

Abstract
Background
Studies using monocyte-derived macrophages (MDM) and animal models have suggested a role for alternatively-activated (M2) macrophages in asthmatic inflammation, but in vivo evidence for this phenotype in human asthma is lacking.
Objective
Phenotypically to characterize lung macrophages from asthmatic patients in relation to disease severity and treatment.
Methods
M2 biomarkers were first identified using MDM exposed to Th2 cytokines and then used to phenotype sputum and bronchoalveolar lavage (BAL) macrophages from 12 healthy controls, 12 mild and 14 moderate asthmatics and to assess the effects of corticosteroids and phosphatidylinositol-3-kinase (PI3K) inhibitors.
Results
Sputum macrophages from asthmatics expressed significantly more CCL17 mRNA but less CD163 than macrophages from healthy individuals. However, none of the other M2 biomarkers were differentially expressed in asthma and ex vivo BAL cells spontaneously produced similar amounts of M2 cytokine/chemokines (IL-10, CCL17 and CCL22). CCL17 mRNA over-expression correlated weakly but significantly with sputum eosinophilia (p=0.0252) and was also observed in macrophages from moderate asthmatics treated with inhaled steroids, suggesting relative insensitivity to inhibition by corticosteroids. The PI3Kinase inhibitor LY294002 inhibited basal CCL17 release from BAL cells and IL-4-stimulated release from MDM.
Conclusions
This study does not support the existence in human asthma of the full M2 phenotype described to date, but points to upregulation of CCL17 in both mild and moderate asthma, providing a further source for this ligand of CCR4+ cells that contribute to airways inflammation. CCL17 expression is corticosteroid resistant but is suppressed by PI3Kinase enzyme inhibitors.
Introduction
Asthma is a complex airways inflammatory disease involving several cell types, with most research focusing on eosinophils and CD4+ T helper (Th)-2 cells1, 2, the latter being an important source of cytokines IL-4, IL-5 and IL-13, key drivers of responses to allergen3. The role of macrophages in driving allergic airways disease has been largely overlooked4 even though they are the prevalent immune cell type in the lungs.
Macrophages have been broadly characterised as either classically activated (M1) or alternatively activated (M2), based on phenotypes observed when macrophages are cultured in vitro in the presence of LPS and IFNγ (M1) or IL-4 or IL-13 (M2)5. M2 macrophages generally express increased levels of receptors involved in phagocytosis, such as CD2066, CD1637 and macrophage galactose C-type lectin (CLEC10A/CD301)8, as well as important Th2 cell chemokines, including the CCR4 ligands CCL17 and CCL225. Recent studies using animal models5, 9 and human monocytes10 have suggested a role for M2 macrophages in allergic lung inflammation, but evidence of a similar phenotype being relevant to human asthma has been lacking and it is recognised that there are differences between human and murine M2 expression profiles11.
Given that macrophages are the most numerous inflammatory cell in the airways where Th2 cytokines are increased, we postulated that lung macrophages from asthmatics are of the M2 phenotype. We also hypothesised that macrophages could be a major source of chemokines that attract CCR4+ cells which we and others have shown as potentially playing a role in asthma12,13. Our previous work has demonstrated that CCR4+ T lymphocytes are a major source of Th2 cytokines12 and that their recruitment into asthmatic airways is controlled by the CCR4 ligands, CCL17 and CCL22. However, the source of these chemokines has not been fully elucidated, with airway dendritic cells and epithelial cells being implicated to date14-17.
In the current study we first identified a panel of M2 biomarkers using macrophages derived from monocytes (MDM) cultured in M2-polarising conditions; these biomarkers were then used to phenotype sputum and BAL macrophages from mild, steroid-naive and moderate, steroid-treated asthmatics and nonatopic controls. In addition to studying M1 (CD14, TNF) and M2 (Ccl17, Clec10a, Arg1, IL-10, IL-13, CD163, CD206, CLEC10A/CD301) biomarkers at the level of mRNA, intracellular protein and cell surface expression we also assessed the potential of airway macrophages recovered by BAL to produce a series of cytokines/chemokines that have been implicated in asthma pathogenesis and have been reported as defining the M1 (TNF, IL-12, IL-6 and IL-8) and M2 (CCL17, CCL22, IL-10) phenotypes5. Having found that the frequency of lung macrophages expressing CCL17 is increased in both steroid-naive and steroid-treated asthmatics, we studied the effects of the corticosteroid, fluticasone propionate, and inhibitors of phosphatidylinositol-3-kinase (PI3Kinase) on ex vivo CCL17 production by MDM and BAL macrophages to explore their therapeutic potential.
Methods
Subjects
12 mild atopic asthmatics taking short-acting β-agonists alone (MA), 14 moderate atopic asthmatics requiring inhaled corticosteroids for disease control (MO), classified according to GINA criteria (www.ginasthma.org), and 12 healthy non-atopic control subjects (HC) were studied (Table 1). All subjects were non-smokers with no respiratory infections for 6 weeks prior to the study. Atopy was assessed using skin tests to common aeroallergens. The study was approved by the Southampton and South West Hampshire Research Ethics Committee (reference: 08/H0504/138).
Table 1. Baseline characteristics of healthy control (HC), mild asthmatic (MA) and moderate asthmatic (MO) volunteers.
Data are expressed as median values (IQr) to 2 d. p. N.D. indicates not determined. Data were analysed using a Kruskal-Wallis test followed by a Dunn’s Multiple Comparisons test. PC20 and ACQ data were analysed using a Mann-Whitney U test.
| HC | MA | MO | |
|---|---|---|---|
| N | 12 | 12 | 14 |
| Sex (M/F) | 5/7 | 6/6 | 6/8 |
| Age | 23.5 (22 – 31) | 24 (21 – 29.5) | 44 (31.5 – 49)*** ¶ |
| Atopy | No | Yes | Yes |
| FEV1, % predicted | 110.5 (102.3 – 120.3) | 99.75 (88.6 – 109.7) | 92.10 (80.35 – 105)*** |
| FEV1/FVC | 0.85 (0.79 – 0.88) | 0.80 (0.72 – 0.83) | 0.78 (0.70 – 0.85) |
| Methacholine PC20 | Not Achieved | 2.59 (0.57 – 5.22) | 1.14 (0.36 – 5.69) |
| Inhaled corticosteroids | No | No | Yes |
| ACQ score | N.D. | 0.43 (0.15 – 0.71) | 1.07 (0.43 – 1.57)† |
| Sputum cells, % | |||
| Macrophages | 59.25 (46.78 – 73.15) | 48.8 (33.5 – 61.9) | 48.8 (42.9 – 76.05) |
| Neutrophils | 34.9 (17.75 – 44) | 34.15 (19.4 – 54.65) | 36.0 (18.28 – 50.25) |
| Eosinophils | 0.38 (0.0 – 0.98) | 1.38 (0.75 – 5.5)** | 2.5 (0.63 – 8.15)** |
| Lymphocytes | 0.25 (0.0 – 0.50) | 0.25 (0.0 – 0.75) | 0.25 (0.0 – 0.5) |
| Epithelial cells | 3.80 (0.75 – 8.30) | 4.65 (1.0 – 7.75) | 2.13 (1.0 – 9.25) |
| BAL cells, % | |||
| Macrophages | 86.55 (74.9 – 90.15) | 77.15 (74.55 – 84.65) | 66.25 (52.75 – 80.75)** |
| Neutrophils | 3.05 (1.63 – 5.25) | 4.65 (2.38 – 5.15) | 8.90 (5.4 – 16.05)*** ‡ |
| Eosinophils | 0.25 (0.0 – 0.88) | 1.75 (1.0 – 3.38)** | 2.40 (0.63 – 5.38)*** |
| Lymphocytes | 1.0 (0.63 – 1.38) | 1.13 (0.70 – 2.13) | 1.0 (0.38 – 1.38) |
| Epithelial cells | 9.30 (4.15 – 17.05) | 12.15 (5.70 – 19.88) | 17.15 (10.0 – 26.05) |
p<0.01 versus HC,
p<0.001 versus HC,
p<0.05 versus MA,
p<0.01 versus MA,
p<0.001 versus MA.
Benchmarking of M2 biomarkers using macrophages derived from blood monocytes
In order to establish a panel of biomarkers that reflect M2 macrophages for subsequent phenotyping of lung macrophages from asthmatic and control subjects, initial experiments were conducted on MDMs derived from circulating monocytes. Monocytes were isolated to >95% purity by magnetic cell sorting (MACS) and anti-CD14 antibodies and differentiated over 12 days in culture in RPMI-1640 (plus 10% foetal bovine serum – FBS) into macrophages using 2 ng/ml GM-CSF, a protocol shown to produce macrophages with phenotypic and functional characteristics similar to lung macrophages18, 19. The GM-CSF containing media were then removed and replaced with media containing 10 ng/ml rhIL-4 or rhIL-13, and culture continued for a further 24 h. In order to check whether the differentiated cells displayed a M2 phenotype, culture supernatants were analysed by ELISA for CCL17 release whilst the resuspended cells were analysed for a panel of M2 biomarkers (CD206, CD163, CLEC10A/CD301, Ccl17 and Arg1) using a combination of flow cytometry and RT-PCR (see online supplement).
Sampling of lung macrophages
Bronchoalveolar lavage (BAL) and sputum induction were performed as described20. Cells were isolated and immediately processed in FACS Buffer containing 2 mg/ml human IgG for flow cytometry and RT-PCR and aliquots were collected for culture.
Analysis of lung macrophage biomarkers
Flow cytometry
Phenotypic characterisation of lung macrophages was performed by flow cytometry using a 9-colour FACSAria cell sorter and FACSDiva Software (v5.0.3). Cells were incubated in reagents from the Live/Dead Violet Viability/Vitality kit and the macrophage detection cocktail, consisting of anti-human CD45 PE-AF610, anti-human CD3 PE-Cy7 and anti-human HLA-DR APC-Cy7. Using this protocol, CD45+CD3-HLA-DR+ cells were >90% pure macrophages, as shown by cytological analysis of sorted cells (Figure E1 in online supplement).
Separate aliquots were additionally incubated in a cocktail of antibodies specific for surface biomarkers (anti-CD206 APC and anti-CD14 PerCP-Cy5.5 or anti-CD301 AF647 and anti-CD163 PerCP-Cy5.5) or isotype control antibodies. For intracellular staining, cell aliquots that had been incubated in the macrophage identification cocktail were first washed with FACS buffer and then incubated in Cytofix/Cytoperm. These were further washed in 1 × Perm/Wash and incubated with anti-human CD68-PE, anti-human TNF APC, anti-IL-10 AF647 or isotype control.
RT-PCR
RNA was extracted from FACS sorted sputum and BAL macrophages using a Stratagene Microprep Kit. Reverse transcription was carried out using a nanoScript Reverse Transcriptase kit. PCR amplifications for M2 biomarkers (Ccl17, Clec10A, Arg1) were performed on a BioRad iCycler using Precision 2× qPCR Mastermix and PerfectProbe® primers (for full sequences see online supplement). Gene expression was normalized to β2-microglobulin gene expression and quantified using the ΔΔ CT method21.
Analysis of cytokine release by MDM and BAL cells
MDM from 5 subjects were cultured in RPMI-1640 (+10% FBS) for 24 h and stimulated with 10 ng/ml IL-4 in the presence or absence of a range of concentrations of fluticasone propionate or PI3Kinase inhibitors. Similarly, BAL cells (from 13 asthmatics), composed of a median of 75% macrophages (for full differential cell counts see online supplement), were cultured in AIM V medium without additional stimulants or in the presence of 10 μM LY294002. The medium was FBS-free so as to avoid skewing of the macrophage phenotype by factors in serum. After 24 h, supernatants were retained for cytokine and chemokine analysis.
Culture supernatants were analysed by a combination of ELISA and Luminex assay for IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12p70, IL-13, TNFα, IFNγ, CCL17 and CCL22 (for full details see online supplement).
Statistics
Statistical analyses were performed using a Kruskal-Wallis or Friedman ANOVA test with a Dunn’s Multiple Comparison test or a Mann-Whitney U test or Wilcoxon’s signed rank test as appropriate (GraphPad Prism v3, GraphPad Software Inc., San Diego, USA). Results were considered significant if p<0.05.
Results
Assessment of M2 biomarkers using monocyte-derived macrophages (MDM)
Both IL-4 and IL-13 significantly increased Ccl17 and Clec10a gene expression (Figure 1A), with a trend towards increased Arg1 gene expression (data not shown) induced by IL-13. Th2 cytokine-treated MDM (i.e. M2 MDM) also expressed significantly more HLA-DR and CD206 (Figure 1B) and less CD14 and CD163 on their surface (Figure 1B), with no difference in CLEC10A (CD301) or CD36 expression (data not shown). Small amounts of CCL17 were released from unstimulated MDM (80.3 ± 39 pg/ml) and this was greatly increased by IL-4 (390.3 ± 204.9 pg/ml) and IL-13 (281.4 ± 257.5 pg/ml). These experiments provided a panel of M2 biomarkers previously reported by others5, 6, 8, 22.
FIGURE 1. Expression of biomarkers on MDMs stimulated with IL-4 or IL-13.
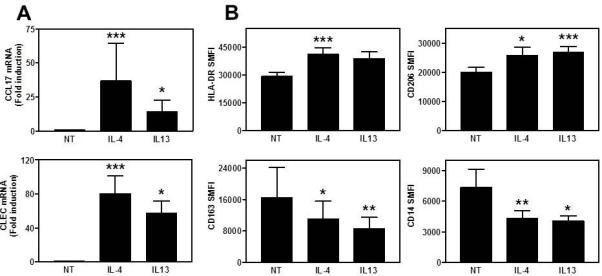
A) Gene expression of Ccl17 and Clec10A are expressed as fold induction compared to NT (mean±SE, n=11). B) Cell surface expression data are expressed as specific mean fluorescent intensity (SMFI - mean±SE, n=12). Data analysed using Friedman ANOVA and Dunn’s multiple comparison test compared to NT. * p<0.05, ** p<0.01, *** p<0.001
M2 cell surface marker expression on sputum and BAL macrophages
The M2 cell surface biomarkers identified above were applied to sputum and BAL macrophages from asthmatic patients and controls and analysed by multichromatic flow cytometry. The only surface biomarker that was differentially expressed was CD163 (Figure 2 and Table 2). Similar to M2 MDM, its expression was reduced in asthma when compared to health on macrophages in sputum (Kruskal-Wallis p=0.0346) but not in BAL (Kruskal-Wallis p=0.2893). Intracellular TNF and IL-10 expression was also not different (Table 2), although BAL macrophages appeared to express more TNF than sputum macrophages. Intracellular IL-13 protein was not detected.
FIGURE 2. Analysis of lung macrophage M2 marker expression by flow cytometry.
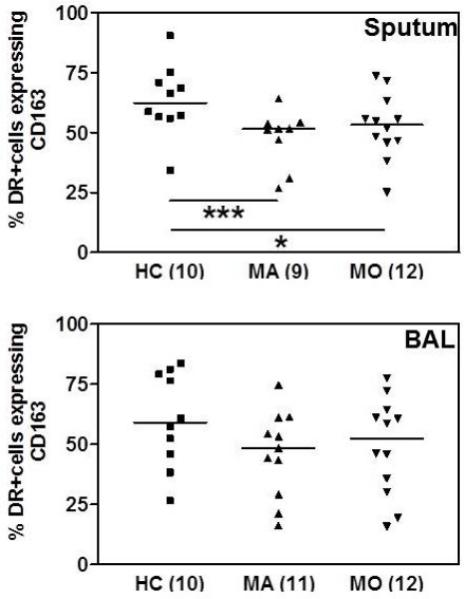
Expression of cell surface CD163 on sputum and BAL macrophages of healthy controls (HC), mild asthmatics (MA) or moderate asthmatics (MO). Bars indicate median values, numerals in parentheses indicate n. Data are expressed as percentage of Live+CD45+CD3-HLA-DR+ cells and analysed by Kruskal-Wallis ANOVA and Dunn’s Multiple Comparison test. * p<0.05, *** p<0.001.
Table 2. Cell surface and intracellular protein expression by macrophages from healthy controls (HC), mild asthmatics (MA) and moderate asthmatics (MO).
HLA-DR, CD14, CD301, TNF and IL10 are represented as specific mean fluorescence intensity (SMFI) and CD206 is shown as percentage of cells staining positively.. Data are expressed as median values (IQR).
| Sputum | BAL | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| HC | MA | MO | HC | MA | MO | |
| Extracellular | ||||||
| HLA-DR (SMFI) |
59210 (44120 – 75520) |
57050 (40780 – 76880) |
46280 (42470 – 63470) |
49140 (34100 – 56390) |
46470 (36650 – 57300) |
44470 (30760 – 61870) |
| CD14 (SMFI) | 1920 (1498 – 3244) |
1729 (1520 – 1993) |
1932 (1489 – 2694) |
1260 (662.5 – 1612) |
1328 (717.5 – 1808) |
1049 (660 – 1922) |
| CD206 (%) | 62.4 (54.7 – 67.5) | 47.7 (28.9 – 62.5) | 56.8 (46.0) – 76.5) | 57.9 (44.1 – 72.7) | 51.6 (42.0 – 64.4) | 57.5 (45.4 – 70.2) |
| CD301 (SMFI) | 2785 (1439 – 5921) |
2993 (1674 – 3852) |
3208 (2008 – 3909) |
2727 (1397 – 5295) |
3314 (2917 – 4640) |
2540 (1198 – 5318) |
| Intracellular | ||||||
| >TNF (SMFI) | 3781 (3174 – 4000) |
4464 (3383 – 5366) |
3867 (3379 – 4831) |
6675 (5232 – 8380) |
6717 (5172 – 8141) |
6120 (5473 – 7148) |
| IL-10 (SMFI) | 1296 (1005 – 1735) |
1242 (1001 – 1900) |
1615 (814.5 – 2472) |
1324 (722 – 2229) | 1976 (1700 – 2179) |
1300 (920 – 2014) |
Gene expression analysis of lung macrophages
The 90% purity of macrophages obtained using the above method of selection was considered insufficient for gene expression analysis as any small contamination with other cell types could be amplified by PCR. Therefore, we added to the macrophage detection protocol the Lineage 1 (Lin) antibody cocktail (BD Biosciences) to exclude dendritic cells from the analysis. Cells sorted in this way (CD45+CD3-Lin+HLA-DR+) (Figure 3) from both sputum and BAL were on average 98% pure macrophages as ascertained by histochemical and morphological analysis of cytospins. Up to 100,000 events were sorted into RNA lysis buffer and stored at −80°C for RNA extraction and RT-PCR.
FIGURE 3. Sorting of lung macrophages for RT-PCR analysis.

Sputum and BAL cells were stained according to the method outlined. Live+CD45+CD3-HLA-DR+Lin1+ events were sorted into PBS using a FACSAria and analysed by cytospin (representative micrograph shown × 40 original magnification). Data are expressed as a percentage of total cells, presented as mean±SE (n=6) and analysed using a one-tailed Wilcoxon signed rank test. ** p<0.01
Sputum macrophages from healthy controls expressed little or no Ccl17 mRNA (median value 0), while significantly greater expression was observed in both mild (median value 3.421) and moderate asthmatics (median value 7.63). The Ccl17 expression correlated weakly (r2=0.1493) but significantly (p=0.0252) with sputum eosinophilia (Figure 4) but not with neutrophil counts (r2 = 0.04). When these analyses were performed on individual asthma groups (MA r2=0.279 p=0.063, MO r2=0.016 p=0.7149) and healthy controls (r2=0.061 p=0.1456) separately, none of these correlations was significant, nor was there a significant correlation when asthmatics were analysed as a group in the absence of controls (r2=0.109, p=0.1331), suggesting the correlation is probably “pulled” into significance because of the inclusion of low CCL17 expressing, low eosinophil healthy controls. Although a significant increase in Ccl17 mRNA expression by BAL cells was observed (Kruskal-Wallis p=0.0449), in contrast to the sputum data, this increase was only significant when comparing mild and moderate asthmatics by Dunn’s post-hoc test. There was a trend towards increased expression of Clec10a mRNA in both sputum and BAL macrophages from mild but not moderate asthmatics, although this was not statistically significant (data not shown).
FIGURE 4. CCL17 mRNA levels (normalised to β2-microglobulin) and association with sputum eosinophil counts.
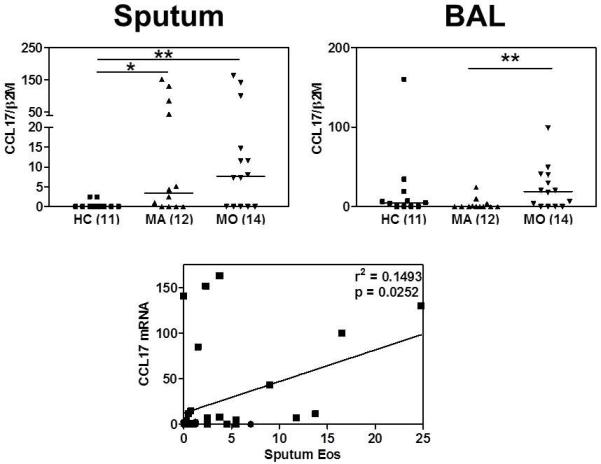
Bars indicate median values and numerals in parentheses indicate n. Data were analysed using Kruskal-Wallis ANOVA followed by Dunn’s Multiple Comparison test. * p<0.05, ** p<0.01 The association between CCL17 mRNA expression and percentage sputum eosinophils was analysed by Spearman rank correlation test (● HC ■ Asthma).
Cytokine production by BAL cells
To ascertain whether increased expression of Ccl17 mRNA in macrophages translated into increased release of CCL17 protein, freshly isolated BAL cells (containing a median of 75% macrophages) were cultured for 24 h and supernatants analysed for CCL17 by Luminex assay. The lack of difference in CCL17 ex vivo release by BAL cells observed (online supplement Figure E4) suggested no correlation between the in vivo gene expression and the ex vivo expression of CCL17. Additional cytokine measurements were also performed in order to assess whether macrophages released a predominance of Th1 or Th2 cytokines. None of the Th2 (IL-4, IL-5 and IL-13) or Th1 (IFN-γ, IL-2 and IL-12) cytokines were detected, while the previously reported M1 (TNF, IL-6, IL-8) and M2 (IL-10 and CCL22) biomarkers5 were released at similar levels in all groups (online supplement Figure E4).
Inhibition of CCL17 release by the PI3Kinase inhibitor, LY294002, but not corticosteroids
The increased Ccl17 mRNA expression observed in lung macrophages from moderate asthmatics on inhaled steroids suggested that the production of this chemokine may not be fully responsive to corticosteroids. We therefore investigated whether the ex vivo CCL17 release from macrophages could be inhibited by steroid treatment. Initial experiments were conducted with M2 MDM where fluticasone propionate had no effect on the CCL17 release in response to 10 ng/ml IL-4 (Figure 5A). We then studied whether inhibition of PI3Kinase, the enzyme suggested to be required for differentiation of monocytes into M2 macrophages23, could reduce CCL17 release. The pan-PI3Kinase inhibitor, LY294002, caused a concentration-dependent inhibition (EC50 = 4.2 μM) that was significant at 10 μM (Figure 5A) with no significant LDH release (data not shown). Similar results were obtained with 10 ng/ml IL-13 (data not shown). In contrast, the specific PI3Kinase-δ inhibitor, IC81174, had little effect on the amount of CCL17 released by IL-4 stimulated-MDM except at the highest concentration used (Figure 5A) that caused an increase in LDH release (data not shown). Similarly, the spontaneous release of CCL17 by BAL cells from asthmatics (n=13) was significantly inhibited by 10 μM LY294002 from mean 14.82 pg/ml to 7.49 pg/ml (Figure 5B).
FIGURE 5.
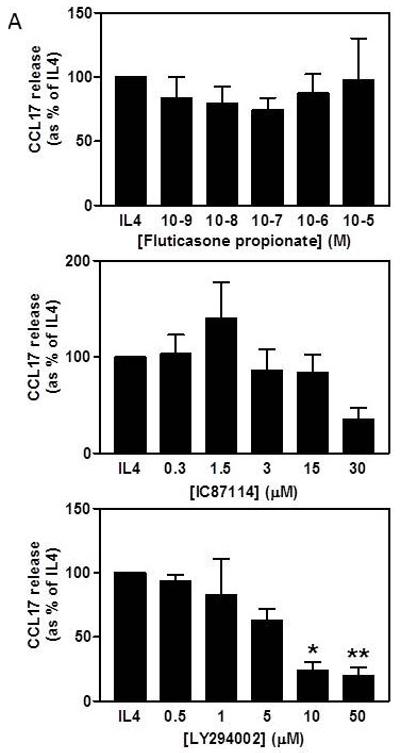
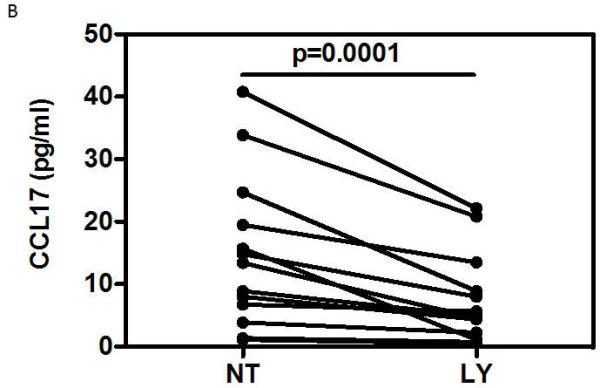
A) Effects of fluticasone propionate and PI3Kinase inhibitors on CCL17 release by IL-4-stimulated MDM. Data expressed as percentage of the maximum IL-4 stimulation (mean±SE, n=5). Data analysed by Friedman ANOVA and Dunn’s multiple comparison test. B) Effect of LY294002 on spontaneous release of CCL17 from BAL cells (medians, n=13). Data analysed using one-tailed Wilcoxon signed rank test. * p<0.05, ** p<0.01, *** p<0.001
Discussion
In contrast to animal models of asthma, this study has demonstrated only a partial M2 macrophage phenotype in asthmatic airways, consisting of increased Ccl17 mRNA expression and reduced CD163 cell surface expression when compared to health. Whilst blood monocytes did not express detectable amounts of Ccl17 mRNA (data not shown), raised expression was evident in lung macrophages regardless of asthma severity and treatment. The expression of Ccl17 mRNA in sputum correlated weakly with sputum eosinophilia, a hallmark of asthma and a marker of Th2 type inflammation24, providing a mechanism for increased recruitment of CCR4+ cells (eosinophils and Th2 lymphocytes) for which CCL17 is a ligand. The finding of persistently raised Ccl17 mRNA in macrophages from both sputum and BAL of moderate asthmatics on inhaled corticosteroids and the inability of corticosteroid to inhibit in vitro CCL17 release by M2 MDM suggests that CCL17 production is relatively corticosteroid insensitive. Finally, our study suggests that the production of CCL17 is regulated by PI3Kinase.
Studies of macrophages in asthma to date have reported on functional readouts such as suppression of T cell proliferation and phagocytic function25, 26, cell surface marker expression27-29, and gene expression30-32. To our knowledge, this is the most comprehensive investigation of M2 lung macrophage biomarkers in asthma using combined cell surface and gene expression analysis and measuring cytokine release. Two previous studies, applying gene expression analysis32 and immunocytochemistry29 respectively, showed no evidence for M2 macrophages in asthma. Another study of M1/M2 macrophages in COPD, using a combination of transcriptomic analysis, RT-PCR and flow cytometry33, showed that smoking was associated with increased M2 phenotype gene expression, with further upregulation in COPD smokers and a reciprocal decrease of M1 phenotype gene expression33. However, the profile of differentially expressed M2 markers (e.g. Adenosine A3 receptor, MMP2, MMP7) was different from our study, while CCL17 and CD163 were not differentially expressed in either healthy smokers or COPD smokers.
Interpretation of the literature is complicated by differences in the definition of M2 phenotypes based on the use of different M2-polarising protocols, applying either IL-4/IL-135, IL-107 or M-CSF34, all of which produce slightly different macrophage phenotypes. Atopic asthma is considered to involve Th2 cytokines2, so in keeping with the M2 phenotype described by Martinez et al (2009)5, we have used IL-4 and IL-13 stimulated MDM as our M2 benchmark.
In the current study, CCL17 and CD163 expression varied considerably between the subjects studied, consistent with the notion that asthma involves heterogeneous inflammatory processes despite similar clinical phenotypes. Differences in CCL17 expression were more pronounced in sputum than in BAL, which sample the central airways and the distal airways and alveoli, respectively35, suggesting that the changes reflect the proximal airways processes. Similarly, only the sputum samples showed reduced levels of CD163.
The role of CCL17 in allergic inflammation has been the subject of a number of studies, showing increased release following allergen exposure12, 13, 36. Applying flow cytometry to induced sputum and bronchial biopsy explants, we have previously shown an important role for CCR4 and its ligand CCL17 in Th2 T cell recruitment12. Stimulation of bronchial tissue explants by allergen induced CCL17 release in steroid-naive asthmatics but, of more relevance to the current study, spontaneous release was raised in explants from moderate asthmatics on inhaled corticosteroids. Furthermore, the numbers of sputum and blood CCR4+ T cells were significantly raised in asthmatics on high-dose inhaled and oral corticosteroids. Taken together with the current findings that Ccl17 mRNA expression is persistently high in corticosteroid-treated asthmatics and that in vitro IL-4-induced CCL17 release by M2 MDM cannot be suppressed by corticosteroids, this study adds to accumulating evidence that CCL17 is an important chemokine in asthma whose production is relatively corticosteroid insensitive.
Increased expression of CD163, a haptoglobin-haemoglobin scavenger receptor37, has been associated with the M2 phenotype in vitro5, 7, 10, 38. While most studies showed a role for IL-4 and IL-13 in driving this phenotype, Tiemessen et al. showed it to depend on IL-10 and not IL-4/IL-137. In contrast to the published in vitro studies, we have found that CD163 expression is decreased in asthma. The finding that MDM exposed to IL-4 and IL-13 reduced CD163 expression suggests that Th2 cytokines may be responsible in asthma. In agreement with our study, Van den Heuvel et al. (1999) demonstrated that IL-4 treatment decreases CD163 expression, although when added to cells pre-treated with glucocorticoids, it increases the expression22. In contrast, in our study the reduced expression of CD163 on asthmatic sputum macrophages was evident despite treatment with inhaled steroids, providing additional evidence of relative corticosteroid insensitivity. Thus, our observations are consistent with the notion that airway macrophages are relatively corticosteroid insensitive39.
Having found the macrophages to be insensitive to corticosteroids, we sought an alternative therapy targeting PI3Kinase, the enzyme postulated to be required for the differentiation of M2 macrophages23. For this purpose, LY294002 was selected on the basis of its relative selectivity for PI3Kinase. At a concentration of 10 μM this compound was effective at inhibiting CCL17 release from both cultured BAL cells and IL-4-stimulated MDM. Although this suggests the involvement of PI3Kinase, it is recognised that at concentrations. >10 μM LY294002 inhibits other kinases40. Previous studies have suggested that inhibiting the PI3Kinase-δ isoform is effective at reducing airway inflammation and hyperresponsiveness in murine models of asthma41. Furthermore, inhibition of PI3Kinase-δ using theophylline reverses the steroid insensitivity of COPD macrophages42. However, the specific PI3Kinase-δ inhibitor, IC87114, had little effect on IL4-induced CCL17 release from MDM, suggesting that, in contrast to the murine model23, an isoform other than PI3Kinase-δ is involved in IL-4-induced CCL17 production in human macrophages. In summary, this study suggests involvement of the PI3Kinase pathway but definitive proof of this being involved will require more specific inhibition using such tools as inhibitory RNAs.
This study raises a number of questions and opportunities for further research. Previous studies8 have shown that CLEC10a mRNA expression is increased on M2 MDM and there have been reports of increases on macrophages in a number of diseases43-45. However, to our knowledge there have been no studies of both mRNA and protein expression. In our study, there was a trend towards increased CLEC10a mRNA but not CD301 (the protein marker of CLEC10a) in macrophages in asthma. Future studies will need to look for internalized or intra-cellular CD301 expression. The difference in Ccl17 mRNA expression was greater in sputum than in BAL and the reasons for that are unclear. This discrepancy could, at least partly, account for the lack of difference in CCL17 protein release by BAL cells ex vivo. Other cell types, although they constituted on average 30% of total BAL cells, may have bound some of the cytokines/chemokines released in culture (e.g CCL17 by CCR4+ cells). Furthermore, contact with plastic may have altered the BAL macrophage phenotype46. Similarly, the in vivo conditions required for the phenotype to be detected may not be sustained ex vivo. However, it is unlikely that either of these would have offset any intrinsic differences between macrophages from patients and healthy individuals. When studying macrophage function, stimuli are often used, but we chose not to expose the BAL cells to any exogenous stimuli such as LPS because this could skew responses towards a M1 profile5. Finally, the biomarker panel in this study was limited to biomarkers published at the time the study was conceived; further studies should focus on additional M2 biomarkers, such as MMP2 and MMP733.
The driver of the macrophage phenotype in asthma remains unclear. It is possible that IL-4 and IL-13 do still provoke this phenotype but that our MDM model does not encapsulate the full complexity of macrophage interactions within the lung environment. IL-4/IL-13 and activation of Stat6 may not be involved in the development of the CCL17-producing macrophages. The promoter for CCL17 also contains NF-κB motifs47, 48 and the archetypal NF-κB activator, TNF, induces expression of Ccl17 mRNA in human bronchial epithelial cells49. Infection of murine lung epithelium with respiratory syncytial virus (RSV) activates the NF-κB pathway to produce CCL1750 and switches macrophages to a M2 phenotype51. Moreover, the house dust mite allergen, Dermatophagoides pteronyssinus (Der p) 1, also induces CCL17 mRNA and protein expression in cultured primary bronchial epithelial cells via an EGFR/NF-κB-dependent pathway16. As both RSV and Der p 1 are implicated in asthma52, 53, these agents may activate macrophages via NF-κB-dependent pathways to produce CCL17 in a steroid-independent manner. This study has involved a mix of eosinophilic and non-eosinophilic asthma. The increasing recognition that phenotypes based on eosinophilic and neutrophilic inflammation are different clinically and in respect of response to corticosteroids will require further study of the identified biomarkers in these phenotypes.
Supplementary Material
Key message.
Lung macrophages in asthma do not express biomarkers associated with the alternative, M2 phenotype reported in studies of monocyte-derived macrophages and animal models.
Lung macrophages of asthmatic patients express more CCL17 and less CD163 and the former is associated, albeit weakly, with sputum eosinophilia and cannot be suppressed by corticosteroids but can be attenuated by PI3Kinase inhibition.
Macrophage expression of CCL17 may contribute to the airway inflammation observed in asthma through recruitment of cells expressing CCR4, the cell surface receptor for CCL17.
Table 3. Differences in expression of IL-4/IL-13 induced M2 markers in different settings.
| Marker | Murine MDM2+ | Human MDM2+ | M2 MDM model in current study@ |
Asthma macrophages@ |
|---|---|---|---|---|
|
| ||||
| Gene expression | ||||
| Arg1 | ↑ | No change | No change | No expression |
| Clec10a# | ↑ | ↑ | ↑ | No change |
| Fizzl | ↑ | No human homolog | n.t. | n.t. |
| Ym1 | ↑ | No human homolog | n.t. | n.t. |
| Cell surface expression | ||||
| CD23 | ↑ | ↑ | n.t. | n.t. |
| CD163 | ↑ 38 | ↓ 22 | ↓ | ↓ |
| CD206 | ↑ | ↑ | ↑ | No change |
| MHCII/HLA-DR | ↑ | ↑ | ↑ | No change |
| Chemokine expression | ||||
| CCL13* | ↑ | ↑ | n.t. | n.t. |
| CCL14 | No murine homolog | ↑ | n.t. | n.t. |
| CCL17 | ↑ | ↑ | ↑ | ↑ |
| CCL18 | No murine homolog | ↑ | n.t. | n.t. |
| CCL22 | ↑ | ↑ | ↑ £ | No change |
| CCL24 | ↑ | ↑ | n.t. | n.t. |
Information from Martinez et al.5 except where referenced differently.
Data all from the current study. n.t. = not tested
Data from the pilot of this manuscript obtained using a slightly different culture model (see online supplement).
Murine equivalent is MGL1.
Murine homolog of CCL13 is CCL2.
Acknowledgements
The authors wish to thank Richard Jewell and Dr Carolann MacGuire of the University of Southampton Faculty of Medicine Flow Cytomtery Unit. We would also like to express our appreciation to Drs Peter Adura, Sahantha De Silva, Paddy Dennison, Valia Kehagia and Tom Wilkinson for clinical support as well as all the staff of the NIHR Wellcome Trust Clinical Research Facility and the Southampton NIHR Respiratory Biomedical Research Unit. We extend our gratitude to all the volunteers who gave of their time and enthusiasm to make this research possible.
This work was supported by a project grant from Asthma UK (08/026). TSCH is a Wellcome Trust Clinical Research Fellow (088365/z/09/z).
Abbreviations
- ACQ
Asthma control questionnaire
- AF647
Alexa Fluor 647
- ANOVA
Analysis of variance
- APC
Allophycocyanin
- APC-Cy7
Allophycocyanin-Cyanine 7
- Arg1
Arginase 1
- BAL
Bronchoalveolar lavage
- CCL
CC Chemokine Ligand
- CCR
CC Chemokine Receptor
- CD
Cluster of differentiation
- CLEC10A
Macrophage galactose C-type lectin (CD301)
- COPD
Chronic obstructive pulmonary disease
- Der p
Dermatophagoides pteronyssinus
- EGFR
Epithelial Growth Factor receptor
- ELISA
Enzyme-linked immunosorbent assay
- FACS
Fluorescence-activated cell sorting
- FBS
Foetal Bovine Serum
- FEV1
Forced expiratory volume in 1 second
- FITC
Fluorescein isothiocyanate
- FVC
Forced vital capacity
- GINA
Global Initiative for Asthma
- GM-CSF
Granulocyte/Macrophage Colony Stimulating Factor
- IFN
Interferon
- IL
Interleukin
- Ig
Immunoglobulin
- HC
Healthy control
- HLA-DR
Human leukocyte antigen DR
- Lin 1
Lineage 1 antibody cocktail
- LPS
Lipopolysaccharide
- M1
Classically-activated macrophages
- M2
Alternatively-activated macrophages
- MA
Mild asthmatic
- MACS
Magnetic-assisted cell sorting
- M-CSF
Macrophage Colony Stimulating Factor
- MDM
Monocyte-derived macrophage
- MO
Moderate asthmatic
- mRNA
messenger Ribonucleic Acid
- NF-κB
Nuclear factor κB
- PC20
Provocative concentration that induces a 20% drop in FEV1
- PE
Phycoerythrin
- PE-AF610
Phycoerythrin-Alexa Fluor 610
- PE-Cy7
Phycoerythrin-Cyanine 7
- PerCP-Cy5.5
Peridinin Chlorophyll Protein-Cyanine 5.5
- PI3Kinase
phosphatidylinositol-3-kinase
- rh
recombinant human
- RSV
Respiratory Syncytial Virus
- RT-PCR
Reverse transcription-polymerase chain reaction
- Stat
Signal transducer and activator of transcription
- Th
T helper
- TNF
Tumor Necrosis Factor
References
- 1.Bousquet J, Chanez P, Lacoste JY, Barneon G, Ghavanian N, Enander I, et al. Eosinophilic inflammation in asthma. N Engl J Med. 1990;323:1033–9. doi: 10.1056/NEJM199010113231505. [DOI] [PubMed] [Google Scholar]
- 2.Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, et al. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 3.Robinson D, Hamid Q, Bentley A, Ying S, Kay AB, Durham SR. Activation of CD4+ T cells, increased TH2-type cytokine mRNA expression, and eosinophil recruitment in bronchoalveolar lavage after allergen inhalation challenge in patients with atopic asthma. J Allergy Clin Immunol. 1993;92:313–24. doi: 10.1016/0091-6749(93)90175-f. [DOI] [PubMed] [Google Scholar]
- 4.Peters-Golden M. The alveolar macrophage: the forgotten cell in asthma. Am J Respir Cell Mol Biol. 2004;31:3–7. doi: 10.1165/rcmb.f279. [DOI] [PubMed] [Google Scholar]
- 5.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–83. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 6.Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176:287–92. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci U S A. 2007;104:19446–51. doi: 10.1073/pnas.0706832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raes G, Brys L, Dahal BK, Brandt J, Grooten J, Brombacher F, et al. Macrophage galactose-type C-type lectins as novel markers for alternatively activated macrophages elicited by parasitic infections and allergic airway inflammation. J Leukoc Biol. 2005;77:321–7. doi: 10.1189/jlb.0304212. [DOI] [PubMed] [Google Scholar]
- 9.Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol. 2009;183:6469–77. doi: 10.4049/jimmunol.0901575. [DOI] [PubMed] [Google Scholar]
- 10.Prasse A, Germann M, Pechkovsky DV, Markert A, Verres T, Stahl M, et al. IL-10-producing monocytes differentiate to alternatively activated macrophages and are increased in atopic patients. J Allergy Clin Immunol. 2007;119:464–71. doi: 10.1016/j.jaci.2006.09.030. [DOI] [PubMed] [Google Scholar]
- 11.Murray PJ, Wynn TA. Obstacles and opportunities for understanding macrophage polarization. J Leukoc Biol. 2011;89:557–63. doi: 10.1189/jlb.0710409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vijayanand P, Durkin K, Hartmann G, Morjaria J, Seumois G, Staples KJ, et al. Chemokine receptor 4 plays a key role in T cell recruitment into the airways of asthmatic patients. J Immunol. 2010;184:4568–74. doi: 10.4049/jimmunol.0901342. [DOI] [PubMed] [Google Scholar]
- 13.Panina-Bordignon P, Papi A, Mariani M, Di Lucia P, Casoni G, Bellettato C, et al. The C-C chemokine receptors CCR4 and CCR8 identify airway T cells of allergen-challenged atopic asthmatics. J Clin Invest. 2001;107:1357–64. doi: 10.1172/JCI12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sekiya T, Miyamasu M, Imanishi M, Yamada H, Nakajima T, Yamaguchi M, et al. Inducible expression of a Th2-type CC chemokine thymus- and activation-regulated chemokine by human bronchial epithelial cells. J Immunol. 2000;165:2205–13. doi: 10.4049/jimmunol.165.4.2205. [DOI] [PubMed] [Google Scholar]
- 15.Ying S, O’Connor B, Ratoff J, Meng Q, Mallett K, Cousins D, et al. Thymic stromal lymphopoietin expression is increased in asthmatic airways and correlates with expression of Th2-attracting chemokines and disease severity. J Immunol. 2005;174:8183–90. doi: 10.4049/jimmunol.174.12.8183. [DOI] [PubMed] [Google Scholar]
- 16.Heijink IH, Marcel Kies P, van Oosterhout AJ, Postma DS, Kauffman HF, Vellenga E. Der p, IL-4, and TGF-beta cooperatively induce EGFR-dependent TARC expression in airway epithelium. Am J Respir Cell Mol Biol. 2007;36:351–9. doi: 10.1165/rcmb.2006-0160OC. [DOI] [PubMed] [Google Scholar]
- 17.Perros F, Hoogsteden HC, Coyle AJ, Lambrecht BN, Hammad H. Blockade of CCR4 in a humanized model of asthma reveals a critical role for DC-derived CCL17 and CCL22 in attracting Th2 cells and inducing airway inflammation. Allergy. 2009;64:995–1002. doi: 10.1111/j.1398-9995.2009.02095.x. [DOI] [PubMed] [Google Scholar]
- 18.Tudhope SJ, Finney-Hayward TK, Nicholson AG, Mayer RJ, Barnette MS, Barnes PJ, et al. Different mitogen-activated protein kinase-dependent cytokine responses in cells of the monocyte lineage. J Pharmacol Exp Ther. 2008;324:306–12. doi: 10.1124/jpet.107.127670. [DOI] [PubMed] [Google Scholar]
- 19.Taylor AE, Finney-Hayward TK, Quint JK, Thomas CM, Tudhope SJ, Wedzicha JA, et al. Defective macrophage phagocytosis of bacteria in COPD. Eur Respir J. 2010;35:1039–47. doi: 10.1183/09031936.00036709. [DOI] [PubMed] [Google Scholar]
- 20.Vijayanand P, Seumois G, Pickard C, Powell RM, Angco G, Sammut D, et al. Invariant natural killer T cells in asthma and chronic obstructive pulmonary disease. N Engl J Med. 2007;356:1410–22. doi: 10.1056/NEJMoa064691. [DOI] [PubMed] [Google Scholar]
- 21.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 22.Van den Heuvel MM, Tensen CP, van As JH, Van den Berg TK, Fluitsma DM, Dijkstra CD, et al. Regulation of CD 163 on human macrophages: cross-linking of CD163 induces signaling and activation. J Leukoc Biol. 1999;66:858–66. doi: 10.1002/jlb.66.5.858. [DOI] [PubMed] [Google Scholar]
- 23.Rauh MJ, Ho V, Pereira C, Sham A, Sly LM, Lam V, et al. SHIP represses the generation of alternatively activated macrophages. Immunity. 2005;23:361–74. doi: 10.1016/j.immuni.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 24.Pizzichini E, Pizzichini MM, Efthimiadis A, Evans S, Morris MM, Squillace D, et al. Indices of airway inflammation in induced sputum: reproducibility and validity of cell and fluid-phase measurements. Am J Respir Crit Care Med. 1996;154:308–17. doi: 10.1164/ajrccm.154.2.8756799. [DOI] [PubMed] [Google Scholar]
- 25.Spiteri MA, Knight RA, Jeremy JY, Barnes PJ, Chung KF. Alveolar macrophage-induced suppression of peripheral blood mononuclear cell responsiveness is reversed by in vitro allergen exposure in bronchial asthma. Eur Respir J. 1994;7:1431–8. doi: 10.1183/09031936.94.07081431. [DOI] [PubMed] [Google Scholar]
- 26.Fitzpatrick AM, Holguin F, Teague WG, Brown LA. Alveolar macrophage phagocytosis is impaired in children with poorly controlled asthma. J Allergy Clin Immunol. 2008;121:1372–8. doi: 10.1016/j.jaci.2008.03.008. 8 e1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Viksman MY, Liu MC, Bickel CA, Schleimer RP, Bochner BS. Phenotypic analysis of alveolar macrophages and monocytes in allergic airway inflammation. I. Evidence for activation of alveolar macrophages, but not peripheral blood monocytes, in subjects with allergic rhinitis and asthma. Am J Respir Crit Care Med. 1997;155:858–63. doi: 10.1164/ajrccm.155.3.9117017. [DOI] [PubMed] [Google Scholar]
- 28.Lensmar C, Katchar K, Eklund A, Grunewald J, Wahlstrom J. Phenotypic analysis of alveolar macrophages and lymphocytes following allergen inhalation by atopic subjects with mild asthma. Respir Med. 2006;100:918–25. doi: 10.1016/j.rmed.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 29.St-Laurent J, Turmel V, Boulet LP, Bissonnette E. Alveolar macrophage subpopulations in bronchoalveolar lavage and induced sputum of asthmatic and control subjects. J Asthma. 2009;46:1–8. doi: 10.1080/02770900802444211. [DOI] [PubMed] [Google Scholar]
- 30.Baines KJ, Simpson JL, Wood LG, Scott RJ, Gibson PG. Transcriptional phenotypes of asthma defined by gene expression profiling of induced sputum samples. J Allergy Clin Immunol. 2011;127:153–60. doi: 10.1016/j.jaci.2010.10.024. 60 e1-9. [DOI] [PubMed] [Google Scholar]
- 31.Madore AM, Perron S, Turmel V, Laviolette M, Bissonnette EY, Laprise C. Alveolar macrophages in allergic asthma: an expression signature characterized by heat shock protein pathways. Hum Immunol. 2010;71:144–50. doi: 10.1016/j.humimm.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, et al. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180:388–95. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shaykhiev R, Krause A, Salit J, Strulovici-Barel Y, Harvey BG, O’Connor TP, et al. Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J Immunol. 2009;183:2867–83. doi: 10.4049/jimmunol.0900473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leidi M, Gotti E, Bologna L, Miranda E, Rimoldi M, Sica A, et al. M2 macrophages phagocytose rituximab-opsonized leukemic targets more efficiently than m1 cells in vitro. J Immunol. 2009;182:4415–22. doi: 10.4049/jimmunol.0713732. [DOI] [PubMed] [Google Scholar]
- 35.Alexis NE, Hu SC, Zeman K, Alter T, Bennett WD. Induced sputum derives from the central airways: confirmation using a radiolabeled aerosol bolus delivery technique. Am J Respir Crit Care Med. 2001;164:1964–70. doi: 10.1164/ajrccm.164.10.2104051. [DOI] [PubMed] [Google Scholar]
- 36.Bochner BS, Hudson SA, Xiao HQ, Liu MC. Release of both CCR4-active and CXCR3-active chemokines during human allergic pulmonary late-phase reactions. J Allergy Clin Immunol. 2003;112:930–4. doi: 10.1016/j.jaci.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 37.Van Gorp H, Delputte PL, Nauwynck HJ. Scavenger receptor CD163, a Jack-of-all-trades and potential target for cell-directed therapy. Mol Immunol. 2010;47:1650–60. doi: 10.1016/j.molimm.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 38.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 39.Bhavsar P, Hew M, Khorasani N, Torrego A, Barnes PJ, Adcock I, et al. Relative corticosteroid insensitivity of alveolar macrophages in severe asthma compared with non-severe asthma. Thorax. 2008;63:784–90. doi: 10.1136/thx.2007.090027. [DOI] [PubMed] [Google Scholar]
- 40.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee KS, Lee HK, Hayflick JS, Lee YC, Puri KD. Inhibition of phosphoinositide 3-kinase delta attenuates allergic airway inflammation and hyperresponsiveness in murine asthma model. FASEB J. 2006;20:455–65. doi: 10.1096/fj.05-5045com. [DOI] [PubMed] [Google Scholar]
- 42.To Y, Ito K, Kizawa Y, Failla M, Ito M, Kusama T, et al. Targeting phosphoinositide-3-kinase-delta with theophylline reverses corticosteroid insensitivity in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182:897–904. doi: 10.1164/rccm.200906-0937OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prokop S, Heppner FL, Goebel HH, Stenzel W. M2 polarized macrophages and giant cells contribute to myofibrosis in neuromuscular sarcoidosis. Am J Pathol. 2011;178:1279–86. doi: 10.1016/j.ajpath.2010.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Desai B, Mattson J, Paintal H, Nathan M, Shen F, Beaumont M, et al. Differential expression of monocyte/macrophage-selective markers in human idiopathic pulmonary fibrosis. Exp Lung Res. 2011;37:227–38. doi: 10.3109/01902148.2010.538132. [DOI] [PubMed] [Google Scholar]
- 45.Semnani RT, Mahapatra L, Moore V, Sanprasert V, Nutman TB. Functional and phenotypic characteristics of alternative activation induced in human monocytes by interleukin-4 or the parasitic nematode Brugia malayi. Infect Immun. 2011;79:3957–65. doi: 10.1128/IAI.05191-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sikkeland LI, Kongerud J, Stangeland AM, Haug T, Alexis NE. Macrophage enrichment from induced sputum. Thorax. 2007;62:558–9. doi: 10.1136/thx.2006.073544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wirnsberger G, Hebenstreit D, Posselt G, Horejs-Hoeck J, Duschl A. IL-4 induces expression of TARC/CCL17 via two STAT6 binding sites. Eur J Immunol. 2006;36:1882–91. doi: 10.1002/eji.200635972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Komine M, Kakinuma T, Kagami S, Hanakawa Y, Hashimoto K, Tamaki K. Mechanism of thymus- and activation-regulated chemokine (TARC)/CCL17 production and its modulation by roxithromycin. J Invest Dermatol. 2005;125:491–8. doi: 10.1111/j.0022-202X.2005.23840.x. [DOI] [PubMed] [Google Scholar]
- 49.Berin MC, Eckmann L, Broide DH, Kagnoff MF. Regulated production of the T helper 2-type T-cell chemoattractant TARC by human bronchial epithelial cells in vitro and in human lung xenografts. Am J Respir Cell Mol Biol. 2001;24:382–9. doi: 10.1165/ajrcmb.24.4.4360. [DOI] [PubMed] [Google Scholar]
- 50.Monick MM, Powers LS, Hassan I, Groskreutz D, Yarovinsky TO, Barrett CW, et al. Respiratory syncytial virus synergizes with Th2 cytokines to induce optimal levels of TARC/CCL17. J Immunol. 2007;179:1648–58. doi: 10.4049/jimmunol.179.3.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shirey KA, Pletneva LM, Puche AC, Keegan AD, Prince GA, Blanco JC, et al. Control of RSV-induced lung injury by alternatively activated macrophages is IL-4R alpha-, TLR4-, and IFN-beta-dependent. Mucosal Immunol. 2010;3:291–300. doi: 10.1038/mi.2010.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sigurs N, Aljassim F, Kjellman B, Robinson PD, Sigurbergsson F, Bjarnason R, et al. Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax. 2010;65:1045–52. doi: 10.1136/thx.2009.121582. [DOI] [PubMed] [Google Scholar]
- 53.Pilette C, Francis JN, Till SJ, Durham SR. CCR4 ligands are up-regulated in the airways of atopic asthmatics after segmental allergen challenge. Eur Respir J. 2004;23:876–84. doi: 10.1183/09031936.04.00102504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.