

Abstract
The extracellular matrix (ECM) receptor dystroglycan (DG) serves as a cellular receptor for the highly pathogenic arenavirus Lassa virus (LASV) that causes a hemorrhagic fever with high mortality in man. In the host cell, DG provides a molecular link between the ECM and the actin cytoskeleton via the adapter proteins utrophin or dystrophin. Here we investigated post-translational modifications of DG in the context of LASV cell entry. Using the tyrosine kinase inhibitor genistein, we found that tyrosine kinases are required for efficient internalization of virus particles, but not virus-receptor binding. Engagement of cellular DG by LASV envelope glycoprotein (LASV GP) in human epithelial cells induced tyrosine phosphorylation of the cytoplasmic domain of DG. LASV GP binding to DG further resulted in dissociation of the adapter protein utrophin from virus-bound DG. This virus-induced dissociation of utrophin was affected by genistein treatment, suggesting a role of receptor tyrosine phosphorylation in the process.
INTRODUCTION
The Old World arenavirus Lassa virus (LASV) is the causative agent of a severe viral hemorrhagic fever in humans with several hundred thousand infections per year in Africa and thousands of deaths annually (McCormick and Fisher-Hoch, 2002). Fatal LASV infection is characterized by rapid viral replication and spread, resulting in uncontrolled viral infection with progressive signs and symptoms of hemorrhagic disease and shock (Geisbert and Jahrling, 2004). The death toll of LASV infection among hospitalized patients can reach 15-30%. There is no licensed vaccine against LASV and current therapeutic options are limited, making LASV arguably one of the most neglected tropical pathogens.
Arenaviruses are enveloped negative-strand RNA viruses with a bi-segmented genome, whose replication takes place in the cytoplasm (de la Torre, 2009; Buchmeier et al., 2007). The two viral RNA segments, L and S, include each two open reading frames. The S segment encodes the envelope glycoprotein precursor (GPC) and the nucleoprotein (NP) and the L segment codes for the matrix protein (Z) and the viral polymerase (L). LASV GPC is synthesized as a single polypeptide and undergoes processing by the host cell protease subtilisin kexin isozyme-1(SKI-1)/site-1 protease (S1P) (Rojek et al., 2008b; Beyer et al., 2003; Lenz et al., 2001), yielding the N-terminal GP1 and the transmembrane GP2. LASV GP1 is involved in binding to cellular receptors (Borrow and Oldstone, 1992), whereas the GP2 part mediates fusion and resembles the membrane-proximal regions of other viral fusion proteins (Igonet et al., 2011; Eschli et al., 2006).
Binding of a virus to its cellular receptor(s) and subsequent entry into target cells are the first steps of virus infection and a fundamental aspect of the virus-host cell interaction. These initial steps of infection are also promising targets to block the pathogen before it can take control over the host cell. Therapeutic intervention at the level of cell entry is of particular interest for highly pathogenic viruses like LASV. The first cellular receptor discovered for LASV and the prototypic Old World arenavirus lymphocytic choriomeningitis virus (LCMV) is dystroglycan (DG) a ubiquitous receptor for extracellular matrix (ECM) proteins (Oldstone and Campbell, 2011; Cao et al., 1998). More recently, additional candidate receptors for LASV have been reported, including the C-type lectins DC-SIGN, LSECtin, and the Tyro3/Axl/Mer (TAM) receptor tyrosine kinases Axl and Tyro3 (Shimojima et al., 2012). However, in DG-expressing cells, DG appears to be the preferred receptor for LASV (Shimojima et al., 2012).
Dystroglycan is a highly conserved protein initially translated as a single polypeptide chain that is cleaved into the extracellular α-DG, and transmembrane β-DG (Barresi and Campbell, 2006). DG is expressed in most developing and adult tissues in cells that adjoin basement membranes (Durbeej et al., 1998) and is crucial for normal cell-matrix interactions (Henry et al., 2001; Henry and Campbell, 1998). Alpha-DG has a central, highly glycosylated mucin-type domain that connects the globular N-and C-terminal domains. At the extracellular site, α-DG undergoes high-affinity interactions with the ECM proteins laminin, agrin, perlecan, and neurexins (Barresi and Campbell, 2006). Alpha-DG is non-covalently associated with β-DG, which binds intracellular to the adaptor proteins dystrophin or utrophin that link DG to the actin cytoskeleton. DG is also associated with a number of signaling molecules, including the adaptor molecule grb2 (Yang et al., 1995), the canonical MAP kinases MEK and ERK (Spence et al., 2004), and the focal adhesion kinase (Cavaldesi et al., 1999). Upon receptor binding, LASV and LCMV are internalized via an endocytotic pathway that is independent of clathrin, caveolin, and dynamin and bypasses classical Rab5-dependent incoming routes of vesicular trafficking (Quirin et al., 2008; Rojek et al., 2008c). During cell entry, LASV and LCMV pass through the multivesicular endosome, where the virus-receptor complex undergoes sorting by the endosomal sorting complex required for transport (ESCRT) delivering the viruses to late endosomes, where low pH-induced fusion occurs after 20-30 minutes (Pasqual et al., 2011; Quirin et al., 2008; Rojek et al., 2008c).
In the host cell, DG appears as a structural component of cell-matrix contacts, which suggest a rather static role. Considering the receptor dynamics expected in the context of LASV cell entry, we hypothesized that virus binding may alter DG trafficking, e.g. by inducing post-translational modifications that target the DG complex towards rapid endocytosis. Studies in prototypic primate and human cells had demonstrated that cell adhesion to ECM proteins can induce phosphorylation of tyrosine Y892 located within a PPxY motif present at the C-terminus of the cytoplasmic domain of β-DG by non-receptor tyrosine kinases of the src family (Sotgia et al., 2003; Sotgia et al., 2001; James et al., 2000). This tyrosine phosphorylation of Y892 prevented the association of β-DG with the cytoskeletal adaptor protein utrophin (James et al., 2000) and resulted in a redistribution of the DG complex from the plasma membrane to intracellular compartments (Sotgia et al., 2003), linking tyrosine phosphorylation of β-DG to receptor internalization. More recent studies confirmed a role of tyrosine phosphorylation of β-DG at Y892 for the endocytosis of DG in myoblasts and provide evidence for a role of tyrosine phosphorylation of DG in the development of muscle pathophysiology in an animal model for muscular dystrophy (Miller et al., 2012). In the present study we investigated the role of receptor tyrosine phosphorylation for cell entry of LASV.
RESULTS
Tyrosine kinases are involved in endocytosis of rLCMV-LASVGP
Recent studies using the broadly specific tyrosine kinase inhibitor genistein revealed a role for tyrosine kinases in cell entry of LASV (Kolokoltsov et al., 2012). However, the specific step(s) of the viral entry process that depend on tyrosine kinases had not yet been defined. In a first step, we sought to confirm and extend these earlier studies and tried to distinguish effects of genistein on virus-cell attachment from endocytosis. Since LASV is a BSL4 pathogen, work with live virus is restricted to laboratories with high security containment. To circumvent these biosafety restrictions, we used a recombinant form of the prototypic LCMV expressing the envelope GP of LASV (rLCMV-LASVGP) (Rojek et al., 2008c). The chimera rLCMV-LASVGP does not show significant attenuation in vitro when compared to the parental LCMV strain and grows to robust titers. Since receptor binding and host cell entry of arenaviruses are mediated exclusively by the viral GP, rLCMV-LASVGP adopts the receptor binding characteristics of LASV (Rojek et al., 2008c) and represents a suitable BSL2 surrogate for our studies on LASV-receptor interaction and cell entry. As a cell culture model, we chose the human lung epithelial cell line WI-26 VA4, which had previously been utilized for studies on DG signaling (Ferletta et al., 2003) and the interaction of LASV with its receptor DG (Rojek et al., 2012).
First, we verified that cell attachment and entry of LASV into WI-26 VA4 cells was mediated by DG. Cells were pre-treated with increasing concentration of the monoclonal antibody (mAb) IIH6 that recognizes a functional glycan epitope on α-DG (Kanagawa et al., 2004) and competes with virus binding (Kunz et al., 2005b). Cells were then infected with rLCMV-LASVGP and infected cells detected after 16 hours by immunofluorescence (IF) staining of the viral nucleoprotein (NP). Pre-treatment with mAb IIH6, but not an IgM isotype control significantly blocked infection with rLCMV-LASVGP in a dose-dependent manner, confirming DG as a major receptor for LASV in these cells (Fig. 1A), as shown previously (Rojek et al., 2012).
Fig. 1. Genistein inhibits internalization of rLCMV-LASVGP.
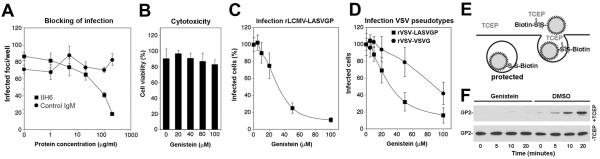
(A) LASV infection in WI-26 VA4 cells is mediated by DG. Monolayers of WI-26 VA4 cells in M96 plates were blocked with MAb IIH6 or an unrelated mouse IgM (Control IgM) at the indicated concentrations for 2 h at 4°C. Next, 200 PFU of rLCMV-LASVGP was added for 45 min. Infection was assessed after 16 hours by immunofluorescence (IF) staining for LCMV NP. Infected foci were counted in each well (means ± SD, n = 3). (B) Cytotoxicity of genistein. WI-26 VA4 cells were treated with the indicated concentrations of the drug for 4 hours, followed by a wash out and incubation for a total of 16 hours. Cell viability was assessed by Cell Titer Glo® assay. Data are triplicates ± SD. (C) Inhibition of rLCMV-LASVGP infection with genistein. WI-26 VA4 cells were treated with the indicated concentrations of genistein for one hour, followed by infection with rLCMV-LASVGP at multiplicity of 1, followed by wash out of the drug at 4 hours. After 12 hours of culture in presence of 20 mM ammonium chloride, cells were fixed and infection detected by IF for LCMV NP. Data are triplicates ± SD. (D) Inhibition of infection of VSV pseudotypes with genistein. WI-26 VA4 cells were treated with the indicated concentrations of genistein for one hour, followed by infection with rVSV-LASVGP and rVSV-VSVG (200 PFU/well). After 24 hours, infection was assessed by detection of GFP positive cells in direct fluorescence microscopy (n = 3 ± SD). (E) Schematic of the virus internalization assay (for details, please see text). (F) Genistein treatment prevents internalization of rLCMV-LASVGP. WI-26 VA4 cells were pre-treated with 50 μM of genistein or vehicle control (DMSO). After one hour, cells were chilled on ice and incubated with biotin-S-S-labeled rLCMV-LASVGP (100 particles/cell) for one hour in the cold. Unbound virus was removed, cells shifted to 37 °C in presence or absence of genistein. After the indicated time points, cells were chilled on ice and treated with TCEP (+TCEP) or reaction buffer only (−TCEP). After quenching of residual TCEP, cells were lysed, viral GP isolated by IP with mAb 83.6 to GP2. Biotinylated GP2 was detected with streptavidine-HRP in Western-blot under nonreducing conditions using enhanced chemiluminescence (ECL). The Upper blot (+TCEP) was exposed for 10 minutes, the lower blot (−TCEP) was exposed for 1 minute.
To exclude unspecific cytotoxicity caused by genistein, WI-26 VA4 cells were treated with increasing concentrations of the drug for 4 hours, followed by a wash out and incubation for a total of 16 hours. Cell viability was assessed by Cell Titer Glo® assay, which measures intracellular levels of ATP. WI-26 VA4 cells tolerated genistein up to a concentration of 100 μM with only mild loss of cell viability (Fig. 1B). To test the effect of genistein on early infection with LCMV-LASVGP, WI-26 VA4 cells were pre-treated with increasing concentrations of genistein for one hour and infected with rLCMV-LASVGP at multiplicity of 1. At four hours post infection, the drug was removed by wash out. To prevent further infection, the lysosomotropic agent ammonium chloride was added to the fresh medium. When added to cells ammonium chloride raises the endosomal pH rapidly and blocks low pH-dependent membrane fusion without causing overall cytotoxicity (Ohkuma and Poole, 1981; Ohkuma and Poole, 1978). After 12 hours in presence of ammonium chloride, cells were fixed and infection detected by IF staining for LCMV NP. As shown in Fig. 1C, genistein blocked infection with rLCMV-LASVGP in a dose-dependent manner.
Using expression of LCMV NP in infected cells as readout for infection did not allow discriminating between effects of genistein on LASVGP-mediated cell entry and post-entry steps of early viral infection. To specifically validate the effects of genistein on LASV cell entry in our system, we used recombinant vesicular stomatitis virus (VSV) pseudotyped with the envelope GPs of LASV (rVSV-LASVGP) and VSV (rVSV-VSVG). These pseudotypes are replications deficient and contain a green fluorescent protein (GFP) reporter in their genome. Previous studies demonstrated that rVSV-LASVGP closely mimic the receptor binding and entry characteristics of LASV (Pasqual et al., 2011; Kunz et al., 2005a). Increasing concentrations of genistein blocked infection of rVSV-LASVGP more efficient than rVSV-VSVG (Fig. 1D), consistent with previous reports (Kolokoltsov et al., 2012). Notably, the dose-response characteristic of rVSV-LASVGP closely matched the one of rLCMV-LASVGP (Fig. 1C, D), indicating that at least some of the inhibitory effect of genistein on early infection with rLCMV-LASVGP was due to perturbation of LASVGP-mediated cell entry.
To differentiate between effects of genistein on LASVGP-mediated cell attachment from perturbation of endocytosis, we used a well-established assay previously used to study virus internalization (Rojek et al., 2008a; Pelkmans et al., 2002), schematically shown in Fig. 1E. Briefly, rLCMV-LASVGP was purified over a renografin gradient and purified virus labeled with the reagent NHS-SS-biotin, resulting in a biotin label that is cleavable by reducing agents. As long as the virus stays bound to the cell surface, the biotin label can be cleaved efficiently with the potent, membrane-impermeable reducing agent Tris(2-carboxyethyl)phosphine (TCEP) (Fig. 1E). Once internalized via endocytosis, the biotin-labeled virus is protected from TCEP and retains its biotin moiety after exposure of cells to TCEP. To assess possible effects of genistein on virus cell attachment and internalization, WI-26 VA4 cells were pre-treated with 50 μM of genistein or vehicle control. After one hour, cells were cooled on ice and incubated with biotin-labeled rLCMV-LASVGP (100 particles/cell) for one hour in the cold. Unbound virus was removed and cells shifted to 37 °C in presence of genistein. After the indicated time points, cells were rapidly chilled on ice and immediately treated with cold TCEP or reaction buffer only. After quenching of residual TCEP with iodacetamide, cells were lysed and the viral GP isolated by immunoprecipitation (IP) with mAb 83.6 to GP2. Proteins were separated by SDS-PAGE and biotinylation of GP2 detected by Western-blot under non-reducing conditions. In specimens treated with reaction buffer only, similar amounts of cell-associated biotinylated virus were detected in presence and absence of genistein (Fig. 1F), indicating that genistein treatment did not affect virus attachment to the receptor. In control samples treated with TCEP, biotinylated virus became detectable after circa 5 min, with an increase over the next 20 min. In cells treated with genistein and TCEP, the signals for biotinylated GP2 were markedly reduced, indicating a block in an early step of virus internalization in presence of the inhibitor. In sum, our studies revealed that genistein does not affect virus-cell attachment, but inhibits the subsequent early steps of virus internalization.
Binding of LASV to cellular DG induces tyrosine phosphorylation of β-DG
Since the binding of LASV to DG is of high affinity and virtually irreversible under neutral pH (Kunz et al., 2005a), we speculated that virus-bound DG may be internalized during endocytosis of the virus. The apparent block of virus internalization in presence of genistein at an early time point (5-20 min) (Fig. 1E) opened therefore the possibility that uptake of the virus-receptor complex could involve tyrosine phosphorylation of the receptor and/or receptor-associated cellular factors. Since phosphorylation of β-DG at residue Y892 had previously been linked to internalization of the DG complex (Miller et al., 2012; Sotgia et al., 2003), we monitored phosphorylation of DG at Y892 during LASV cell entry. For this purpose, we applied mAb cl14a that specifically recognizes β-DG phosphorylated at Y892 (Sotgia et al., 2003). In a first step, we confirmed the specificity of mAb cl14a in our system. To this end, we co-expressed recombinant full-length DG containing a C-terminal HA-tag (DGHA, Fig. 2A) with recombinant c-src in HEK293 cells. Previous studies demonstrated that C-terminal tagging of β-DG had no influence on the biosynthesis, transport, and function of DG (Rojek et al., 2007b). Cells were either treated with the specific src tyrosine kinase family inhibitor PP2 (20 μM) or mock treated. After 48 hours, DGHA was isolated by IP with HA matrix and phosphorylation of Y892 detected with mAb cl14a in Western-blot. As expected, DGHA isolated from cells over-expressing c-src was specifically recognized by mAb cl14a, whereas treatment with PP2 markedly reduced the signal, confirming the specificity of the assay (Fig. 2B).
Fig. 2. Binding of LASV to cellular DG induces tyrosine phosphorylation of β-DG by src-family kinases.

(A) Schematic representation of C-terminally tagged DG (DGHA). The N-terminal domain (white), the mucin-type domain (black) and the C-terminal domain (gray) of α-DG, β-DG, and the C-terminal HA tag are indicated. (B). Detection of tyrosine phosphorylation at residue Y892 with mAb cl14a. DGHA was transiently expressed either alone or in combination with c-src. Parallel specimens were pretreated with 20 μM PP2 or mock treated with vehicle (DMSO). After 48 hours, DGHA was isolated by pull-down with HA matrix. Proteins were separated and probed in Western blot with an antibody to HA (anti-HA) or mAb cl14a to β-DG phosphorylated at tyrosine 892 (anti-β-DG PY892). Apparent molecular masses and the positions of β-DG are indicated. (C) Attachment of rLCMVLASVGP to cells induces tyrosine phosphorylation of β-DG. Monolayers of WI-26 VA4 cells were incubated with rLCMV-LASVGP or PICV (100 particles/cell) for 1 hour in the cold. Unbound virus was removed and cells shifted to 37°C. At the indicated time points, cells were lysed and DG enriched by WGA affinity purification. WGA-bound glycoproteins were probed in Western-blot with mAb cl14a (anti-β-DG PY892) and antibody 8D5 to β-DG. The positions of β-DG and β-DG PY892 are indicated. (D) Virus induced tyrosine phosphorylation of β-DG is blocked by PP2. Monolayers of WI-26 VA4 cells were pretreated with 20 μM PP2 or DMSO (Control) for 1 hour prior to exposure to rLCMV-LASVGP. Virus-induced phosphorylation of β-DG at Y892 was assessed as in (C). (E) The phosphorylation of β-DG at PY892 is not required for LASV cell entry. Monolayers of WI-26 VA4 cells were pretreated with 20 μM PP2 or DMSO (Control) for 1 hour as in (D), followed by incubation with rLCMV-LASVGP (MOI = 1) in the cold in presence of the drug. After 1 hour, unbound virus was removed and pre-warmed (37 °C) medium containing the drug added. At the indicated time points, 20 mM ammonium chloride was added and left throughout the experiment. At 16 hours post infection, cells were fixed and infection detected by intracellular staining for LCMV NP (means ± SD, n = 3). The apparent differences in infection at 60 minutes were not statistically significant.
To examine the effect of LASV binding on tyrosine phosphorylation of DG at Y892, monolayers of WI-26 VA4 cells were incubated with rLCMV-LASVGP (100 particles per cell). Virus attachment was performed in the cold to allow virus binding but preventing lateral movement of receptor molecules in the membrane. As a control, we used the New World arenavirus Pichinde (PICV) that does not bind to α-DG (Spiropoulou et al., 2002). After removal of unbound virus, cells were rapidly shifted to 37°C to restore membrane fluidity. At the indicated time points, cells were lysed in presence of the phosphatase inhibitor sodium orthovanadate. Cleared cell lysates were subjected to lectin purification with wheat germ agglutinin (WGA). Precipitated proteins were separated by SDS-PAGE and probed with mAb cl14a anti-β-DGPY892 and mAb 8D5 to β-DG that binds independently of phosphorylation. Binding of rLCMV-LASVGP, but not PICV resulted in transient phosphorylation of β-DG at Y892 (Fig. 2C) with maximal signals observed after 5-10 minutes. To confirm the role of src family kinases in the apparent virus-induced receptor phosphorylation at Y892, we treated cells with the src kinase inhibitor PP2 for 30 minutes prior to addition of virus. As shown in Fig. 2D, pretreatment with PP2 markedly reduced virus-induced tyrosine phosphorylation of β-DG at Y892, indicating a direct or indirect involvement of src kinases. In some experiments, we observed an apparent decrease in total β-DG at later time points (Fig. 2C, D). However, this was not observed consistently.
To address a possible role of the observed virus-induced receptor phosphorylation at Y892 for viral entry, we monitored the entry kinetics of LASV in presence of PP2. Upon receptor binding, LASV is taken up by clathrin- and caveolin-independent endocytosis and rapidly delivered to late endosomes, where low pH-dependent membrane fusion occurs (Quirin et al., 2008; Rojek et al., 2008a; Rojek et al., 2008c; Borrow and Oldstone, 1994). To assess how fast receptor-bound rLCMV-LASVGP trafficked to late endosomes in presence and absence of PP2, we determined the time required for the virus to become resistant to ammonium chloride. Briefly, WI-26 VA4 cells were either pretreated with PP2 for one hour, or mock treated with vehicle (DMSO) only. Cells were then incubated with rLCMVLASVGP in the cold, allowing virus attachment without internalization. Unbound virus was removed and cells quickly shifted to 37 °C to allow virus internalization in presence or absence of PP2. After different time points, 20 mM ammonium chloride was added to cells and kept throughout the experiment. At 16 h post infection, cells were fixed and infection assessed by IF detection NP. As shown in Fig. 2E pretreatment of cells with PP2 had no significant effect on the entry kinetics of rLCMV-LASVGP, suggesting that tyrosine phosphorylation of β-DG at Y892 was dispensable for virus cell entry. To complement these inhibitor studies, we examined the entry kinetics of rLCMV-LASVGP in murine embryonic fibroblasts (MEFs) derived from mice deficient for the src-family kinases src, fyn, and yes (Newsome et al., 2006). When compared to wild-type MEFs, src/fyn/yes-deficient cells were infected with similar kinetics (data not shown), in line with our src kinase inhibitor studies (Fig. 2E).
Despite the marked inhibition of rLCMV-LASVGP internalization by genistein (Fig. 1F), treatment of cells with the src inhibitor PP2 did not affect LASV cell entry, which was rather unexpected. Examination of the sequence of the cytoplasmic tail of β-DG revealed the presence of four putative sites of tyrosine phosphorylation, in addition to Y892 (Fig. 3A). To assess tyrosine phosphorylation of β-DG in response to LASV binding at sites other than Y892, WI-26 VA4 cells were pretreated with PP2 or vehicle, followed by exposure to rLCMV-LASVGP as described above. After the indicated time points, β-DG was isolated by IP using mAb 8D5 that does not discriminate between phosphorylated and unphosphorylated β-DG. Immunocomplexes were separated by SDS-PAGE and probed in Western blot using mAb cl14a to β-DGPY892 and mAb 4G10 to tyrosine phosphate. Total β-DG was detected with polyclonal antibody AP83. As expected, treatment with PP2 prevented virus-induced phosphorylation at Y892 (Fig. 3B). However, immunoblotting with the broadly specific anti-phosphotyrosine mAb 4G10 revealed significant tyrosine phosphorylation of β-DG in response to virus binding that was not affected by PP2 (Fig. 3B). This suggested that binding of rLCMV-LASVGP to cellular DG induced tyrosine phosphorylation of β-DG at sites other than Y892, possibly implicating non-src family tyrosine kinases. Pre-treatment of cells with genistein (50 μM) for 30 minutes reduced virus-induced tyrosine phosphorylation of β-DG altogether (Fig. 3C).
Fig. 3. Binding of LASV to cellular DG induces tyrosine phosphorylation of β-DG by non-src tyrosine kinases.
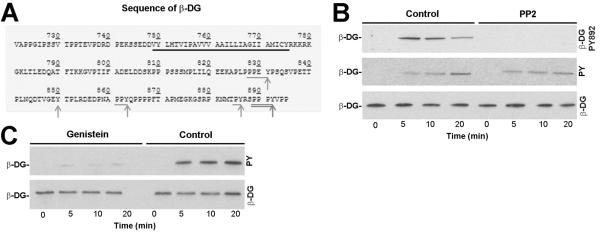
(A) Sequence of the cytoplasic domain of human β-DG. The putative transmembrane domain is underlined in black and tyrosine residues indicated with gray arrows. Putative phosphorylation sites for src-family kinases are underlined and Y residues marked (arrows). The PPxY motif including Y892 is double underlined. (B) Detection of virus-induced tyrosine phosphorylation β-DG in presence of PP2. WI-26 VA4 cells were pretreated with 20 μM PP2 or vehicle, followed by exposure to rLCMV-LASVGP as in Fig. 2D). After the indicated time points, β-DG was isolated by IP using mAb 8D5 against β-DG covalently coupled to Sepharose 4B. Immunocomplexes were eluted under non-reducing conditions, separated by SDS-PAGE and probed in Western blot using mAb cl14a to β-DGPY892 and mAb 4G10 to tyrosine phosphate (pY). For detection of bound mouse IgG a TrueBlot® detection system was used to avoid cross-reaction with the murine IgG heavy and light chains. Total β-DG was detected with rabbit polyclonal antibody AP83. (C) Genistein blocks virus-induced tyrosine phosphorylation of β-DG. Cells were pre-treated with 50 μM genistein or vehicle (DMSO) only. After 30 minutes, rLCMV-LASVGP was added (100 particles/cell) for the indicated time points and tyrosine phosphorylation of β-DG was assessed as in (B).
Engagement of DG by LASV detaches the DG complex from the adaptor utrophin
In the host cell, DG provides a molecular link between the ECM and the actin cytoskeleton by anchorage of the cytoplasmic domain of β-DG to the cytoskeletal adapter proteins dystrophin or utrophin. Cell entry of LASV occurs independently of the dynamics and stability of the actin cytoskeleton (Rojek et al., 2008c). We hypothesized that virus binding, possibly involving receptor clustering and signaling, may somehow detach DG from the cytoskeletal adaptors, allowing subsequent actin-independent endocytosis. A major challenge to test this hypothesis was to assess specific changes in utrophin binding to the relatively small fraction of virus-bound DG as compared to total cellular DG. To overcome this problem, we used recombinant retroviruses bearing the recombinant GP of LASV. Retroviral pseudotypes containing the GP of the New World virus Amapari (AMPV), which does not use DG as a receptor (Spiropoulou et al., 2002), served as a negative control. These arenavirus pseudotypes, which have been previously generated and extensively characterized in our laboratory, adopt the receptor binding characteristics and cellular tropism of the viruses from which the GPs are derived (Rojek et al., 2007a). Importantly, the use of retroviral pseudotypes allowed the insertion of a C-terminal FLAG-tag into LASV GP and AMPV GP (Fig. 4A) allowing co-IP of the viral GP with associated cellular receptor proteins. The C-terminal FLAG-tag had no adverse effect on the function of the viral GPs in host cell attachment and entry (Rojek et al., 2008b). Retroviral pseudotypes containing FLAG-tagged LASV GP and AMPV GP were produced and purified as described (Rojek et al., 2008b; Rojek et al., 2007a) and detection of GP in purified pseudotypes by ELISA revealed more efficient incorporation of LASV GP when compared to AMPV GP (Fig. 4B). As previously shown, infection of cells with LASV pseudotypes, but not AMPV pseudotypes or pseudotypes of VSV depended on DG (Fig. 4C), confirming their receptor specificity.
Fig. 4. Retroviral pseudotypes.
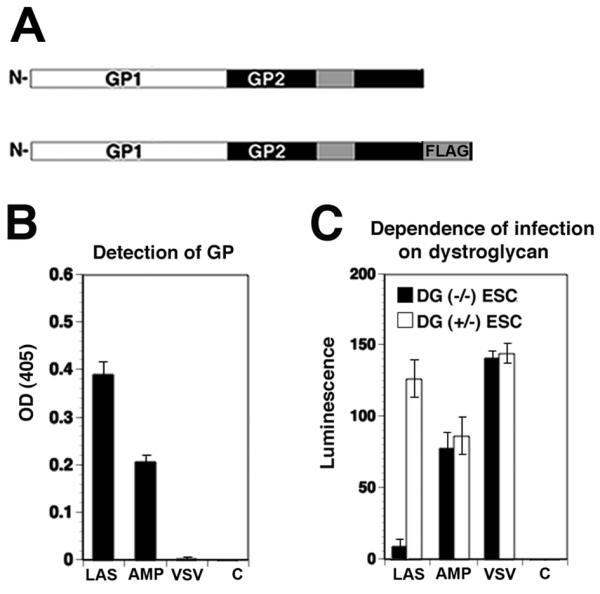
(A) Schematic representation of Flag-tagged LASV GP. The receptor-binding GP1 and transmembrane GP2 parts are indicated. The transmembrane domain of GP2 is represented as a grey box and the C-terminal FLAG-tag indicated. (B) Detection of GP in retroviral pseudotypes. Retroviral pseudotypes were generated by cotransfection of packaging cell line GP2293 expressing retroviral Gag/Pol with a GP expression plasmid and an expression plasmid for a packable retroviral genome bearing a luciferase and a GFP reporter gene. Pseudotypes were purified by ultracentrifugation through a sucrose cushion and re-suspended in HBSS. Purified pseudotypes of LASV (LAS), AMPV (AMP), and VSV, as well as pseudotypes lacking GP (C) were immobilized in microtiter plates and the viral GP detected with mAb 83.6, combined with an HRP-conjugated secondary antibody in a colour reaction (means ± SD, n = 3). (C) Infection of cells with LASV pseudotypes depends on DG. DG (−/−) murine embryonic stem (ES) cells and parental DG (+/−) ES cells were infected with pseudotypes of LASV (LAS), AMPV (AMP), and VSV, as well as pseudotypes lacking GP (C). After 48 hours, infection was detected by luciferase assay (means ± SD, n = 3).
To assess the impact of LASV GP binding on the association of cellular DG with utrophin, we incubated LASV or AMPV pseudotypes with monolayers of WI-26 VA4 cells. A parallel set of cells was incubated with medium only. This step was carried out in the cold to allow receptor binding without clustering or internalization. Cells were either kept on ice or shifted to 37 °C for 10 minutes, followed by cell lysis in the cold. Lysates prepared from cells incubated with pseudotypes were subjected to IP with FLAG matrix. Lysates of untreated control cells were incubated with mAb 16G4 to α-DG, combined with protein G sepharose. Immunocomplexes and total cell lysates were probed for β-DG and utrophin in Western blot. As shown in Fig. 5A, IP anti-FLAG in specimens incubated with LASV, but not AMPV pseudotypes resulted in the detection of β-DG and co-IP of utrophin, suggesting specific pull-down of LASV GP-associated DG complex. As expected, IP of α-DG resulted in robust co-IP of utrophin under all conditions. To quantify possible changes in the ratio of utrophin/β-DG upon exposure of cells to LASV pseudotypes, we performed densitometric analysis of the signals for both conditions and normalized to the utrophin/β-DG detected in the IP of α-DG (Fig. 5B). In cells kept at 4°C, we consistently observed a significantly lower utrophin/β-DG ratio in the LASV GP-associated receptor fraction when compared to the DG-utrophin complexes isolated by IP with anti-α-DG antibody. This suggested that the LASV pseudotypes associate preferentially with cellular DG that shows a weaker or more transient association with utrophin. As shown in Fig. 5B, the temperature shift to 37 °C, which allows clustering of the receptor and signaling to occur, resulted in a reduction of the utrophin/β-DG ratio in LASV GP-associated DG when compared to cells kept in the cold. This suggests that virus-induced receptor clustering and/or signaling promotes the dissociation of the DG complex from utrophin.
Fig. 5. Effect of LASV pseudotype binding on the association of DG with utrophin.

Monolayers of WI-26 VA4 cells were chilled on ice and incubated with either LASV or AMPV pseudotypes (LASV-PS, AMPV-PS) at a multiplicity of infection (MOI) of 50 transforming units (TU)/cell. Parallel specimens were incubated with mAb 16G4 to α-DG (anti-DG). After one hour, unbound virus or mAb were removed by washing. Cells were either kept on ice (virus binding 4°C) or shifted to 37 °C for 10 minutes (temperature shift 37°C). Cells were quickly chilled on ice, lysed and subjected to IP using FLAG matrix or protein G-conjugated Sepharose 4B. Immunocomplexes were separated by SDS-PAGE using 100% of the IPs anti-FLAG and 5% of the IP anti-DG. Beta-DG and utrophin were detected on Western-blot using monoclonal antibodies 8D5 and combined with HRP-conjugated secondary antibodies in a TrueBlot® detection system to avoid cross-reaction with the IgG heavy chain. For the detection of total protein in cell lysates, 1/20 of the lysate were separated by SDS-PAGE and subjected to Western-blot detection. (B) Quantification of the signals in (A). Blots were scanned in a densitometer and the ratios of the signals for utrophin normalized to β-DG (utrophin/β-DG) for the IPs anti-FLAG (LASV pseudotypes only) and the IP anti-DG. For each series, the utrophin/β-DG ratio detected in the IP anti-DG was defined as 1.0. (C) Pre-treatment with genistein, but not PP2 reduced virus-induced dissociation of utrophin from DG. Monolayers of WI-26 VA4 cells were pre-treated with DMSO only (control), 20 μM PP2 and 50 μM genistein for one hour. Cells were then chilled on ice and incubated with LASV pseudotypes (LASV-PS) for one hour in the cold in presence of drugs. Cells were then quickly shifted to 37 °C, lysed, and subjected to IP with FLAG matrix as in (A). Precipitated β-DG and utrophin were detected in Western blot and the ratios utrophin/β-DG determined as in (B). (D) Quantification of the data in (C). (E) The 15 C-terminal amino acids of β-DG are dispensable for LASV cell entry. Murine ES cells expressing either wild-type DG (DG wt) or DG lacking the C-terminal 15 amino acids of β-DG (DGΔC) were infected with rLCMV-LASVGP or rLCMV-VSVG at a multiplicity of 0.1. Infection of the cells expressing wild-type DG was set at 100% (means ± SD, n = 3).
To address the role of tyrosine phosphorylation in the observed virus-induced dissociation of utrophin from DG, we tested the effect of genistein and PP2. To this end, cells were pre-treated with genistein and PP2 for one hour, followed by exposure to LASV pseudotypes. After removal of unbound virus, cells were shifted to 37 °C in presence of the drugs. Virus-associated DG was isolated by IP with FLAG matrix and the ratio of utrophin/β-DG assessed as described above. Treatment with genistein significantly reduced virus-induced dissociation of utrophin from DG, whereas PP2 had only a weak effect (Fig. 5C, D). This suggested that the phosphorylation of β-DG at Y892 was dispensable for the virus-induced dissociation of utrophin from the DG complex.
To complement our inhibitor studies, we examined LASV cell entry into murine ES cells expressing a DG variant lacking the last 15 C-terminal amino acids of β-DG (DGΔC), including the PPxY motif containing Y892. Briefly, ES cells expressing either wild type DG or DGΔC were infected with rLCMV-LASVGP at low multiplicity. To exclude differences at the level of post-entry steps in LCMV replication, a recombinant LCMV expressing VSVG was used as a control. At four hours of infection, 20 mM ammonium chloride was added to prevent secondary infection. After 16 hours, cells were fixed and infection detected by IF for LCMV NP. As shown in Fig. 5E, ES cells expressing DGΔC were as permissive as wild-type cells, suggesting that the last 15 amino acids of β-DG are dispensable for LASV cell entry.
DISCUSSION
In the present study we investigated the role of tyrosine phosphorylation for cell entry of LASV. We show that tyrosine kinases are required for endocytosis of the virus-receptor complex, but dispensable for virus-receptor binding. Binding of LASV to cellular dystroglycan (DG) induced phosphorylation of DG at Y892 by src family kinases and other tyrosine residues of β-DG by non-src kinases. Virus-induced receptor phosphorylation was accompanied by dissociation of DG from the cytoskeletal adaptor protein utrophin, which might facilitate virus endocytosis.
In the host cell, DG provides a molecular link between the ECM and the actin-based cytoskeleton and has a slow turnover. However, engagement of DG by LASV results in rapid internalization of the virus and delivery to the late endosome (Quirin et al., 2008; Rojek et al., 2008c), suggesting marked changes in trafficking dynamics of DG induced by virus binding. In our study, we investigated if attachment of LASV affects post-translational modifications of DG, altering receptor trafficking in the membrane. Extending previous studies (Kolokoltsov et al., 2012; Vela et al., 2008), we found that treatment of cells with the broadly-specific tyrosine kinase inhibitor genistein prevented LASV cell entry at an early step of virus internalization without affecting virus-receptor binding, providing a first link between tyrosine phosphorylation and viral endocytosis. Since tyrosine phosphorylation of β-DG at Y892 by src family kinases has previously been implicated in internalization of DG (Miller et al., 2012; Sotgia et al., 2003), we tested the effect of LASV binding on phosphorylation of the receptor at Y892. Engagement of cellular DG by rLCMV-LASVGP rapidly induced phosphorylation of β-DG at Y892 that was blocked by the src kinase inhibitor PP2. The observed kinetics of Y892 phosphorylation was compatible with a role in viral endocytosis. However, rather unexpected, treatment of cells with PP2 had no effect on the entry kinetics of the virus. Since the cytoplasmic tail of β-DG contains four additional putative sites of tyrosine phosphorylation, in addition to Y892, we examined virus-induced phosphorylation at tyrosines. We found that LASV binding induced tyrosine phosphorylation of β-DG at sites other than Y892 with similar kinetics. These tyrosine phosphorylation events were not sensitive to PP2 and likely involved non-src tyrosine kinases. Virus-induced tyrosine phosphorylation of β-DG was entirely abrogated by genistein.
Using a pseudotype platform, we assessed the composition of DG complexes associated with LASV GP upon virion attachment. We found that LASV pseudotypes associated with a fraction of cellular DG that showed less association with utrophin. The reasons for this are currently unclear, but may be related to different accessibility of DG as a function of its association with utrophin and the actin cytoskeleton. In cells, DG associated with cell-substrate adhesion structures at the lower plasma membrane tends to co-localize with utrophin (Belkin and Smalheiser, 1996). Due to the close association with the substratum at the basal face of the cell, DG located in such substrate-adhesion complexes may be less accessible to the virus.
Our analysis further revealed that engagement of DG by LASV GP results in dissociation of utrophin from β-DG. This virus-induced dissociation of utrophin could be perturbed by genistein, but not PP2, suggesting that phosphorylation of β-DG at Y892 by src kinases was dispensable. The efficient virus-induced dissociation of utrophin from DG in presence of PP2 is in line with the inability of the inhibitor to perturb viral entry. However, our findings are different from reports supporting a role for phosphorylation of β-DG at Y892 by src kinases in the regulation of utrophin binding and endocytosis of DG in prototypic primate cells (Sotgia et al., 2003; James et al., 2000) and myoblasts (Miller et al., 2012) in response to endogenous substrates. The reasons for this discrepancy are currently unclear. One possibility is that, contrary to our initial assumption, DG may not stay associated with the virus during the entry process. In this scenario, DG would serve as an attachment factor rather than a true entry receptor. One would have to postulate the existence of another, yet unknown receptor molecule that would mediate viral endocytosis. The recently discovered alternative LASV receptors Axl and Dtk (Shimojima et al., 2012) appear as interesting candidates in this context. Alternatively binding of the multivalent virion particle to cellular DG may induce extensive clustering and activate signaling pathways that are not triggered by DG’s ECM ligands, allowing the virus to “bypass” a block in Y892 phosphorylation. To address these possibilities, studies aiming at tracking the virus-DG complex in live cells are currently launched in our laboratory.
Since virus-induced phosphorylation of β-DG is blocked by genistein, which inhibits internalization of the virus, it is tempting to speculate that virus-induced phosphorylation of β-DG is necessary for virus entry. However, in the host cell, DG is found associated with a number of cellular proteins that are required for the correct assembly and stability of the DG complex in the membrane, as well as its anchorage to the actin cytoskeleton (Barresi and Campbell, 2006). Such “pre-formed” DG complexes represent the functional units of virus attachment. Our data have shown that virus binding to DG results in receptor signaling. Such virus-induced signaling may affect the composition of the virus-receptor complex by recruiting new proteins into the virus-DG complex and/or excluding others. During the entry process, the “interactome” of the virus-DG complex may therefore change in a dynamic manner resulting in sorting at the plasma membrane required for subsequent cell entry. Candidate cellular proteins that interact with the virus-DG complex during the entry process and are part of this “interactome” would represent potential substrates for tyrosine phosphorylation. We cannot exclude the possibility that tyrosine phosphorylation of such receptor-associated proteins, and not β-DG itself, is the actual target of genistein in the viral entry process.
In sum, the data at hand suggest that attachment of LASVGP to cellular DG induces tyrosine phosphorylation of β-DG at Y892 and other tyrosine residues accompanied by the dissociation of DG from utrophin. The consequent detachment of virus-bound DG from the actin-based cytoskeleton may facilitate subsequent endocytosis of the virus-receptor complex, providing a possible link between virus-induced post-translational modification of DG and virus entry.
EXPERIMENTAL PROCEDURES
Cell lines and viruses
WI-26 VA4 cells (ATCC CCL-95.1) were cultured in DMEM, 10 % (vol/vol) FBS, supplemented with glutamine, and penicillin/streptomycin. Embryonic stem (ES) cells DG (+/−), DG (−/−) have been described (Henry and Campbell, 1998). Transgenic ES cells expressing DG lacking the last 15 amino acids (DGΔC) were generated through introduction of a triple premature stop codon affecting all possible reading frames via targeted homologous recombination (gift from Kevin P. Campbell).
The recombinant virus rLCMV-LASVGP has been described elsewhere (Rojek et al., 2008c) and was produced and the titers determined as previously described (Dutko and Oldstone, 1983). Recombinant LASV GP and AMPV GP containing a C-terminal FLAG-tag have been described (Rojek et al., 2008b). Retroviral pseudotypes expressing GFP and luciferase reporters were produced and concentrated, and titers determined as described (Rojek et al., 2006). Concentrated pseudotypes were diluted in HBSS at 107 transforming units per ml. For detection of viral GP in ELISA, purified pseudotypes were immobilized in microtiter plates at 106 TU/ml and the viral GP detected as described (Rojek et al., 2008a). Recombinant VSV pseudotyped with LASV GP (rVSVΔG-LASVGP), and VSV GP (rVSVΔG-VSVG) were generated as reported previously (Kunz et al., 2005a). Virus titers were determined by the infection of Vero E6 cell monolayers and detection of GFP-positive cells by fluorescence microscopy.
Antibodies and reagents
Monoclonal antibodies (mAbs) 113 (anti-LCMVNP) and 83.6 (anti-LCMVGP) have been described (Weber and Buchmeier, 1988; Buchmeier et al., 1981), as has mAb IIH6 anti-α-DG (Ervasti and Campbell, 1991). Other mAbs included mouse IgG 8D5 anti-β-DG (Novocastra) and mouse IgG 16C4 to α-DG (provided by Kevin P. Campbell), mouse IgG anti-utrophin from St. Cruz Biotechnology (St. Cruz, CA), mAb cl14a to phospho-β-DG PY982 (BD Bioscience), and mAb 4G10 to phospho-tyrosine (St. Cruz, CA). Rabbit anti-influenza HA (Y11) was from St. Cruz Biotechnology (St. Cruz, CA). Polyclonal rabbit anti-mouse secondary antibodies conjugated to HRP were from Dako (Glostrup, Denmark) and goat anti-mouse antibody conjugated with Rhodamin Red X were from Jackson Immuno Research Laboratories. Genistein and PP2 were purchased from Calbiochem. The Bright-Glo® luciferase assay and Cell Titer Glo® assay systems were obtained from Promega (Madison WI).
Immunoblotting
Proteins were separated by gel electrophoresis using 12% polyacrylamide gels and transferred to nitrocellulose. After blocking in 5% (wt/vol) skim milk in PBS, membranes were incubated with Abs used at following concentrations: mAb 8D5, mAb cl14a, mAb 4G10, and polyclonal Abs AP83, and Y11 (10 μg/ml) in 2% (wt/vol) skim milk, PBS for 12 h at 6 °C. Secondary Abs coupled to HRP were applied 1: 5, 000 in PBS, 0.1 % (wt/vol) Tween for 1 hour at room temperature. Blots were developed by enhanced chemiluminescence (ECL) using Super Signal West Pico ECL Substrate or TrueBlot® detection system (Pierce), where indicated.
Infection of cells with retroviral pseudotypes
Cells were plated in 96-well plates in a density of 104 cells/well. After 24 hours, retroviral pseudotypes were added at the indicated MOI and incubated for 1 hour at 37°C. The viral particles were removed, cells washed twice with DMEM, and fresh medium added. Infection was quantified by Bright Glo® luciferase assay. Luminescence was calculated as fold-increase over background signals obtained from uninfected cells.
Infection of cells with rLCMV-LASVGP
Cells were plated in 96-well plates in a density of 104 cells/well. After 24 hours, cells were pre-treated with genistein or PP2 as indicated, followed by infection with rLCMV-LASVGP at the indicated MOI for 1 hour at 37°C. Unbound virus were removed, cells washed twice with DMEM, and fresh medium added. Infection was quantified by detection of LCMV NP in IF as described (Kunz et al., 2004). Cell entry kinetics of rLCMV-LASV in presence and absence of PP2 were performed as described (Rojek et al., 2008c). Blocking of infection with mAb IIH6 was done as reported (Kunz et al., 2005b).
Virus internalization assay
Purification of rLCMV-LASVGP was performed by ultracentrifugation on a renografin gradient. Purified LCMV was labeled with the thiol-cleavable reagent NHS-SS-biotin (Pierce). The cleavage of the biotin label was verified by reaction with the membrane-impermeable reducing agent Tris(2-carboxyethyl)phosphine (TCEP) (10 mM) (Pierce) for 30 min, which resulted in a loss of >95% of the biotin label. Internalization assay was performed as described previously (Rojek et al., 2008a). Briefly, cells were cultured in 10-cm dishes to obtain closed monolayers. Medium was removed, and cells were washed twice with cold HBSS and chilled on ice for 5 min. Cold solution containing NHS-SS-biotinylated rLCMVLASVGP (107 PFU/ml) in HBSS was added. After incubation for 1 h on ice, unbound virus was removed and cells were washed with cold HBSS. For internalization, cells were shifted to 37°C. After the indicated incubation times, medium was removed and cells chilled on ice. TCEP (15 mM) in 50 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2 was added (5 ml/dish) and applied twice for 30 min on ice. Cells were washed three times with cold HBSS, and the remaining TCEP was quenched with 100 mM iodoacetamide for 10 min, and cells were lysed immediately. LCMV GP2 was isolated by IP as described (Rojek et al., 2008a). Immunocomplexes were separated by nonreducing SDS-PAGE. Biotinylated LCMV GP2 was detected by Western blotting with HRP-conjugated streptavidin as described above.
Detection of DG-utrophin complexes associated with LASV GP
Triplicate cultures of WI-26 VA4 cells were cultured in 10 cm plates over night to obtain closed monolayers. Retroviral pseudotypes of LASV and AMPV (107 TU/ml in HBSS) were added at 50 TU/cell. Parallel specimens were incubated with 10 μg/ml mAb 16C4 to α-DG in HBSS. After incubation for one hour in the cold, cells were washed 3 times with cold HBSS. One series of cultures was kept on ice whereas the other was quickly shifted to 37°C in a water bath. After 10 minutes, cells were chilled on ice. Cells were lyzed for 30 minutes in 1% (wt/vol) β-octylglucoside, 1 mM CaCl2, 1 mM MgCl2, 150 mM NaCl, 50 mM Hepes pH 7.5, 10% (wt/vol) glycerin supplemented with protease inhibitor cocktail Complete® (Roche) and 1 mM PMSF. Cleared lysates were subjected to IP with FLAG matrix (Sigma) or protein G-conjugates sepharose 4B (Sigma) for 2 hours at 6°C. After three short washes in lysis buffer, the matrix was eluted with non-reducing SDS-PAGE sample buffer to minimize elution of IgG from the FLAG matrix. After addition of 100 mM DTT (final concentration) and boiling for another 5 minutes Western blot performed as described above. For quantification, X-ray films were scanned with a Storm densitometer, and acquired data were processed using Image Quant software.
ACKNOWLEDGEMENTS
The authors thank Jillian M. Rojek, Alexandra von Siebenthal, and Zoe Enderlin for their contributions to the experiments. This research was supported by Swiss National Science Foundation grant FN 310030_132844 (S.K.), the Prix Leenaards 2009 pour la promotion de la recherche scientifique (2009) (S.K.), and the Marie Curie International Reintegration Grant Nr. 224780 of the European Community (S.K.). K.P.C. is an investigator of the Howard Hughes Medical Institute. The retroviral construct pLZRs-Luc-gfp was kindly provided by Dr. Gary Nabel.
REFERENCES
- Barresi R, Campbell KP. Dystroglycan: from biosynthesis to pathogenesis of human disease. J Cell Sci. 2006;119:199–207. doi: 10.1242/jcs.02814. [DOI] [PubMed] [Google Scholar]
- Belkin AM, Smalheiser NR. Localization of cranin (dystroglycan) at sites of cell-matrix and cell-cell contact: recruitment to focal adhesions is dependent upon extracellular ligands. Cell Adhes Commun. 1996;4:281–296. doi: 10.3109/15419069609010772. [DOI] [PubMed] [Google Scholar]
- Beyer WR, Popplau D, Garten W, von Laer D, Lenz O. Endoproteolytic processing of the lymphocytic choriomeningitis virus glycoprotein by the subtilase SKI-1/S1P. J Virol. 2003;77:2866–2872. doi: 10.1128/JVI.77.5.2866-2872.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrow P, Oldstone MB. Characterization of lymphocytic choriomeningitis virus-binding protein(s): a candidate cellular receptor for the virus. J Virol. 1992;66:7270–7281. doi: 10.1128/jvi.66.12.7270-7281.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrow P, Oldstone MB. Mechanism of lymphocytic choriomeningitis virus entry into cells. Virology. 1994;198:1–9. doi: 10.1006/viro.1994.1001. [DOI] [PubMed] [Google Scholar]
- Buchmeier MJ, de la Torre JC, Peters CJ. Arenaviridae: the viruses and their replication. In: Knipe DL, Howley PM, editors. Fields Virology. Lippincott-Raven; Philadelphia: 2007. pp. 1791–1828. [Google Scholar]
- Buchmeier MJ, Lewicki HA, Tomori O, Oldstone MB. Monoclonal antibodies to lymphocytic choriomeningitis and pichinde viruses: generation, characterization, and cross-reactivity with other arenaviruses. Virology. 1981;113:73–85. doi: 10.1016/0042-6822(81)90137-9. [DOI] [PubMed] [Google Scholar]
- Cao W, Henry MD, Borrow P, Yamada H, Elder JH, Ravkov EV, et al. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus [see comments] Science. 1998;282:2079–2081. doi: 10.1126/science.282.5396.2079. [DOI] [PubMed] [Google Scholar]
- Cavaldesi M, Macchia G, Barca S, Defilippi P, Tarone G, Petrucci TC. Association of the dystroglycan complex isolated from bovine brain synaptosomes with proteins involved in signal transduction. J Neurochem. 1999;72:1648–1655. doi: 10.1046/j.1471-4159.1999.721648.x. [DOI] [PubMed] [Google Scholar]
- de la Torre JC. Molecular and cell biology of the prototypic arenavirus LCMV: implications for understanding and combating hemorrhagic fever arenaviruses. Ann N Y Acad Sci. 2009;1171(Suppl 1):E57–64. doi: 10.1111/j.1749-6632.2009.05048.x. [DOI] [PubMed] [Google Scholar]
- Durbeej M, Henry MD, Ferletta M, Campbell KP, Ekblom P. Distribution of dystroglycan in normal adult mouse tissues. J Histochem Cytochem. 1998;46:449–457. doi: 10.1177/002215549804600404. [DOI] [PubMed] [Google Scholar]
- Dutko FJ, Oldstone MB. Genomic and biological variation among commonly used lymphocytic choriomeningitis virus strains. J Gen Virol. 1983;64:1689–1698. doi: 10.1099/0022-1317-64-8-1689. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. Membrane organization of the dystrophin glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- Eschli B, Quirin K, Wepf A, Weber J, Zinkernagel R, Hengartner H. Identification of an N-terminal trimeric coiled-coil core within arenavirus glycoprotein 2 permits assignment to class I viral fusion proteins. J Virol. 2006;80:5897–5907. doi: 10.1128/JVI.00008-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferletta M, Kikkawa Y, Yu H, Talts JF, Durbeej M, Sonnenberg A, et al. Opposing roles of integrin alpha6Abeta1 and dystroglycan in laminin-mediated extracellular signal-regulated kinase activation. Mol Biol Cell. 2003;14:2088–2103. doi: 10.1091/mbc.E03-01-0852. Epub 2003 Feb 2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisbert TW, Jahrling PB. Exotic emerging viral diseases: progress and challenges. Nat Med. 2004;10:S110–121. doi: 10.1038/nm1142. [DOI] [PubMed] [Google Scholar]
- Henry MD, Campbell KP. A role for dystroglycan in basement membrane assembly. Cell. 1998;95:859–870. doi: 10.1016/s0092-8674(00)81708-0. [DOI] [PubMed] [Google Scholar]
- Henry MD, Satz JS, Brakebusch C, Costell M, Gustafsson E, Fassler R, Campbell KP. Distinct roles for dystroglycan, beta1 integrin and perlecan in cell surface laminin organization. J Cell Sci. 2001;114:1137–1144. doi: 10.1242/jcs.114.6.1137. [DOI] [PubMed] [Google Scholar]
- Igonet S, Vaney MC, Vonhrein C, Bricogne G, Stura EA, Hengartner H, et al. X-ray structure of the arenavirus glycoprotein GP2 in its postfusion hairpin conformation. Proc Natl Acad Sci U S A. 2011;108:19967–19972. doi: 10.1073/pnas.1108910108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James M, Nuttall A, Ilsley JL, Ottersbach K, Tinsley JM, Sudol M, Winder SJ. Adhesion-dependent tyrosine phosphorylation of (beta)-dystroglycan regulates its interaction with utrophin. J Cell Sci. 2000;113:1717–1726. doi: 10.1242/jcs.113.10.1717. [DOI] [PubMed] [Google Scholar]
- Kanagawa M, Saito F, Kunz S, Yoshida-Moriguchi T, Barresi R, Kobayashi YM, et al. Molecular recognition by LARGE is essential for expression of functional dystroglycan. Cell. 2004;117:953–964. doi: 10.1016/j.cell.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Kolokoltsov AA, Adhikary S, Garver J, Johnson L, Davey RA, Vela EM. Inhibition of Lassa virus and Ebola virus infection in host cells treated with the kinase inhibitors genistein and tyrphostin. Arch Virol. 2012;157 doi: 10.1007/s00705-011-1115-8. [DOI] [PubMed] [Google Scholar]
- Kunz S, Sevilla N, Rojek JM, Oldstone MB. Use of alternative receptors different than alpha-dystroglycan by selected isolates of lymphocytic choriomeningitis virus. Virology. 2004;325:432–445. doi: 10.1016/j.virol.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Kunz S, Rojek JM, Perez M, Spiropoulou CF, Oldstone MB. Characterization of the interaction of lassa fever virus with its cellular receptor alpha-dystroglycan. J Virol. 2005a;79:5979–5987. doi: 10.1128/JVI.79.10.5979-5987.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz S, Rojek JM, Kanagawa M, Spiropoulou CF, Barresi R, Campbell KP, Oldstone MB. Posttranslational modification of alpha-dystroglycan, the cellular receptor for arenaviruses, by the glycosyltransferase LARGE is critical for virus binding. J Virol. 2005b;79:14282–14296. doi: 10.1128/JVI.79.22.14282-14296.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz O, ter Meulen J, Klenk HD, Seidah NG, Garten W. The Lassa virus glycoprotein precursor GP-C is proteolytically processed by subtilase SKI-1/S1P. Proc Natl Acad Sci U S A. 2001;98:12701–12705. doi: 10.1073/pnas.221447598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick JB, Fisher-Hoch SP. Lassa fever. Curr Top Microbiol Immunol. 2002;262:75–109. doi: 10.1007/978-3-642-56029-3_4. [DOI] [PubMed] [Google Scholar]
- Miller G, Moore CJ, Terry R, La Riviere T, Mitchell A, Piggott R, et al. Preventing phosphorylation of dystroglycan ameliorates the dystrophic phenotype in mdx mouse. Hum Mol Genet. 2012 doi: 10.1093/hmg/dds293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newsome TP, Weisswange I, Frischknecht F, Way M. Abl collaborates with Src family kinases to stimulate actin-based motility of vaccinia virus. Cell Microbiol. 2006;8:233–241. doi: 10.1111/j.1462-5822.2005.00613.x. [DOI] [PubMed] [Google Scholar]
- Ohkuma S, Poole B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci U S A. 1978;75:3327–3331. doi: 10.1073/pnas.75.7.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkuma S, Poole B. Cytoplasmic vacuolation of mouse peritoneal macrophages and the uptake into lysosomes of weakly basic substances. J Cell Biol. 1981;90:656–664. doi: 10.1083/jcb.90.3.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldstone MB, Campbell KP. Decoding arenavirus pathogenesis: essential roles for alpha-dystroglycan-virus interactions and the immune response. Virology. 2011;411:170–179. doi: 10.1016/j.virol.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqual G, Rojek JM, Masin M, Chatton JY, Kunz S. Old world arenaviruses enter the host cell via the multivesicular body and depend on the endosomal sorting complex required for transport. PLoS Pathog. 2011;7:e1002232. doi: 10.1371/journal.ppat.1002232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkmans L, Puntener D, Helenius A. Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science. 2002;296:535–539. doi: 10.1126/science.1069784. [DOI] [PubMed] [Google Scholar]
- Quirin K, Eschli B, Scheu I, Poort L, Kartenbeck J, Helenius A. Lymphocytic choriomeningitis virus uses a novel endocytic pathway for infectious entry via late endosomes. Virology. 2008;378:21–33. doi: 10.1016/j.virol.2008.04.046. [DOI] [PubMed] [Google Scholar]
- Rojek JM, Spiropoulou CF, Kunz S. Characterization of the cellular receptors for the South American hemorrhagic fever viruses Junin, Guanarito, and Machupo. Virology. 2006;349:476–491. doi: 10.1016/j.virol.2006.02.033. [DOI] [PubMed] [Google Scholar]
- Rojek JM, Perez M, Kunz S. Cellular entry of lymphocytic choriomeningitis virus. J Virol. 2008a;82:1505–1517. doi: 10.1128/JVI.01331-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojek JM, Spiropoulou CF, Campbell KP, Kunz S. Old World and Clade C New World Arenaviruses Mimic the Molecular Mechanism of Receptor Recognition Used by {alpha}-Dystroglycan’s Host-Derived Ligands. J Virol. 2007a;81:5685–5695. doi: 10.1128/JVI.02574-06. Epub 2007 Mar 5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojek JM, Campbell KP, Oldstone MB, Kunz S. Old World Arenavirus Infection Interferes with the Expression of Functional {alpha}-Dystroglycan in the Host Cell. Mol Biol Cell. 2007b;29:29. doi: 10.1091/mbc.E07-04-0374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojek JM, Lee AM, Nguyen N, Spiropoulou CF, Kunz S. Site 1 protease is required for proteolytic processing of the glycoproteins of the South American hemorrhagic fever viruses Junin, Machupo, and Guanarito. J Virol. 2008b;82:6045–6051. doi: 10.1128/JVI.02392-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojek JM, Sanchez AB, Nguyen NT, de la Torre JC, Kunz S. Different mechanisms of cell entry by human-pathogenic Old World and New World arenaviruses. J Virol. 2008c;82:7677–7687. doi: 10.1128/JVI.00560-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojek JM, Moraz ML, Pythoud C, Rothenberger S, Van der Goot FG, Campbell KP, Kunz S. Binding of Lassa virus perturbs extracellular matrix-induced signal transduction via dystroglycan. Cell Microbiol. 2012;14:1122–1134. doi: 10.1111/j.1462-5822.2012.01784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojima M, Stroher U, Ebihara H, Feldmann H, Kawaoka Y. Identification of cell surface molecules involved in dystroglycan-independent lassa virus cell entry. J Virol. 2012;86:2067–2078. doi: 10.1128/JVI.06451-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotgia F, Lee H, Bedford MT, Petrucci T, Sudol M, Lisanti MP. Tyrosine phosphorylation of beta-dystroglycan at its WW domain binding motif, PPxY, recruits SH2 domain containing proteins. Biochemistry. 2001;40:14585–14592. doi: 10.1021/bi011247r. [DOI] [PubMed] [Google Scholar]
- Sotgia F, Bonuccelli G, Bedford M, Brancaccio A, Mayer U, Wilson MT, et al. Localization of phospho-beta-dystroglycan (pY892) to an intracellular vesicular compartment in cultured cells and skeletal muscle fibers in vivo. Biochemistry. 2003;42:7110–7123. doi: 10.1021/bi0271289. [DOI] [PubMed] [Google Scholar]
- Spence HJ, Dhillon AS, James M, Winder SJ. Dystroglycan, a scaffold for the ERK-MAP kinase cascade. EMBO Rep. 2004 doi: 10.1038/sj.embor.7400140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiropoulou CF, Kunz S, Rollin PE, Campbell KP, Oldstone MB. New World arenavirus clade C, but not clade A and B viruses, utilizes alpha-dystroglycan as its major receptor. J Virol. 2002;76:5140–5146. doi: 10.1128/JVI.76.10.5140-5146.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vela EM, Bowick GC, Herzog NK, Aronson JF. Genistein treatment of cells inhibits arenavirus infection. Antiviral Res. 2008;77:153–156. doi: 10.1016/j.antiviral.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber EL, Buchmeier MJ. Fine mapping of a peptide sequence containing an antigenic site conserved among arenaviruses. Virology. 1988;164:30–38. doi: 10.1016/0042-6822(88)90616-2. [DOI] [PubMed] [Google Scholar]
- Yang B, Jung D, Motto D, Meyer J, Koretzky G, Campbell KP. SH3 domain-mediated interaction of dystroglycan and Grb2. J Biol Chem. 1995;270:11711–11714. doi: 10.1074/jbc.270.20.11711. [DOI] [PubMed] [Google Scholar]