

This article discusses the current approaches available for the diagnosis, evaluation, and management of patients and their family members with suspected medullary thyroid cancer. A review of recent guidelines on the extent and timing of surgical excision is discussed. There are not very many effective systemic treatment options for MTC, but several emerging therapeutic targets have promise.
Keywords: Medullary Thyroid Cancer (MTC), Multiple Endocrine Neoplasia Syndrome, Familial Medullary Thyroid Cancer (FMTC), RET proto-oncogene, Serum calcitonin, Prophylactic surgery in MTC
Learning Objectives
Identify and evaluate a patient with a diagnosis of medullary thyroid cancer.
Utilize genetic testing for the RET proto-oncogene and explain how the location of the mutation affects the risks for the patient.
Select among the surgical treatment options for patients with medullary thyroid cancer, including prophylactic surgery in genetic carriers.
Abstract
Medullary thyroid cancer (MTC) typically accounts for 3%–4% of all thyroid cancers. Although the majority of MTCs are sporadic, 20% of cases are hereditary. Hereditary MTC can be found in multiple endocrine neoplasia 2A or 2B or as part of familial MTC based on a specific germline mutation in the RET proto-oncogene. This article discusses the current approaches available for the diagnosis, evaluation, and management of patients and their family members with suspected MTC. The disease is predominantly managed surgically and typically requires a total thyroidectomy and lymph node dissection. A review of recent guidelines on the extent and timing of surgical excision is discussed. There are not very many effective systemic treatment options for MTC, but several emerging therapeutic targets have promise.
Implications for Practice:
Medullary thyroid cancer (MTC) typically accounts for approximately 4% of all thyroid cancers. Of these, 20% of cases are hereditary and can be found in multiple endocrine neoplasia syndrome or as part of familial MTC based on a specific germline mutation in the RET proto-oncogene. This article summarizes the current approaches and guidelines available for the diagnosis, evaluation, and management of patients and their family members with suspected MTC. Surgery is the standard of care, and prophylactic surgeries are performed in genetic carriers. Tyrosine kinase receptor inhibitors vandetanib (ZD6474) and cabozantinib (XL184) were recently approved by the U.S. Food and Drug Administration as promising systemic therapy for advanced MTC.
Introduction
Medullary thyroid cancer (MTC) accounts for 3%–4% of all thyroid cancers [1, 2]. The clinical course of MTC can be indolent, remaining unchanged for years, or it can be aggressive, associated with high mortality. Although the majority of MTC cases are sporadic, approximately 20% are hereditary because of a germline mutation in the rearranged during transfection proto-oncogene (RET) [3–7]. Hereditary MTC can present in isolation (familial medullary thyroid cancer [FMTC]) or as part of the multiple endocrine neoplasia syndrome type 2 (MEN2; MEN2A or MEN2B).
MTC arises from the neural crest, specifically the parafollicular C cells of the thyroid gland. Although the C cells are located throughout the thyroid gland, they are predominant at the junction of the upper third and lower two-thirds, which is where the majority of MTCs are found. C cells secrete a variety of peptides and hormones, and MTC is characterized by the secretion of calcitonin, which is used as a diagnostic and prognostic marker in MTC.
Diagnosis
Clinical Features
The typical age of presentation of sporadic MTC is in the fifth or sixth decade, with a slight female preponderance [8, 9]. In contrast, MEN2A and FMTC typically present in the third decade of life, and MEN2B usually presents in those younger than age 20.
The most common presentation in sporadic MTC is a palpable neck mass, either a solitary thyroid nodule or an enlarged lymph node (35%–50%) [8]. These tumors are generally unifocal and tend to arise in the posterior thyroid, often invading the surrounding structures and causing dysphagia, hoarseness, and/or respiratory difficulty [8]. High levels of calcitonin secreted from these tumors can also lead to symptoms such as flushing, diarrhea, and/or weight loss in patients with MTC. In addition, 10%–15% of patients can present with distant metastases at the time of diagnosis. The most common locations for metastatic MTC are the mediastinum, liver, lungs, and bone.
Patients with MTC in MEN2 commonly present with multifocal and bilateral disease. They can present with diarrhea, flushing, or weight loss caused by excessive secretion of calcitonin by the tumor or can be diagnosed after presenting with an associated disease such as pheochromocytoma or hyperparathyroidism. In uncommon cases, MTC can also present as Cushing syndrome because of ectopic corticotropin production by the tumor [10, 11]. Currently, most patients with either FMTC or MEN2 are identified by genetic testing of at-risk family members. Patients with a positive family history of germline mutation of the RET gene have a 50% chance of inheriting the same mutation. Once identified as genetic carriers, there is a nearly 100% lifetime risk of developing malignancy.
Currently, most patients with either FMTC or MEN2 are identified by genetic testing of at-risk family members. Patients with a positive family history of germline mutation of the RET gene have a 50% chance of inheriting the same mutation. Once identified as genetic carriers, there is a nearly 100% lifetime risk of developing malignancy.
Laboratory Evaluation
The diagnosis of MTC is most commonly obtained from fine-needle aspiration (FNA) of a new thyroid nodule. The accuracy of FNA is 50%–80% [12, 13] and is characterized by the presence of stromal amyloid and the absence of thyroid follicles (Fig. 1); however, FNA cannot always distinguish MTC based on the appearance of the cells alone. Higher accuracy can be obtained by the addition of immunohistochemical staining for calcitonin, chromogranin A, or carcinoembryonic antigen (CEA) when there is clinical suspicion of MTC [14]. More recently, measurement of calcitonin in the washout fluid from FNA has been described as a very sensitive method of detection [15, 16]. If FNA suggests MTC or if there is clinical suspicion, a preoperative calcitonin level can be very useful for both diagnosis and staging [17, 18]. In cases of nodal metastases, basal calcitonin levels can be in the range of 10–40 pg/mL (normal range, <10 pg/mL), whereas distant metastases are typically associated with a calcitonin level >150 pg/mL and often >1,000 pg/mL. The recent guideline by the American Thyroid Association (ATA) does not recommend for or against calcitonin screening, but if testing is done, a serum calcitonin value >100 pg/mL should be considered suspicious for medullary cancer [19, 20]. Unfortunately, a diagnosis of MTC cannot always be excluded by a normal preoperative calcitonin level.
Figure 1.

Gross view and histology of medullary thyroid cancer. (A): Operative specimen of left thyroid and associated lymph nodes. (B, C): Histology of medullary thyroid cancer shown by hematoxylin and eosin (B) and Congo red stain (C) emphasizing the characteristic stromal amyloid. From [41] with permission from AlphaMed Press.
CEA is another useful tumor marker found in >50% of patients with MTC, especially when preoperative serum calcitonin values are negative. A preoperative serum CEA level >30 ng/mL is suggestive of a poor prognosis and indicates that surgical intervention will likely not cure the patient [21]. A CEA level >100 ng/mL suggests extensive lymph node involvement and distant metastasis. An increasing CEA level in the presence of a stable calcitonin level can be a sign of dedifferentiation of the tumor and is associated with a worse prognosis. Conversely, a rapid decrease in serum CEA predicts cure by surgery [14, 22]. Chromogranin A is another useful diagnostic and prognostic marker in MTC [14].
Radiological Evaluation
Imaging studies are critical in the detection and management of patients with MTC, especially in sporadic cases. Most patients with sporadic MTC will present with a thyroid mass or cervical lymphadenopathy that can be evaluated by a neck ultrasound (U/S) [14]. U/S can be used to characterize the mass, to identify additional thyroid lesions or suspicious lymph nodes, and to perform FNA to confirm the diagnosis of MTC. Cross-sectional imaging (computed tomography [CT] or magnetic resonance imaging [MRI]) of the chest and the neck is recommended because lymph node involvement may be deep in the neck or in the mediastinum and not captured by U/S alone. If the preoperative serum basal calcitonin is >400 pg/mL or there are local lymph node metastases, these patients should undergo three-phase contrast-enhanced liver CT or contrast-enhanced liver MRI [20]. In patients with suspicion of skeletal metastases, MRI is superior to other imaging modalities [23]. 18F-Fludeoxyglucose positron emission tomography or somatostatin receptor imaging as initial screening imaging studies for metastatic disease are not recommended because of variable sensitivity [24, 25], although the sensitivity improves when calcitonin levels are >1,000 pg/mL [26]. In addition, small liver metastases can be visualized by laparoscopy [27].
Pathological Features
Grossly, MTC is white or gray in color and very firm to palpation. Histologically, MTC is arranged as nests of uniform cells that are characterized by the deposition of stromal amyloid (Fig. 1). “C-cell hyperplasia” is defined as >6 C cells per follicle or >50 C cells per low-power field and is often found in patients with hereditary disease. Even though C-cell hyperplasia is considered a precursor of malignant transformation in hereditary disease, its implication in nonhereditary disease is less well defined.
Familial Disease
The majority of the MTCs are sporadic; however, approximately 20% present as hereditary MEN2. MEN2 is further subclassified into three distinct syndromes, MEN2A, MEN2B, and FMTC, which are inherited in an autosomal dominant fashion with a high but variable penetrance.
MEN2A
MEN2A is the most common MEN2 disorder and includes MTC, pheochromocytoma, and primary hyperparathyroidism. A few variants of MEN2A are also linked with other disorders. Cutaneous lichen amyloidosis (CLA) is linked to mutations on codon 634, and a mutation on codon 620 carries a significant risk for developing Hirschsprung disease [28–32].
MTC is the predominant disorder (>95%) in MEN2A and accounts for 75% of hereditary MTC. It is characteristically multifocal and bilateral. The age at presentation can vary with the specific genetic mutation but typically manifests in early adulthood. Mortality associated with MEN2A is primarily from MTC; therefore, prompt recognition and intervention are necessary. MTC can be cured or even prevented by early thyroidectomy, with screening of at-risk family members.
Pheochromocytomas occur in up to 50% of cases and are generally multifocal but can also present with adrenal medullary hyperplasia. Screening can be performed using either plasma metanephrines or a 24-hour urine specimen for catecholamines and metanephrines. Once detected, pheochromocytomas should be appropriately managed by treatment with selective α-blockers for blood pressure control, followed by resection with a laparoscopic adrenalectomy. Resection of the adrenal gland should be performed before proceeding with a neck operation for MTC.
Hyperparathyroidism occurs in up to 20%–35% of patients with MEN2A. Screening for hyperparathyroidism is done with serum calcium and parathyroid hormone levels. The disease may manifest as hyperplasia or may present as a single, enlarged parathyroid gland. Treatment is the same as that for an isolated case of primary hyperparathyroidism: Only the malfunctioning parathyroid gland (or glands) should be removed by surgery, with autotransplantation when needed.
MEN2B
MEN2B affects 8%–15% of all MEN2 patients. Although MEN2B has the hereditary predisposition to MTC and pheochromocytoma, hyperparathyroidism is not seen in these patients. Patients with MEN2B have mucosal neuromas of the lips and tongue and intestinal ganglioneuromas. Disorders of the colon such as chronic constipation and megacolon are also common. Moreover, many patients have developmental abnormalities that include a decreased upper-to-lower-body ratio, skeletal deformations such as kyphoscoliosis or lordosis, joint laxity, Marfanoid habitus (without ectopia lentis or aortic abnormalities), everted eyelids, and myelinated corneal nerves [33].
In MEN2B, almost 100% of patients develop MTC at a very young age, even at infancy and has a more aggressive course. Because of the younger age at onset and often a delay in diagnosis, patients with MEN2B are seldom cured of this disease and many die at a young age. Therefore, prophylactic or curative thyroidectomy during the neonatal period is required in patients with MEN2B following identification by genetic screening.
FMTC
FMTC closely mimics MEN2A with a strong predisposition to MTC in various family members but lacks other clinical manifestations of MEN2A (or MEN2B) such as pheochromocytoma or hyperparathyroidism. It may be difficult to distinguish FMTC from MEN2A, and identical genetic mutations can lead to either disease [4]. Because MTC is often the first manifestation of MEN2A and diagnosis of pheochromocytoma or hyperparathyroidism is often made later, the definition of FMTC is strict. In FMTC, there must not be any diagnosis of either pheochromocytoma or hyperparathyroidism in >10 carriers, and presentation must be after the age of 50 in multiple family members affected by the disease. Once a family is labeled as FMTC, there is a higher chance of missing a pheochromocytoma because these patients are not screened for the disease. If there is any uncertainty, it is much safer to label the family as MEN2 instead of FMTC.
Genetic Testing
MTC in MEN2 is inherited in an autosomal dominant pattern with very high penetrance. The genetic defect in these disorders involves the RET proto-oncogene on chromosome 10q11.2 [34]. Currently known RET mutations account for >95% of cases of hereditary MTC. Consequently, the 2009 ATA guidelines for MTC recommend that all patients with C-cell hyperplasia or MTC be offered germline RET testing. It is important to provide appropriate genetic counseling to patients prior to screening for RET mutations. The risks and benefits of genetic testing must be discussed with patients and their families. In addition, once a positive RET mutation is detected, the patient must be carefully advised regarding the risks for other members of the family. Other clinical presentations such as Hirschsprung disease and CLA should also prompt genetic testing for RET mutations [20]. Ideally, in the event that a RET mutation is identified, at-risk family members should be offered a prophylactic thyroidectomy prior to the development of MTC. On occasion, a germline RET mutation may not be detected in a family with a clinical diagnosis of MEN2. In these at-risk relatives, periodic screening for MTC should be performed with neck U/S and serum calcitonin levels, screening for pheochromocytoma should be done by measurement of plasma or 24-hour urine metanephrines and normetanephrines, and hyperparathyroidism should be screened with albumin-corrected calcium or ionized calcium and parathyroid hormone levels [20].
The germline RET mutations in MTC in MEN2 and FMTC lead to a gain of function, and only a single-point mutation is adequate for malignant transformation. These mutations lead to the activation of major intracellular pro-oncogenic pathways (e.g., RAS/MAPK, JUN kinase, PI3K/AKT, and nuclear factor-κB). Inactivating mutations of RET are also possible and have been associated with Hirschsprung disease.
RET Mutations in the Extracellular Domain
There are dissimilarities and similarities in the specific RET gene mutations underlying MEN2A and FMTC [4]. In contrast, MEN2B is associated with specific RET mutations [3]. The mutations in MEN2A and FMTC affect one of the cysteine residues in the cysteine-rich region of the RET extracellular domain encoded by exon 10 (codons 609, 611, 618, and 620) or exon 11 (codons 630 or 634). Mutation in MEN2A is frequently found in codon 634 (up to 80%), whereas the most common mutations in FMTC are detected in codons 620, 630, and 634. These extracellular MEN2A/FMTC cysteine mutations are constitutively active and act in a ligand-independent manner leading to dimerization and activation of RET-mediated intracellular signaling pathways.
RET Mutations in the Intracellular Domain
Germline mutations in the intracellular tyrosine kinase (TK) domain of the RET protein can be found in FMTC and in MEN2B. Mutations in codons 768, 790, 791 (exon 13), 804 (exon 14), and 891 (exon 15) are detected only in FMTC and account for a minority of cases. The codon most frequently (95%) mutated in MEN2B is 918 (exon 16, p.M918T) of the intracellular TK domain of RET, and it is extremely useful during molecular diagnosis in this disorder. Other mutations found in MEN2B are in codon 883 (exon 15) in addition to dual mutations involving codons 804 and 806 or 804 and 904.
Although the MTC in MEN2 results from germline mutations, sporadic MTC can harbor somatic mutations of the RET proto-oncogene. Typically these mutations are detected in exon 16 (codon 918), but exons 13, 14, or 15 can also be affected. These mutations are found only in the patients' tumor cells and are not genetically transmitted to offspring. Consequently, it is important for physicians to properly identify and distinguish MTC presentations of MEN2 from presentations of true sporadic MTC.
Genotype-Phenotype Correlation in MTC
Genotype-phenotype association in inherited neoplasia syndromes were first identified between RET and MEN2 [3]. The gain-of-function mutations affected several codons of RET, most frequently exons 10 and 11. Mutations of codon 634 (exon 11) are highly associated with the classical phenotype of MEN2A: the triad of MTC, pheochromocytoma, and hyperparathyroidism. The most common mutation on codon 634, p.C634R, has the highest penetrance and is the most aggressive variant associated with a higher probability of metastatic disease at diagnosis of MTC than other codon 634 mutations. Although 25% of FMTC patients carry a mutation in codon 634, p.C634Y is the most common type seen in these patients; p.C634R mutations are not detected in FMTC. In addition, codon 634 mutations are also known to be associated with development of CLA, and mutations of the cysteine residues on codons 609, 618, and 620 in exon 10 of RET in MEN2A or FMTC are coinherited with Hirschsprung disease. Codon 918 mutation (exon 16, p.M918T) is the most specific for MEN2B.
The aggressiveness of the MTC varies depending on specific RET mutations. Currently, the ATA guidelines task force has classified these mutations into four groups based on the level of risk and aggressiveness for MTC (Table 1). These guidelines are useful for predicting phenotype and supporting recommendations for the earliest age when prophylactic thyroidectomy should be performed for MTC and for beginning biochemical screening for pheochromocytoma and hyperparathyroidism in MEN2 [5, 20].
Table 1.
Risk of aggressive medullary thyroid cancer based on RET mutations and American Thyroid Association recommendations
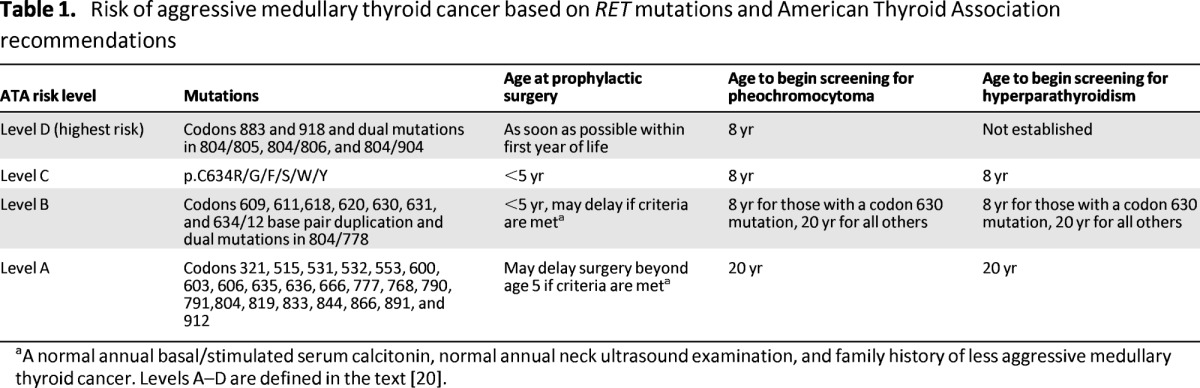
aA normal annual basal/stimulated serum calcitonin, normal annual neck ultrasound examination, and family history of less aggressive medullary thyroid cancer. Levels A–D are defined in the text [20].
Level D mutations (ATA-D, codons 883 and 918 and dual mutations in 804/805, 804/806, and 804/904) carry the highest risk, have the highest penetrance, and have the most aggressive course, with metastatic disease presenting in the first year of life. Consequently, thyroidectomy is recommended as soon as the diagnosis is made or within the first year of life. Level C mutations (ATA-C, codon 634 [p.C634R/G/F/S/W/Y]) are considered to be high risk for MTC, and the ATA recommends that these patients undergo thyroidectomy before the age of 5 years. Level B mutations (ATA-B, codons 609, 611,618, 620, 630, 631, 634/12 base pair duplication, and dual mutations in 804/778) are still considered high risk for MTC but are lower risk for RET mutations. Thyroidectomy is recommended before age 5 but can be delayed based on a normal serum calcitonin, normal annual neck U/S, less aggressive family history of MTC, and family preference. Level A mutations (ATA-A, codons 321, 515, 531, 532, 553, 600, 603, 606, 635, 636, 666, 777, 768, 790, 791,804, 819, 833, 844, 866, 891, and 912) carry the least high risk of MTC. Compared with ATA-B of the same age, ATA-A mutant carriers have lower serum calcitonin, lower tumor stage, and a higher rate of biochemical cure when prophylactic thyroidectomy is performed in those older than age 4 [35]. Because early detection and intervention in MTC leads to significant improvement in patient outcome, there is an ongoing debate regarding the timing of prophylactic thyroidectomy. There is interpatient and interfamily variability as well as unpredictability within families. Many surgeons prefer to treat all patients with MEN2A the same way and to perform prophylactic surgery by age 5 whenever possible. Recent data also suggest that prophylactic surgery in patients who are past the recommended age is still beneficial and improves disease-free survival compared with surgery performed in patients who are at long past the recommended age [36].
Because early detection and intervention in MTC leads to significant improvement in patient outcome, there is an ongoing debate regarding the timing of prophylactic thyroidectomy. There is interpatient and interfamily variability as well as unpredictability within families. Many surgeons prefer to treat all patients with MEN2A the same way and to perform prophylactic surgery by age 5 whenever possible.
Surgical Treatment
Treatment of Clinically Evident Disease
Standard treatment for MTC requires surgical removal of the thyroid with regional lymph node dissection because conventional measures such as chemotherapy and radiation therapy are not effective [20]. Patients with clinically evident disease should be treated with a total thyroidectomy and bilateral central neck dissection at a minimum. Central nodal disease is present in up to 81% of patients with palpable tumors, and addition of a central neck dissection results in a higher cure rate than thyroidectomy alone [37, 38]. The need for routine lateral neck dissection is controversial. Because of the risks associated with a bilateral lateral neck dissection, many surgeons perform it selectively when the primary tumor is large (>1 cm) or when there is evidence of nodal disease on preoperative neck U/S. Even with aggressive surgical removal of all neck lymph nodes, biological cure may not be achieved postoperatively [39].
Removal and autotransplantation of parathyroid glands are usually not performed along with thyroidectomy unless there is evidence of hyperparathyroidism [20]. If patients develop hyperparathyroidism after a total thyroidectomy, then they should undergo preoperative imaging and should repeat surgery as indicated for treatment of their primary disease. Medical management of primary hyperparathyroidism should be considered for patients with a high risk of surgical mortality, limited life expectancy, or persistent or recurring primary hyperparathyroidism after one surgical exploration or more [20].
If a parathyroid gland is devascularized during surgery, then an autotransplant can be performed. In patients with sporadic MTC, FMTC, or MEN2B, the autotransplant can be placed in the sternocleidomastoid; however, in patients with MEN2A, the parathyroid tissue should be autotransplanted into the nondominant forearm because of the risk for developing hyperparathyroidism in the remnant tissue at a later stage. It is important to keep in mind that the autotransplanted parathyroid tissue usually takes about 4–8 weeks to become functional, so calcium and vitamin D should be supplemented during this time. For MEN2A patients, it is important to exclude coexistence of pheochromocytomas before the thyroid surgery [20]. When presented with pheochromocytoma of a single gland, most authorities recommend unilateral adrenalectomy or a cortical-sparing surgery. In cases of bilateral pheochromocytoma or in patients with a single adrenal gland, the recommendation is cortical-sparing adrenal surgery on at least one side, with close monitoring of the residual tissue [20].
Prophylactic Surgery
Prophylactic thyroidectomy is recommended in at-risk patients before the onset of clinically significant disease. It is imperative to determine whether the risk of clinically significant disease outweighs the risks of prophylactic intervention. In hereditary MTC, there is a strong age-related progression from C-cell hyperplasia to MTC and progression to nodal disease. The RET mutations associated with hereditary MTC and the current guidelines for prophylactic thyroidectomy and screening for each mutation are listed in Table 1. If intervention is performed before the development of a primary tumor and nodal involvement, then a total thyroidectomy alone is adequate treatment.
Postoperative Surveillance
In patients with disease restricted to the thyroid gland and without nodal involvement, the risk of recurrence and mortality is very low [40]. When MTC presents with nodal disease, patients are at a very high risk of developing recurrent or persistent disease. Close and adequate follow-up should start 2–3 months postoperatively by obtaining new baseline calcitonin and CEA levels. Patients with undetectable calcitonin levels postoperatively should be followed with annual measurements of serum calcitonin and CEA. A rise in serum biochemical markers should prompt imaging such as CT or MRI. Thyroid hormone replacement is essential after a total thyroidectomy without thyroid-stimulating hormone suppression. In addition, patients with hereditary disease should be screened for the development of pheochromocytoma and hyperparathyroidism annually by routine biochemical markers.
Prognosis
The prognosis for patients with MTC has a 10-year survival rate of 75%–85%, and when disease is localized to the thyroid gland, the 10-year survival rate reaches 95.6% [41]. Patients with regional lymph node disease have a 5-year overall survival rate of 75.5%. Distant metastases are seen in 13% of patients at initial diagnosis, and these patients have a poor prognosis, with a 10-year survival rate of only 40% [41].
Recurrence
Recurrent disease develops in approximately 50% of patients with MTC. Calcitonin levels are very sensitive for detecting either residual or recurrent disease. Patients with near-normal values can be followed, but values >100 pg/mL indicate either residual resectable disease in the neck or the presence of metastases. Patients with basal serum calcitonin values >1,000 pg/mL and no obvious MTC in the neck and upper mediastinum probably have distant metastases, most likely in the liver [27, 41]. Because of the significant risks associated with reoperative neck surgery, elevated postoperative calcitonin levels should be combined with a careful metastatic evaluation by imaging studies prior to considering surgical exploration [27, 41].
Because of the significant risks associated with reoperative neck surgery, elevated postoperative calcitonin levels should be combined with a careful metastatic evaluation by imaging studies prior to considering surgical exploration.
Radiation Therapy
External-beam radiation therapy is considered ineffective in the treatment of patients with MTC, although some reports have described partial remission and stability of the disease as well as improved symptoms, quality of life, and survival [42].
Medical Therapy
Patients with metastatic disease can have significant symptoms such as diarrhea from calcitonin excess and may benefit from initial medical treatment with antimotility agents. These patients may also benefit from somatostatin analogs and local therapies such as resection and ablation as alternative treatments to provide symptomatic relief from tumor burden [20].
Conventional chemotherapy has limited efficacy in patients with MTC. Single-agent regimens with doxorubicin, dacarbazine, capecitabine, and 5-fluorouracil have partial response rates up to 24%–29% [41]. A new class of therapies targeting the RET receptor TK family has been developed because of its role in the pathogenesis of MTC. The first commercially available receptor TK inhibitor, imatinib mesylate (Gleevec; Novartis International, Basel, Switzerland, http://www.novartis.com), showed limited efficacy in patients with MTC [43]. More recently, vandetanib (ZD6474), which simultaneously targets KDR (also known as VEGFR), RET, and the epidermal growth factor receptor, was approved by the U.S. Food and Drug Administration for the treatment of adults with symptomatic or progressive MTC [44–47]. Cabozantinib (XL184) is another oral, small-molecule inhibitor of VEGFR2, hepatocyte growth factor receptor (MET) and RET that was recently approved by the U.S. Food and Drug Administration for the treatment of progressive, metastatic medullary thyroid cancer [48–50]. Other TK inhibitors, such as sorafenib (targets VEGFR2 and VEGFR3, RET, and BRAF), sunitinib (an inhibitor of VEGF1–3, RET, and RET/PTC subtypes 1 and 3), motesanib (targets all three VEGF receptors), and axitinib (inhibits VEGFRs but not RET) demonstrated partial response to stable disease in phase II clinical trials [51–55].
Potential Emerging Therapy
Emerging research indicates that several signal transduction pathways contribute to the growth and hormone production in MTC [41, 56–61]. These include the PI3K/AKT, MAPK, and Notch pathways and the glycogen synthase kinase-3 signaling pathways. Small molecule inhibitors targeting one or more of these pathways are also currently being investigated [62–65]. The goal is to manipulate these various cellular signaling pathways and discover novel therapeutic strategies to improve patient outcome in MTC.
Conclusion
Both the diagnosis and management of MTC can be challenging. Therefore, the practicing surgeon must always consider the possibility of a familial syndrome during the evaluation of patients with thyroid disease. Genetic testing following the established guidelines should be performed whenever there is a diagnosis of MTC. Due to limited adjuvant treatment options, adequate initial surgical management is essential to the successful treatment of MTC. In cases of distant or recurrent disease not amenable to surgery, there are promising new treatment options.
This article is available for continuing medical education credit at CME.TheOncologist.com.
Author Contributions
Conception/Design: Madhuchhanda Roy, Herbert Chen, Rebecca S. Sippel
Collection and/or assembly of data: Madhuchhanda Roy
Manuscript writing: Madhuchhanda Roy, Herbert Chen, Rebecca S. Sippel
Final approval of manuscript: Herbert Chen, Rebecca S. Sippel
Disclosures
Herbert Chen: Digirad (C/A). Rebecca S. Sippel: Novartis [research grant for a clinical trial on MTC, results not published or included] (RF). The other author indicated no financial relationships.
Section editors: Herbert Chen: None; Stan Sidhu: None
Reviewer “A”: None
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Hundahl SA, Cady B, Cunningham MP, et al. Initial results from a prospective cohort study of 5583 cases of thyroid carcinoma treated in the united states during 1996. US and German Thyroid Cancer Study Group An American College of Surgeons Commission on Cancer Patient Care Evaluation study Cancer. 2000;89:202–217. doi: 10.1002/1097-0142(20000701)89:1<202::aid-cncr27>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 2.Thyroid Cancer Treatment (PDQ®) Medullary Thyroid Cancer. [Accessed July 29, 2013]. Available at http://www.cancer.gov/cancertopics/pdq/treatment/thyroid/HealthProfessional/page7.
- 3.Eng C, Clayton D, Schuffenecker I, et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA. 1996;276:1575–1579. [PubMed] [Google Scholar]
- 4.Mulligan LM, Eng C, Healey CS, et al. Specific mutations of the RET proto-oncogene are related to disease phenotype in MEN 2A and FMTC. Nat Genet. 1994;6:70–74. doi: 10.1038/ng0194-70. [DOI] [PubMed] [Google Scholar]
- 5.Moline J, Eng C. Multiple endocrine neoplasia type 2: An overview. Genet Med. 2011;13:755–764. doi: 10.1097/GIM.0b013e318216cc6d. [DOI] [PubMed] [Google Scholar]
- 6.Hofstra RM, Landsvater RM, Ceccherini I, et al. A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature. 1994;367:375–376. doi: 10.1038/367375a0. [DOI] [PubMed] [Google Scholar]
- 7.Hazard JB, Hawk WA, Crile G. Medullary (solid) carcinoma of the thyroid; a clinicopathologic entity. J Clin Endocrinol Metab. 1959;19:152–161. doi: 10.1210/jcem-19-1-152. [DOI] [PubMed] [Google Scholar]
- 8.Saad MF, Ordonez NG, Rashid RK, et al. Medullary carcinoma of the thyroid. A study of the clinical features and prognostic factors in 161 patients. Medicine (Baltimore) 1984;63:319–342. [PubMed] [Google Scholar]
- 9.Dottorini ME, Assi A, Sironi M, et al. Multivariate analysis of patients with medullary thyroid carcinoma. Prognostic significance and impact on treatment of clinical and pathologic variables. Cancer. 1996;77:1556–1565. doi: 10.1002/(SICI)1097-0142(19960415)77:8<1556::AID-CNCR20>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 10.Mure A, Gicquel C, Abdelmoumene N, et al. Cushing's syndrome in medullary thyroid carcinoma. J Endocrinol Invest. 1995;18:180–185. doi: 10.1007/BF03347800. [DOI] [PubMed] [Google Scholar]
- 11.Smallridge RC, Bourne K, Pearson BW, et al. Cushing's syndrome due to medullary thyroid carcinoma: Diagnosis by proopiomelanocortin messenger ribonucleic acid in situ hybridization. J Clin Endocrinol Metab. 2003;88:4565–4568. doi: 10.1210/jc.2002-021796. [DOI] [PubMed] [Google Scholar]
- 12.Chang TC, Wu SL, Hsiao YL. Medullary thyroid carcinoma: Pitfalls in diagnosis by fine needle aspiration cytology and relationship of cytomorphology to RET proto-oncogene mutations. Acta Cytol. 2005;49:477–482. doi: 10.1159/000326191. [DOI] [PubMed] [Google Scholar]
- 13.Papaparaskeva K, Nagel H, Droese M. Cytologic diagnosis of medullary carcinoma of the thyroid gland. Diagn Cytopathol. 2000;22:351–358. doi: 10.1002/(sici)1097-0339(200006)22:6<351::aid-dc5>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 14.Chen H, Sippel RS, O'Dorisio MS, et al. The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: Pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010;39:775–783. doi: 10.1097/MPA.0b013e3181ebb4f0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kudo T, Miyauchi A, Ito Y, et al. Diagnosis of medullary thyroid carcinoma by calcitonin measurement in fine-needle aspiration biopsy specimens. Thyroid. 2007;17:635–638. doi: 10.1089/thy.2006.0338. [DOI] [PubMed] [Google Scholar]
- 16.Boi F, Maurelli I, Pinna G, et al. Calcitonin measurement in wash-out fluid from fine needle aspiration of neck masses in patients with primary and metastatic medullary thyroid carcinoma. J Clin Endocrinol Metab. 2007;92:2115–2118. doi: 10.1210/jc.2007-0326. [DOI] [PubMed] [Google Scholar]
- 17.Cohen R, Campos JM, Salaün C, et al. Preoperative calcitonin levels are predictive of tumor size and postoperative calcitonin normalization in medullary thyroid carcinoma. Groupe d'Etudes des Tumeurs a Calcitonine (GETC) J Clin Endocrinol Metab. 2000;85:919–922. doi: 10.1210/jcem.85.2.6556. [DOI] [PubMed] [Google Scholar]
- 18.Barbet J, Campion L, Kraeber-Bodéré F, et al. Prognostic impact of serum calcitonin and carcinoembryonic antigen doubling-times in patients with medullary thyroid carcinoma. J Clin Endocrinol Metab. 2005;90:6077–6084. doi: 10.1210/jc.2005-0044. [DOI] [PubMed] [Google Scholar]
- 19.Cooper DS, Doherty GM, Haugen BR, et al. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2009;19:1167–1214. doi: 10.1089/thy.2009.0110. [DOI] [PubMed] [Google Scholar]
- 20.Kloos RT, Eng C, Evans DB, et al. Medullary thyroid cancer: Management guidelines of the American Thyroid Association. Thyroid. 2009;19:565–612. doi: 10.1089/thy.2008.0403. [DOI] [PubMed] [Google Scholar]
- 21.Machens A, Dralle H. Pretargeted anti-carcinoembryonic-antigen radioimmunotherapy for medullary thyroid carcinoma. J Clin Oncol. 2006;24:e37. doi: 10.1200/JCO.2006.06.8171. author reply e38. [DOI] [PubMed] [Google Scholar]
- 22.Giovanella L, Crippa S, Cariani L. Serum calcitonin-negative medullary thyroid carcinoma: Role of CgA and CEA as complementary markers. Int J Biol Markers. 2008;23:129–131. doi: 10.1177/172460080802300212. [DOI] [PubMed] [Google Scholar]
- 23.Mirallié E, Vuillez JP, Bardet S, et al. High frequency of bone/bone marrow involvement in advanced medullary thyroid cancer. J Clin Endocrinol Metab. 2005;90:779–788. doi: 10.1210/jc.2004-1500. [DOI] [PubMed] [Google Scholar]
- 24.Oudoux A, Salaun PY, Bournaud C, et al. Sensitivity and prognostic value of positron emission tomography with F-18-fluorodeoxyglucose and sensitivity of immunoscintigraphy in patients with medullary thyroid carcinoma treated with anticarcinoembryonic antigen-targeted radioimmunotherapy. J Clin Endocrinol Metab. 2007;92:4590–4597. doi: 10.1210/jc.2007-0938. [DOI] [PubMed] [Google Scholar]
- 25.Giraudet AL, Vanel D, Leboulleux S, et al. Imaging medullary thyroid carcinoma with persistent elevated calcitonin levels. J Clin Endocrinol Metab. 2007;92:4185–4190. doi: 10.1210/jc.2007-1211. [DOI] [PubMed] [Google Scholar]
- 26.Ong SC, Schöder H, Patel SG, et al. Diagnostic accuracy of 18F-FDG PET in restaging patients with medullary thyroid carcinoma and elevated calcitonin levels. J Nucl Med. 2007;48:501–507. doi: 10.2967/jnumed.106.036681. [DOI] [PubMed] [Google Scholar]
- 27.Tung WS, Vesely TM, Moley JF. Laparoscopic detection of hepatic metastases in patients with residual or recurrent medullary thyroid cancer. Surgery. 1995;118:1024–1029. doi: 10.1016/s0039-6060(05)80109-6. discussion 1029–1030. [DOI] [PubMed] [Google Scholar]
- 28.Moore SW, Zaahl M. Familial associations in medullary thyroid carcinoma with Hirschsprung disease: The role of the RET-C620 “Janus” genetic variation. J Pediatr Surg. 2010;45:393–396. doi: 10.1016/j.jpedsurg.2009.10.080. [DOI] [PubMed] [Google Scholar]
- 29.Fialkowski EA, DeBenedetti MK, Moley JF, et al. RET proto-oncogene testing in infants presenting with Hirschsprung disease identifies 2 new multiple endocrine neoplasia 2A kindreds. J Pediatr Surg. 2008;43:188–190. doi: 10.1016/j.jpedsurg.2007.09.043. [DOI] [PubMed] [Google Scholar]
- 30.Bütter A, Gagné J, Al-Jazaeri A, et al. Prophylactic thyroidectomy in pediatric carriers of multiple endocrine neoplasia type 2A or familial medullary thyroid carcinoma: Mutation in C620 is associated with Hirschsprung's disease. J Pediatr Surg. 2007;42:203–206. doi: 10.1016/j.jpedsurg.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 31.Donovan DT, Levy ML, Furst EJ, et al. Familial cutaneous lichen amyloidosis in association with multiple endocrine neoplasia type 2A: A new variant. Henry Ford Hosp Med J. 1989;37:147–150. [PubMed] [Google Scholar]
- 32.Verga U, Fugazzola L, Cambiaghi S, et al. Frequent association between MEN 2A and cutaneous lichen amyloidosis. Clin Endocrinol (Oxf) 2003;59:156–161. doi: 10.1046/j.1365-2265.2003.01782.x. [DOI] [PubMed] [Google Scholar]
- 33.O'Riordain DS, O'Brien T, Crotty TB, et al. Multiple endocrine neoplasia type 2B: More than an endocrine disorder. Surgery. 1995;118:936–942. doi: 10.1016/s0039-6060(05)80097-2. [DOI] [PubMed] [Google Scholar]
- 34.Donis-Keller H, Dou S, Chi D, et al. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet. 1993;2:851–856. doi: 10.1093/hmg/2.7.851. [DOI] [PubMed] [Google Scholar]
- 35.Frank-Raue K, Buhr H, Dralle H, et al. Long-term outcome in 46 gene carriers of hereditary medullary thyroid carcinoma after prophylactic thyroidectomy: Impact of individual RET genotype. Eur J Endocrinol. 2006;155:229–236. doi: 10.1530/eje.1.02216. [DOI] [PubMed] [Google Scholar]
- 36.Shepet K, Alhefdhi A, Lai N, et al. Hereditary medullary thyroid cancer: Age-appropriate thyroidectomy improves disease-free survival. Ann Surg Oncol. 2013;20:1451–1455. doi: 10.1245/s10434-012-2757-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moley JF, DeBenedetti MK. Patterns of nodal metastases in palpable medullary thyroid carcinoma: Recommendations for extent of node dissection. Ann Surg. 1999;229:880–887. doi: 10.1097/00000658-199906000-00016. discussion 887–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Greenblatt DY, Elson D, Mack E, et al. Initial lymph node dissection increases cure rates in patients with medullary thyroid cancer. Asian J Surg. 2007;30:108–112. doi: 10.1016/S1015-9584(09)60141-X. [DOI] [PubMed] [Google Scholar]
- 39.Scollo C, Baudin E, Travagli JP, et al. Rationale for central and bilateral lymph node dissection in sporadic and hereditary medullary thyroid cancer. J Clin Endocrinol Metab. 2003;88:2070–2075. doi: 10.1210/jc.2002-021713. [DOI] [PubMed] [Google Scholar]
- 40.Machens A, Hauptmann S, Dralle H. Increased risk of lymph node metastasis in multifocal hereditary and sporadic medullary thyroid cancer. World J Surg. 2007;31:1960–1965. doi: 10.1007/s00268-007-9185-1. [DOI] [PubMed] [Google Scholar]
- 41.Sippel RS, Kunnimalaiyaan M, Chen H. Current management of medullary thyroid cancer. The Oncologist. 2008;13:539–547. doi: 10.1634/theoncologist.2007-0239. [DOI] [PubMed] [Google Scholar]
- 42.Brierley J, Tsang R, Simpson WJ, et al. Medullary thyroid cancer: Analyses of survival and prognostic factors and the role of radiation therapy in local control. Thyroid. 1996;6:305–310. doi: 10.1089/thy.1996.6.305. [DOI] [PubMed] [Google Scholar]
- 43.de Groot JW, Zonnenberg BA, van Ufford-Mannesse PQ, et al. A phase II trial of imatinib therapy for metastatic medullary thyroid carcinoma. J Clin Endocrinol Metab. 2007;92:3466–3469. doi: 10.1210/jc.2007-0649. [DOI] [PubMed] [Google Scholar]
- 44.Almeida MQ, Hoff AO. Recent advances in the molecular pathogenesis and targeted therapies of medullary thyroid carcinoma. Curr Opin Oncol. 2012;24:229–234. doi: 10.1097/CCO.0b013e328351c71a. [DOI] [PubMed] [Google Scholar]
- 45.Wells SA, Gosnell JE, Gagel RF, et al. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Oncol. 2010;28:767–772. doi: 10.1200/JCO.2009.23.6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robinson BG, Paz-Ares L, Krebs A, et al. Vandetanib (100 mg) in patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Endocrinol Metab. 2010;95:2664–2671. doi: 10.1210/jc.2009-2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wells SA, Robinson BG, Gagel RF, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J Clin Oncol. 2012;30:134–141. doi: 10.1200/JCO.2011.35.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cabozantinib approved for rare thyroid cancer. Cancer Discov. 2013;3:OF7. [Google Scholar]
- 49.Hart CD, De Boer RH. Profile of cabozantinib and its potential in the treatment of advanced medullary thyroid cancer. Onco Targets Ther. 2013;6:1–7. doi: 10.2147/OTT.S27671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Traynor K. Cabozantinib approved for advanced medullary thyroid cancer. Am J Health Syst Pharm. 2013;70:88. doi: 10.2146/news130005. [DOI] [PubMed] [Google Scholar]
- 51.Ahmed M, Barbachano Y, Riddell A, et al. Analysis of the efficacy and toxicity of sorafenib in thyroid cancer: A phase II study in a UK based population. Eur J Endocrinol. 2011;165:315–322. doi: 10.1530/EJE-11-0129. [DOI] [PubMed] [Google Scholar]
- 52.Lam ET, Ringel MD, Kloos RT, et al. Phase II clinical trial of sorafenib in metastatic medullary thyroid cancer. J Clin Oncol. 2010;28:2323–2330. doi: 10.1200/JCO.2009.25.0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carr LL, Mankoff DA, Goulart BH, et al. Phase II study of daily sunitinib in FDG-PET-positive, iodine-refractory differentiated thyroid cancer and metastatic medullary carcinoma of the thyroid with functional imaging correlation. Clin Cancer Res. 2010;16:5260–5268. doi: 10.1158/1078-0432.CCR-10-0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schlumberger MJ, Elisei R, Bastholt L, et al. Phase II study of safety and efficacy of motesanib in patients with progressive or symptomatic, advanced or metastatic medullary thyroid cancer. J Clin Oncol. 2009;27:3794–3801. doi: 10.1200/JCO.2008.18.7815. [DOI] [PubMed] [Google Scholar]
- 55.Cohen EE, Rosen LS, Vokes EE, et al. Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: Results from a phase II study. J Clin Oncol. 2008;26:4708–4713. doi: 10.1200/JCO.2007.15.9566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kunnimalaiyaan M, Chen H. Tumor suppressor role of Notch-1 signaling in neuroendocrine tumors. The Oncologist. 2007;12:535–542. doi: 10.1634/theoncologist.12-5-535. [DOI] [PubMed] [Google Scholar]
- 57.Ning L, Greenblatt DY, Kunnimalaiyaan M, et al. Suberoyl bis-hydroxamic acid activates Notch-1 signaling and induces apoptosis in medullary thyroid carcinoma cells. The Oncologist. 2008;13:98–104. doi: 10.1634/theoncologist.2007-0190. [DOI] [PubMed] [Google Scholar]
- 58.Vaccaro A, Chen H, Kunnimalaiyaan M. In-vivo activation of Raf-1 inhibits tumor growth and development in a xenograft model of human medullary thyroid cancer. Anticancer Drugs. 2006;17:849–853. doi: 10.1097/01.cad.0000217424.36961.47. [DOI] [PubMed] [Google Scholar]
- 59.Kunnimalaiyaan M, Vaccaro AM, Ndiaye MA, et al. Inactivation of glycogen synthase kinase-3beta, a downstream target of the raf-1 pathway, is associated with growth suppression in medullary thyroid cancer cells. Mol Cancer Ther. 2007;6:1151–1158. doi: 10.1158/1535-7163.MCT-06-0665. [DOI] [PubMed] [Google Scholar]
- 60.Kunnimalaiyaan M, Ndiaye M, Chen H. Apoptosis-mediated medullary thyroid cancer growth suppression by the PI3K inhibitor LY294002. Surgery. 2006;140:1009–1014. doi: 10.1016/j.surg.2006.06.040. [DOI] [PubMed] [Google Scholar]
- 61.Kunnimalaiyaan M, Vaccaro AM, Ndiaye MA, et al. Overexpression of the NOTCH1 intracellular domain inhibits cell proliferation and alters the neuroendocrine phenotype of medullary thyroid cancer cells. J Biol Chem. 2006;281:39819–39830. doi: 10.1074/jbc.M603578200. [DOI] [PubMed] [Google Scholar]
- 62.Adler JT, Cook M, Luo Y, et al. Tautomycetin and tautomycin suppress the growth of medullary thyroid cancer cells via inhibition of glycogen synthase kinase-3beta. Mol Cancer Ther. 2009;8:914–920. doi: 10.1158/1535-7163.MCT-08-0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adler JT, Hottinger DG, Kunnimalaiyaan M, et al. Inhibition of growth in medullary thyroid cancer cells with histone deacetylase inhibitors and lithium chloride. J Surg Res. 2010;159:640–644. doi: 10.1016/j.jss.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cook MR, Luo J, Ndiaye M, et al. Xanthohumol inhibits the neuroendocrine transcription factor achaete-scute complex-like 1, suppresses proliferation, and induces phosphorylated ERK1/2 in medullary thyroid cancer. Am J Surg. 2010;199:315–318. doi: 10.1016/j.amjsurg.2009.08.034. discussion 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Truong M, Cook MR, Pinchot SN, et al. Resveratrol induces Notch2-mediated apoptosis and suppression of neuroendocrine markers in medullary thyroid cancer. Ann Surg Oncol. 2011;18:1506–1511. doi: 10.1245/s10434-010-1488-z. [DOI] [PMC free article] [PubMed] [Google Scholar]