

Abstract
Purpose
Pediatric gastrointestinal stromal tumors (GIST) are rare and occur preferentially in females as multifocal gastric tumors, typically lacking mutations in KIT and PDGFRA. As KIT oncoprotein is consistently overexpressed in pediatric GIST, we sought to investigate the activation of KIT downstream targets and alterations of KIT/PDGFRA gene copy number, mine novel therapeutic targets by gene expression, and test tyrosine kinase receptor activation by proteomic profiling.
Experimental Design
Seventeen pediatric GISTs were investigated for KIT/PDGFRA genotype and biochemical activation of KIT downstream targets. The transcriptional profile of 13 nodules from 8 pediatric patients was compared with 8 adult wild-type (WT) GISTs, including 3 young adults. The drug sensitivity of second-generation kinase inhibitors was tested in murine Ba/F3 cells expressing human WT KIT, as well as in short-term culture of explants of WT GIST cells.
Results
A KIT/PDGFRA WT genotype was identified in all 12 female patients, whereas two of five males had either a KIT exon 11 or PDGFRA exon 18 mutation. KIT downstream targets were consistently activated. Pediatric GISTs showed a distinct transcriptional signature, with overexpression of BAALC, PLAG1, IGF1R, FGF4, and NELL1. In vitro studies showed that nilotinib, sunitinib, dasatinib, and sorafenib are more effective than imatinib against WT KIT.
Conclusions
Rare cases of pediatric GIST may occur in male patients and harbor activating KIT/PDGFRA mutations. Pediatric GISTs show distinct transcriptional signature, suggesting a different biology than WT GIST in adults. In vitro drug screening showed that second-generation kinase inhibitors may provide greater clinical benefit in pediatric GIST.
Gastrointestinal stromal tumors (GIST), the most common mesenchymal tumors of the gastrointestinal tract, typically occur in adults over the age of 40 years. GISTs in the pediatric age group are rare and account for 1% to 2% of all GIST cases (1, 2). Pediatric GISTs are preferentially located in the stomach as multiple nodules and histologically have either an epithelioid or a mixed spindle and epithelioid morphology (1, 2). Of interest is that unlike in adults, the majority of GISTs in pediatric patients follow an indolent course, in spite of the high rate of metastasis to the peritoneal cavity and liver. Furthermore, metastasis to locoregional lymph nodes is common in pediatric GIST patients and rare in adults (2). Also, in contrast with adult GISTs, tumors in the pediatric age group often lack activating mutations in KIT or PDGFRA. In spite of the wild-type (WT) genotype, KIT oncoprotein is consistently overexpressed in these tumors. Certain clinicopathologic features, such as female predisposition, multifocal gastric location, and WT genotype suggest a relationship with Carney's triad.
Although imatinib mesylate achieves a clinical response in >80% of adult patients with metastatic or advanced GIST, the efficacy of selective kinase inhibition in pediatric GIST population has not been well defined. This question remains unresolved due to the rarity of pediatric GIST and its indolent natural history, both of which preclude large clinical trials.
In this study, we investigated the activation of KIT downstream targets and alterations of KIT/PDGFRA gene copy number, mined novel therapeutic targets by gene expression, and tested the activation status of receptor tyrosine kinase (RTK) by proteomic profiling. In addition, we compared the sensitivity of WT KIT oncoprotein to imatinib and second-generation kinase inhibitors in vitro.
Materials and Methods
Patient selection and clinicopathologic features
Patients with a diagnosis of GIST who were 18 years of age or younger were identified from the Memorial Sloan-Kettering Cancer Center database and the personal consultation files of one of the authors (C.R.A.). Patient demographics, treatment data, and follow-up information were obtained from chart review. The pathologic diagnosis was confirmed using standard H&E staining and immuno-reactivity for CD117 on formalin-fixed paraffin-embedded tissue. In addition, we searched for GISTs in young adult patients (older than 18 years but younger than 30 years) with tissue available for molecular analysis, to include as a control group for gene expression and protein analysis. The study was approved by the institutional review board.
KIT/PDGFRA genotyping and gene copy number analysis
Genomic DNA was isolated either from snap-frozen tumor tissue in eight patients or from paraffin-embedded tissue in nine patients, as described previously (3). Adequate DNA for mutational analysis was obtained in all 17 patients (see Table 1). All cases were tested for the known sites of KIT (exons 9, 11, 13, and 17) and PDGFRA (exons 12, 14, and 18) mutations. In seven cases, good-quality total RNA was obtained for full-length sequencing of KIT and PDGFRA cDNA. Primer sequences and annealing temperatures were as described (3, 4). Direct sequencing of PCR products was done for all exons tested and each ABI sequence was compared with the National Center for Biotechnology Information human KIT and PDGFRA gene sequences.
Table 1.
Clinicopathologic features and molecular data in 17 pediatric GISTs
| No. | Age/sex | Site | Multifocal | Type | MF/50 HPF | KIT/PDGFRA | Mets | Imatinib response/mo/dose(mg/d) | Sunitinib response/mo/dose(mg/d) | Follow-up (mo) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1*†‡ | 10F | Stomach | Yes | E | 6 | WT | P | POD/18/800 | SD/8/25 | 69/AWD |
| 2*†§ | 12F | Stomach | Yes | S+E | 2 | WT | LN,P,L | No | No | 78/AWD |
| 3*‡ | 8F | Stomach | Yes | S+E | 15 | WT | LN | No | No | 23/NED |
| 4 | 16F | Stomach | Yes | E | 3 | WT | P | NA | NA | 6/NED |
| 5∥ | 16F | Stomach | Yes | S+E | 4 | WT | LN | DI/3/600 | DI/1/37.5 | 60/AWD |
| 6*§‡ | 14F | Stomach | Yes | S+E | 10 | WT | P | SD+POD/6/800 | POD/5/25 | 36/AWD |
| 7*†‡ | 12F | Stomach | Yes | E | 3 | WT | LN,P,L | No | No | 80/NED |
| 8*†‡ | 15F | Stomach | Yes | E | 48 | WT | P,L | POD/4 | No | 138/DOD |
| 9† | 10F | Stomach | Yes | E | 6 | WT | P, L | SD/9/300 | No | 188/AWD |
| 10 | 18F | Omentum | NA | E | 76 | WT | P,L | POD/3/400 | PR/8/50 | 48/AWD |
| 11*‡ | 13F | Stomach | Yes | E | 20 | WT | No | NA | NA | 3/NED |
| 12 | 14F | Stomach | Yes | S+E | 40 | WT | no | A/24 | No | 30/AWD |
| 13*‡ | 12M | Stomach | No | S | 3 | WT | P | No | No | 49/AWD |
| 14 | 18M | Stomach | Yes | S | 6 | WT | No | NA | NA | 24/NED |
| 15 | 15M | Stomach | Yes | S+E | 1 | WT | No | NA | NA | NA |
| 16 | 14M | Stomach | No | E | 1 | D842V | No | NA | NA | NA |
| 17 | 17M | SB | No | S | 1 | 558K del | No | No | No | 22/NED |
Abbreviations: S, spindle; E, epithelioid, NA, not available, SB, small bowel; SD, stable disease; POD, progression of disease; DI, drug intolerance; PR, partial response; A, adjuvant therapy; NED, no evidence of disease; AWD, alive with disease; DOD, dead of disease; P, peritoneum; L, liver; LN, perigastric lymph nodes; MF, mitotic figure; HPF, high-power field; Mets, metastasis.
Gene expression analysis.
Cases 1, 2, and 7 to 9 were previously reported in ref. 2.
Full-length KIT and PDGFRA cDNA sequencing.
Cases with Carney's triad.
Suspected of Carney's triad.
Fluorescence in situ hybridization was used for detection of KIT/PDGFRA gene copy number on formalin-fixed paraffin-embedded sections of tumors from 9 pediatric and 3 young adults. Briefly, paraffin sections were dewaxed in xylene and then microwaved in 10 mmol/L sodium citrate (pH 6–6.5) solution for 5 to 10 min, cooled to room temperature, rinsed, and dehydrated. The slides were then denatured in 70% formamide at 68°C for 2 to 4 min, quenched, dehydrated, and air dried. The KIT probes used were two overlapping BAC clones: CTD-3180G20 and RP11-722F21 (Invitrogen), labeled by nick translation with Spectrum Green (Vysis, Abbott Laboratories). The PDGFRA probe used included two BAC clones that spanned ~290 kb around the gene: RP11-117E8 and RP11-231C18. A chromosome 4 centromeric probe labeled with Spectrum Orange (CEP 4, Vysis) was used as reference. The probe mix, 50 to 80 ng of each KIT or PDGFRA BAC and 2 μL Cot-1 DNA (Invitrogen), was ethanol precipitated and resuspended in hybridization buffer. The KIT or PDGFRA probe mix was denatured at 70°C for 10 min, followed by preannealing at 37°C for 30 min. The KIT or PDGFRA probe was then combined with the denatured CEP 4 probe on the slide, coverslipped, and incubated overnight at 37°C. After posthybridization washes, the slides were stained with 4′,6-diamidino-2-phenylindole and mounted in Antifade (Vectashield, Vector Laboratories). Analysis was done using a Nikon E800 epifluorescence microscope with MetaSystems Isis 3 imaging software. A minimum of 100 cells was scanned over separate regions for each slide. Image z-stacks were captured using a Zeiss Axioplan 2 motorized microscope controlled by Isis 5 software (Metasystems).
Biochemical analysis for KIT and downstream targets
Whole-cell lysates were prepared from tumors of five pediatric patients by grinding 1 g of snap-frozen tumor tissue, using a PowerGen 700 Homogenizer (Omni International). The results were compared with a control group of WT GISTs from 3 adult patients, including 1 young adult. The ground tissue was resuspended in radioimmuno-precipitation assay lysis buffer (Upstate) containing a cocktail of protease and phosphatase inhibitors (Sigma), sodium fluoride, sodium orthovanadate, and phenylmethylsulfonyl fluoride. Antibodies tested included rabbit polyclonal anti–phospho-KIT Y721, anti–phosphorylated signal transducer and activator of transcription 1 (STAT1), anti-STAT1, anti-STAT3, anti–phospho-STAT5, anti-STAT5 (Zymed Lab, Inc.), rabbit anti-KIT (Oncogene Science), mouse anti-actin (Santa Cruz Biotechnology), anti–phospho-AKT (Thr308), anti-AKT, anti–phospho-p44/p42 mitogen-activated protein kinase (Thr202/Tyr204), anti–mitogen-activated protein kinase, anti–phospho-mTOR (Ser2448), anti-mTOR, anti–phospho-PDK1 (Ser241), anti-PDK1, anti–phospho-S6 ribosomal protein (Ser235/236), anti-S6 ribosomal protein, anti–phospho-PDGFRA (Tyr754), anti-PDGFRA, anti – phospho-PDGFRB (Tyr751), anti-PDGFRB, anti–phospho-STAT3 (Tyr705), anti–phospho-epidermal growth factor receptor (EGFR; Tyr1068), and anti-EGFR (Cell Signaling Technology, Inc.). The secondary antibodies used included donkey–anti-mouse (Santa Cruz Biotechnology) and anti-rabbit (Calbiochem).
Phospho-RTK array
Human phospho-RTK array kit (R&D Systems, Inc.), containing 42 different anti-RTK antibodies per array, was used to detect phosphorylated RTKs in the samples. Each array was hybridized using 500 μg of tumor lysate and subsequent steps were done as per the manufacturer's protocol. Validation of the results was done by Western blotting analysis.
U133A chip Affymetrix microarray analysis and expression data analysis
Microarray analysis was done and analyzed, as described previously (5), on 13 nodules from 8 pediatric GIST patients with fresh-frozen tissue available for RNA extraction.
The data were clustered using two different methods. In the first analysis, the filtering included only genes scored present in at least 25% of the samples, which gave 14,371 genes. The data were then clustered using hierarchical clustering with the Pearson correlation metric and average linkage. To assess the robustness of the clustering result, bootstrap resampling was done (6). A parametric method was used to resample the data to simulate noise. This was done 1,000 times and each replica of the data was clustered. The 1,000 trees were then combined using a majority rule algorithm (6) to give a consensus tree. Each node was scored by how many times it appeared in the 1,000 bootstrap trees.
The second hierarchical clustering analysis was done using Genespring GX 7.3.1 software. After filtering for flags and expression values, a gene list that selected for genes with significant fold changes between the groups was identified. Hierarchical clustering with the Pearson metric and centroid linkage was used.
Validation of differentially expressed genes by real-time PCR
One microgram of total RNA was reverse-transcribed using the Thermoscript reverse transcription-PCR system (Invitrogen) at 52°C for 1 h. Twenty nanograms of resultant cDNA was used in a quantitative PCR reaction using an iCycler (Bio-Rad Laboratories) and predesigned TaqMan ABI Gene expression Assays. Primers were chosen based on their ability to span the most 3′ exon-exon junction. Amplification was carried for 40 cycles (95°C for 15 s, 60°C for 1 min). To calculate the efficiency of the PCR reaction, and to assess the sensitivity of each assay, a 7-point standard curve (5, 1.7, 0.56, 0.19, 0.062, 0.021, and 0.0069 ng) was done. Triplicate CT values were averaged, and amounts of target were interpolated from the standard curves and normalized to hypoxanthine phosphoribosyltransferase.
Screening for activating mutation in overexpressed candidate genes
Selected candidate genes found to be either up-regulated on transcriptional profiling (BAALC), activated by phospho-RTK array (EGFR) or overexpressed by immunohistochemistry (P53), were investigated for activating mutations by PCR and direct sequencing. Hotspot mutation areas of EGFR (exon 19–21) and p53 (exon 5–8) were examined. Based on prior evidence that KIT and BRAF mutations are mutually exclusive in melanomas, we also investigated the hotspot mutations in BRAF (exon 15).
Establishing Ba/F3 WT KIT and KITV559D stable transformant cell line
A WT KIT cDNA was cloned into the retroviral expression vector pMSCV (a generous gift from Dr. Gary Gilliland, Harvard Medical School, Boston, MA). Site Directed Mutagenesis PCR using Quick-Change II XL site-directed Mutagenesis Kit (Qiagen, Inc.) was used to obtain KIT V559D mutation, an imatinib-sensitive juxtamembrane mutation, used as control for in vitro drug testing. The two pMSCV-KIT cDNA constructs were introduced into Ba/F3 cells (German Collection of Microorganisms and Cell Cultures-Human and Animal Cell Cultures, Braunshweig, Germany) by electroporation. Twenty-five micrograms of each pMSCV-KIT cDNA construct were cotransfected into 10 million Ba/F3 with 1 μg of linear hygromycin (Clonetech) marker using GenPulser (Bio-Rad). The electroporated cells were first grown in the presence of hygromycin and interleukin-3 for a week and the selected cells were sorted based on green fluorescent protein positivity. The selected green fluorescent protein–positive cells were first grown in the presence of interleukin-3 (10 ng/mL) for 2 wk and subsequently interleukin-3 was withdrawn and cells were grown in the presence of the KIT ligand (20 ng/mL). The transforming activity was determined by interleukin-3–independent growth curve.
In vitro drug testing of Ba/F3 WT KIT cells
Sunitinib, dasatinib, nilotinib, sorafenib, and imatinib were tested in Ba/F3 WT KIT cell line and the efficacy was compared with Ba/F3 KITV559D, an imatinib-sensitive KIT juxtamembrane domain mutation. Imatinib, sunitinib, and sorafenib were purchased commercially. Dasatinib and nilotinib were synthesized and generously provided by Dr. Bayard Clarkson's laboratory, Sloan-Kettering Institute. Each drug was tested at similar concentrations of 10, 100, 1,000, and 5,000 nmol/L. Each cell line with no drug treatment served as baseline control. The drug response was monitored by measurements in cell proliferation and apoptosis, as well as inhibition of KIT activation, biochemically.
Cell proliferation assays
To determine growth-inhibitory drug effects, Ba/F3 transformants containing KIT mutants were starved from growth factor for 4 h before drug administration. Ten minutes before adding the drug, growth factors were added: 10 ng/mL of interleukin-3 for the negative control BaF3 cells and 20 ng/mL of KL for Ba/F3 cell lines expressing KIT isoforms. Cells were incubated with the drug at 37°C for 48 h. Bromodeoxyuridine was then added to the cell culture and incubated for 6 h before harvesting. Cells were fixed and stained as per the protocol recommended for the bromodeoxyuridine-APC kit (PharMingen). Bromodeoxyuridine incorporation was determined by flow cytometry. A minimum of 2 × 105 events were acquired and the data were analyzed using FlowJo software (version 5.7.2). Cell growth inhibition curves were plotted and IC50 values were calculated with GraphPad Prism software, version 4.03.
Apoptosis assays
Induction of apoptosis on Ba/F3 KIT transformants was evaluated by flow cytometry using Annexin V-Phycoerythrin Apoptosis Detection kit (PharMingen). Cells at a density of 1 × 106 were cultured and starved from growth factors for 4 h before drug delivery. Ten minutes before adding the drug, growth factors were added. Cells were incubated with the drug at the same concentrations indicated above for 48 h. Cells were then harvested and stained with anti–Annexin V-phycoerythrin antibody as per the provider's protocol. A minimum of 10,000 events were analyzed by FACScan (Becton Dickinson) within an hour after staining and the results were analyzed by FlowJo.
Immunoprecipitation and Western blotting
The Ba/F3 cells expressing WT KIT and KITV559D were treated with the indicated doses of each drug. Cells were starved from serum and growth factors for 2 h; only growth factors were added back to medium 15 min before drug administration. Drugs were then incubated at 37°C in the absence of serum for 90 min. After treatment, cells were harvested and subjected to protein extraction. Whole-cell lysate (200 μg) was incubated with 2 μg of anti-KIT antibody (Assay Designs, Inc.) for an hour, and then the mixture was incubated with Magna beads (Pierce Biotechnology) overnight at 4°C. The beads were washed and isolated using a magnetic column and resuspended in LDS sample buffer (Invitrogen Life Technologies). Electrophoresis and immunoblotting was done on the protein extracts using the standard protocol. Phosphorylated KIT was detected with anti–phospho-tyrosine antibodies PY20 and PY99 (Santa Cruz Biotechnology), and total KIT was detected by mouse monoclonal anti–c-KIT (Santa Cruz Biotechnology). The secondary antibody used was donkey–anti-mouse secondary (Santa Cruz Biotechnology).
Short-term culture of WT GIST cells
Five to 10 grams of tumor tissue were minced and incubated with collagenase type 1A (Sigma) overnight in the presence of DMEM HGF12 medium containing 15% FCS, 30 mg/L BPE (BD Biosciences), and 1 mg/L of Mito+ (BD Biosciences). The next day, cells were washed, resuspended in fresh medium, and plated onto 60-mm Petri dishes for 1 wk. Cells were treated with two protein kinase inhibitors, imatinib and dasatinib (1 μmol/L), when cell culture was 50% confluent. Before the treatment, cells were starved from serum and growth factors for 2 h; growth factors and serum were added to the culture 15 min before the treatment. Cells were incubated with drugs for 90 min before harvesting and then lysed in protein lysis buffer to get cell lysate. The response to each drug was monitored by immunoprecipitation and Western blotting for phosphorylation inhibition of KIT.
Results
Pediatric GIST is not restricted to female patients or gastric location
We identified 17 patients who were 18 years of age or younger with the confirmed diagnosis of GIST. There were 12 females and 5 males, ranging from 8 to 18 years (mean 13.6, median 14). All except two patients had their tumors located in the stomach. The only female patient with extragastric involvement was found to have multifocal intra-abdominal disease, with no identifiable relationship to the adjacent gastrointestinal tract. One male patient had a solitary small-bowel tumor. Most gastric GISTs showed a distinct multifocal nodular growth, with histologically normal tissue separating individual nodules. Thus, the majority of patients who underwent a partial or distal gastrectomy had positive surgical margins. Microscopically, the morphology was predominantly epithelioid in 8 tumors, spindle in 3 tumors, and mixed epithelioid and spindle in 6 tumors. The predominantly spindle cell GISTs occurred only in male patients (Table 1). Interstitial cell of Cajal hyperplasia was not identified near the tumors or at distant sites. The mitotic activity varied widely (1–76 mitotic figures/50 high-power fields), and, surprisingly, did not correlate with the metastatic potential or clinical behavior. Five of the six patients with a low proliferation rate (<5 mitotic figures/50 high-power fields) and in whom longer follow-up was available developed either locoregional lymph node, peritoneal, or liver metastasis.
The incidence of Carney's triad among pediatric GIST patients may be underestimated
Although all patients included in this series had no other stigmata of Carney's triad at initial presentation, 2 of the 12 female patients subsequently developed a second neoplasm diagnostic of this syndrome. The first patient (patient 2) developed an extra-adrenal paraganglioma, 66 months from the initial GIST diagnosis. This lesion was detected during periodic follow-up computed tomography/positron emission tomography imaging and surgical exploration undertaken to exclude metastatic disease. The second patient (patient 6) developed bilateral pulmonary chondromas 30 months following the diagnosis of GIST (Fig. 1A–C). A third patient (patient 5) is presently being followed for a stable lung lesion, which has the clinical appearance of a pulmonary chondroma.
Fig. 1.
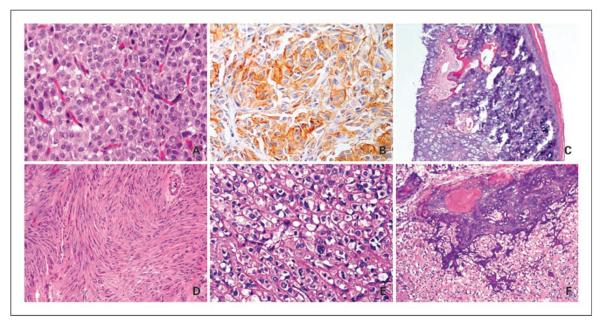
Histologic appearance of pediatric GIST. Epithelioid gastric GIST in a 14-year-old female patient showing a uniform plasmacytoid appearance (A, H&E, ×200); immunoreactive with CD117 antibody (B, ×200); and who developed bilateral pulmonary chondromas, diagnostic of Carney's triad (C, patient 6, ×40). D, KIT exon 11 – mutated small-bowel GIST showing a pure spindle cell morphology in a 17-year-old male (H&E, ×100, patient 17). E and F, PDGFRA D842V – mutated gastric GIST showing an epithelioid phenotype, with focal nuclear pleomorphism and binucleated forms (H&E, ×200) and distinct areas of calcification and ossification (H&E, ×100, patient 16).
Pediatric GISTs lack gene copy number alterations of KIT/PDGFRA and rarely show KIT/PDGFRA mutations in a subset of male patients
Sequencing analysis showed a WT KIT and PDGFRA genotype in all except two male patients. Patient 17, a 17-year-old male with a solitary small-bowel spindle cell GIST, revealed a KIT exon 11 K557 deletion (Fig. 1D). Patient 16, a 14-year-old male with a large gastric tumor, showed a PDGFRA exon 18, D842V, mutation. Histologically, the latter tumor had a pure epithelioid morphology, with large cells containing abundant clear to eosinophilic cytoplasm, focal marked nuclear pleomorphism, and distinct areas of dystrophic calcification and ossification (Fig. 1E and F). Both these mutation-positive GISTs had a low mitotic index (<5 mitotic figures/50 high-power fields).
Fluorescence in situ hybridization analysis was done on 12 WT GIST tumor nodules from 9 pediatric and 3 young adult patients. Tumor samples were analyzed for alterations in KIT and PDGFRA copy number with reference to a centromeric probe on chromosome 4. All samples showed a 1:1 KIT/CEP4 or PDGFRA/CEP4 ratio, similar to control samples.
Pediatric GIST has a distinct gene expression signature
The transcriptional profile of 13 tumor nodules derived from 8 pediatric GIST patients was investigated on the U133A Affymetrix platform. The 13 tumor nodules included 3 tumor nodules from the 2 patients with Carney's triad. The expression values were first compared with a control group of 5 adult WT GISTs, with available array data. By unsupervised hierarchical clustering, pediatric GIST samples formed a tight cluster distinct from the adult GIST tumors (Fig. 2). The tumors from the pediatric patients, with and without Carney's triad, clustered together and showed no difference in gene expression profiles. In a supervised analysis using a ≥2-fold change cutoff, 1,532 genes were differentially expressed between these two groups.
Fig. 2.

Unsupervised hierarchical clustering of 13 pediatric GISTs and 5 adult WT GISTs showing distinct clustering of the two groups (Genespring GX 7.3).
Because all 13 pediatric GIST samples originated in the stomach, it is possible that the expression profile may be skewed by the anatomic location. Thus, we carried out a second analysis using as control group 19 primary gastric GISTs from imatinib-naBve adult patients. The genotype of this second control group is as follows: 12 KIT exon 11 mutation, 4 PDGFRA mutation, and 3 WT. Using a ≥2-fold change cutoff, 1,335 differentially expressed genes were identified between these two groups.
Within the adult GIST group with available array data, we identified 3 patients, younger than age of 30 years, with clinical and pathologic features more in keeping with the pediatric GIST subset, such as multifocal gastric location, epithelioid morphology, and indolent course. Two of these cases had a WT genotype, whereas the third showed a KIT exon 9 mutation. When included in the unsupervised hierarchical clustering, these three samples clustered together with the pediatric rather than the WT adult tumors.
Using a Venn diagram to identify genes in common between the above two analyses, 814 genes were differentially expressed in pediatric GISTs. The top-ranked genes found to be overexpressed in the pediatric group compared with adult tumors included FGF4 (fibroblast growth factor 4), BAALC (brain and acute leukemia, cytoplasmic), IGF1R (insulin-like growth factor I receptor), NELL1 (NEL-like 1), CRLF1 (cytokine receptor-like factor 1), PLAG1 (pleomorphic adenoma gene 1), and FGF3 (fibroblast growth factor 3; Table 2). A subset of these genes, such as IGF1R, BAALC, FGF4, PLAG1, and NELL1, was validated using quantitative PCR (Table 3). Full-length sequencing of the coding region was done for BAALC but no mutations were identified.
Table 2.
Differentially expressed genes in pediatric GISTs in comparison with adult WT GISTs
| Gene symbol | Gene title | Fold change | Chromosomal location | Gene ontology biological process |
|---|---|---|---|---|
| CRLF1 | Cytokine receptor-like factor 1 | 186.2 | 19p12 | Antimicrobial humoral response |
| BAALC | Brain and acute leukemia, cytoplasmic | 40.93 | 8q22.3 | — |
| FGF4 | Fibroblast growth factor 4 | 18.88 | 11q13.3 | Cell proliferation |
| PLAG1 | Pleomorphic adenoma gene 1 | 16.63 | 8ql2 | — |
| IGF1R | Insulin-like growth factor 1 receptor | 10.96 | 15q25 | Positive regulation of cell proliferation |
| FGF3 | Fibroblast growth factor 3 | 9.913 | 11q13 | Cell proliferation |
| GLRB | Glycine receptor, β | 17.94 | 4q31.3 | Receptor linked signal transduction |
| NEFL | Neurofilament, light polypeptide 68 kDa | 15.84 | 8p21 | — |
| NRCAM | Neuronal cell adhesion molecule | 14.95 | 7q31.1 | Neuronal migration |
| NELL1 | NEL-like 1 (chicken) | 12.67 | 11p15.2 | Cell adhesion, neurogenesis |
| RTN1 | Reticulon 1 | 11.87 | 14q21 | Neuron differentiation |
| MAGEA3 | Human MAGE-6 antigen (MAGE6) | 11.51 | Xq28 | — |
| RELN | Reelin | 8.574 | 7q22 | Cell adhesion, development |
| FGF18 | Fibroblast growth factor 18 | 5.589 | 5q34 | Regulation of transcription |
Table 3.
Validation by quantitative reverse transcription-PCR of selective overexpressed genes
| Gene symbol | Gene title | Fold change* |
|
|---|---|---|---|
| Reverse transcription-PCR | Microarray | ||
| IGF1R | Insulin-like growth factor 1 receptor | 6.9 | 10.9 |
| BAALC | Brain and acute leukemia, cytoplasmic | 29.1 | 40.9 |
| FGF4 | Fibroblast growth factor 4 | 5.8 | 18.9 |
| PLAG1 | Pleomorphic adenoma gene 1 | 13.5 | 16.6 |
| NELL1 | NEL-like 1 (chicken) | 32.6 | 12.7 |
Fold change is the expression in pediatric GISTs relative to that in the adult WT tumors.
IGF1R was highly overexpressed in pediatric GIST. One of the explanations for this finding could be an autocrine loop mechanism, secondary to IGF1 up-regulation. Based on the gene expression analysis, IGF1 expression was 5-fold higher in pediatric GIST compared with adult WT GIST. We speculate that this may be one of the mechanisms that induce the expression of IGF1R. Another explanation for this increase in IGF1R could be alterations of p53/MDM (7–10). Werner et al. (10) have shown that mutations of p53, usually associated with malignant states, activate the IGF1R promoter leading to an overexpression of IGF1 receptor. Idelman et al. (8), in a later study, identified that WT1 interacts with p53 in the regulation of IGF1R. Following this latter hypothesis, we have investigated the p53 pathway in pediatric GIST. Using immunohistochemical analysis as method of screening for p53 abnormalities, we identified four of six pediatric GIST tumors to show p53 protein expression. The four positive tumors were further screened for p53 gene mutations, but no mutations were identified by DNA PCR genotyping of the exons 5 to 8 “hotspot” regions (4).
KIT and its downstream signaling targets are activated in pediatric GIST similar to WT adult GIST
Five pediatric WT gastric tumors were studied biochemically for activation (phosphorylation) of KIT and its downstream signaling pathways. The results were compared with the activation pattern seen in three samples from adult WT GISTs (data not shown) and two samples of KIT exon 11 mutated adult GISTs. Consistent activation of KIT and its downstream targets, including AKT, PDK1, mTOR, S6 kinase, and ribosomal S6, were noted in the pediatric GISTs (Fig. 3). Mitogen-activated protein kinase was activated in three of the five tumors. An important observation was that none of the tumors showed activation of PDGFRA or PDGFRB proteins, thereby indicating that the signaling pathway in these cases is through the constitutively active KIT receptor. STAT3 was activated in all cases, whereas STAT1 and STAT5 were not. Essentially, there was no difference in the activation pattern of these downstream targets of receptor tyrosine kinases when compared with adult WT and KIT-mutated GISTs.
Fig. 3.

Western blotting of 5 pediatric tumors and 2 adult KIT exon 11-mutant GIST. A and B, tumors show activation of KIT and its downstream signaling targets. PDGFRA and PDGFRB are not activated in any of the pediatric cases tested.
Screening for alternative activated RTKs showed EGFR phosphorylation
Using a proteome array, containing 42 phospho-RTK antibodies, we analyzed 4 pediatric and 2 adult WT GISTs, including 1 from a young adult. Only 1 of the 4 pediatric tumors was successfully analyzed and showed, in addition to phosphorylated KIT, a weakly phosphorylated EGFR (Fig. 4A). The young adult case also showed phosphorylation of EGFR, in addition to KIT protein (Fig. 4B). The adult tumor revealed phosphorylation of PDGFRB, EGFR, and FGFR2a but no KIT activation (Fig. 4C). We further validated these findings by Western blot analysis of these three GISTs for phosphorylated and total EGFR and KIT expression. An EGFR-mutated lung cancer was used as a positive control (Fig. 4D). The Western blotting results also showed that the WT young adult GIST had the strongest EGFR phosphorylation compared with the other two cases. Furthermore, it confirmed that the WT adult tumor lacked both total and phosphorylated KIT. These results were also in concordance with the mRNA expression obtained on microarray (Fig. 4). As the adult patient with a WT GIST showed no KIT activation and low level of KIT mRNA expression, but activated PDGFRB and up-regulated PDGFRB gene expression, we sequenced exons 7 to 21 of PDGFRB of this tumor, but did not identify any mutations. Because EGFR phosphorylation was noted in all three WT tumors, we further analyzed the EGFR mutation status in these tumors. Sequencing analysis for the known mutations in EGFR, located within exons 19 to 21, did not reveal any mutations (11).
Fig. 4.

The phospho-RTK array highlights phosphorylated kinases in tumor lysates from a pediatric (A), young adult (B), and an adult WT GIST (C). Western blotting of the same three GIST tumors (D) showing similar phosphorylation patterns of KIT and EGFR (an EGFR-mutated lung cancer sample used as positive control). The columns on the right show the mRNA raw expression values of the phosphorylated tyrosine kinases, obtained from the DNA microarray studies on the same three tumors.
Pediatric GISTs do not exhibit BRAF mutations
As KIT mutations have been recently described in a subset of malignant melanoma, a tumor that commonly exhibits BRAF mutations (12), we postulated that WT GIST, including the pediatric group, may harbor BRAF mutations. Sequencing analysis for the hotspot mutation, V600E in exon 15, was analyzed in 10 pediatric patients, but no BRAF mutations were identified.
Ba/F3 WT KIT cells are more sensitive to nilotinib, sunitinib, dasatinib, and sorafenib than to imatinib
In adults, patients with WT tumors are the least sensitive to imatinib mesylate. To assess the efficacy of second-generation tyrosine kinase inhibitors on WT KIT protein, a retroviral expression vector, containing the full-length cDNA WT KIT, was transfected by electroporation into murine pro-B Ba/F3 cells. The cells were then treated, in the presence of KIT ligand, with imatinib, dasatinib, sunitinib, sorafenib, and nilotinib. The drug response was assessed by measurements of proliferation inhibition, induction of apoptosis, and inhibition of KIT phosphorylation by immunoprecipitation/Western blot assays (Table 4; Fig. 5). Nilotinib showed the highest efficacy for the proliferation inhibition of Ba/F3WT KIT with an IC50 of 35 nmol/L. Sunitinib, dasatinib, and sorafenib were also effective with IC50s of 245, 316, and 910 nmol/L, respectively. In contrast, imatinib-treated Ba/F3WT KIT–transfected cells showed a significantly higher IC50 of 3,132 nmol/L (Table 4). This finding was also reflected in the apoptosis assay where imatinib induced significant apoptosis only at 5,000 nmol/L. All the remaining drugs induced overt apoptosis at ~1,000 nmol/L, whereas nilotinib and dasatinib induced apoptosis of 40% of transfectant cells at 100 nmol/L (Fig. 5A).
Table 4.
IC50 values obtained by proliferation inhibition studies
| Imatinib (nmol/L) | Dasatinib (nmol/L) | Sorafenib (nmol/L) | Nilotinib (nmol/L) | Sunitinib (nmol/L) | |
|---|---|---|---|---|---|
| V559D | 63 | 27 | 66 | 44 | 276 |
| WT KIT | 3,132 | 316 | 910 | 35 | 245 |
Fig. 5.

Apoptosis assay (A) and biochemical assays (B) on WT KIT – transfected Ba/F3 cells showed nilotinib- and dasatinib-induced apoptosis of ~40% of cells at 100 nmol/L, whereas the remaining drugs, except imatinib, showed significant apoptosis at only 1,000 nmol/L. Biochemical assays (B) showed nilotinib- and dasatinib-induced loss of KIT activation, whereas KIT activation persisted with imatinib. In contrast, all inhibitors were effective against KITV559D – transfected Ba/F3 cells, with dasatinib being the most potent. This finding is also reflected by apoptosis (C) and biochemical (D) assays.
In comparison, Ba/F3 KITV559D, the imatinib-sensitive juxtamembrane mutation, was sensitive to all four inhibitors, with most sensitivity to dasatinib (Table 4). Its kinase activity was distinctly inhibited at lower than 10 nmol/L of dasatinib, whereas imatinib, nilotinib, sunitinib, and sorafenib showed the same results at 100 nmol/L. Similarly, dasatinib inhibited the cell growth with an IC50 of 27 nmol/L, whereas imatinib, nilotinib, sorafenib, and sunitinib had IC50s of 63, 44, 66, and 276 nmol/L, respectively. All drugs, including imatinib, induced overt apoptosis at ~100 nmol/L, whereas dasatinib induced apoptosis of >40% of transfectant cells at 10 nmol/L (Fig. 5C).
These results were further confirmed in a short-term culture experiment of explanted WT GIST cells from a young adult patient, whose tumor had a similar transcriptional profile as the pediatric group. The WT GIST cells were tested against imatinib and dasatinib at a dose of 1 μmol/L. Dasatinib completely inhibited the phosphorylation of KIT when compared with imatinib (Fig. 6).
Fig. 6.
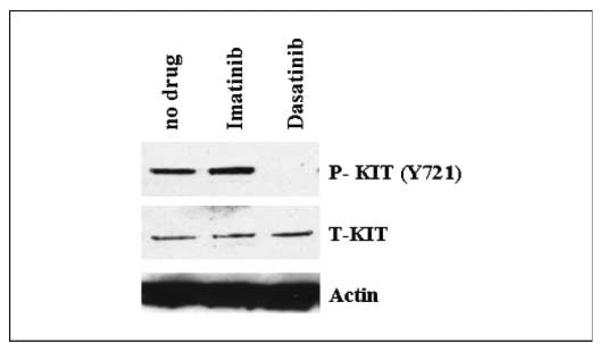
Western blot assay on short-term culture of WT GIST cells shows loss of KIT activation with dasatinib treatment, which is retained in the imatinib-treated cells.
The clinical response to imatinib is limited in pediatric GIST patients
Seven patients were treated with imatinib, 6 for metastatic disease and 1 in the adjuvant setting (Table 1). The 6 patients treated for measurable disease were on the drug for 3 to 18 months (median 5 months) and the best response was stable disease seen in 1 patient, no response in 4 patients, whereas the remaining patient showed a mixed response—stable in some nodules and slow progression in others. Two of the seven children treated with imatinib developed severe muscle, joint, and/or bone pain, which required pain palliation and discontinuation of the drug at 3 and 6 months. In one of these patients, the pain disappeared once off imatinib and reappeared while on sunitinib (patient 6). The only patient treated in the adjuvant setting was a 14-year-old girl (patient 12) who, after undergoing partial gastrectomy with microscopic positive margins for multiple gastric tumors, received imatinib for 24 months. Two months after she discontinued therapy due to periorbital edema, she was found to have positron emission tomography–positive uptake in her gastric remnant.
A total of four patients who either developed progression or intolerance on imatinib were then treated with sunitinib. Two developed drug intolerance and required drug interruption 1 and 5 months, respectively, after initiation. One patient (patient 1) has remained on sunitinib for 8 months and shows stable disease. The fourth patient (patient 10) was initially started on imatinib after an incomplete debulking of intra-abdominal disease. Imatinib was stopped after <3 months due to disease progression as evidenced by computed tomography/positron emission tomography imaging (0.5 cm increase in each of the masses). The patient was then started on sunitinib, 37.5 mg/d for 28 days, followed by 2-week break. After the first 28 days of treatment, computed tomography/positron emission tomography showed a mixed response with a minimal growth in most lesions, except for the pelvic lesion, which decreased in size. For the second cycle, the sunitinib dose was increased to 50 mg/d and a repeat computed tomography showed resolution of liver metastases and decreased size (1 cm or more) of all abdominal masses. Sunitinib therapy was continued for five cycles (8 months) after which the patient developed disease progression. She was then switched to compassionate nilotinib for 9 months with some reduction of her tumors/stable disease. Nilotinib was interrupted after the unexplained development of widespread patchy bowel necrosis, for which she underwent emergency surgery. She is now 7 months off therapy and she is alive with disease.
Overall follow-up data was available in 15 patients and ranged from 3 to 188 months (Table 1). In 12 patients, the follow-up was >6 months (22–188 months, median 60 months), whereas 3 patients were recently diagnosed. Ten (83%) of 12 patients with longer follow-up developed metastases: 4 (36%) to perigastric lymph nodes, 8 (72%) to peritoneum, and 5 (45%) to the liver. One of the two patients in this group, who did not develop metastases, is alive with disease due to local recurrence/persistent disease at the gastric stump (patient 12). The only patient who did not recur locally or distantly (patient 17) after the 22-month follow-up was a 17-year-old male whose tumor had distinctive features, such as pure spindle cell morphology, intestinal location, and a KIT exon 11 mutation, recapitulating the adult GIST phenotype. One patient with low-volume indolent recurrence within the peritoneal cavity and liver opted for repeated surgical removal of these implants and denied therapy with selective kinase inhibitors. During a recent abdominal exploration, she was diagnosed with an extra-adrenal paraganglioma, stigmata of Carney's triad. This patient is alive and well 78 months after diagnosis. The second patient with Carney's triad is presently alive with disease after surgical removal of multiple peritoneal implants and bilateral pulmonary chondromas. At last follow-up, 8 patients were alive with disease, 3 patients had no evidence of disease, and 1 patient had died of disease.
Discussion
In our previous study, we showed that pediatric GIST is a distinct subset of this rare type of sarcoma, with strong predominance for females, gastric location, and WT genotype for KIT and PDGFRA (2). In this present work, we have expanded our molecular analysis to 17 children, including 5 male patients 1 of whom had a solitary small-bowel tumor. This larger cohort confirms that pediatric GIST is more prevalent in females, who develop multiple tumors within the stomach, without associated interstitial cell of Cajal hyperplasia. Microscopically, these tumors often show an epithelioid morphology, with a variable proliferation index. All but 2 male patients lacked activating mutations in KIT/PDGFRA. Similarly, Miettinen et al. (1) showed a WT KIT/PDGFRA genotype in all 13 pediatric GISTs analyzed, although sequencing analysis was not done in the only pediatric male patient. Two of the 5 male patients in the present study had mutations, one in the juxtamembrane domain of KIT receptor and the other in the kinase domain of PDGFRA. Thus, this is the first report of a pediatric GIST patient to harbor either a KIT exon 11 or PDGFRA mutation. In the study by Price et al. (13), 2 of their 5 pediatric patients were males, and one of them harbored a novel point mutation in codon 456 of KIT exon 9. In spite of the overwhelming prevalence of WT genotype, pediatric GIST tumors consistently overexpressed KIT protein, as evidenced by the strong immunostaining for CD117 (2, 3) and KIT phosphorylation on biochemical assays. This finding is further confirmed by the high expression levels of KIT mRNA on transcriptional profiling. The mechanism of constitutive activation of the KIT protein in these cases remains unclear. Although KIT/PDGFRA gene copy alterations have been implicated as a potential mechanism of KIT activation, specifically in the setting of developing imatinib resistance (14), we did not detect KIT/PDGFRA gene amplification in 9 pediatric and 3 young adult GISTs by fluorescence in situ hybridization analysis.
The association of multifocal gastric GIST with paragangliomas and pulmonary chondromas affecting mostly females is diagnostic of Carney's triad (15). Although mostly sporadic, a few familial cases were included in the original cohort of Carney's triad (16). More recently, it was recognized that the autosomal dominant inheritance of the dyad “paraganglioma and gastric GIST,” or the “Carney-Stratakis syndrome,” represents a separate condition that affects both males and females and lacks the association with pulmonary chondromas (17). Mutations of the genes coding for succinate dehydrogenase subunits, typically associated with familial paragangliomas, are most likely implicated in the pathogenesis of Carney-Stratakis syndrome (18). Once cases of Carney-Stratakis syndrome are eliminated, there are no inherited cases of the Carney's triad. The significant overlap between clinicopathologic features of pediatric GIST and Carney's triad, such as female predisposition, multifocal gastric location requiring multiple gastric operations, and relatively long survival even in the presence of lymph node or peritoneal/liver metastatic disease, suggests a pathogenetic link. Thus, at least some of the pediatric GIST patients may represent a form fruste of Carney's triad, because longer follow-up in our study identified two children who developed a second neoplasm, diagnostic of this syndrome. Furthermore, in a recent comprehensive genetic analysis of 41 tumors from 37 patients with Carney's triad, sequencing analysis for the entire coding region of KIT, PDGFRA, SDHA, SDHB, SDHC, and SDHD failed to identify any activating mutations (19). This result parallels the findings of a WT genotype in the majority of pediatric GIST patients.
The transcriptional signature of pediatric GIST is distinct from adult WT or gastric GISTs. The top-ranked genes overexpressed in the pediatric subset include BAALC, FGF4, PLAG1, IGF1R, NEFL, NELL1, RTN1, and CRLF1. BAALC is a recently cloned gene located on human chromosome 8q22.3, which is normally expressed in tissues of neuroectodermal origin. It was initially identified in patients with acute myeloid leukemia and further studies have shown that high expression of BAALC is an independent risk factor of poor prognosis in acute myeloid leukemia, particularly when it is associated with a normal karyotype (20–23). In spite of its overexpression, sequencing of its full coding region did not identify any mutations. PLAG1 is a proto-oncogene whose ectopic expression can trigger the development of salivary gland pleomorphic adenomas and soft tissue lipoblastoma. Oncogenic activation of this gene located on chromosome 8q12 occurs mainly as a result of promoter swapping between PLAG1 and other genes. A few translocation partners have been identified in salivary gland pleomorphic adenomas and lipoblastomas (CTNNB1, LIFR, CHCHD7, TCEA1; refs. 24–26). Additional studies in salivary gland tumors have shown that PLAG1 induces the expression of a number of growth factors that eventually promote tumor growth and development (24–28). Recently, it has been speculated that PLAG1 could be one of the regulators of CRLF1 (cytokine receptor-like factor 1) expression in the developing salivary gland (28). This could also explain the high expression of CRLF1 in pediatric GISTs.
GIST in young adults is a heterogeneous entity, with some cases resembling the clinicopathologic features of pediatric disease, whereas most are more in keeping with the adult counterpart. Upon review of our files, we identified 19 patients younger than age of 30 years. Although there was still a female sex and gastric location predominance, 5 tumors occurred in male patients and 7 tumors occurred in an extragastric location. Sequencing analysis available in 16 cases showed the presence of KIT/PDGFRA mutation in 10 (62.5%) tumors: 6 KIT exon 11, 3 exon 9, and 1 PDGFRA exon 12. All 6 WT tumors were located in the stomach and 5 of them occurred in female patients. From this group, we identified 3 patients with available frozen tissue, who had clinicopathologic features indistinguishable from pediatric GIST. By gene expression analysis, these tumors clustered together with the pediatric GIST group and not with the adult WT GIST group. The same distinctive set of genes described in pediatric GIST above was also overexpressed in the GISTs from young adults. These findings suggest that a subset of GISTs occurring in patients younger than 30 years old may be biologically related to pediatric GIST.
Biochemically, pediatric GISTs showed consistent activation (phosphorylation) of KIT and its downstream signaling targets like PDK1, AKT, mTOR, S6 kinase, and ribosomal S6 protein. Mitogen-activated protein kinase was phosphorylated in some but not all tumors. These results closely matched the activation pattern seen in the adult WT group, indicating that KIT oncogene addiction is essential for the tumor development. Given the fact that pediatric GISTs showed a characteristic gene expression profile distinct from WT or gastric adult GIST tumors, it is likely that pediatric GISTs are associated with alternative mechanisms of KIT activation. The consistent KIT-dependent signaling is significant from a therapeutic standpoint, suggesting that inhibition of the KIT signaling pathway may prevent tumor growth.
There are a few controversial points regarding the prediction of outcome in pediatric GIST. The conventional criteria for assessing risk of malignancy, such as tumor size, mitotic activity, and anatomic location, are not reliable in pediatric GIST. These patients frequently present with multiple nodules within the stomach, and thus the largest tumor dimension cannot be easily defined. Furthermore, we noted a wide range of variability in proliferation index between patients and even among multiple tumors from the same patient. Our findings are consistent with those of Miettinen (1), who noted that some pediatric patients with GIST developed metastasis despite being classified as low risk by criteria established in adult GIST. Similarly, in our study, 5 of 6 patients with a low proliferation index (<5 mitotic figures/50 high-power fields) eventually developed recurrent disease within the perigastric lymph nodes, peritoneal cavity or liver. These findings suggest that GISTs in children are unpredictable, being more prone to metastasis than comparable gastric tumors in adults. Second, the biology of pediatric GIST seems to be more indolent than the adult counterpart, with long-term survival even in the presence of metastatic disease and without kinase inhibition therapy. Among the 12 pediatric patients with >6 months follow-up, all except 2 (83%) developed metastatic disease. In spite of such a high metastatic rate, only one patient has died thus far, 138 months after initial diagnosis.
Imatinib mesylate (Gleevec, Novartis), a selective KIT, PDGFRA, PDGFRB, and BCR-ABL small-molecule inhibitor, has been extensively used as the first-line agent in adult patients with metastatic/advanced GIST. Imatinib induces a stable disease or a partial response in >80% of patients. Furthermore, an unequivocal relationship between genotype and imatinib sensitivity has been proven in large multi-institutional trials, with KIT exon 11 mutated GISTs being more sensitive to imatinib inhibition than WT GISTs. Patients carrying KIT exon 9-mutated tumors have an intermediate response to imatinib and may benefit from higher initial doses (29, 30). In contrast, this subset shows a superior response to sunitinib (Pfizer), a broad-based kinase inhibitor, with activity against KIT, PDGFRA, PDGFRB, and VEGFR (31). Extrapolating from the adult experience, in which the WT genomic subset is the least sensitive to imatinib inhibition, the question still remains if pediatric GISTs, which typically lack KIT/PDGFRA mutations, will respond to imatinib. Anecdotal evidence mainly based on case reports shows poor clinical responses to imatinib (32). Also of interest is that the pattern of excruciating somatic soft tissue and bone pain seen in 2 of the 7 children treated with imatinib and/or sunitinib has not been previously described in imatinib-treated children with other diseases, such as pediatric chronic myelogenous leukemia and acute lymphocytic leukemia (33).
Effective agents for pediatric GIST are needed. Thus, we conducted an in-depth in vitro analysis of second-generation kinase inhibitors, such as sunitinib, nilotinib, sorafenib, and dasatinib. Nilotinib showed the highest potency in inhibiting cell growth with an IC50 of 35 nmol/L. Dasatinib and sorafenib showed IC50s 10 and 30 times higher than nilotinib, respectively, but were still more effective than imatinib against WT KIT–transfected cells. The IC50 of imatinib exceeded 3,000 nm. These results are consistent with the findings by Casteran et al. (34), who showed that juxtamembrane domain mutations are more sensitive to imatinib inhibition than the WT form of the KIT receptor. The efficacy of dasatinib was further confirmed by the complete inhibition of KIT phosphorylation in a short-term primary culture experiment of a WT GIST explanted from a young adult patient with similar clinicopathologic and genomic features as the pediatric group. Based on these in vitro results, second-generation tyrosine kinase inhibitors have a superior activity to imatinib and might prove to be effective in WT GIST patients, including pediatric GIST patients. Although we cannot specifically address the therapeutic response of pediatric GISTs based on this in vitro study, we speculate that these drugs will be equally effective in both pediatric GISTs and the adult WT tumors.
Although initially regarded as a homogenous clinical and genetic subset with a predilection for females, gastric location, and WT genotype, our study expands the entity of pediatric GIST. Up to one third of cases may occur in males and these can harbor activating KIT/PDGFRA mutations and occur in the small bowel. With longer follow-up, it is now becoming clear that some pediatric patients eventually develop secondary neoplasms diagnostic for Carney's triad, obscuring the distinction from the more common form of pediatric GIST. However, the gene expression profiles of pediatric tumors, including tumors from patients with Carney's triad, is distinct from the WT adult GIST and includes overexpression of BAALC, IGF1R, FGF4, PLAG1, and NELL1. A subset of GISTs occurring in young adults shares clinicopathologic features as well as a similar gene expression profile with the pediatric counterpart. The in vitro data presented in this study are compelling that second-generation kinase inhibitors are superior to imatinib therapy against WT KIT–transfected cells or short-term culture of WT GIST cells. This work sets the stage for future clinical trial design for WT GIST patients. It remains to be determined if these newer-generation, broad-based inhibitors will prove efficacious in pediatric GIST patients as well.
Acknowledgments
We thank Agnes Viale and Genomic Core Laboratory for assistance with microarray technology; Margaret Leversha, Lei Zhang, and the Molecular Cytogenetic Core for assistance with fluorescence in situ hybridization; Allyne Manzo for photographic assistance; and Diann Desantis for obtaining follow-up information.
Grant support: ACS MRSG CCE-106841 (C.R. Antonescu), P01CA47179 (C.R. Antonescu and R.G. Maki), Life Raft Group (C.R. Antonescu), GIST Cancer Research Fund (C.R. Antonescu), Shuman Family Fund for GIST Research (C.R. Antonescu and R.G. Maki), CA102613 (R.P. DeMatteo), CA102774 (P. Besmer), and HL/DK55748 (P. Besmer).
Footnotes
Disclosure of Potential Conflicts of Interest
R. DeMatteo is a consultant to and on the advisory board of Novartis. He is also a speaker for Novartis and has received honoraria from Novartis.
References
- 1.Miettinen M, Lasota J, Sobin LH. Gastrointestinal stromal tumors of the stomach in children and young adults: a clinicopathologic, immunohistochemical, and molecular genetic study of 44 cases with long-term follow-up and review of the literature. Am J Surg Pathol. 2005;29:1373–81. doi: 10.1097/01.pas.0000172190.79552.8b. [DOI] [PubMed] [Google Scholar]
- 2.Prakash S, Sarran L, Socci N, et al. Gastrointestinal stromal tumors in children and young adults: a clinicopathologic, molecular, and genomic study of 15 cases and review of the literature. J Pediatr Hematol Oncol. 2005;27:179–87. doi: 10.1097/01.mph.0000157790.81329.47. [DOI] [PubMed] [Google Scholar]
- 3.Antonescu CR, Sommer G, Sarran L, et al. Association of KITexon 9 mutations with nongastric primary site and aggressive behavior: KIT mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin Cancer Res. 2003;9:3329–37. [PubMed] [Google Scholar]
- 4.Agaram NP, Besmer P, Wong GC, et al. Pathologic and molecular heterogeneity in imatinib-stable or imatinib-responsive gastrointestinal stromal tumors. Clin Cancer Res. 2007;13:170–81. doi: 10.1158/1078-0432.CCR-06-1508. [DOI] [PubMed] [Google Scholar]
- 5.Antonescu CR, Viale A, Sarran L, et al. Gene expression in gastrointestinal stromal tumors is distinguished by KIT genotype and anatomic site. Clin Cancer Res. 2004;10:3282–90. doi: 10.1158/1078-0432.CCR-03-0715. [DOI] [PubMed] [Google Scholar]
- 6.Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–91. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 7.Girnita L, Girnita A, Brodin B, et al. Increased expression of insulin-like growth factor I receptor in malignant cells expressing aberrant p53: functional impact. Cancer Res. 2000;60:5278–83. [PubMed] [Google Scholar]
- 8.Idelman G, Glaser T, Roberts CT, Jr., Werner H. WT1–53 interactions in insulin-like growth factor-I receptor gene regulation. J Biol Chem. 2003;278:3474–82. doi: 10.1074/jbc.M211606200. [DOI] [PubMed] [Google Scholar]
- 9.Larsson O, Girnita A, Girnita L. Role of insulin-like growth factor 1 receptor signalling in cancer. Br J Cancer. 2005;92:2097–101. doi: 10.1038/sj.bjc.6602627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Werner H, Karnieli E, Rauscher FJ, LeRoith D. Wild-type and mutant p53 differentially regulate transcription of the insulin-like growth factor I receptor gene. Proc Natl Acad Sci U S A. 1996;93:8318–23. doi: 10.1073/pnas.93.16.8318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Irmer D, Funk JO, Blaukat A. EGFR kinase domain mutations—functional impact and relevance for lung cancer therapy. Oncogene. 2007;26:5693–701. doi: 10.1038/sj.onc.1210383. [DOI] [PubMed] [Google Scholar]
- 12.Willmore-Payne C, Holden JA, Tripp S, Layfield LJ. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol. 2005;36:486–93. doi: 10.1016/j.humpath.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 13.Price VE, Zielenska M, Chilton-MacNeill S, Smith CR, Pappo AS. Clinical and molecular characteristics of pediatric gastrointestinal stromal tumors (GISTs) Pediatr Blood Cancer. 2005;45:20–4. doi: 10.1002/pbc.20377. [DOI] [PubMed] [Google Scholar]
- 14.Miselli FC, Casieri P, Negri T, et al. c-Kit/PDGFRA gene status alterations possibly related to primary imatinib resistance in gastrointestinal stromal tumors. Clin Cancer Res. 2007;13:2369–77. doi: 10.1158/1078-0432.CCR-06-1745. [DOI] [PubMed] [Google Scholar]
- 15.Carney JA, Sheps SG, Go VL, Gordon H. The triad of gastric leiomyosarcoma, functioning extra-adrenal paraganglioma and pulmonary chondroma. N Engl J Med. 1977;296:1517–8. doi: 10.1056/NEJM197706302962609. [DOI] [PubMed] [Google Scholar]
- 16.Carney JA. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc. 1999;74:543–52. doi: 10.4065/74.6.543. [DOI] [PubMed] [Google Scholar]
- 17.Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet. 2002;108:132–9. doi: 10.1002/ajmg.10235. [DOI] [PubMed] [Google Scholar]
- 18.Pasini B, McWhinney SR, Bei T, et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet. 2007;16:79–88. doi: 10.1038/sj.ejhg.5201904. [DOI] [PubMed] [Google Scholar]
- 19.Matyakhina L, Bei TA, McWhinney SR, et al. Genetics of Carney triad: recurrent losses at chromosome 1 but lack of germline mutations in genes associated with paragangliomas and gastrointestinal stromal tumors. J Clin Endocrinol Metab. 2007;92:2938–43. doi: 10.1210/jc.2007-0797. [DOI] [PubMed] [Google Scholar]
- 20.Baldus CD, Tanner SM, Kusewitt DF, et al. BAALC, a novel marker of human hematopoietic progenitor cells. Exp Hematol. 2003;31:1051–6. [PubMed] [Google Scholar]
- 21.Baldus CD, Tanner SM, Ruppert AS, et al. BAALC expression predicts clinical outcome of de novo acute myeloid leukemia patients with normal cytogenetics: a Cancer and Leukemia Group B Study. Blood. 2003;102:1613–8. doi: 10.1182/blood-2003-02-0359. [DOI] [PubMed] [Google Scholar]
- 22.Baldus CD, Thiede C, Soucek S, Bloomfield CD, Thiel E, Ehninger G. BAALC expression and FLT3 internal tandem duplication mutations in acute myeloid leukemia patients with normal cytogenetics: prognostic implications. J Clin Oncol. 2006;24:790–7. doi: 10.1200/JCO.2005.01.6253. [DOI] [PubMed] [Google Scholar]
- 23.Tallman MS. New strategies for the treatment of acute myeloid leukemia including antibodies and other novel agents. Hematology Am Soc Hematol Educ Program. 2005;1:143–50. doi: 10.1182/asheducation-2005.1.143. [DOI] [PubMed] [Google Scholar]
- 24.Asp J, Persson F, Kost-Alimova M, Stenman G. CHCHD7-1 and TCEA1-1 gene fusions resulting from cryptic, intrachromosomal 8q rearrangements in pleomorphic salivary gland adenomas. Genes Chromosomes Cancer. 2006;45:820–8. doi: 10.1002/gcc.20346. [DOI] [PubMed] [Google Scholar]
- 25.Gisselsson D, Hibbard MK, Dal Cin P, et al. PLAG1 alterations in lipoblastoma: involvement in varied mesenchymal cell types and evidence for alternative oncogenic mechanisms. Am J Pathol. 2001;159:955–62. doi: 10.1016/S0002-9440(10)61771-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stenman G. Fusion oncogenes and tumor type specificity—insights from salivary gland tumors. Semin Cancer Biol. 2005;15:224–35. doi: 10.1016/j.semcancer.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 27.Debiec-Rychter M, Van Valckenborgh I, Van den Broeck C, et al. Histologic localization of PLAG1 (pleomorphic adenoma gene 1) in pleomorphic adenoma of the salivary gland: cytogenetic evidence of common origin of phenotypically diverse cells. Lab Invest. 2001;81:1289–97. doi: 10.1038/labinvest.3780342. [DOI] [PubMed] [Google Scholar]
- 28.Voz ML, Mathys J, Hensen K, et al. Microarray screening for target genes of the proto-oncogene PLAG1. Oncogene. 2004;23:179–91. doi: 10.1038/sj.onc.1207013. [DOI] [PubMed] [Google Scholar]
- 29.Debiec-Rychter M, Sciot R, Le Cesne A, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42:1093–103. doi: 10.1016/j.ejca.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 30.Debiec-Rychter M, Dumez H, Judson I, et al. Use of c-KIT/PDGFRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2004;40:689–95. doi: 10.1016/j.ejca.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 31.Prenen H, Cools J, Mentens N, et al. Efficacy of the kinase inhibitor SU11248 against gastrointestinal stromal tumor mutants refractory to imatinib mesylate. Clin Cancer Res. 2006;12:2622–7. doi: 10.1158/1078-0432.CCR-05-2275. [DOI] [PubMed] [Google Scholar]
- 32.Bond M, Bernstein ML, Pappo A, et al. A phase II study of imatinib mesylate in children with refractory or relapsed solid tumors: A Children's Oncology Group study. Pediatr Blood Cancer. 2007;50:254–8. doi: 10.1002/pbc.21132. [DOI] [PubMed] [Google Scholar]
- 33.Champagne MA, Capdeville R, Krailo M, et al. Imatinib mesylate (STI571) for treatment of children with Philadelphia chromosome-positive leukemia: results from a Children's Oncology Group phase 1 study. Blood. 2004;104:2655–60. doi: 10.1182/blood-2003-09-3032. [DOI] [PubMed] [Google Scholar]
- 34.Casteran N, De Sepulveda P, Beslu N, et al. Signal transduction by several KIT juxtamembrane domain mutations. Oncogene. 2003;22:4710–22. doi: 10.1038/sj.onc.1206587. [DOI] [PubMed] [Google Scholar]