Mutations. J Clin Oncol. 2013 doi: 10.1200/JCO.2012.44.2806. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 17.Wu YL, Zhou C, Hu CP, et al. LUX-Lung 6: A randomized, open-label, phase III study of afatinib versus gemcitabine/cisplatin as first-line treatment for Asian patients with EGFR mutation-positive advanced adenocarcinoma of the lung. J Clin Oncol 2013;31:abstr 8016.
- 18.Fukuoka M, Wu YL, Thongprasert S, et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J Clin Oncol 2011;29:2866-74 [DOI] [PubMed] [Google Scholar]
- 19.Inoue A, Kobayashi K, Maemondo M, et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann Oncol 2013;24:54-9 [DOI] [PubMed] [Google Scholar]
- 20.Mitsudomi T, Morita S, Yatabe Y, et al. Updated overall survival results of WJTOG 3405, a randomized phase III trial comparing gefitinib with cisplatin plus docetaxel as the first-line treatment for patients with non-small cell lung cancer harboring mutations of the epidermal growth factor rece. J Clin Oncol 2012;30:abstr 7521.
- 21.Keedy VL, Temin S, Somerfield MR, et al. American Society of Clinical Oncology provisional clinical opinion: epidermal growth factor receptor (EGFR) Mutation testing for patients with advanced non-small-cell lung cancer considering first-line EGFR tyrosine kinase inhibitor therapy. J Clin Oncol 2011;29:2121-7 [DOI] [PubMed] [Google Scholar]
- 22.Dogan S, Shen R, Ang DC, et al. Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin Cancer Res 2012;18:6169-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peters S, Adjei AA, Gridelli C, et al. Metastatic non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012;23Suppl 7:vii56-64 [DOI] [PubMed] [Google Scholar]
- 24.Ettinger DS, Akerley W, Bepler G, et al. Non-small cell lung cancer. J Natl Compr Canc Netw 2010;8:740-801 [DOI] [PubMed] [Google Scholar]
- 25.Mok T, Yang JJ, Lam KC. Treating patients with EGFR-sensitizing mutations: first line or second line--is there a difference? J Clin Oncol 2013;31:1081-8 [DOI] [PubMed] [Google Scholar]
- 26.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 2005;353:123-32 [DOI] [PubMed] [Google Scholar]
- 27.Thatcher N, Chang A, Parikh P, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 2005;366:1527-37 [DOI] [PubMed] [Google Scholar]
- 28.Garassino M, Martelli O, Bettini A, et al. TAILOR: A phase III trial comparing erlotinib with docetaxel as the second-line treatment of NSCLC patients with wild-type EGFR. J Clin Oncol 2012;30:abtract LBA7501.
- 29.Kim ES, Hirsh V, Mok T, et al. Gefitinib versus docetaxel in previously treated non-small-cell lung cancer (INTEREST): a randomised phase III trial. Lancet 2008;372:1809-18 [DOI] [PubMed] [Google Scholar]
- 30.Cappuzzo F, Ciuleanu T, Stelmakh L, et al. Erlotinib as maintenance treatment in advanced non-small-cell lung cancer: a multicentre, randomised, placebo-controlled phase 3 study. Lancet Oncol 2010;11:521-9 [DOI] [PubMed] [Google Scholar]
- 31.Ciuleanu T, Stelmakh L, Cicenas S, et al. Efficacy and safety of erlotinib versus chemotherapy in second-line treatment of patients with advanced, non-small-cell lung cancer with poor prognosis (TITAN): a randomised multicentre, open-label, phase 3 study. Lancet Oncol 2012;13:300-8 [DOI] [PubMed] [Google Scholar]
- 32.Gridelli C, Ciardiello F, Gallo C, et al. First-line erlotinib followed by second-line cisplatin-gemcitabine chemotherapy in advanced non-small-cell lung cancer: the TORCH randomized trial. J Clin Oncol 2012;30:3002-11 [DOI] [PubMed] [Google Scholar]
- 33.Miller VA, Hirsh V, Cadranel J, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol 2012;13:528-38 [DOI] [PubMed] [Google Scholar]
- 34.Ramalingam SS, Blackhall F, Krzakowski M, et al. Randomized phase II study of dacomitinib (PF-00299804), an irreversible pan-human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol 2012;30:3337-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kurai J, Chikumi H, Hashimoto K, et al. Antibody-dependent cellular cytotoxicity mediated by cetuximab against lung cancer cell lines. Clin Cancer Res 2007;13:1552-61 [DOI] [PubMed] [Google Scholar]
- 36.Pirker R, Pereira JR, Szczesna A, et al. Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX): an open-label randomised phase III trial. Lancet 2009;373:1525-31 [DOI] [PubMed] [Google Scholar]
- 37.Lynch TJ, Patel T, Dreisbach L, et al. Cetuximab and first-line taxane/carboplatin chemotherapy in advanced non-small-cell lung cancer: results of the randomized multicenter phase III trial BMS099. J Clin Oncol 2010;28:911-7 [DOI] [PubMed] [Google Scholar]
- 38.Pirker R, Pereira JR, von Pawel J, et al. EGFR expression as a predictor of survival for first-line chemotherapy plus cetuximab in patients with advanced non-small-cell lung cancer: analysis of data from the phase 3 FLEX study. Lancet Oncol 2012;13:33-42 [DOI] [PubMed] [Google Scholar]
- 39.Khambata-Ford S, Harbison CT, Hart LL, et al. Analysis of potential predictive markers of cetuximab benefit in BMS099, a phase III study of cetuximab and first-line taxane/carboplatin in advanced non-small-cell lung cancer. J Clin Oncol 2010;28:918-27 [DOI] [PubMed] [Google Scholar]
- 40.Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 2007;104:20932-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 2011;3:75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arcila ME, Oxnard GR, Nafa K, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res 2011;17:1169-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res 2013;19:2240-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chmielecki J, Foo J, Oxnard GR, et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci Transl Med 2011;3:90ra59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316:1039-43 [DOI] [PubMed] [Google Scholar]
- 46.Ohashi K, Sequist LV, Arcila ME, et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A 2012;109:E2127-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takezawa K, Pirazzoli V, Arcila ME, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov 2012;2:922-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Z, Lee JC, Lin L, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet 2012;44:852-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li D, Ambrogio L, Shimamura T, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008;27:4702-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Janjigian YY, Groen HJM, Horn L, et al. Activity and tolerability of afatinib (BIBW 2992) and cetuximab in NSCLC patients with acquired resistance to erlotinib or gefitinib. J Clin Oncol 2011;29:abstr 7525.
- 51.Sequist L V, Soria JC, Gadgeel SM, et al. First-in-human evaluation of CO-1686, an irreversible, selective, and potent tyrosine kinase inhibitor of EGFR T790M. J Clin Oncol 2013;31:abstr 2524.
- 52.Rivera VM, Wang F, Anjum R, et al. AP26113 is a dual ALK/EGFR inhibitor: Characterization against EGFR T790M in cell and mouse models of NSCLC. Cancer Res 2012;72:abstr 1794.
- 53.Wang W, Li Q, Takeuchi S, et al. Met kinase inhibitor E7050 reverses three different mechanisms of hepatocyte growth factor-induced tyrosine kinase inhibitor resistance in EGFR mutant lung cancer. Clin Cancer Res 2012;18:1663-71 [DOI] [PubMed] [Google Scholar]
- 54.Nakagawa T, Takeuchi S, Yamada T, et al. Combined therapy with mutant-selective EGFR inhibitor and Met kinase inhibitor for overcoming erlotinib resistance in EGFR-mutant lung cancer. Mol Cancer Ther 2012;11:2149-57 [DOI] [PubMed] [Google Scholar]
- 55.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561-6 [DOI] [PubMed] [Google Scholar]
- 56.Rikova K, Guo A, Zeng Q, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007;131:1190-203 [DOI] [PubMed] [Google Scholar]
- 57.Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994;263:1281-4 [DOI] [PubMed] [Google Scholar]
- 58.McDermott U, Iafrate AJ, Gray NS, et al. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res 2008;68:3389-95 [DOI] [PubMed] [Google Scholar]
- 59.Soda M, Takada S, Takeuchi K, et al. A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci U S A 2008;105:19893-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koivunen JP, Mermel C, Zejnullahu K, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res 2008;14:4275-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cui JJ, Tran-Dubé M, Shen H, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J Med Chem 2011;54:6342-63 [DOI] [PubMed] [Google Scholar]
- 62.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010;363:1693-703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol 2012;13:1011-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shaw AT, Yeap BY, Solomon BJ, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol 2011;12:1004-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009;27:4247-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rodig SJ, Mino-Kenudson M, Dacic S, et al. Unique clinicopathologic features characterize ALK-rearranged lung adenocarcinoma in the western population. Clin Cancer Res 2009;15:5216-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wong DW, Leung EL, So KK, et al. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer 2009;115:1723-33 [DOI] [PubMed] [Google Scholar]
- 68.Zhang YG, Jin ML, Li L, et al. Evaluation of ALK rearrangement in Chinese non-small cell lung cancer using FISH, immunohistochemistry, and real-time quantitative RT-PCR on paraffin-embedded tissues. PLoS One 2013. May;8:e64821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus Chemotherapy in Advanced ALK-Positive Lung Cancer. N Engl J Med 2013;368:2385-94 [DOI] [PubMed] [Google Scholar]
- 70.Lindeman NI, Cagle PT, Beasley MB, et al. Molecular Testing Guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Mol Diagn 2013;15:415-53 [DOI] [PubMed] [Google Scholar]
- 71.Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med 2012;4:120ra17. [DOI] [PMC free article] [PubMed]
- 72.Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res 2012;18:1472-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gainor JF, Varghese AM, Ou SH, et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res 2013;19:4273-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim S, Kim TM, Kim DW, et al. Heterogeneity of genetic changes associated with acquired crizotinib resistance in ALK-rearranged lung cancer. J Thorac Oncol 2013;8:415-22 [DOI] [PubMed] [Google Scholar]
- 75.Shaw AT, Engelman JA. ALK in lung cancer: past, present, and future. J Clin Oncol 2013;31:1105-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Seto T, Kiura K, Nishio M, et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1-2 study. Lancet Oncol 2013;14:590-8 [DOI] [PubMed] [Google Scholar]
- 77.Shaw AT, Mehra R, Kim DW, et al. Clinical activity of the ALK inhibitor LDK378 in advanced, ALK-positive NSCLC. J Clin Oncol 2013;31:abstr 8010.
- 78.Graziano SL, Gamble GP, Newman NB, et al. Prognostic significance of K-ras codon 12 mutations in patients with resected stage I and II non-small-cell lung cancer. J Clin Oncol 1999;17:668-75 [DOI] [PubMed] [Google Scholar]
- 79.Slebos RJ, Kibbelaar RE, Dalesio O, et al. K-ras oncogene activation as a prognostic marker in adenocarcinoma of the lung. N Engl J Med 1990;323:561-5 [DOI] [PubMed] [Google Scholar]
- 80.Linardou H, Dahabreh IJ, Kanaloupiti D, et al. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol 2008;9:962-72 [DOI] [PubMed] [Google Scholar]
- 81.Mao C, Qiu LX, Liao RY, et al. KRAS mutations and resistance to EGFR-TKIs treatment in patients with non-small cell lung cancer: a meta-analysis of 22 studies. Lung Cancer 2010;69:272-8 [DOI] [PubMed] [Google Scholar]
- 82.Sun JM, Hwang DW, Ahn JS, et al. Prognostic and predictive value of KRAS mutations in advanced non-small cell lung cancer. PLoS One 2013;8:e64816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zimmermann G, Papke B, Ismail S, et al. Small molecule inhibition of the KRAS-PDEδ interaction impairs oncogenic KRAS signalling. Nature 2013;497:638-42 [DOI] [PubMed] [Google Scholar]
- 84.Wang Y, Kaiser CE, Frett B, et al. Targeting Mutant KRAS for Anticancer Therapeutics: A Review of Novel Small Molecule Modulators. J Med Chem. 2013 doi: 10.1021/jm3017706. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yeh TC, Marsh V, Bernat BA, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res 2007;13:1576-83 [DOI] [PubMed] [Google Scholar]
- 86.Jänne PA, Shaw AT, Pereira JR, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol 2013;14:38-47 [DOI] [PubMed] [Google Scholar]
- 87.Khan KH, Yan L, Mezynski J, et al. A phase I dose escalation study of oral MK-2206 (allosteric Akt inhibitor) with oral selumetinib (AZD6244; ARRY-142866) (MEK 1/2 inhibitor) in patients with advanced or metastatic solid tumors. J Clin Oncol 2012;30:abstr e13599.
- 88.Barlesi F, Blons H, Beau-Faller M, et al. Biomarkers France: Results of routine EGFR, HER2, KRAS, BRAF, PI3KCA mutations detection and EML4-ALK gene fusion assessment on the first 10,000 non-small cell lung cancer patients. J Clin Oncol 2013;31:abstr 8000.
- 89.Kim ES, Herbst RS, Wistuba II, et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov 2011;1:44-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Imielinski M, Berger AH, Hammerman PS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012;150:1107-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol 2012;30:863-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Takeuchi K, Soda M, Togashi Y, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med 2012;18:378-81 [DOI] [PubMed] [Google Scholar]
- 93.Awad MM, Katayama R, McTigue M, et al. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med 2013;368:2395-401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Planchard D, Mazieres J, Riely GJ, et al. Interim results of phase II study BRF113928 of dabrafenib in BRAF V600E mutation-positive non-small cell lung cancer (NSCLC) patients. J Clin Oncol 2013;31:abstr 8009.
- 95.Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 2011;29:2046-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Marchetti A, Felicioni L, Malatesta S, et al. Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol 2011;29:3574-9 [DOI] [PubMed] [Google Scholar]
- 97.Shigematsu H, Takahashi T, Nomura M, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res 2005;65:1642-6 [DOI] [PubMed] [Google Scholar]
- 98.Clamon G, Herndon J, Kern J, et al. Lack of trastuzumab activity in nonsmall cell lung carcinoma with overexpression of erb-B2: 39810: a phase II trial of Cancer and Leukemia Group B. Cancer 2005;103:1670-5 [DOI] [PubMed] [Google Scholar]
- 99.Kwak E.The role of irreversible HER family inhibition in the treatment of patients with non-small cell lung cancer. Oncologist 2011;16:1498-507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.De Grève J, Teugels E, Geers C, et al. Clinical activity of afatinib (BIBW 2992) in patients with lung adenocarcinoma with mutations in the kinase domain of HER2/neu. Lung Cancer 2012;76:123-7 [DOI] [PubMed] [Google Scholar]
- 101.Lipson D, Capelletti M, Yelensky R, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med 2012;18:382-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wells SA, Jr, Robinson BG, Gagel RF, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol 2012;30:134-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 2009;9:550-62 [DOI] [PubMed] [Google Scholar]
- 104.Garnett MJ, Edelman EJ, Heidorn SJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012;483:570-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chaft JE, Arcila ME, Paik PK, et al. Coexistence of PIK3CA and other oncogene mutations in lung adenocarcinoma-rationale for comprehensive mutation profiling. Mol Cancer Ther 2012;11:485-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yamamoto H, Shigematsu H, Nomura M, et al. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res 2008;68:6913-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Weiss J, Sos ML, Seidel D, et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med 2010;2:62ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dutt A, Ramos AH, Hammerman PS, et al. Inhibitor-sensitive FGFR1 amplification in human non-small cell lung cancer. PLoS One 2011;6:e20351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wolf J, LoRusso PM, Camidge RD, et al. A phase I dose escalation study of NVP-BGJ398, a selective pan FGFR inhibitor in genetically preselected advanced solid tumors. Cancer Res 2012;72:abstr nr LB-122.
- 110.Hammerman PS, Sos ML, Ramos AH, et al. Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discov 2011;1:78-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Haura EB, Tanvetyanon T, Chiappori A, et al. Phase I/II study of the Src inhibitor dasatinib in combination with erlotinib in advanced non-small-cell lung cancer. J Clin Oncol 2010;28:1387-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Johnson FM, Bekele BN, Feng L, et al. Phase II study of dasatinib in patients with advanced non-small-cell lung cancer. J Clin Oncol 2010;28:4609-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Folkman J.Anti-angiogenesis: new concept for therapy of solid tumors. Ann Surg 1972;175:409-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646-74 [DOI] [PubMed] [Google Scholar]
- 115.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med 2003;9:669-76 [DOI] [PubMed] [Google Scholar]
- 116.Paz-Ares LG, Biesma B, Heigener D, et al. Phase III, randomized, double-blind, placebo-controlled trial of gemcitabine/cisplatin alone or with sorafenib for the first-line treatment of advanced, nonsquamous non-small-cell lung cancer. J Clin Oncol 2012;30:3084-92 [DOI] [PubMed] [Google Scholar]
- 117.Scagliotti G, Novello S, von Pawel J, et al. Phase III study of carboplatin and paclitaxel alone or with sorafenib in advanced non-small-cell lung cancer. J Clin Oncol 2010;28:1835-42 [DOI] [PubMed] [Google Scholar]
- 118.Scagliotti GV, Krzakowski M, Szczesna A, et al. Sunitinib plus erlotinib versus placebo plus erlotinib in patients with previously treated advanced non-small-cell lung cancer: a phase III trial. J Clin Oncol 2012;30:2070-8 [DOI] [PubMed] [Google Scholar]
- 119.Lee JS, Hirsh V, Park K, et al. Vandetanib Versus placebo in patients with advanced non-small-cell lung cancer after prior therapy with an epidermal growth factor receptor tyrosine kinase inhibitor: a randomized, double-blind phase III trial (ZEPHYR). J Clin Oncol 2012;30:1114-21 [DOI] [PubMed] [Google Scholar]
- 120.De Boer RH, Arrieta Ó, Yang CH, et al. Vandetanib plus pemetrexed for the second-line treatment of advanced non-small-cell lung cancer: a randomized, double-blind phase III trial. J Clin Oncol 2011;29:1067-74 [DOI] [PubMed] [Google Scholar]
- 121.Natale RB, Thongprasert S, Greco FA, et al. Phase III trial of vandetanib compared with erlotinib in patients with previously treated advanced non-small-cell lung cancer. J Clin Oncol 2011;29:1059-66 [DOI] [PubMed] [Google Scholar]
- 122.Scagliotti GV, Vynnychenko I, Park K, et al. International, randomized, placebo-controlled, double-blind phase III study of motesanib plus carboplatin/paclitaxel in patients with advanced nonsquamous non-small-cell lung cancer: MONET1. J Clin Oncol 2012;30:2829-36 [DOI] [PubMed] [Google Scholar]
- 123.Reck M, Kaiser R, Mellemgaard A, et al. Nintedanib (BIBF 1120) plus docetaxel in NSCLC patients progressing after first-line chemotherapy: LUME Lung 1, a randomized, double-blind phase III trial. J Clin Oncol 2013;31:abstr LBA8011.
- 124.Hanna NH, Kaiser R, Sullivan RN, et al. Lume-lung 2: A multicenter, randomized, double-blind, phase III study of nintedanib plus pemetrexed versus placebo plus pemetrexed in patients with advanced nonsquamous non-small cell lung cancer (NSCLC) after failure of first-line chemotherapy. J Clin Oncol 2013;31:abstr 8034.
- 125.Ferrara N, Hillan KJ, Gerber HP, et al. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov 2004;3:391-400 [DOI] [PubMed] [Google Scholar]
- 126.Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med 2006;355:2542-50 [DOI] [PubMed] [Google Scholar]
- 127.Reck M, von Pawel J, Zatloukal P, et al. Overall survival with cisplatin-gemcitabine and bevacizumab or placebo as first-line therapy for nonsquamous non-small-cell lung cancer: results from a randomised phase III trial (AVAiL). Ann Oncol 2010;21:1804-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 2011;331:1565-70 [DOI] [PubMed] [Google Scholar]
- 129.Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010;363:411-22 [DOI] [PubMed] [Google Scholar]
- 131.Al-Shibli KI, Donnem T, Al-Saad S, et al. Prognostic effect of epithelial and stromal lymphocyte infiltration in non-small cell lung cancer. Clin Cancer Res 2008;14:5220-7 [DOI] [PubMed] [Google Scholar]
- 132.Kawai O, Ishii G, Kubota K, et al. Predominant infiltration of macrophages and CD8(+) T Cells in cancer nests is a significant predictor of survival in stage IV nonsmall cell lung cancer. Cancer 2008;113:1387-95 [DOI] [PubMed] [Google Scholar]
- 133.Petersen RP, Campa MJ, Sperlazza J, et al. Tumor infiltrating Foxp3+ regulatory T-cells are associated with recurrence in pathologic stage I NSCLC patients. Cancer 2006;107:2866-72 [DOI] [PubMed] [Google Scholar]
- 134.Tao H, Mimura Y, Aoe K, et al. Prognostic potential of FOXP3 expression in non-small cell lung cancer cells combined with tumor-infiltrating regulatory T cells. Lung Cancer 2012;75:95-101 [DOI] [PubMed] [Google Scholar]
- 135.Nemunaitis J, Dillman RO, Schwarzenberger PO, et al. Phase II study of belagenpumatucel-L, a transforming growth factor beta-2 antisense gene-modified allogeneic tumor cell vaccine in non-small-cell lung cancer. J Clin Oncol 2006;24:4721-30 [DOI] [PubMed] [Google Scholar]
- 136.Sienel W, Varwerk C, Linder A, et al. Melanoma associated antigen (MAGE)-A3 expression in Stages I and II non-small cell lung cancer: results of a multi-center study. Eur J Cardiothorac Surg 2004;25:131-4 [DOI] [PubMed] [Google Scholar]
- 137.Vansteenkiste J, Zielinski M, Linder A, et al. Adjuvant MAGE-A3 Immunotherapy in Resected Non-Small-Cell Lung Cancer: Phase II Randomized Study Results. J Clin Oncol 2013;31:2396-403 [DOI] [PubMed] [Google Scholar]
- 138.Gendler SJ, Lancaster CA, Taylor-Papadimitriou J, et al. Molecular cloning and expression of human tumor-associated polymorphic epithelial mucin. J Biol Chem 1990;265:15286-93 [PubMed] [Google Scholar]
- 139.Rochlitz C, Figlin R, Squiban P, et al. Phase I immunotherapy with a modified vaccinia virus (MVA) expressing human MUC1 as antigen-specific immunotherapy in patients with MUC1-positive advanced cancer. J Gene Med 2003;5:690-9 [DOI] [PubMed] [Google Scholar]
- 140.Ho SB, Niehans GA, Lyftogt C, et al. Heterogeneity of mucin gene expression in normal and neoplastic tissues. Cancer Res 1993;53:641-51 [PubMed] [Google Scholar]
- 141.Butts C, Murray N, Maksymiuk A, et al. Randomized phase IIB trial of BLP25 liposome vaccine in stage IIIB and IV non-small-cell lung cancer. J Clin Oncol 2005;23:6674-81 [DOI] [PubMed] [Google Scholar]
- 142.Butts CA, Socinski MA, Mitchell P, et al. START: A phase III study of L-BLP25 cancer immunotherapy for unresectable stage III non-small cell lung cancer. J Clin Oncol 2013;31:abstr 7500.
- 143.Ramlau R, Quoix E, Rolski J, et al. A phase II study of Tg4010 (Mva-Muc1-Il2) in association with chemotherapy in patients with stage III/IV Non-small cell lung cancer. J Thorac Oncol 2008;3:735-44 [DOI] [PubMed] [Google Scholar]
- 144.Quoix E, Ramlau R, Westeel V, et al. Therapeutic vaccination with TG4010 and first-line chemotherapy in advanced non-small-cell lung cancer: a controlled phase 2B trial. Lancet Oncol 2011;12:1125-33 [DOI] [PubMed] [Google Scholar]
- 145.Lynch TJ, Bondarenko I, Luft A, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol 2012;30:2046-54 [DOI] [PubMed] [Google Scholar]
- 146.Wolchok JD, Hoos A, O’Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res 2009;15:7412-20 [DOI] [PubMed] [Google Scholar]
- 147.Reck M, Bondarenko I, Luft A, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol 2013;24:75-83 [DOI] [PubMed] [Google Scholar]
- 148.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455-65 [DOI] [PMC free article] [PubMed] [Google Scholar]