

Diterpenoids are an important and structurally diverse class of natural products. In 1987, Hashimoto and co-workers reported a new diterpenoid with a unique bridged bicyclic core densely decorated with eight stereocenters.1 The authors named this new natural product vinigrol. As evident from the three structural perspectives shown in Figure 1, vinigrol's architecture is quite striking. Most notable is the bis-axial four carbon tether that bridges the two six-membered rings of the decalin in such a way that a very rigid and compact framework results. Vinigrol has been evaluated in numerous biological assays and shown to impact platelet aggregation2 and act as a tumor necrosis factor (TNF) antagonist,3 as well as displaying other interesting properties. 4 Vinigrol's unprecedented structure and intriguing biological profile have prompted numerous attempts at its synthesis.5 In 2009, Baran6 and co-workers reported the first total synthesis of vinigrol and a few years later Barriault7 published a formal synthesis of vinigrol.
FIgure 1.
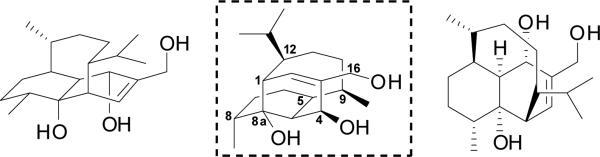
Vinigrol Structural Perspectives.
Although details of our strategy have evolved over time,8 we have remained faithful to several design features. The following reactions have been and still are critical to our vinigrol synthesis plan: 1) one-pot oxidative dearomatization/Diels-Alder reaction, 2) tandem 6-exo-trig cyclizations to form the carbocyclic core, 3) fragmentation to unravel the bridged bicyclic architecture and 4) use of the rigid carbocyclic core to install stereocenters in a substrate-controlled manner. Our retrosynthetic analysis for vinigrol is outlined in Figure 2. Initial plans called for a late-stage carbanion mediated fragmentation of the rigid cage-like structure 2, wherein the functional groups for this key reaction would originate from ketal 3. The cage architecture (4) would direct installation of the C8 and C9-methyl stereocenters, and would be assembled in two steps by dearomatization of pyrogallol ether 5. The components for the oxidative dearomatization precursor (5) would be synthesized from known compounds 79 and 8.10
Figure 2.

Vinigrol Retrosynthesis.
Synthesis of the oxidative dearomatization precursor is presented in Scheme 1. Following iodination of 8, the terminal olefin was selectively dihydroxylated (9) and oxidatively cleaved to afford aldehyde 10. Horner-Wadsworth-Emmons olefination with phosphonate 1111 yielded enoate 12, which was then reduced to the corresponding allylic alcohol. Attachement of the aromatic moiety (7) was accomplished using a Mitsunobu reaction. To set the stage for the key oxidative dearomatization reaction, the carboxylate needed to be converted to a free phenol and the adjacent hydroxyl group protected in such a way that it electronically deactivated to guide the oxidative dearomatization reaction towards the more hindered site and suppress unwanted opening of the intermediate acetal. These goals were accomplished by first treating 14 with DibalH and then protecting the free phenol as β,β,β,-trifluoroethyl ether (15). Dakin oxidation then converted 15 to phenol 16.12
Scheme 1.

Synthesis of Oxidative Dearomatization Precursor.
Following optimization of the oxidative dearomatization reaction, we were rewarded with high yields of intramolecular Diels-Alder cycloadduct 17 (Scheme 2). A Heck cyclization cascade afforded the carbocyclic core of vinigrol (18) in only two steps from 16. During the Heck cyclization 11% of the product isomerized to a trisubstituted olefin isomer of 18. This was inconsequential for the next step as both isomers were expected to afford 19 in the subsequent step. We next turned our attention to the installation of the congested C9 and C8-stereocenters. Hydrogenation of the carbocyclic cage predictably afforded the C9-methyl stereocenter (19). The C8-methyl stereocenter proved far more challenging to install. All attempts at converting the C8-ketone of 19 to a methylene group (22) using either direct olefination or addition/dehydration strategies failed. This problem was finally solved by first converting 19 to 20, derivatizing the resulting alcohol13 as a xanthate (21), which was then subjected to Chugaev elimination (22). Unfortunately, when the exo-olefin of 22 was subjected to classical heterogeneous hydrogenation conditions only the undesired C8-epimer was obtained. Remarkably, this problem could be solved by using iridium catalyzed directed hydrogenation (23).14 Presumably, the iridium catalyst first coordinates to the furan oxygen prior to hydrogen transfer.
Scheme 2.

Synthesis of the Carbocyclic Core of Vinigrol.
We next turned our attention to opening up the tetracyclic cage via fragmentation (Scheme 3). After a few unsuccessful attempts at carbanion fragmentation, we chose to pursue a Wharton fragmentation (27 to 28) strategy. Towards that end, acetal 23 was opened and the resulting alcohol oxidized to an aldehyde (24), which we found could be oxidized selectively using a Baeyer-Villiger reaction and then exhaustively reduced (25). Although diol 25 is potentially competent for fragmentation, the C7-epimer would be better aligned. Inversion 15 and derivatization of the secondary alcohol as a mesylate (27) was straight forward. We were delighted to find that the desired Wharton fragmentation16 reaction proceeded in high yield to produce the vinigrol core (28).
Scheme 3.

Fragmentation – Synthesis of the Vinigrol Core.
To complete the total synthesis of vinigrol we needed to address three final challenges: 1) convert the C12 ketone to an isopropyl group, 2) add the C4-hydroxyl group and 3) deprotect the C8atrifluoroethyl ether. We started our endgame journey by first tackling the C12-isopropyl group installation (Scheme 4).
Scheme 4.

Endgame – Total Synthesis of Vinigrol
Following hydrogenation of 28, we added a vinyl cerium reagent17 to ketone 29. The complete selectivity in this addition step is the results of steric control. Deoxygenation or dehydration of the tertiary hydroxyl (30) proved tricky as unwanted Grob-fragmentation of the trifluoromethyl ether proved quite facile. This problem was finally alleviated by employing Burgess’ reagent for the dehydration (31).18 Conjugate additions and reductions of the enone afforded primarily the undesired C12-epimer of the methyl ketone. By performing a hydrogenation in the presence of potassium hydroxide the desired thermodynamically more stable C12-epimer (32) could be accessed selectively. 19 Following a Wittig olefination (33) and reduction the installation of the C12-isopropyl group was completed (34). We next turned out attention to the C4-hydroxyl group. Deprotection of the methyl ether was accomplished using selenium dioxide (35),20 which set the stage for substrate controlled installation of the C4-hydoxyl group via a directed epoxidation (36). Conversion of the primary alcohol to an iodide followed by a reductive zinc mediated21 opening of the epoxide yielded allylic alcohol 37. Isomerization of the allylic alcohol was challenging, but could be done forcefully with selenium dioxide and the aid of oxidative workup conditions. With trifluoroethyl ether protected vinigrol (38) in hand, the final challenge we were confronted with was deprotection of the C8a-tertiary alcohol. With no reported examples of removing this stable “protecting” group being used in synthesis, we needed to develop conditions that were compatible with the rest of the vinigrol architecture. Finally, we found that we could convert 38 to 39 by treatment with LDA 22 and then selectively dihydroxylate 23 the difluorovinyl ether to afford vinigrol (1).24
In summary, a total synthesis of vinigrol has been accomplished. Our synthesis highlights the rapid complexity-generating power of a strategic oxidative dearomatization/Diels-Alder reaction, which coupled with a Heck cyclization cascade, affords the carbocyclic core of vinigrol in only two steps from a simple precursor. Efforts are underway to render the oxidative transformation asymmetric, which would provide access to vinigrol enantiomers. The synthesis features a number of notable transformations such as: 1) a directed hydrogenation in a very complex and hindered setting, 2) selenium dioxide-mediated deprotection and olefin isomerization, 3) Wharton fragmentation and 4) unique strategic applications and deprotection of a trifluoroethyl ether. We are currently using the lessons learned from this synthetic journey to stream-line the synthesis.
Supplementary Material
Footnotes
We thank the NIH-NIGMS (RO1 GM086584) for generous financial support of this program.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author
Contributor Information
Qingliang Yang, Department of Chemistry and Chemical Biology Cornell University, Baker Laboratory Ithaca, NY 14853-1301 (USA).
Jon T. Njardarson, Department of Chemistry and Biochemistry University of Arizona 1306 E. University Blvd., Tucson AZ 85721 (USA).
Cristian Draghici, Department of Chemistry and Chemical Biology Cornell University, Baker Laboratory Ithaca, NY 14853-1301 (USA).
Fang Li, Department of Chemistry and Biochemistry University of Arizona 1306 E. University Blvd., Tucson AZ 85721 (USA).
References
- 1.Uchida I, Ando T, Fukami N, Yoshida K, Hashimoto M, Tada T, Koda S, Morimoto Y. J. Org. Chem. 1987;52:5292–5293. [Google Scholar]
- 2.a Ando T, Tsurumi Y, Ohata N, Uchida I, Yoshida K, Okahura M. J. Antibiot. 1988;41:25–30. doi: 10.7164/antibiotics.41.25. [DOI] [PubMed] [Google Scholar]; b Ando T, Yoshida K, Okahura M. J. Antibiot. 1998;41:31–35. [Google Scholar]
- 3.Norris DB, Depledge P, Jackson AP. PCT Int. Appl. 1991 WO 9107 953.
- 4.a Nakajima H, Yamamoto N, Kaizu T, Kino T. Jpn. Kokai Tokkyo Koho. 1995 JP 07206668.; b Keane JT. PCT Int. Appl. 2001 WO 20011000229.; c Onodera H, Ichimura M, Sakurada K, Kawabata A, Ota T. PCT Int. Appl. 2006 WO 2006077954.; b Onodera H, Ichimura M, Sakurada K, Kawabata A, Ota T. PCT Int. Appl. WO 2006077954.
- 5.Devaux J-F, Hanna I, Lallemand J-Y. J. Org. Chem. 1993;58:2349–2350.Devaux J-F, Hanna I, Fraisse P, Lallemand J-Y. Tetrahedron Lett. 1995;36:9471–9474.Devaux J-F, Hanna I, Lallemand J-Y, Prange T. J. Chem. Res. Synth. 1996:32–33.Mehta G, Reddy KS. Synlett. 1996:625–627.Kito M, Sakai T, Haruta N, Shirahama H, Matsuda F. Synlett. 1996:1057–1060.Devaux J-F, Hanna I, Lallemand J-Y. J. Org. Chem. 1997;62:5062–5068.Kito M, Sakai T, Shirahama H, Miyashita M, Matsuda F. Synlett. 1997:219–220.Matsuda F, Sakai T, Okada N, Miyashita M. Tetrahedron Lett. 1998;39:863–864.Matsuda F, Kito M, Sakai T, Okada N, Miyashita M, Shirahama H. Tetrahedron. 1999;55:14369–14380.Gentric L, Hanna I, Ricard L. Org. Lett. 2003;5:1139–1142. doi: 10.1021/ol034217k.Gentric L, Hanna I, Huboux A, Zaghdoudi R. Org. Lett. 2003;5:3631–3634. doi: 10.1021/ol035283p.Paquette LA, Guevel R, Sakamoto S, Kim IH, Crawford J. J. Org. Chem. 2003;68:6096–6107. doi: 10.1021/jo0301301.Morency L, Barriault L. Tetrahedron Lett. 2004;45:6105–6107.Paquette LA, Efremov I, Liu Z. J. Org. Chem. 2005;70:505–509. doi: 10.1021/jo048458x.Paquette LA, Efremov I. J. Org. Chem. 2005;70:510–513. doi: 10.1021/jo0484575.Paquette LA, Liu Z, Efremov I. J. Org. Chem. 2005;70:514–518. doi: 10.1021/jo048456c.Morency L, Barriault L. J. Org. Chem. 2005;70:8841–8853. doi: 10.1021/jo051318i.Grise CM, Tessier G, Barriault L. Org. Lett. 2007;9:1545–1548. doi: 10.1021/ol0702977.Souweha MS, Enright GD, Fallis AG. Org. Lett. 2007;9:5163–5166. doi: 10.1021/ol702202b.Maimone TJ, Voica A-F, Baran PS. Angew. Chem. Int. Ed. 2008;47:3054–3056. doi: 10.1002/anie.200800167. For reviews on vinigrol approaches, consult: Tessier G, Barriault L. Org. Prep. Proc. Int. 2007;37:313–353.Harmata M, Calkins NL. Chemtracts. 2009;22:205–209.Lu J-Y, Hall DG. Angew. Chem. Int. Ed. 2010;49:2286–2288. doi: 10.1002/anie.200906826.Huters AD, Garg NK. Chem. Eur. J. 2010;16:8586–8595. doi: 10.1002/chem.201000916.
- 6.Maimone TJ, Shi J, Ashida S, Baran PS. J. Am. Chem. Soc. 2009;131:17066–17067. doi: 10.1021/ja908194b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poulin J, Grise-Bard CM, Barriault L. Angew. Chem. Int. Ed. 2012;51:2111–2114. doi: 10.1002/anie.201108779. [DOI] [PubMed] [Google Scholar]
- 8.a Morton JGM, Kwon LD, Freeman JD, Njardarson JT. Synlett. 2009:23–27. [Google Scholar]; b Morton JGM, Kwon LD, Freeman JD, Njardarson JT. Tetrahedron Lett. 2009;50:1684–1686. [Google Scholar]; c Morton JGM, Draghici C, Njardarson JT. Org. Lett. 2009;11:4492–4495. doi: 10.1021/ol901519k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hadfield A, Schweitzer H, Trova MP, Green K. Synth. Commun. 1994;24:1025–1028. [Google Scholar]
- 10.Compound 8 can be accessed in two steps from commercially available 3-(Trimethylsilyl)propargyl alcohol: Labaudiniere L, Hanaizi J, Normant J-F. J. Org. Chem. 1992;57:6903–6908.Duboudin JG, Jousseaume B. J. Organomet. Chem. 1979;168:1–11.
- 11.Petter RC, Banerjee S, Englard S. J. Org. Chem. 1990;55:3088–3097. [Google Scholar]
- 12.Using boric acid as an additive was critical to the success of the Dakin oxidation: Roy A, Reddy KR, Mohanta PK, Ila H, Junjappa H. Synth. Commun. 1999;29:3781–3791.
- 13.The outcome of this Grignard addition was very sensitive to solvents. For example when ether was replaced with THF or MgBr2 was omitted the diastereoselectivity plummeted.
- 14.Wustenberg B, Pfaltz A. Adv. Synth. Catal. 2008;350:174–178. We are grateful to Professor Andreas Pfaltz for sending us samples of his iridium catalysts.
- 15.Evans DA, Chapman KT. Tetrahedron Lett. 1986;27:5939–5942. [Google Scholar]
- 16.Wharton PS. J. Org. Chem. 1961;26:4781–4782. [Google Scholar]
- 17.Imamoto M, Sugiura Y, Takiyama N. Tetrahedron Lett. 1984;25:4233–4236. [Google Scholar]
- 18.Atkins GM, Burgess EM. J. Am. Chem. Soc. 1968;90:4744–4745. [Google Scholar]
- 19.Wilds AL, Johnson JA, Sutton RE. J. Am. Chem. Soc. 1950;72:5524–5529. [Google Scholar]
- 20.a Bulman PC, McCarthy TJ. Comp. Org. Synth. 1991;7:83. [Google Scholar]; b Hanessian S, Szychowski J, Maianti JP. Org. Lett. 2009;11:429–432. doi: 10.1021/ol802421d. [DOI] [PubMed] [Google Scholar]
- 21.Sarandeses LA, Mourino A, Luche J-L. J. Chem. Soc. Chem. Commun. 1991:818–820. [Google Scholar]
- 22.a Nakai T, Tanaka K, Ishikawa N. Chem. Lett. 1976:1263–1266. [Google Scholar]; b Tanaka K, Shiraishi S, Nakai T, Ishikawa N. Tetrahedron Lett. 1978;19:3103–3106. [Google Scholar]
- 23.Herrmann WA, Eder SJ, Scherer W. Angew. Chem. Int. Ed. 1992;31:1345–1347. [Google Scholar]
- 24.Our spectral and physical data excellently matched that of a sample of vinigrol graciously shared with us by Professor Phil S. Baran (See supplementary section for more details).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.