

Abstract
Overexpression of ABCG2, a membrane-bound multidrug transporter, can make tumor cells resistant to treatment with conventional chemotherapeutic agents. A high-throughput screening effort with the NCI repository of natural product extracts revealed that eight tropical plant extracts significantly inhibited the function of ABCG2. This activity was tracked throughout the extract fractionation process to a series of ABCG2 inhibitory flavonoids (1–13). Their structures were identified by a combination of NMR, mass spectrometry, and circular dichroism studies, and this resulted in the elucidation of (2S)-5,7,3′-trihydroxy-4′-methoxy-8-(3″-methylbut-2″-enyl)-flavonone (1), (2S)-5,7,3′,5′-tetrahydroxy-8-[3″,8″ -dimethylocta-2″(E),7″-dienyl]flavonone (3), and 5,7,3′-trihydroxy-3,5′-dimethoxy-2′-(3′-methylbut-2-enyl)flavone (12) as new compounds.
ABCG2 (also known as breast cancer resistance protein, BCRP) is a member of the ATP binding cassette (ABC) family of multidrug transporters.1 It is a membrane-bound protein that can extrude cytotoxic agents from cells using the energy of ATP hydrolysis, and it has been implicated in the absorption, distribution, and excretion of a variety of drugs and cytotoxins. Overexpression of this cellular transporter is observed in a wide variety of human solid tumors, and an increase in ABCG2 has been associated with the development of resistance by tumor cells to chemotherapeutic agents such as topotecan and mitoxantrone.2,3 ABCG2 can confer drug resistance independent of the well-known multidrug resistance protein P-glycoprotein 1 (Pgp-1), and it has been demonstrated that inhibition of ABCG2 function increases the effectiveness of topotecan treatment in cancer patients.4 Therefore, new inhibitors of ABCG2 could be used to potentially enhance the efficacy and reduce the associated toxicity of several commonly administered anticancer drugs.
Flavonoids, a class of natural products ubiquitously present in higher plants, are reported to have an inhibitory effect on the activity of ABCG2.5–8 Several in vitro studies performed using different classes of flavonoids in combination with cytotoxic drugs have confirmed their ability to inhibit ABCG2 and thereby enhance the anticancer activity of mitoxantrone and topotecan.7,8 These studies have suggested some plausible structure—activity relationships for flavonoids as well.3 Nearly 5000 flavonoids have been reported from terrestrial plants, and many of them occur in fruits, seeds, vegetables, spices, herbs, wines, and teas that are commonly consumed by humans.9,10 Flavonoids are potent antioxidants and anti-inflammatory agents, and they have been reported to confer numerous health benefits.9–11 It has been postulated that their ability to function as effective free-radical scavengers in a cellular environment contributes to the prevention of several diseases, including cancer and cardiovascular diseases. There are numerous reports about potential cancer chemopreventive properties of flavonoids, and some of these compounds have been investigated extensively in the past few years.9,11 Based on these studies, there is considerable evidence to suggest that flavonoids may help prevent the occurrence of cancer in humans through a variety of different mechanisms.
In a previous study, a high-throughput screening campaign to identify natural product inhibitors of ABCG2 was conducted with the NCI natural products extract repository, and it was revealed that liphophilic extracts of eight tropical plants, Anonna reticulata L. (Annonaceae), Artocarpus odoratissimus Blanco (Moraceae), Calycopteris floribunda Lam. (Combretaceae), Evodia confusa Merr. (Rutaceae), E. elleryana F. Muell. (Rutaceae), Macaranga bicolor Muell. Arg. (Euphorbiaceae), M. conifera (Zoll.) Muell. Arg. (Euphorbiaceae), and Tabernaemontana macrocarpa Jack (Apocynaceae) showed significant ABCG2 inhibition. Bioassay-guided fractionation of these extracts yielded 13 flavonoids, and, in this paper, we report the isolation, structural characterization, and ABCG2-inhibitory activity of these compounds.
RESULTS AND DISCUSSION
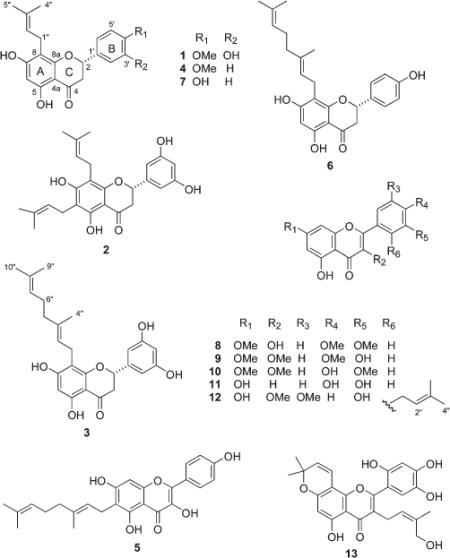
Organic solvent extracts of the eight plant species were fractionated by passing each individual extract through a diol solid-phase extraction column, followed by size-exclusion chromatography using Sephadex LH-20, while tracking the ABCG2-inhibitory activity via a cell-based fluorescence assay.12 Further purification of the ABCG2-active fractions by HPLC on C18 afforded flavonoids 1–13 (Table 1). Characterization of these metabolites by NMR and mass spectrometry revealed that compounds 1, 3, and 12 are new, while compounds 2, 4–11, and 13 are known. The latter were identified by comparison of their spectroscopic data with previously published values.13–23
Table 1.
ABCG2 Inhibitory Flavonoids Present in Eight Different Tropical Plant Extracts
| species | compounds isolated (w/w % yield from extract) |
|---|---|
| Macaranga conifera | 1 (3.56), 213 (5.03) |
| Macaranga bicolor | 3 (0.63), 414 (4.23), 515 (0.33), 616 (0.90), 717,18 (2.20) |
| Evodia confusa | 819 (0.18) |
| Evodia elleryana | 920 (0.42), 1021 (0.32) |
| Anonna reticulata | 1122 (1.06) |
| Calycopteris floribunda | 920 (3.30) |
| Tabernaemontana macrocarpa | 12 (0.18) |
| Artocarpus odoratissimus | 1323 (10.00) |
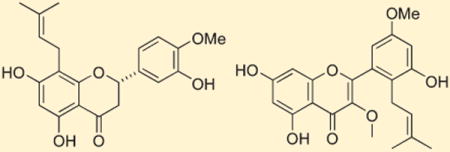
Analysis of the HRESIMS of compound 1 showed a [M + H]+ ion at m/z 371.1506, consistent with a molecular formula of C21H22O6. The 1H and 13C NMR data for compound 1 (Table 2) were indicative of a flavanone structure, while two 3H singlets at δH 1.63 and 1.59 were characteristic of two vinyl methyls of an isoprene unit (H3-4″ and H3-5″). This was supported by HMBC correlations from H3-4″ to C-5″ (δC 26.1) and from H3-5″ to C-4″ (δC 18.0), C-3″, and C-2″. H-1″ showed a COSY correlation with H-2″, and HMBC correlations with C-7 and C-8a, which established the connectivity of the isoprene moiety to the flavonoid skeleton at C-8. The singlet observed at δH 5.93 (H-6) had HMBC correlations with C-4a, C-5, and C-8 that further defined the A ring of a flavonone. Strong COSY correlations revealed two vicinal aromatic protons at δH 6.93 (H-6′) and 6.95 (H-5′), while HMBC correlations from H-5′ to C-4′, from H-6′ to C-2 and C-4′, and from an aromatic singlet δH 6.98 (H-2′) to C-1′ were consistent with a 1,3,4-trisubstituted benzene (ring B) attached to C-2 (δC 80.3). The H-2 oxymethine (δH 5.30) showed COSY correlations with both H-3a (δH 3.03) and H-3b (δH 2.73), while the C-4 resonance (δC 198.1) was assigned as a conjugated ketone to complete the flavanone C ring. A 3H singlet at δH 3.87 (δC 56.6) established the presence of a methoxy group, and its HMBC correlation with C-4′ confirmed it was substituted on this carbon. Both the 1H and 13C NMR chemical shifts assigned for rings A, B, and C of 1 were in good agreement with the values reported for related compounds.6–9 The absolute stereochemistry at C-2 was assigned by the analysis of the CD spectrum. Compound 1 showed a negative Cotton effect resulting from a π–π* transition at 290 nm, and this was highly indicative of a 2S configuration.24 The structure of 1 was thus assigned as (2S)-5,7,3′-trihydroxy-4′-methoxy-8-(3″-methylbut-2″-enyl)flavanone.
Table 2.
1H and 13C NMR Data (500 MHz, CD3OD) for Compounds 1, 3, and 12
| compound 1
|
compound 3
|
compound 12
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| position | δC | δH (J in Hz) | HMBCa | δC | δH (J in Hz) | HMBCa | δC | δH (J in Hz) | HMBCa |
| 2 | 80.3 | 5.30 1H, dd (12.5, 2.5) | 80.6 | 5.25 1H, dd (12.5, 3.0) | 158.3 | ||||
| 3 | 44.2 | 3.03 1H, dd (17.0, 12.5) | 2, 1′ | 44.4 | 3.05 1H, dd (17.0, 12.5) | 2 | 139.7 | ||
| 2.73 1H, dd (17.0, 2.5) | 2.69 1H, dd (17.0, 3.0) | ||||||||
| 4 | 198.1 | 197.9 | 180.1 | ||||||
| 4a | 103.5 | 103.3 | 106.0 | ||||||
| 5 | 166.2 | 162.7 | 163.2 | ||||||
| 6 | 96.6 | 5.93 1H, s | 4a, 5, 8 | 95.7 | 5.94 1H, s | 4a, 5, 7, 8 | 99.9 | 6.19 1H, s | 4a, 5, 7, 8 |
| 7 | 163.3 | 166.1 | 166.0 | ||||||
| 8 | 109.2 | 109.9 | 94.9 | 6.38 1H, s | 4a, 6, 8a | ||||
| 8a | 161.6 | 162.6 | 158.5 | ||||||
| 1′ | 133.6 | 132.2 | 122.0 | ||||||
| 2′ | 114.7 | 6.98 1H, d (1.3) | 1′ | 119.4 | 6.79 1H, s | 2, 1′, 3′ | 129.6 | ||
| 3′ | 147.9 | 147.0 | 148.8 | ||||||
| 4′ | 149.4 | 114.8 | 6.91 1H, s | 3′,5′,6′ | 110.2 | 7.54 1H, s | 3′,6′ | ||
| 5′ | 112.7 | 6.95 1H, d (8.5) | 4′ | 146.7 | 148.7 | ||||
| 6′ | 119.0 | 6.93 1H, dd (8.5, 1.3) | 2, 4′ | 116.4 | 6.79 1H, s | 2, 1′, 3′ | 124.1 | 7.54 1H, s | 2, 1′, 4′, 5′ |
| 1″ | 22.6 | 3.17 1H, dd (7.6, 14.4) | 7, 8a | 22.0 | 3.21 2H, d (7.3) | 7, 8, 8a, 2″, 3″ | 29.0 | 3.36 2H, d (7.0) | 1′,2′,3′,2″ |
| 3.20 1H, dd (7.6, 14.4) | 7, 8a | ||||||||
| 2″ | 124.1 | 5.15 1H, t (7.5) | 124.2 | 5.19 1H, br t (7.0) | 123.5 | 5.35 1H, br t (7.0) | 1″,4″,5″ | ||
| 3″ | 131.8 | 135.4 | 133.9 | ||||||
| 4″ | 18.0 | 1.63 3H s | 5″ | 16.3 | 1.75 3H, s | 2″,3″ | 18.9 | 1.75 3H, s | 3″,5″ |
| 5″ | 26.1 | 1.59 3H s | 2″,3″,4″ | 41.0 | 1.95 2H, t (7.2) | 2″,3″,6″ | 26.1 | 1.77 3H, s | 4″,3″ |
| 6″ | 27.9 | 2.06 2H, dt (6.8, 7.4) | 5″,8″ | ||||||
| 7″ | 125.6 | 5.06 1H, br t (7.4) | |||||||
| 8″ | 132.1 | ||||||||
| 9″ | 17.9 | 1.56 3H, s | 8″, 10″ | ||||||
| 10″ | 26.0 | 1.62 3H, s | 8″,9″ | ||||||
| 3-OCH3 | 60.7 | 3.77 3H, s | 3 | ||||||
| 4′-OCH3 | 56.6 | 3.87 3H s | 4′ | ||||||
| 5′-OCH3 | 56.8 | 3.93 3H, s | 5′ | ||||||
HMBC correlations are from the proton(s) stated to the indicated carbons.
Compound 3 displayed a [M + H]+ ion at m/z 425.1963 by HRESIMS, which established a molecular formula of C25H28O6. It also had 1H and 13C NMR data that were consistent with a substituted flavonone structure (Table 2). The A ring displayed only one aromatic singlet at δH 5.94 (H-6) that showed HMBC correlations with four quaternary carbons assigned as C-4a, C-5, C-7, and C-8. A doublet at δH 3.14 (H-1″) had HMBC correlations with C-8 and two oxygen-substituted aromatic carbons at δC 166.12 (C-7) and 162.6 (C-8a). These observations and the respective carbon chemical shift values were characteristic of a pentasubstituted flavanone ring A subunit with the C-1″ methylene attached at C-8. This side chain was extended to be a bisisoprene unit partly on the basis of HMBC correlations from H-1″ to C-2″ and C-3″ and from H3-4″ to C-2″ and C-3″. The upfield chemical shift of C-4″ (δC 16.3) was indicative of E geometry for the C-2″, C-3″ double bond. The side chain was further elaborated by HMBC correlations observed from H-5″ to C-3″ and from H-6″ to C-5″ and C-8″. Finally, 1H–1H COSY coupling data and HMBC correlations from both H3-10″ and H3-9″ to C-8″ confirmed the attachment of a second isoprene unit to C-5″, which fully defined the geranyl side chain in compound 3.
Ring B was assigned as a 1,3,5-trisubstituted benzene ring on the basis of a lack of 1H–1H coupling observed for the three ring-associated aromatic protons and HMBC correlations from H-6′ to C-1′, C-2, and C-5′, from H-4′ to C-3′, C-5′, and C-6′, and from H-2′ to C-2, C-1′, and C-3′. The chemical shifts for C-3′ (δC 147.0) and C-5′ (δC 146.7) were consistent with the substitution of hydroxy groups at these positions. While ring B is symmetric in its substitution pattern, the nonequivalence in chemical shift of these two carbons as well as C-2′ (δC 119.4) and C-6′ (δC 116.4) indicates there is restricted rotation of this ring. Similar NMR data have been reported for other flavonoids with the same substituted B ring.25,26 The connectivity of ring B to ring C was established by the HMBC correlations from H-6′ and H-2′ to the oxymethine C-2 (δC 80.6). Vicinal coupling between H-2 and both H-3 methylene protons in conjunction with 13C chemical shift comparisons27 defined the flavanone ring C in 3, which completed the planar structural assignment. Observation of a negative Cotton effect at 295 nm in the CD spectrum allowed assignment of S absolute stereochemistry for C-2, and thus compound 3 was defined as (2S)-5,7,3′,5′-tetrahydroxy-8-[3″,8″-dimethylocta-2″ (E),7″-dienyl]flavanone.
HRESIMS analysis of compound 12 provided a [M +H]+ion at m/z 399.1456 consistent with a molecular formula of C22H22O7. The 1H NMR spectrum contained a singlet at δH 6.38 (H-8), which had HMBC correlations with C-6, C-8a, and C-4a. A second aromatic singlet at δH 6.19 (H-6) showed HMBC correlations with C-5, C-7, C-8, and C-4a that helped define ring A. An overlapped two-proton singlet at δH 7.54 (H-4′/6′) had HMBC correlations with C-2, C-3′, C-4′, C-5′, and C-6′ that were consistent with the substitution pattern assigned for ring B. The chemical shifts of C-3′ (δC 148.8) and C-5′ (δC 148.7) revealed oxygenation at these carbons, and an OMe group appearing at δH 3.93 showed a HMBC correlation with C-5′, which affirmed its attachment at this position. HMBC correlations from a methylene resonance at δH 3.36 (H-1″) to C-2′, C-3′, C-2″, and C-3″ helped establish the attachment of an isoprene substituent at C-2′. The isoprenoid subunit was further defined by HMBC correlations from both H3-4″ (δH 1.77) and H3-5″ (δH 1.75) to C-3″ and C-2″. An OMe group that resonated at δH 3.76 was correlated by HMBC to C-3 (δC 139.7), which secured the C ring of a flavone. The structure of compound 12 could then be assigned as 5,7,3′-trihdroxy-3,5′-dimethoxy-2′-(3″-methylbut-2-enyl)flavone.
A cell-based high-throughput assay developed in our laboratory12 was used to assess the ABCG2 inhibitory activity of flavanoids 1–13 (Table 3). This assay measured cellular accumulation of the fluorescent chlorophyll derivative pheophorbide a (PhA), which is a substrate specific for ABCG2. When the ABCG2 transporter is fully functional, PhA is actively pumped out of cells, and there is little cellular associated fluorescence. However, ABCG2 inhibition results in the accumulation of PhA within the cells and increased cellular fluorescence. The ABCG2 inhibitory activity of test compounds was measured in terms of cell-associated fluorescence intensity (670 nm) relative to the positive control compound fumitremorogin C (FTC).12 DMSO solutions of compounds 1–13 were tested for their ability to inhibit ABCG2, and the results are summarized in Table 3. All of the test materials were active against ABCG2, and IC50 potencies ranged from 3.1 to 22.6 μM. These results are in line with previous reports of ABCG2 inhibitory activity for flavonoids. While there has been some speculation about possible structure—activity relationships regarding the substituents and the substitution patterns for various classes of flavonoids,3 the present ABCG2 test results did not support any clear trends in this regard. Comprehensive testing of a much larger set of related compounds will be required to define what structural parameters are associated with potency, efficacy, and specificity with regard to ABCG2. The fact that prenylated flavonoids with ABCG2 inhibitory properties occur in a wide diversity of plant sources is intriguing and raises important questions about the physiological role and function of these compounds in the host plant.
Table 3.
ABCG2 Inhibitory Activity of Compounds 1–13
| compound | ABCG2 inhibition IC50 (μM) |
|---|---|
| 1 | 6.6 |
| 2 | 4.1 |
| 3 | 15.5 |
| 4 | 22.6 |
| 5 | 5.7 |
| 6 | 12.3 |
| 7 | 3.1 |
| 8 | naa |
| 9 | 5.7 |
| 10 | 17.2 |
| 11 | 8.9 |
| 12 | 8.4 |
| 13 | 6.5 |
| FTCb | 0.8 |
Not tested.
Fumitremorogin C.
EXPERIMENTAL SECTION
General Experimental Procedures
Optical rotations were measured on a Perkin-Elmer 241 polarimeter using a Na lamp at 25°C. Infrared spectra were obtained with a Perkin-Elmer Spectrum 2000 FT-IR spectrometer. Ultraviolet–visible experiments were measured on a Varian Cary 50 Bio UV spectrophotometer. CD spectra were obtained from a JASCO J720 spectropolarimeter. 1H, 13C, gHSQC, gHMBC, and 1H –1H COSY NMR spectra were obtained on a Varian Inova 500 instrument operating at 500 MHz for 1H and 125 MHz or 13C, using residual solvent resonances for internal reference. 1H chemical shifts were recorded relative to δ 7.24 (CDCl3), whereas the 13C shifts were referenced to δ 77.23 (CDCl3). High-resolution mass spectra were recorded on an Agilent Q-TOF 6520 mass spectrometer. Low-resolution mass spectra were recorded on an Agilent Series 1100 LC-MS. HPLC was performed with a Varian Prostar multisolvent delivery system connected to a Varian Prostar photodiode array detector using a (5 μm) Phenomenex C18 column (250 mm × 10 mm).
Plant Material
Specimens of Anonna reticulata L. (Annonaceae) were collected in Belize at 89°04′ W and 17°06′ N on October 13, 1994, and identified by Rosita Arvigo of the Institute of Economic Botany, New York Botanical Garden (voucher number OCJT2026). Artocarpus odoratissimus Blanco (Moraceae) was collected in the Similajan forest in Sarawak, Malaysia, at longitude 113°03′ E and latitude 3°20′ N on September 5, 1987. The sample was identified by D. D. Soerjarto of the University of Illinois at Chicago (voucher number Q6601979). Calycopteris floribunda Lam. (Combretaceae) was collected in the Chittagong district in Bangladesh at longitude 91°05′ and latitude 22°34′, on April 13, 1994, and identified by Ahmed M. Huq (voucher number OFCZ11). Samples of Evodia confusa Merr. (Rutaceae) were collected in Borneo north of Safoda Camp in Telupid on September 8, 1994, and identified by W. Meijer (voucher number OFCZ1145). Evodia elleryana F. Muell (Rutaceae) was collected in Madang Province in Papua New Guinea at longitude 145°58′ andlatitude -5°18′ on January 27, 1989. The sample was identified by W. Takeuchi (voucher number Q6606980). Macaranga bicolor Muell. Arg. (Euphorbiaceae) was collected in Palawan, Philippines, at longitude 118°02′ and latitude 9°51′ on April 14, 1989. The sample was identified by D. D. Soerjarto of the University of Illinois at Chicago (voucher number Q6608058). Macaranga conifera (Zoll.) Muell. Arg. (Euphorbiaceae) was collected in the Sandakan district, Saba, Borneo, near Kebun China on September 22, 1987, and identified by W. Meijer (voucher number Q6605205). Tabernaemontana macrocarpa Jack (Apocynaceae) was collected in Sarawak, Malaysia, at longitude 113°03′ E and latitude 3°20′ N on September 12, 1987. The sample was identified by M. M. J. Van Balgooy (voucher number Q6602165).
Extraction and Isolation
The standardized NCI extraction protocol for terrestrial plant samples has recently been described in detail by McCloud.28 In brief, dried, ground plant material was sequentially extracted with MeOH−CH2Cl2 (1:1) followed by 100% MeOH. These two solutions were combined and evaporated under reduced pressure to give the crude organic solvent extract. An aliquot of each active plant extract was individually loaded onto prepacked diol SPE cartridges (100 mg of extract per 500 mg cartridge) and fractionated by elution with the following solvent combinations: (A) hexane−CH2Cl2 (9:1), (B) CH2Cl2−EtOAc (20:1), (C) EtOAc (100%), (D) EtOAc−MeOH (5:1), (E) MeOH (100%). Chromatography fractions that inhibited ABCG2 were further separated by size-exclusion chromatography on Sephadex LH-20 eluted with hexane−CH2Cl2−MeOH (2:5:1). Compounds 1 and 2 were obtained in pure form directly from the LH-20 separation of fraction C from Macaranga conifera. LH-20 separation of fraction A from M. bicolor provided compound 4 directly, while similar processing of the B fraction gave compounds 6 and 7. Final purification of compounds 3 and 5 was accomplished by HPLC on C18 eluted with CH3CN−water (6:4) followed by 100% CH3CN The B fraction from Evodia confusa was fractionated on LH-20 followed by C18 HPLC eluted with 100% H2O (20 min), CH3CN−H2O (1:1, 15 min), and 100% CH3CN (5 min) to give compound 8. The B fraction from E. elleryana was chromatographed in an identical manner to that described for E. confusa to provide compounds 9 and 10. The B fraction from Calycopteris floribunda was treated identically to give compound 9. The C fraction from Tabernaemontana macrocarpa was passed through LH-20 eluted with CH2Cl2−MeOH (1:1) and then C18 HPLC sequentially eluted with H2O−CH3CN (9:1), followed by 100% CH3CN, to give compound 12. Fraction D from Artocarpus odoratissimus was separated by LH-20 eluted with CH2Cl2−MeOH (1:1) and then C18 HPLC eluted with H2O−CH3CN (55:45) to give compound 13.
(2S)-5,7,3′-Trihydroxy-4′-methoxy-8-(3″-methylbut-2″-enyl)flavanone (1): colorless, amorphous solid; [α]25d −20.6 (c 0.08, MeOH); UV (MeOH) λmax(ɛ) 210 (1510), 225 (3700), 235 (1469), 245 (3700), 275 (3700), 310 (1488), 320 (1223) nm; IR (thin film) νmax3391 (br), 2928, 1634, 1515, 1442, 1385, 1272, 1174, 1131, 1078, 1027, 807, 763, 737 cm−1; 1H and 13C NMR data, see Table 2; HRESIMS m/z [M + H]+ 371.1506 (C21H23O6 requires 371.1489).
(2S)-5,7,3′,5′-Tetrahydroxy-8-(3″,8″-dimethylocta-2″,7″-dienyl)flavanone (3): colorless, amorphous solid; [α]25d −7.2 (c 0.04, MeOH); UV (MeOH) λmax(ɛ) 244 (16920), 260 (16 920), 270 (7598), 295 (16920), 305 (6851) nm; IR (thin film) νmax 3367 (br), 2924,1634, 1455,1284,1159,1131,1087, 815 cm−1; 1Hand 13C NMR data, see Table 2; HRESIMS m/z 425.1963 [M + H]+ (C26H29O6 requires 425.1958).
5,7,3′-Trihdroxy-3,5′-dimethoxy-2′-(3′-methylbut-2-enyl)-flavone (12): colorless, amorphous solid; [α]25d −22.8 (c 0.14, MeOH); UV (MeOH) λmax(ɛ) 208 (16920), 254, 360 (16920), 270 (7598), 295 (16920), 305 (6851) nm; IR (thin film) νmax 3139 (br), 2853,1650,1597,1581, 1494, 1441,1378,1352,1172,1162,1070, 838, 806 cm−1; 1Hand 13C NMR data, see Table 2; HRESIMS m/z 399.1456 [M +H]+ (C22H23O7 requires 399.1438.
Bioactivity Determination
ABCG2 inhibition of the chromatographic fractions and pure compounds was determined using a fluorescence accumulation assay previously developed in our laboratory.12 This assay was performed with NCI-H460 human lung non-small-cell carcinoma cells maintained in RPMI1640 supplemented with penicillin, streptomycin, 10% FBS, and 20 nM mitoxantrone. Inclusion of mitoxantrone results in selection of cells that overexpress ABCG2.29 The cells and the media were prepared according to the previously published protocol and transferred to 384-well assay plates.12 PhA was added to the cells and then immediately followed by the addition of the test compounds, the DMSO vehicle control, or the FTC positive control. After incubation (37 °C, 2–20 h) the medium was removed, and the plates were washed with PBS in order to reduce background fluorescence. Fluorescence was read using a fluorescence plate reader with the emission set at 670 nm.
Supplementary Material
Acknowledgments
We thank D. Newman (NCI) and T. McCloud (SAIC-Frederick) for the plant extracts and M. Dyba and S. Terasova (Biophysics Resource, SBL, NCI-Frederick) for assistance with the HRLCMS studies. MA.V. gratefully acknowledges the Higher Education Commission (HEC), Government of Pakistan, for providing a Post Doctoral scholarship. This research was supported in part by the Intramural Research Program of NIH, National Cancer Institute, Center for Cancer Research. This project was also funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Supporting Information. HRESIMS, 1HNMR, 13CNMR, gHSQC, and gHMBC spectra for (2S)-5,7,3′-trihydroxy-4′-methoxy-8-(3″-methylbut-2″-enyl)flavanone (1), (2S)-5,7,3′,5′-tetrahydroxy-8-[3″,8″-dimethylocta-2″(E),7″-dienyl]flavanone (3), and 5,7,3′-trihdroxy-3,5′-dimethoxy-2′-(3′-methylbut-2-en-yl)flavone (12). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Krishnamurthy P, Schuetz JD. Annu Rev Pharmacol Toxicol. 2006;46:381–410. doi: 10.1146/annurev.pharmtox.46.120604.141238. [DOI] [PubMed] [Google Scholar]
- 2.Rzhetsky DM, Allikmets R. Genome Res. 2001;11:1156–1166. doi: 10.1101/gr.184901. [DOI] [PubMed] [Google Scholar]
- 3.Ahmed-Belkacem A, Pozza A, Munoz-Martínez F, Bates SE, Castanys S, Gamarro A, Di Pietro A, Perez-Victoria JM. Cancer Res. 2005;65:4852–4860. doi: 10.1158/0008-5472.CAN-04-1817. [DOI] [PubMed] [Google Scholar]
- 4.Kruijtzer CJ. Clin Oncol. 2002;20:2943–2950. doi: 10.1200/JCO.2002.12.116. [DOI] [PubMed] [Google Scholar]
- 5.Wand X, Morris ME. Drug Metab Dispos. 2007;35:268–274. doi: 10.1124/dmd.106.011684. [DOI] [PubMed] [Google Scholar]
- 6.Zhang S, Yang X, Coburn R, Morris ME. Biochem Pharmacol. 2005;70:627–639. doi: 10.1016/j.bcp.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 7.Zhang S, Yang X, Morris ME. Mol Pharmacol. 2004;65:1208–1216. doi: 10.1124/mol.65.5.1208. [DOI] [PubMed] [Google Scholar]
- 8.Megumi Y, Yoji I, Kazumi S, Hisahiro Y, Hideyuki M, Seigo S, Toshihisa IJ. Exper Ther Oncol. 2004;4:25–35. [Google Scholar]
- 9.Marchand LL. Biomed Pharmacother. 2002;56:296–301. doi: 10.1016/s0753-3322(02)00186-5. [DOI] [PubMed] [Google Scholar]
- 10.Middleton E, Jr, Kandaswami C, Theoharides TC. Pharmacol Rev. 2000;52:673–751. [PubMed] [Google Scholar]
- 11.Nijveldt RJ, van Nood E, van Hoorn DE, Boelens PG, van Norren K, van Leeuwen PA. Am J Clin Nutr. 2001;74:418–425. doi: 10.1093/ajcn/74.4.418. [DOI] [PubMed] [Google Scholar]
- 12.Henrich CJ, Bokesch HR, Dean M, Bates SE, Robey RW, Goncharowa EI, Wilson JA, McMahon JB. J Biomol Screen. 2006;11:176–183. doi: 10.1177/1087057105284576. [DOI] [PubMed] [Google Scholar]
- 13.Garo E, Wolfender J, Hostettmann K. Helv Chim Acta. 1998;81:754–763. [Google Scholar]
- 14.Parsons IC, Gray AI, Waterman PGJ. Nat Prod. 1993;56:46–53. [Google Scholar]
- 15.Hnawia E, Thoison O, Gueritte-Voegelein F, Bourret D, Sevenet T. Phytochemistry. 1990;29:2367–2368. [Google Scholar]
- 16.Shirataki Y, Yokoe I, Endo M, Komatsu M. Chem Pharm Bull. 1985;33:444–447. doi: 10.1248/cpb.26.3863. [DOI] [PubMed] [Google Scholar]
- 17.McCormick S, Robson K, Bohm B. Phytochemistry. 1986;25:1723–1726. [Google Scholar]
- 18.Jang D, Cuendet M, Hawthorne ME, Kardono LBS, Kawanishi K, Fong H, Mehta RG, Pezzuto JM, Kinghorn AD. Phytochemistry. 2002;61:867–872. doi: 10.1016/s0031-9422(02)00378-3. [DOI] [PubMed] [Google Scholar]
- 19.Rodriguez G, Vander Velde E, Mabry TJ. Phytochemistry. 1972;11:2821–2826. [Google Scholar]
- 20.King FE, King TJ, Sellars KJ. Chem Soc. 1952:92–95. [Google Scholar]
- 21.Ahmed A, Ali A, Mabry TJ. Phytochemistry. 1989;28:665–667. [Google Scholar]
- 22.Imre S, Islimyeli S, Oztunc A, Buyuktimkin N. Planta Med. 1984;50:360. doi: 10.1055/s-2007-969735. [DOI] [PubMed] [Google Scholar]
- 23.Cao S, Butler MS, Buss AD. Nat Prod Res. 2003;17:79–81. doi: 10.1080/1478641031000103641. [DOI] [PubMed] [Google Scholar]
- 24.Slade D, Ferreira D, Marais JP. Phytochemistry. 2005;66:2117–2215. [Google Scholar]
- 25.Wang X-L, Wang N-L, Zhang Y, Gao H, Pang W-Y, Wong M-S, Zhang G, Qin L, Yao X-S. Chem Pharm Bull. 2008;56:46–51. doi: 10.1248/cpb.56.46. [DOI] [PubMed] [Google Scholar]
- 26.Wang XW, Mao Y, Wang N-L, Yao XS. Molecules. 2008;13:2796–2803. doi: 10.3390/molecules13112796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tseng M, Chou C, Chen Y, Kuo YJ. Nat Prod. 2001;64:827–828. doi: 10.1021/np0100338. [DOI] [PubMed] [Google Scholar]
- 28.McCloud TG. Molecules. 2010;15:4526–4563. doi: 10.3390/molecules15074526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robey RW, Honjo Y, van de Laar A, Miyake K, Regis JT, Litman T, Bates SE. Biochim Biophys Acta. 2001;1512:171–182. doi: 10.1016/s0005-2736(01)00308-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.