

Abstract
On activation, naive T cells differentiate into effector T-cell subsets with specific cytokine phenotypes and specialized effector functions1. Recently a subset of T cells, distinct from T helper (TH)1 and TH2 cells, producing interleukin (IL)-17 (TH17) was defined and seems to have a crucial role in mediating autoimmunity and inducing tissue inflammation2–5. We and others have shown that transforming growth factor (TGF)-β and IL-6 together induce the differentiation of TH17 cells, in which IL-6 has a pivotal function in dictating whether T cells differentiate into Foxp3+ regulatory T cells (Treg cells) or TH17 cells6–9. Whereas TGF-β induces Foxp3 and generates Treg cells, IL-6 inhibits the generation of Treg cells and induces the production of IL-17, suggesting a reciprocal developmental pathway for TH17 and Treg cells. Here we show that IL-6-deficient (Il6−/−) mice do not develop a TH17 response and their peripheral repertoire is dominated by Foxp3+ Treg cells. However, deletion of Treg cells leads to the reappearance of TH17 cells in Il6−/− mice, suggesting an additional pathway by which TH17 cells might be generated in vivo. We show that an IL-2 cytokine family member, IL-21, cooperates with TGF-β to induce TH17 cells in naive Il6−/− T cells and that IL-21-receptor-deficient T cells are defective in generating a TH17 response.
We previously proposed a reciprocal relationship between TH17 and Treg cells and suggested that IL-6 is pivotal in determining whether an immune response is dominated by TH17 or Treg cells9,10. We predicted that Il6−/− mice would not develop a TH17 response and should have high numbers of CD4+CD25+Foxp3+ Treg cells in the peripheral repertoire. Thus, we crossed Il6−/− mice with Foxp3gfp ‘knock-in’ (Foxp3gfp.KI) mice9,11 and analysed the presence of Treg versus TH17 cells in Il6−/− × Foxp3gfp.KI mice after immunization with the encephalitogenic myelin oligodendrocyte glycoprotein 35–55 (MOG35–55) peptide emulsified in complete Freund’s adjuvant (CFA). We and others have shown previously that Il6−/− mice are resistant to the development of experimental autoimmune encephalomyelitis (EAE)9,12–15. However, the cellular or molecular basis for this resistance was unclear although some studies suggested that Il6−/− mice might have a defect in T-cell priming12,13,16. On immunization, Il6−/− × Foxp3gfp.KI mice mounted an attenuated T-cell response with a defect in the generation of TH17 cells (Fig. 1a). We found that Il6−/− × Foxp3gfp.KI mice had a significantly elevated fraction of Foxp3+ Treg cells in the draining lymph nodes (Fig. 1b). Thus, the ‘impaired priming’ of antigen-specific T-cell responses reported in Il6−/− mice could simply be due to the expansion of Treg cells at the expense of effector T cells in an environment that is devoid of IL-6. We therefore used MOG35–55/IAb (MHC class II) tetramers to analyse the frequency of antigen-specific (tetramer+) effector T cells (Foxp3/GFP−) and Treg cells (Foxp3/GFP+). In contrast with wild-type mice, Il6−/− mice had more MOG-specific Treg cells than effector T cells and the ratio of antigen-specific Treg to effector T cells was reversed (Fig. 1c, d). Thus, the lack of IL-6 favoured the generation or expansion of antigen-specific Treg cells and inhibited the development of effector T-cell responses. These data are consistent with our hypothesis that IL-6 is crucial in dictating the balance between effector T cells and Treg cells.
Figure 1. In the absence of IL-6, antigen-specific Foxp31 Treg cells expand at the expense of effector T cells (Teff cells)in vivo.
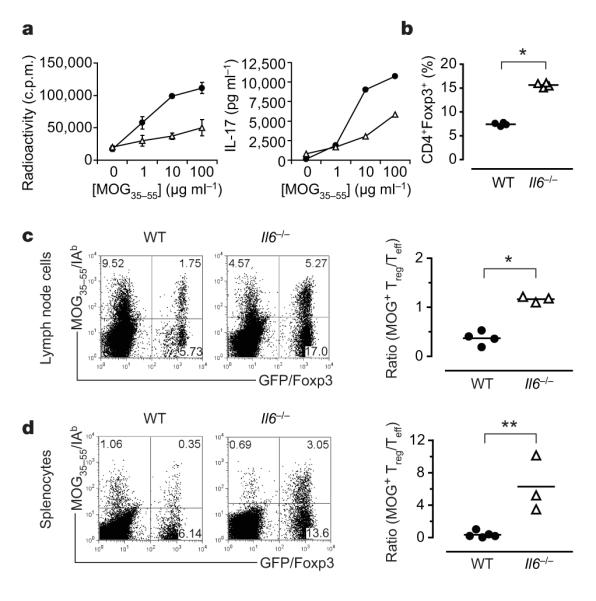
Foxp3gfp.KI (filled circles)or Il6−/− × Foxp3gfp.KI (Il6−/−; open triangles) mice were immunized with MOG35–55/CFA. a, Draining lymph-node cells were tested for MOG-specific proliferation and IL-17 production (means ± s.d. for triplicate determinations). b, The fraction of Foxp3/GFP+ T cells was determined ex vivo by flow cytometry (asterisk, P < 6 × 10−7; t-test). WT, wild type. c, d, Lymphnode cells (c) and splenocytes (d) from MOG35–55/CFA-immunized WT and Il6−/− mice were cultured for four days in the presence of MOG35–55 and stained with a MOG35–55/IAb tetramer. The ratios of antigen-specific Treg cells (MOG tetramer+CD4+Foxp3+) to Teff (MOG tetramer+CD4+Foxp3−) cells are presented (asterisk, P < 0.0003; two asterisks, P < 0.05; t-test).
The deletion of Treg cells from the peripheral repertoire of Il6−/− mice should therefore result in T-cell priming and induction of autoimmunity dominated by interferon (IFN)-γ-producing TH1 cells but not TH17 cells. We depleted Treg cells in Il6−/− mice by using a monoclonal antibody against CD25 (which led to more than 50% depletion of Foxp3+ Treg cells; Supplementary Fig. 1) and then immunized with MOG35–55/CFA. As predicted and in contrast with untreated Il6−/− mice, Treg-depleted Il6−/− mice became susceptible to EAE (Fig. 2a, b and Table 1). However, cytokine analysis by enzyme-linked immunosorbent assay (ELISA) and intracellular cytokine staining of CD4+ T cells from Treg-depleted Il6−/− mice yielded a reappearance of TH17 cells (Fig. 2c, d). IL-17 production in Treg-depleted Il6−/− mice reached almost wild-type levels, suggesting that TH17 cells can be generated in vivo in the absence of IL-6. After confirming that Il6−/− mice did not produce residual amounts of IL-6 (Supplementary Fig. 2), we reasoned that there might be an alternative pathway that induces TH17 cells in the absence of IL-6.
Figure 2. Depletion of Treg cells in Il6−/− mice restores the development of TH17 cells and susceptibility to EAE.

Il6−/− × Foxp3gfp.KI mice were treated with antibody against CD25 to deplete Treg cells or with a control immunoglobulin (rat IgG1) and then immunized with MOG35–55/CFA. a, b, Clinical EAE scores (means and s.e.m.) (a) and linear regression analysis in acute (b, top) and chronic (b, bottom) stages of the disease for wild-type (WT), control Il6−/− and Treg-depleted Il6−/− mice (n.s., not significant). c, Lymph-node (LN) cells and mononuclear cells from the central nervous system (CNS) were recovered on days 6, 14–17 (peak disease) and 29 (recovery) and stained for CD4 and intracellular IL-17 and IFN-γ. The numbers in the quadrants show percentages. d, Splenocytes (day 10) were stimulated with MOG35–55in vitro. Culture supernatants were collected after 48 h, and IL-17 and IFN-γ concentrations were determined. Filled circles, WT (IgG); open triangles, Il6−/− (IgG); open circles, Il6−/− (anti-CD25).
Table 1.
EAE in wild-type and Il6−/− mice
| Group | Incidence | Mean day of onset (mean±s.d.) |
Mean maximum score (mean±s.d.) |
|---|---|---|---|
| Wild type (IgG) | 11 of 13 (85%) | 12.4±1.0* | 2.5±0.7‡ |
| Il6−/− (IgG) | 2 of 7 (29%) | 16.5±3.5† | 1.0±0 § |
| Il6−/− (anti-CD25) | 6 of 11 (55%) | 14.5±1.9*† | 2.9±0.5‡§ |
Mice treated with rat IgG1 (control) or monoclonal antibody against CD25 (PC61) were immunized with MOG35–55 peptide emulsified in complete Freund’s adjuvant. The animals were monitored for EAE development. Statistical analysis was performed by comparing groups using one-way analysis of variance.
P<0.008.
Not significant.
Not significant (mean maximum score of wild-type (IgG) versus Il6−/− (anti-CD25)).
P<0.002 (mean maximum score of Il6−/− (IgG) versus Il6−/− (anti-CD25)).
Our previous studies suggested a reciprocal relationship between Treg and TH17 cells9. Using this logic, we screened several cytokines to identify possible candidates that could substitute for IL-6. We first tested those cytokines that were able to suppress the TGF-β-mediated induction of Foxp3 in sorted naive Foxp3/GFP− T cells derived from Foxp3gfp.KI mice. As well as IL-6, the IL-12 family member IL-27 and the IL-2 family member IL-21 were most efficient in inhibiting TGF-β-driven Foxp3 induction (Fig. 3a, b, and Supplementary Fig. 3). Our analysis confirmed previous results17,18 showing that IL-27 did not induce the differentiation of TH17 cells when combined with TGF-β. In contrast, IL-21 in combination with TGF-β not only suppressed Foxp3 expression but also induced IL-17 production from naive T cells, although not as strongly as TGF-β plus IL-6 (Fig. 3b, c). Retinoic acid receptor-related orphan receptor (ROR)-γt has been shown to be a transcription factor involved in the differentiation of TH17 cells19, and TGF-β plus IL-21 also induced ROR-γt expression in differentiating TH17 cells (Fig. 3d). Thus, IL-21 could substitute for IL-6 in the induction of ROR-γt and IL-17, indicating that in addition to IL-6, IL-21 might cooperate with TGF-β to drive the differentiation of TH17 cells.
Figure 3. Inhibition of induction of Treg cells and generation of TH17 cells by IL-21.

CD4+CD62LhiFoxp3/GFP− T cells from Foxp3gfp.KI mice were stimulated with anti-CD3 and anti-CD28 for three days in the presence of the indicated cytokines. a, The expression of Foxp3/GFP was measured and the fraction of Foxp3+ cells induced by TGF-β was normalized to 100%. b, Individual histograms showing Foxp3/GFP expression. The numbers above the histogram regions (horizontal lines) represent the percentages of Foxp3/GFP+ cells. c, IL-17 production in these cultures after 48 h as measured by ELISA. d, ROR-γt expression as determined by quantitative RT–PCR in naive 2D2 (ref. 30) T cells activated for 48 h with anti-CD3 in the presence of irradiated syngeneic antigen-presenting cells and the indicated cytokines. ROR-γt expression is shown as mean and s.e.m. for duplicate determinations, relative to β-actin.
To test whether IL-21 was responsible for the induction of IL-17 from naive T cells that do not produce IL-6, we compared the differentiation of naive wild-type and Il6−/− T cells in vitro in the presence of TGF-β plus IL-21 or TGF-β plus IL-6. TGF-β plus IL-21 was sufficient to drive Il6−/− T cells into the TH17 pathway, although less efficiently than for wild-type T cells (Fig. 4a). Thus, in an IL-6-deficient environment, TGF-β plus IL-21 could act independently of IL-6 to induce TH17 cells. However, the frequency of TH17 cells induced by TGF-β plus IL-21 was consistently lower than that of the induction of TH17 cells driven by TGF-β plus IL-6.
Figure 4. IL-21-driven TH17 differentiation is independent of IL-6.

Naive (a, b), total (c) or CD44+CD4+ T cells (d, e) were cultured with anti-CD3 plus corresponding irradiated antigen-presenting cells (a, c) or anti-CD3 plus anti-CD28 (b, d, e) and the indicated cytokines. a, c, Percentages of IL-17+ and IFN-γ+ cells in T cells from wild-type (WT) or Il6−/− mice (a) and WT or Il21r−/− mice (c) after four days of culture. b, IL-21 mRNA was determined by quantitative RT–PCR (means and s.e.m. for duplicate determinations). d, e, CD4+CD44+ T cells from WT or Il21r−/− mice were stimulated with or without recombinant IL-23 for 48 h. IL-17 and IFN-γ production were determined by intracellular cytokine staining (d) and ELISA (e; open columns, WT; filled columns, Il21r−/−) (asterisk, P < 0.0005; two asterisks, P < 0.003; t-test). f, WT (filled circles) and Il21r−/− (open triangles) mice were immunized with ovalbumin 323–339 peptide (OVA323–339)/CFA. Draining lymph-node cells were assayed for antigen-specific proliferation and cytokine production (means and s.d. for triplicate cultures).
Next we evaluated whether IL-21 was involved in the induction of TH17 differentiation under ‘standard’ differentiation conditions of IL-6 plus TGF-β and whether the two cytokines (IL-6 and IL-21)cooperated to induce TH17 cells. T cells activated in the presence of IL-6 alone or in combination with TGF-β expressed IL-21 (Fig. 4b). Furthermore, of all CD4+ T-cell subsets, TH17 cells were the highest producers of IL-21 (Fig. 4b). This suggested that there might be an amplification loop in which IL-21 produced by TH17 cells participates in enhancing further differentiation of TH17 cells. We therefore compared the induction of TH17 cells by TGF-β plus IL-6 in wild-type and Il21r−/− T cells20. When IL-6 and TGF-β were used to drive the differentiation of TH17 cells, the frequency of TH17 cells induced in Il21r−/− CD4+ T cells was half that of wild-type T cells (Fig. 4c), suggesting that IL-21 might normally contribute to TH17 differentiation mediated by TGF-β plus IL-6. If these in vitro data were accurate, the TH17 response in Il21r−/− mice should be deficient. We found that the fraction of CD4+CD44+IL-17+ T cells was significantly reduced in Il21r−/− mice in comparison with wild-type controls (data not shown), and this difference was also observed when CD44hi T cells were activated in vitro (Fig. 4d, e). Notably, the addition of recombinant IL-23 could not compensate for the defective TH17 response observed in CD44+Il21r−/− T cells (Fig. 4d, e). On immunization, Il21r−/− mice showed no appreciable defect in T-cell proliferation but a marked decrease in their ability to generate a TH17 response, whereas the production of IFN-γ seemed to be increased (Fig. 4f). These data suggest that IL-21 produced by differentiated TH17 cells must have a function in amplifying TH17 differentiation such that in the absence of IL-21 signalling, the TH17 response is compromised. Whereas IL-6 is predominantly produced by cells of the innate immune system, IL-21 is produced mainly by the adaptive immune system, and activated T cells are the main source of IL-21 (Supplementary Fig. 4). We therefore speculate that TGF-β and IL-6 together initiate the differentiation of TH17 cells, which in turn can produce IL-21 and thus further amplify the TH17 differentiation process. However, in the absence of IL-6, TGF-β together with IL-21 can drive TH17 differentiation on their own. IL-6 and IL-21 cooperate in TH17 differentiation, which is further supported by the observation that in vivo neutralization of IL-21 in Treg-depleted Il6−/− mice essentially abrogated the generation of TH17 cells (Supplementary Fig. 5). This suggests that IL-6 and IL-21 independently and together must cooperate with TGF-β to induce TH17 differentiation.
In summary, together with TGF-β, IL-21 constitutes an additional pathway for the generation of pathogenic TH17 cells. The induction of IL-17 in naive T cells by a combination of TGF-β and IL-21 also suppresses Foxp3 expression, suggesting that similarly to IL-6, IL-21 is able to regulate the reciprocal developmental pathway of generation of Treg and TH17 cells. The importance of the Idd3 genetic locus, which controls susceptibility to both EAE and type 1 diabetes and regulates the function of Treg cells21, might in part be due to the presence of Il21 in addition to Il2 in the Idd3 interval. IL-21 binds to the IL-21 receptor, which consists of a unique IL-21 receptor subunit and the common cytokine receptor γ-chain shared by other cytokine receptors including the receptors for IL-2, IL-4, IL-7, IL-9 and IL-15 (ref. 22). In contrast to these cytokines, binding of IL-21 to its receptor preferentially activates the downstream signalling molecules STAT1 and STAT3 (ref. 23). Because STAT3 is an important signalling molecule in TH17 differentiation24, it is likely that IL-21-induced activation of STAT3, like IL-6-induced activation of STAT3, cooperates with TGF-β signalling to induce the transcription factor ROR-γt and drive TH17 differentiation.
Because differentiated TH17 cells may be the most robust producers of IL-21, we propose that the ability of TH17 cells to produce IL-21 further amplifies the TH17 response. This reverberating pathway is similar to the mechanism in which IL-4 produced by TH2 cells promotes TH2 differentiation25,26. However, TH17 responses also seem to be tightly counter-regulated in that IL-27, an IL-12 family member produced by cells of the innate immune system, and IL-25, an IL-17 family member produced by the innate immune system and activated T cells, can dampen and eventually shut off TH17 responses17,18,27. We suggest that enhanced autoimmunity observed in mice injected with exogenous IL-21 (ref. 28) might be partly due to increased differentiation of TH17 cells and suppression of Treg generation in vivo. Targeting IL-21 in autoimmune diseases may therefore readjust the balance between pathogenic TH17 and Foxp3+ Treg cells, which is believed to be defective in human autoimmune diseases.
In vitro T-cell proliferation and measurement of cytokines
Proliferation was determined by incorporation of [3H]thymidine. Cytokines were measured by ELISA, cytometric bead array (BD Biosciences) or fluorescent bead assay (Luminex). Intracellular cytokine staining and isolation of mRNA for determination of cytokine expression by real-time polymerase chain reaction (PCR) were performed after stimulation with 12-O-tetradecanoylphorbol-13-acetate/ionomycin (see Methods).
T-cell differentiation in vitro
Naive CD4+ T cells (CD4+CD62Lhi or CD4+CD62LhiFoxp3/GFP−) were purified by fluorescence-activated cell sorting and stimulated for three days with plate-bound antibody against CD3 (145-2C11, 4 μg ml−1) plus soluble antibody against CD28 (PV-1, 2 μg ml−1) or by soluble anti-CD3 (2 μg ml−1) plus irradiated syngeneic splenocytes as antigen-presenting cells and recombinant cytokines: human TGF-β1 (3 ng ml−1), mouse IL-6 (30 ng ml−1), mouse IL-21 (100 ng ml−1) or mouse IL-27 (25 ng ml−1; all from R&D Systems). Blocking of IL-21 activity in vitro was performed by the addition of a goat anti-mouse IL-21 antibody (25 μg ml−1; R&D Systems) or by IL-21R-Ig (100 μg ml−1). Polarization of T cells into TH1, TH2 or TH17 cells was performed by solid-phase anti-CD3 (4 μg ml−1) and soluble anti-CD28 (2 μg ml−1) in the presence of recombinant mouse IL-12 (10 ng ml−1; R&D Systems) plus anti-IL-4 (11B.11; 10 μg ml−1) for TH1, mouse IL-4 (10 ng ml−1; R&D Systems) plus anti-IL-12 (C17.8; 10 μg ml−1) for TH2, and TGF-β plus IL-6 for TH17. Monoclonal antibodies against mouse CD3, mouse CD28, mouse IL-4 and mouse IL-12 were purified from the supernatants of hybridomas obtained from the American Type Culture Collection (ATCC).
METHODS
Induction of EAE
EAE was induced by subcutaneous immunization of mice with 100 μl of an emulsion containing 100 μg of MOG35–55 peptide (MEVGWYRSPFSRVVHLYRNGK) and 250 μg of M. tuberculosis H37 Ra (Difco) in incomplete Freund’s adjuvant oil plus an intraperitoneal injection of 200 ng of pertussis toxin (List Biological Laboratories) on days 0 and 2. For Treg depletion, animals were injected intraperitoneally with 500 μg of monoclonal antibody against CD25 (PC61) on days −5 and −3 before immunization. Clinical signs of EAE were assessed as reported11. Linear regression analysis of individual EAE scores was performed for the acute disease phase (until the mean maximum score was reached in each group) and the chronic disease phase (over the complete disease course). Animals were kept in a conventional, pathogen-free facility at the Harvard Institutes of Medicine, and all experiments were performed in accordance with the guidelines prescribed by the standing committee of animals at Harvard Medical School, Boston, Massachusetts.
Generation of IL-21 receptor-Ig (IL-21R-Ig)
The complementary DNA fragment encoding the extracellular domain (amino acids 20–236) of mouse IL-21R (GenBank accession number NM_021887) was amplified by PCR from a mouse splenocyte cDNA library. A second cDNA fragment encoding the Fc portion of human IgG4 was cloned by PCR from a human cDNA library from peripheral blood mononuclear cells. The two cDNA fragments were joined in frame by overlapping PCR and subsequently cloned into the mammalian expression vector pSecTag/FRT/V5-His-TOPO (Invitrogen). The ligated construct (1.0 μg) was co-transfected with pOG44 (10.0 μg; Invitrogen) into the Flp-In CHO cell line (Invitrogen), using GeneJammer transfection reagent (Stratagene), in accordance with the manufacturer’s instructions. Transfectants were selected in 800 μg ml−1 hygromycin B, and maintained in UltraCHO (BioWhittaker). IL-21R-Ig was purified from the culture supernatant by passage through a Protein G–Sepharose column. Bound protein was eluted with 100 mM glycine-HCl pH 3.0 and immediately neutralized with 1.25 M Tris-HCl pH 8.8. The eluted protein was concentrated with an UltraFree-4 centrifugal device (Millipore), and the concentration was determined spectrophotometrically. The reagent was tested in vitro for its ability to neutralize the activity of IL-21-mediated TH17 differentiation.
Preparation of mononuclear cells from the central nervous system
After perfusion through the left cardiac ventricle with cold PBS, forebrain and cerebellum were dissected and spinal cords were flushed out with PBS by hydrostatic pressure followed by digestion with collagenase D (2.5 mg ml−1; Roche Diagnostics) and DNase I (1 mg ml−1; Sigma) at 37 °C for 45 min. Mononuclear cells were isolated by passing the tissue through a cell strainer (70 μm mesh) and Percoll gradient (70%–37%) centrifugation. Mononuclear cells were removed from the interphase, washed and resuspended in culture medium for further analysis.
In vitro T-cell proliferation
Draining lymph-node cells or splenocytes were cultured in DMEM/10% FCS supplemented with 50 μM 2-mercaptoethanol, 1 mM sodium pyruvate, non-essential amino acids, l-glutamine and 100 U ml−1 penicillin/100 μg ml−1 streptomycin. For antigen-specific recall cultures, 2.5 × 106 lymph-node cells or 5 × 106 ml−1 splenocytes were cultured for three days in the presence of MOG35–55 or OVA323–339. During the last 16 h, cells were pulsed with 1 μCi of [3H]thymidine (PerkinElmer). [3H]Thymidine incorporation in triplicate wells was measured with a β-counter (1450 MicroBeta Trilux; PerkinElmer).
Measurement of cytokines
Cell culture supernatants were collected after 48 h and the secreted cytokines were determined by ELISA (antibodies for IL-17 from BD Biosciences), by cytometric bead array (BD Biosciences) or by fluorescent bead assay (Luminex) for the indicated cytokines, in accordance with the manufacturers’ instructions. For quantitative PCR, RNA was extracted from FACS-sorted cells by using RNAeasy columns (Qiagen) after 48 h of stimulation in vitro. cDNA was transcribed as recommended (Applied Biosystems) and used as a template for quantitative PCR. Primer/probe mixtures for mouse ROR-γt and mouse IL-21 were obtained from Applied Biosystems. The Taqman analysis was performed on the AB 7500 Fast System (Applied Biosystems). Gene expression was normalized to the expression of β-actin.
Staining with MOG35–55/IAb tetramers and intracellular cytokine staining
MOG35–55/IAb tetramers were generated as reported11. The procedure for staining ex vivo with MOG35–55/IAb tetramers has been described in detail previously29. In brief, single-cell suspensions were incubated at a density of 107 cells ml−1 with the IAb multimers (30 μg ml−1) in DMEM supplemented with 5 μM IL-2 and 2% FCS (pH 8.0) at room temperature for 2.5 h. After being washed, cells were stained with 7-AAD (Molecular Probes) and CD4 (RM4-5; BD Biosciences). The percentage of tetramer+ cells was determined in the CD4+ gate of live (7-AAD−) cells. To control for non-specific binding, IAs control tetramers were used29. For intracellular cytokine staining, cells were stimulated for 4 h in culture medium containing 12-O-tetradecanoylphorbol-13-acetate (50 ng ml−1; Sigma), ionomycin (1 μg ml−1; Sigma), and monensin (GolgiStop, 1 μl ml−1; BD Biosciences) at 37 °C under 10% CO2. After staining of surface markers, cells were fixed and permeabilized (Cytofix/Cytoperm and Perm/Wash buffer, BD Biosciences) followed by staining with monoclonal antibodies against mouse IL-17 and IFN-γ (BD Biosciences) and fluorocytometric analysis (FACSCalibur).
Supplementary Material
Acknowledgements
We thank M. Collins for providing Il21r−/− mice, and D. Kozoriz, S. Tente, R. Chandwaskar and D. Lee for cell sorting and technical assistance. This work was supported by grants from the National Multiple Sclerosis Society, the National Institutes of Health, the Juvenile Diabetes Research Foundation Center for Immunological Tolerance at Harvard, and the Deutsche Forschungsgemeinschaft. V.K.K. is the recipient of the Javits Neuroscience Investigator Award from the National Institutes of Health.
Footnotes
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Information Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
References
- 1.Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 2.Cua DJ, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 3.Langrish CL, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu. Rev. Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 5.Steinman L. A brief history of TH17, the first major revision in the TH1/TH2 hypothesis of T cell-mediated tissue damage. Nature Med. 2007;13:139–145. doi: 10.1038/nm1551. [DOI] [PubMed] [Google Scholar]
- 6.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Veldhoen M, Hocking RJ, Flavell RA, Stockinger B. Signals mediated by transforming growth factor-β initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nature Immunol. 2006;7:1151–1156. doi: 10.1038/ni1391. [DOI] [PubMed] [Google Scholar]
- 8.Mangan PR, et al. Transforming growth factor-β induces development of the TH17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 9.Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 10.Bettelli E, Oukka M, Kuchroo VK. TH17 cells in the circle of immunity and autoimmunity. Nature Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 11.Korn T, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nature Med. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samoilova EB, Horton JL, Hilliard B, Liu TS, Chen Y. IL-6-deficient mice are resistant to experimental autoimmune encephalomyelitis: roles of IL-6 in the activation and differentiation of autoreactive T cells. J. Immunol. 1998;161:6480–6486. [PubMed] [Google Scholar]
- 13.Okuda Y, et al. IL-6-deficient mice are resistant to the induction of experimental autoimmune encephalomyelitis provoked by myelin oligodendrocyte glycoprotein. Int. Immunol. 1998;10:703–708. doi: 10.1093/intimm/10.5.703. [DOI] [PubMed] [Google Scholar]
- 14.Mendel I, Katz A, Kozak N, Ben-Nun A, Revel M. Interleukin-6 functions in autoimmune encephalomyelitis: a study in gene-targeted mice. Eur. J. Immunol. 1998;28:1727–1737. doi: 10.1002/(SICI)1521-4141(199805)28:05<1727::AID-IMMU1727>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 15.Eugster HP, Frei K, Kopf M, Lassmann H, Fontana A. IL-6-deficient mice resist myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. Eur. J. Immunol. 1998;28:2178–2187. doi: 10.1002/(SICI)1521-4141(199807)28:07<2178::AID-IMMU2178>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 16.Okuda Y, et al. IL-6 plays a crucial role in the induction phase of myelin oligodendrocyte glucoprotein 35–55 induced experimental autoimmune encephalomyelitis. J. Neuroimmunol. 1999;101:188–196. doi: 10.1016/s0165-5728(99)00139-3. [DOI] [PubMed] [Google Scholar]
- 17.Batten M, et al. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nature Immunol. 2006;7:929–936. doi: 10.1038/ni1375. [DOI] [PubMed] [Google Scholar]
- 18.Stumhofer JS, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nature Immunol. 2006;7:937–945. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- 19.Ivanov II, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 20.Kasaian MT, et al. IL-21 limits NK cell responses and promotes antigen-specific T cell activation: a mediator of the transition from innate to adaptive immunity. Immunity. 2002;16:559–569. doi: 10.1016/s1074-7613(02)00295-9. [DOI] [PubMed] [Google Scholar]
- 21.Yamanouchi J, et al. Interleukin-2 gene variation impairs regulatory T cell function and causes autoimmunity. Nature Genet. 2007;39:329–337. doi: 10.1038/ng1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leonard WJ, Spolski R. Interleukin-21: a modulator of lymphoid proliferation, apoptosis and differentiation. Nature Rev. Immunol. 2005;5:688–698. doi: 10.1038/nri1688. [DOI] [PubMed] [Google Scholar]
- 23.Zeng R, et al. The molecular basis of IL-21-mediated proliferation. Blood. 2007;109:4135–4142. doi: 10.1182/blood-2006-10-054973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang XO, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 25.Murphy E, et al. Reversibility of T helper 1 and 2 populations is lost after long-term stimulation. J. Exp. Med. 1996;183:901–913. doi: 10.1084/jem.183.3.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamura T, Kamogawa Y, Bottomly K, Flavell RA. Polarization of IL-4- and IFN-γ-producing CD4+ T cells following activation of naive CD4+ T cells. J. Immunol. 1997;158:1085–1094. [PubMed] [Google Scholar]
- 27.Kleinschek MA, et al. IL-25 regulates Th17 function in autoimmune inflammation. J. Exp. Med. 2007;204:161–170. doi: 10.1084/jem.20061738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vollmer TL, et al. Differential effects of IL-21 during initiation and progression of autoimmunity against neuroantigen. J. Immunol. 2005;174:2696–2701. doi: 10.4049/jimmunol.174.5.2696. [DOI] [PubMed] [Google Scholar]
- 29.Reddy J, et al. Detection of autoreactive myelin proteolipid protein 139–151-specific T cells by using MHC II (IAs) tetramers. J. Immunol. 2003;170:870–877. doi: 10.4049/jimmunol.170.2.870. [DOI] [PubMed] [Google Scholar]
- 30.Bettelli E, et al. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J. Exp. Med. 2003;197:1073–1081. doi: 10.1084/jem.20021603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.