

Type 2 diabetes mellitus patients are at higher cancer risk, probably because of hyperinsulinemia and insulin growth factor 1 pathway activation. The aims of this work are to review preclinical evidence of second-generation sulfonylureas effects on tumor growth, to clarify the potential mechanisms of action, and to identify possible metabolic targets for patient selection.
Keywords: Sulfonylurea compounds, Anticancer agents, Glibenclamide, Apoptosis
Learning Objectives
Describe possible opposite effects on tumor growth of different 2nd generation sulfonylureas and diarylsulfonylureas.
Review preclinical and clinical studies investigating anticancer activity of different sulfonylureas and diarylsulfonylureas.
Abstract
Type 2 diabetes mellitus patients are at higher cancer risk, probably because of hyperinsulinemia and insulin growth factor 1 pathway activation. The effects of antidiabetic drugs on cancer risk have been described and discussed in several studies suggesting opposite effects of the biguanide metformin and sulfonylureas on cancer incidence and mortality. The anticancer mechanisms of metformin have been clarified, and some clinical studies, particularly in breast cancer patients, have been published or are currently ongoing; however, data about the effects of sulfonylureas on cancer growth are less consistent. The aims of this work are to review preclinical evidence of second-generation sulfonylureas effects on tumor growth, to clarify the potential mechanisms of action, and to identify possible metabolic targets for patient selection. Most evidence is on the adenosine triphosphate-sensitive potassium channels inhibitor glibenclamide, which interacts with reactive oxygen species production thus inducing cancer cell death. Among diarylsulfonylureas, next-generation DW2282 derivatives are particularly promising because of the proapoptotic activity in multidrug-resistant cells.
Implications for Practice:
The effects of anti-diabetic drugs on cancer risk have been described in several studies suggesting opposite effects of metformin and sulfonylureas on cancer incidence and mortality, respectively. Although the anticancer mechanisms of metformin have been clarified, no univocal data about sulfonylureas' effects on cancer growth are available. No previous review articles about the same topic have been published; therefore, there is conflicting evidence about the real role of different compounds of the sulfonylurea family on cancer cell growth. This article highlights specific proapoptotic pathways involved in the anticancer effects of these drugs, which might help in the identification of metabolic targets for preclinical and clinical study design and patient selection.
Introduction
Cancer and diabetes are rising causes of morbidity and mortality worldwide and represent significant health care issues. Several reports have highlighted the increased risk of different cancer types in patients affected by type 2 diabetes mellitus [1–3], probably due to chronic inflammation, hyperglycemia, hyperinsulinemia, and enhanced levels of insulin-like growth factor (IGF) with subsequent activation of the related pathway [4] (Fig. 1). Additional studies are underway to confirm and clarify the mechanisms of such correlation because of the heterogeneous features of diabetes mellitus and the potential confounders and shared risk factors such as obesity, metabolic syndrome, diet, and drugs.
Figure 1.
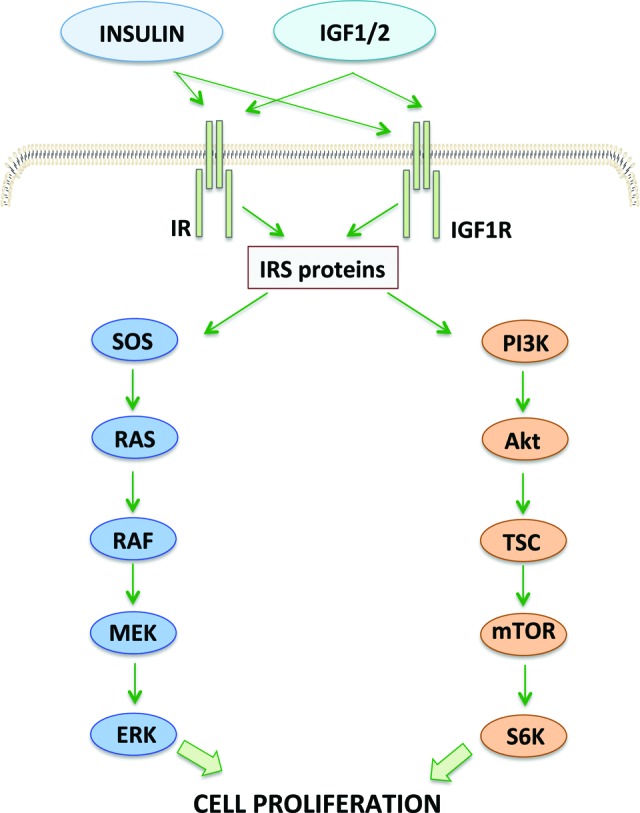
Insulin and IGF pathway. Insulin and IGF bind to insulin receptors and IGF receptors, activating insulin receptor substrates and, subsequently, mitogen-activated protein kinase and PI3K-Akt-mTOR pathways. These signals lead to cell survival and proliferation.
Abbreviations: IGF, insulin growth factor; IR, insulin receptor; IRS, insulin receptor substrates; mTOR, mammalian target or rapamycin; PI3K, phosphoinositide 3 kinase; SOS, son of sevenless; RAS, rat sarcoma; RAF, rapidly accelerated fibrosarcoma; MEK, mitogen-activated protein kinase/ERK kinase; ERK, extracellular signal-regulated kinases; TSC, tuberous sclerosis complex; S6K, S6 protein kinase.
Recently, the effects of antidiabetic drugs on cancer risk were described and discussed in several studies [5–8], suggesting opposite effects of the biguanide metformin and sulfonylureas on cancer incidence and mortality. A primary data meta-analysis analyzed 24 metformin studies and 18 sulfonylurea studies investigating the correlation between antidiabetic drugs and cancer incidence. Case-control and cohort studies confirmed that metformin treatment is associated with reduced cancer incidence, but these results were not supported by randomized controlled trials; regarding sulfonylureas, increased cancer risk emerged only in cohort studies, whereas case-control and randomized controlled trials did not confirm this finding [9].
Two different mechanisms seem to be at the basis of these results. Metformin acts through adenosine monophosphate-activated protein kinase (AMPK)/liver kinase B-1 (LKB1) pathway activation, directly suppressing cell proliferation and indirectly reducing glucose and insulin levels [10]. In contrast, sulfonylureas are insulin secretagogues, and this positive effect on insulin and IGF levels seems to promote tumorigenesis [4], even though heterogeneous effects of different sulfonylureas have been shown in a cohort study including more than 6,000 patients [11]. The precise mechanisms supporting possible protumoral or anticancer effects of sulfonylureas are still unknown.
The aim of this work is to review the preclinical and, when available, clinical evidence of second-generation sulfonylureas and new diarylsulfonylurea (DSU) effects on tumor growth in order to give an updated picture of current knowledge that might contribute to progress in the area of diabetes and cancer.
Materials and Methods
An electronic search was performed using the PubMed database. To optimize the search strategy, we used the advanced search builder with selected Bayesian words: “sulfonylurea” OR “glibenclamide,” “glipizide,” “glicazide,” “glimepiride” AND “anticancer” or “antitumor,” “apoptosis,” “tumor/cancer cell death.”
We also reviewed the reference lists in relevant publications and the abstracts from the meetings of the American Society of Clinical Oncology, the European Society of Medical Oncology, and other main international conferences.
Results
From second-generation sulfonylurea glibenclamide to new DSU derivatives, several compounds were identified as anticancer agents, even if the mechanism of action is still obscure in many cases.
Among new-generation sulfonylureas, at a preclinical level, glibenclamide proved to be a tumor growth inhibitor [12–35]. Derivatives of DSU (S)-(+)-4-phenyl-1-[N-(4-aminobenzoyl)-indoline-5-sulfonyl-4,5-dihydro-2-imidazolone]hydrochloride (DW2282) that combine dual anticancer effects are of more interest in the clinical setting because they might bypass chemoresistance to “classic” anticancer agents such as taxanes or vinca alkaloids.
Glibenclamide
Glibenclamide is a sulfonylurea drug used in type 2 diabetes that acts through sulfonylurea receptors (SURs) on pancreatic β cells. SURs are subunits of adenosine triphosphate-sensitive potassium channels (KATP channels), which are inhibited by glibenclamide with subsequent cell depolarization, opening of voltage-gated calcium channels, calcium influx into the cell, and finally insulin secretion through vesicle exocytosis [36, 37] (Fig. 2). Over the years, some evidence has shown the effects of glibenclamide on tumor growth arrest of different cancer types [12, 22, 23, 30, 31, 33, 34, 38], even though the mechanisms of such antitumor activity were not completely clarified.
Figure 2.
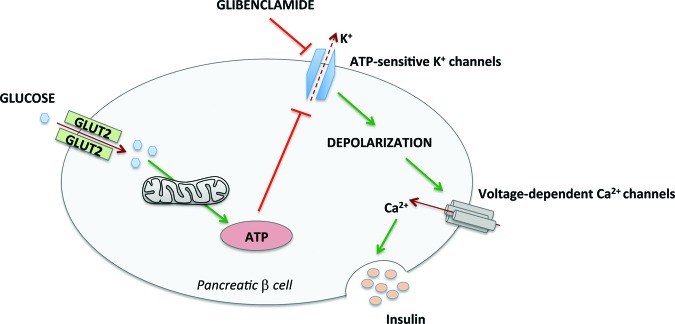
Glibenclamide-dependent secretion of insulin in pancreatic β cells. Physiologically, glucose influx into pancreatic β cell induces ATP synthesis through glycolysis and mitochondrial respiration with subsequent K+ channel closure. In type 2 diabetes mellitus patients, glibenclamide directly closes ATP-sensitive K+ channels, reducing membrane potential and inducing calcium influx, which in turn stimulates insulin secretion.
Abbreviations: ATP, adenosine triphosphate; Ca2+, calcium ion; K+, potassium ion.
A first step toward better knowledge of glibenclamide's mechanism of action was achieved by a French group investigating its role as inhibitor of adenosine triphosphate-binding cassette (ABC) transporters [24].
SUR belongs to the ABC protein superfamily, a group of transmembrane proteins that use ATP to transport a large variety of substrates across extra- and intracellular membranes, including metabolic products, lipids and sterols, and drugs. Multidrug-resistant proteins (MRPs) represent a subfamily of ABC transporters involved in the cellular export of several drugs, including anthracyclines and vinca alkaloids [21]. Consequently, MRP overexpression might confer chemotherapy resistance to tumor cells and thus might represent a promising target for anticancer treatment. Payen and coauthors considered the known properties of glibenclamide as an ABC protein inhibitor (cystic fibrosis transmembrane conductance regulator, ABC1, bile salt export pump, P-glycoprotein) [19, 20, 26, 27] as a premise for investigating its role as an MRP inhibitor in lung cancer cell lines. Glibenclamide induced an increased MRP1 substrate calcein accumulation in lung cancer cell lines overexpressing MRP1, confirming that MRP1 is a target for the sulfonylurea. Furthermore, intracellular accumulation of vincristine, another MRP1 substrate, was shown, and this indicates that glibenclamide might act as a sensitizer of cancer cells to chemotherapeutic agents. Even though the MRP1-inhibiting dosage of the sulfonylurea might preclude its clinical application, these results confirm its role as a general ABC transporter inhibitor [24].
Over the last decade, increasing evidence has supported a regulatory function of potassium ion (K+) channels in cancer cell proliferation and survival, suggesting that these channels might be potential therapeutic targets [15, 16, 18, 29]. It is well known that KATP channels in plasma and mitochondrial membrane are made up of four regulatory SURs and four inwardly rectifying potassium channel subunits Kir6.x and that the opening of KATP channels inhibits apoptosis in ischemia, hypoxia, or oxidative stress, whereas, in contrast, their closure induces cell damage and apoptosis [14]. K+ influx into the cancer cell promotes deregulated tumor growth and allows tumor cells to survive in a hypoxic microenvironment through resting potential depolarization [16].
KATP channels are expressed in different tissues (pancreas, cardiac, smooth and skeletal muscle, and brain) and cancer cells. Anticancer effects of glibenclamide, as well as other KATP channel closers, have been demonstrated in KATP channels expressing cancer cells.
In 2008, KATP channel expression was shown in the gastric cancer cell line MGC-803, in which the anticancer effect of glibenclamide was investigated. Glibenclamide was able to induce reactive oxygen species (ROS) followed by cancer cell apoptosis. The authors investigated the mechanisms and molecular pathways involved in such process and demonstrated that ROS generation could decrease the mitochondrial membrane potential through the activation of proapoptotic c-Jun NH2-terminal kinase and inhibition of antiapoptotic AKT kinase. The subsequent release of mitochondrial cytochrome c and apoptosis-inducing factor to the cytosol finally could lead to caspase-dependent and independent apoptosis [25].
Recently, our group tested the synergic effect of engaging the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) apoptotic pathway and glibenclamide in order to induce cell death in malignant pleural mesothelioma cell lines. We observed a statistically significant increase of caspases activity in all cell lines (three epithelioid, one sarcomatoid, three biphasic) treated with the combination of the two agents compared with untreated controls and compared with TRAIL and glibenclamide used as single agents. We analyzed ROS levels in two cell lines (epithelioid ZL55 and sarcomatoid ZL34), and we observed ROS induction in ZL55 treated with glibenclamide with or without TRAIL compared with no treatment, whereas no higher ROS levels were assessed in ZL34 treated with glibenclamide with or without TRAIL compared with untreated controls. Moreover, preincubation with the ROS scavenger N-acetyl-cysteine resulted in a reduction of ZL55 cell death after treatment with glibenclamide plus TRAIL. We concluded that glibenclamide sensitizes malignant pleural mesothelioma cell lines and primary cultures to TRAIL-mediated apoptosis, probably through different mechanisms of action in the epithelioid and sarcomatoid histotypes [35].
A different proapoptotic pathway was implied in human melanoma cell lines in which glibenclamide induced cell death, while sparing normal melanocytes, through sensitization to a TRAIL-dependent extrinsic apoptotic pathway. A Japanese paper published in 2012 showed that KATP channel inhibitors such as glibenclamide were able to sensitize melanoma cells to TRAIL-induced apoptosis, probably through enhanced plasma membrane potential depolarization, activation of effector caspases 3 and 7, and activation of endoplasmic reticulum stress-induced caspase 12 [28].
Besides the proapoptotic effect of glibenclamide, a recent role of KATP channel closers in neoangiogenesis has been described. Glibenclamide showed an inhibitory effect on ovarian ES-2 cell line invasion and migration through the inhibition of the angiogenic pathway. Decreased secretion of several proangiogenic proteins was observed after treatment with glibenclamide and subsequent KATP channel closure, with particular reference to platelet-derived growth factor AA, which is involved in cell growth, migration, and differentiation. In particular, an inhibitory effect of glibenclamide on the autocrine system through which platelet-derived growth factor promotes ovarian cancer invasiveness was observed [32].
Other Sulfonylureas
No evidence of any effect on tumor growth of glipizide and glimepiride has been reported.
Gliclazide is another commonly used second-generation sulfonylurea, with a mechanism of action similar to that of glibenclamide but with a rapidly reversible binding to SUR1 compared with the prolonged binding of the other sulfonylureas. Antioxidant activity of gliclazide has been reported in type 2 diabetes mellitus patients [17], and increasing evidence of reduced DNA damage induced by ROS was observed [39–41].
It is known that chronic hyperglycemia in type 2 diabetes mellitus patients might generate oxidative stress with subsequent intracellular signaling pathway impairment and genome stability damage [42].
When a protective effect of gliclazide on DNA damage of cancer cells was explored, positive activity on DNA repair in pancreatic cancer cells was demonstrated [43]. Particularly, gliclazide seemed to stimulate nucleotide excision repair and nonhomologous end-joining double-strand-break repair mechanisms in pancreatic cancer cells, whereas no protective effects were observed in human normal cells [43].
Previous evidence of glibenclamide and other sulfonylureas has shown a ROS-inducing effect of such drugs, with subsequent loss of mitochondrial membrane potential, cytosolic calcium rise, and pancreatic cell death. In contrast, a peculiar ROS scavenging effect of gliclazide has been described more recently [44, 45], with antiapoptotic activity in pancreatic and endothelial cells [46–48]. Apparently, the ROS scavenging effect is not the only “protective” mechanism; reversible binding to SUR1 and upregulation of antioxidant enzymes also have been described.
The antiapoptotic effects of gliclazide in pancreatic and breast cancer cells were investigated recently, and a protective effect toward oxidative stress was confirmed; loss of mitochondrial membrane potential was inhibited by gliclazide, with reduced cytosol calcium levels and suppressed cancer cell death [49].
From Glibenclamide to DSUs
Since the end of the 1980s, the anticancer effects of DSUs—compounds structurally similar to oral antidiabetic glibenclamide—have been described in vitro and in vivo [50]. In particular, several authors reported antitumor activity of N-(4-methylphenylsulfonyl)-N′-(4-chlorophenyl)urea (LY181984) and N-(5-indanylsulfonyl)-N′-(4-chlorophenyl)urea (LY186641; sulofenur) in in vitro and in vivo models of solid tumors, with modest activity in hematologic cancer [50–53].
Preclinical studies underscored that sulofenur, a compound with a long half-life and highly protein bound, has a new and not completely defined antitumor mechanism that distinguishes it from other chemotherapeutic agents.
The anticancer effects of sulofenur on solid tumors were also explored in phase I and II clinical trials [50, 54, 55]. On the basis of partial response observed in the phase I trial in a ovarian cancer patient [50], the phase II trial was designed to elucidate response rates and toxicity profile in stage III–IV previously treated ovarian cancer patients [54]. Sulofenur at the daily dose of 800 mg/m2 showed prolonged stable disease (median: 20 weeks) in 42% of the study population and partial response (6.5–18 weeks) in 15% of the patients. In both phase I and phase II clinical trials, major toxicities were methemoglobinemia with decreased red blood cell survival and severe anemia. Toxicity was also the main reason why a different schedule was tested in the phase II trial: 2 days of rest after 5 days of daily treatment for 3 weeks of reduced toxicity, even though dose reduction was still required in 31% of the patients. The toxicity profile of sulofenur precluded further clinical studies in cancer patients, although future study of the mechanism of action of this and other DSUs might be interesting. DSU seemed not to interfere with proteins and nucleic acids synthesis [56, 57], although a mitochondrial effect was suggested as a possible cytotoxic mechanism. Several prior studies showed mitochondrial localization of DSU with subsequent morphological changes and cell death [58, 59]. Similar to glibenclamide and other sulfonylureas, the mitochondrial oxidative phosphorylation uncoupling and the lowering of adenosine triphosphate levels were particularly evident with sulofenur, and this likely represents the new cytotoxic mechanism shown in preclinical cancer models [59].
Chlebowski and colleagues showed that breast cancer incidence in postmenopausal diabetic women, compared with nondiabetic women, can be different according to the type of antidiabetic drug; in fact, a 25% risk reduction of breast cancer was reported among women taking metformin versus a 16% increase in risk for women who received other antidiabetics. The main pitfall of this analysis relates to the fact that diabetes seems to confer about a 30% higher risk of breast cancer.
In order to bypass hematological toxicity of the first-generation DSU sulofenur, new drugs such LY181984 and LY295501 were investigated in preclinical and clinical studies [60–62]. The different metabolism of such compounds compared with sulofenur, particularly no formation of aniline metabolites (a possible cause of methemoglobinemia), makes them less toxic. The higher potency of LY295501 compared with other DSUs [63] led to a phase I clinical trial with advanced solid tumors [62] that set the maximum tolerated doses for further clinical studies at 1,000 mg/m2 per day, administered weekly for 3–4 weeks; as expected, no typical toxicities of sulofenur were shown, with neutropenia and thrombocytopenia as the most common hematological toxicities.
Looking for new DSU derivatives with minimal toxicity and appreciable clinical benefit in cancer patients, further studies led to a novel compound synthesis, DW2282, that is able to suppress tumor growth in vitro and in vivo with an uncertain mechanism [64, 65]. A preclinical study in promyelocytic leukemia cells tried to elucidate the antitumoral mechanism of DW2282; decreased levels of cdc2 with subsequent cell cycle arrest in the G2/M phase seemed to induce apoptosis in treated cell lines. Moreover, reduction of antiapoptotic protein BCL2 and procaspase 3 activation contribute to apoptosis induction [66].
More recently, new DW2282 derivatives methanesulfonates and arylsulfonamides were synthesized as promising anticancer agents. These drugs combine an inhibitory activity against tubulin polymerization, similar to well-known chemotherapeutic agents taxanes and vinca alkaloids, with the ability to target multidrug-resistant tumors. Efflux pumps such as P-glycoprotein and MRP seem to be the basis of resistance to taxanes and vinca alkaloids. Some compounds in this new DSU family were tested in different cancer cell lines (breast, colon, non-small cell lung cancer) and showed antiproliferative effects correlated with tubulin polymerization inhibition and the G2/M phase of cell cycle arrest; finally, antimitotic activity was confirmed in multidrug-resistant cell lines treated with these compounds [67].
Discussion
Cancer and diabetes are the second and seventh causes of death worldwide, respectively, and both diseases are multifactorial and heterogeneous. Increased cancer incidence in diabetic patients has been described in several cohort studies [68–74] and confirmed in some meta-analyses [1, 75], although it is difficult to provide a precise risk estimate because of confounding factors, shared medical conditions, and pathophysiological pathways. Diabetes is present in 8%–18% of cancer patients, and the relative risk of cancer in diabetic patients ranges between 1.12 (non-Hodgkin lymphoma) and 2.51 (liver cancer). Higher relative risks of liver, pancreatic, or endometrial cancer have been reported in diabetic patients, whereas the increase of relative risks of breast, bladder, colorectal, kidney, and biliary neoplasms is less evident; moreover, the relative risk of prostate cancer seems even lower in diabetic patients [2, 76].
Apart from the epidemiologic association between diabetes and cancer, the observation that some antidiabetic drugs seem to modify the risk of cancer is of great interest [9, 77]. Observational studies, however, have some limitations because they consider various metabolic conditions and different indications on antidiabetic treatment in affected patients.
Metformin is the first-line antidiabetic drug and belongs to the biguanide family, which lowers glucose levels in type 2 diabetes patients. Recently, metformin was associated with a 10%–30% decreased risk of cancer in case-control studies (24,829 patients) and cohort studies (355,420 patients); however, this protective effect was not confirmed in the only two available randomized clinical trials (6,578 patients) [9].
The bulk of the evidence on metformin and cancer incidence has been produced in breast cancer. Epidemiologic studies showed a 23%–56% decreased risk of breast cancer in diabetic women who received metformin compared with those not on metformin [78, 79]. The association between metformin treatment and breast cancer incidence has been explored recently in the Women's Health Initiative clinical trial setting. Chlebowski and colleagues showed that breast cancer incidence in postmenopausal diabetic women, compared with nondiabetic women, can be different according to the type of antidiabetic drug; in fact, a 25% risk reduction of breast cancer was reported among women taking metformin versus a 16% increase in risk for women who received other antidiabetics [80]. The main pitfall of this analysis relates to the fact that diabetes seems to confer about a 30% higher risk of breast cancer [81], and this represents a confounding factor when comparing nondiabetic women and diabetic women using metformin. It has become clear that diabetes and breast cancer share some clinical conditions, such as obesity and metabolic syndrome, and this indicates a higher risk when hyperinsulinemia and insulin resistance occur [82].
The antiproliferative effects of metformin were explored in some clinical studies. A retrospective analysis of 2,529 early stage breast cancer patients showed a higher rates of pathological complete response after neoadjuvant chemotherapy in diabetic women taking metformin compared with those not using metformin (24% vs. 8%, p = .07) and in nondiabetic women compared with diabetic patients not taking metformin (16% vs. 8%, p = .04). Moreover, metformin was identified as an independent predictive factor of pathological complete response [83].
The effect of metformin on Ki-67 change between biopsy and surgical samples was investigated in a window-of-opportunity randomized study including 200 nondiabetic operable breast cancer patients. Ki-67 was not significantly affected by metformin treatment compared with placebo in the overall population, whereas a trend toward Ki-67 decrease was observed in patients with insulin resistance and high body mass index, and this decrease was more evident in luminal B tumors [84].
It is known that metformin targets a specific molecular pathway and directly produces antitumor activity through mammalian target of rapamycin downregulation in an AMPK-dependent manner (insulin independent) [10, 85]. In addition, metformin downregulates the mammalian target of rapamycin pathway through decrease of insulin levels (insulin-dependent, AMPK-independent mechanism). Available clinical data from breast cancer patients underscore higher antitumor activity in patients with insulin resistance and metabolic disorder subsequent to type 2 diabetes, suggesting that anticancer effects of metformin are related to improvement in metabolic disorder rather than an insulin-independent mechanism [83, 84].
An intergroup phase III clinical trial of metformin versus placebo in early stage breast cancer is currently ongoing. This trial has recurrence-free and overall survival as study endpoints and will explore whether the anticancer effect of metformin is related to an insulin-dependent or -independent mechanism (ClinicalTrials.gov identifier NCT01101438).
Available clinical data from breast cancer patients underscore higher antitumor activity in patients with insulin resistance and metabolic disorder subsequent to type 2 diabetes, suggesting that anticancer effects of metformin are related to improvement in metabolic disorder rather than an insulin-independent mechanism.
Although a protective effect of metformin against cancer has been described, data about sulfonylureas and cancer risk are conflicting. Six cohort studies (296,904 patients) reported higher cancer risk in diabetic patients taking sulfonylureas compared with nonsulfonylurea users; however, these results were not confirmed in 10 case-control studies (12,040 patients) and 2 randomized clinical studies (6,573 patients) [9].
No specific mechanism supporting an antitumoral rather than a protumoral effect of sulfonylureas was identified. Sulfonylureas are insulin secretagogues, and the increased levels of insulin and activation of the IGF1–IGF-1R pathway was hypothesized as a possible protumoral mechanism. Several compounds belong to the sulfonylureas family, and the correlation with cancer incidence should be studied for each individual compound. Recently, gliclazide and glibenclamide were related to a 35% reduced risk of cancer, whereas glipizide was related to a 16% increased cancer risk [11]. In contrast, another case-control study reported increased cancer risk in patients treated with glibenclamide and a lower risk in patients who received gliclazide [5].
In this review, we have reported conflicting epidemiologic evidence about cancer incidence in diabetic patients receiving sulfonylureas [9, 77], and we have described the mechanism of action of these drugs. In particular, we reviewed preclinical and clinical evidence of proposed mechanisms of action supporting antitumoral or antiapoptotic activity.
Most evidence of antitumor activity of sulfonylureas concerns glibenclamide, which was tested in different cancer types at the preclinical level. The role of glibenclamide as a KATP channel closer and its interaction with ROS production seem to underlie the antitumor effect of this compound (Fig. 3).
Figure 3.

Proposed model for antitumor activity of glibenclamide. Glibenclamide increases NADPH oxidase and mitochondrial respiratory chain ROS production followed by release of proapoptotic factors and caspase activation. Membrane depolarization by glibenclamide and TRAIL induces ER stress-mediated apoptosis. Glibenclamide sensitizes tumor cells to TRAIL-dependent apoptosis through membrane depolarization and ROS production.
Abbreviations: Cas, caspase; ER, endoplasmic reticulum; K+, potassium ion; rc, respiratory chain; ROS, reactive oxygen species; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand.
ROS are implied in several signaling transduction pathways and regulate different biological activities such as cell growth, survival, and angiogenesis. ROS increase is associated with abnormal cancer cell growth and reflects a disruption of redox homeostasis related to either an elevation of ROS production or a decline of ROS-scavenging capacity. Cancer cells exhibit a higher ROS set point compared with normal cells because of increased metabolic activity and thus are likely to be more vulnerable to damage by further ROS insults [86, 87]. This is the reason why several antitumor treatments (e.g., chemotherapy, radiotherapy) are based on increased and irreversible oxidative stress.
The main cell sites of ROS production are cytosol, through NADPH oxidase, and redox centers of the mitochondrial respiratory chain. The interaction between mitochondrial potassium channels and ROS induction has not been clarified, paving the way for investigation of different K+ channel modulators. Uncertainty about the role of potassium flux effects on ROS production has emerged, and conflicting results about ROS increase or decrease through K+ mitochondrial influx have been reported. Probably these discrepancies derive from a tissue-dependent contribution of different respiratory chain complexes [88].
Glibenclamide closes KATP channels in both plasma and mitochondrial membrane, and other potassium channel modulators showed some off-target effects, such as mitochondrial uncoupling [89].
Oxidative stress subsequent to K+ efflux probably activates signaling pathways and leads to apoptosis directly or through sensitization to other extrinsic triggers [25, 28, 35] (Fig. 3).
Another target of glibenclamide is the MRP, which is often overexpressed by cancer cells and mediates the efflux of chemotherapeutic agents, resulting in chemoresistance. Glibenclamide seems to directly bind and block MRP at higher concentrations than those permitted in affected patients without toxicity [24].
The main limitation of glibenclamide and other sulfonylureas is the lack of clinical studies, probably because the principal mechanism of action has not been identified yet. First-generation DSUs, which are structurally similar to glibenclamide with an unknown anticancer mechanism, reached clinical application in some cases but with poor tolerability and disappointing results [50, 54, 55].
New-generation DW2282 derivatives, developed to solve the dose-limiting toxicity of first-generation DSUs, are more promising because of the proapoptotic activity in multidrug-resistant cells. Two promising antimitotic mechanisms have been described. First, these new agents showed strong antiproliferative activity in vitro through tubulin polymerization inhibition. This mechanism is already known for traditional chemotherapeutic agents such as taxanes and vinca alkaloids. As described above, tumor cells could develop resistance to these drugs through MRP and other efflux pumps overexpression. The second property of DW2282 derivatives is their anticancer activity, even in presence of MRPs on cancer cells, probably because they are not substrates of the efflux pumps [67]. Further investigation of these novel DSUs is currently ongoing.
Conclusion
Specific metabolic targets in different tissues should be identified in order to clarify the anticancer mechanisms of sulfonylureas and to select cancer patients for treatment. Considering their use as antidiabetic agents, a better knowledge of sulfonylureas might make them a serendipitous discover in oncology.
This article is available for continuing medical education credit at CME.TheOncologist.com.
Acknowledgments
Our preliminary results reported in the present review are part of work supported by a European Society of Medical Oncology translational research fellowship awarded in 2010 to Giulia Pasello.
Author Contributions
Conception/Design: Giulia Pasello
Provision of study material or patients: Giulia Pasello, Loredana Urso
Collection and/or assembly of data: Giulia Pasello, Loredana Urso
Data analysis and interpretation: Giulia Pasello, Pierfranco Conte, Adolfo Favaretto
Manuscript writing: Giulia Pasello
Final approval of manuscript: Giulia Pasello, Pierfranco Conte, Adolfo Favaretto
Disclosures
The authors indicated no financial relationships.
Section editors: Jan Schellens: oral taxanes (IP); Merck, AstraZeneca, Lilly, Roche (C/A); Roche, AstraZeneca (RF); Roche, Merck, AstraZeneca, MerckSerono, Eisai, Pfizer, Bayer, Novartis, GlaxoSmithKline (PI of sponsored trials); Eddie Reed: BristolMeyers Squibb (IP); Ortho Biotech (H); Protea Biosciences (O); William D. Figg: None
Reviewer “A”: None
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Noto H, Tsujimoto T, Sasazuki T, et al. Significantly increased risk of cancer in patients with diabetes mellitus: A systematic review and meta-analysis. Endocr Pract. 2011;17:616–628. doi: 10.4158/EP10357.RA. [DOI] [PubMed] [Google Scholar]
- 2.Vigneri P, Frasca F, Sciacca L, et al. Diabetes and cancer. Endocr Relat Cancer. 2009;16:1103–1123. doi: 10.1677/ERC-09-0087. [DOI] [PubMed] [Google Scholar]
- 3.Gandini S, Guerrieri-Gonzaga A, Puntoni M, et al. Metformin and breast cancer risk. J Clin Oncol. 2013;31:973–974. doi: 10.1200/JCO.2012.46.3596. [DOI] [PubMed] [Google Scholar]
- 4.Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8:915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 5.Monami M, Lamanna C, Balzi D, et al. Sulphonylureas and cancer: A case-control study. Acta Diabetol. 2009;46:279–284. doi: 10.1007/s00592-008-0083-2. [DOI] [PubMed] [Google Scholar]
- 6.Bowker SL, Majumdar SR, Veugelers P, et al. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin: Response to Farooki and Schneider. Diabetes Care. 2006;29:1990–1991. doi: 10.2337/dc06-0997. [DOI] [PubMed] [Google Scholar]
- 7.Currie CJ, Poole CD, Gale EA. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia. 2009;52:1766–1777. doi: 10.1007/s00125-009-1440-6. [DOI] [PubMed] [Google Scholar]
- 8.Evans JM, Donnelly LA, Emslie-Smith AM, et al. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thakkar B, Aronis KN, Vamvini MT, et al. Metformin and sulfonylureas in relation to cancer risk in type II diabetes patients: A meta-analysis using primary data of published studies. Metabolism. 2013;62:922–934. doi: 10.1016/j.metabol.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 10.Martin M, Marais R. Metformin: A diabetes drug for cancer, or a cancer drug for diabetics? J Clin Oncol. 2012;30:2698–2700. doi: 10.1200/JCO.2012.42.1677. [DOI] [PubMed] [Google Scholar]
- 11.Yang X, So WY, Ma RC, et al. Use of sulphonylurea and cancer in type 2 diabetes—the Hong Kong Diabetes Registry. Diabetes Res Clin Pract. 2010;90:343–351. doi: 10.1016/j.diabres.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 12.Abdul M, Hoosein N. Expression and activity of potassium ion channels in human prostate cancer. Cancer Lett. 2002;186:99–105. doi: 10.1016/s0304-3835(02)00348-8. [DOI] [PubMed] [Google Scholar]
- 13.Abdul M, Hoosein N. Voltage-gated sodium ion channels in prostate cancer: Expression and activity. Anticancer Res. 2002;22:1727–1730. [PubMed] [Google Scholar]
- 14.Ardehali H, O'Rourke B. Mitochondrial K(ATP) channels in cell survival and death. J Mol Cell Cardiol. 2005;39:7–16. doi: 10.1016/j.yjmcc.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonnet S, Archer SL, Allalunis-Turner J, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 16.Conti M. Targeting K+ channels for cancer therapy. J Exp Ther Oncol. 2004;4:161–166. [PubMed] [Google Scholar]
- 17.Fava D, Cassone-Faldetta M, Laurenti O, et al. Gliclazide improves anti-oxidant status and nitric oxide-mediated vasodilation in type 2 diabetes. Diabet Med. 2002;19:752–757. doi: 10.1046/j.1464-5491.2002.00762.x. [DOI] [PubMed] [Google Scholar]
- 18.Felipe A, Vicente R, Villalonga N, et al. Potassium channels: New targets in cancer therapy. Cancer Detect Prev. 2006;30:375–385. doi: 10.1016/j.cdp.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 19.Golstein PE, Boom A, van Geffel J, et al. P-glycoprotein inhibition by glibenclamide and related compounds. Pflugers Arch. 1999;437:652–660. doi: 10.1007/s004240050829. [DOI] [PubMed] [Google Scholar]
- 20.Hamon Y, Luciani MF, Becq F, et al. Interleukin-1beta secretion is impaired by inhibitors of the Atp binding cassette transporter, ABC1. Blood. 1997;90:2911–2915. [PubMed] [Google Scholar]
- 21.Lautier D, Canitrot Y, Deeley RG, et al. Multidrug resistance mediated by the multidrug resistance protein (MRP) gene. Biochem Pharmacol. 1996;52:967–977. doi: 10.1016/0006-2952(96)00450-9. [DOI] [PubMed] [Google Scholar]
- 22.Malhi H, Irani AN, Rajvanshi P, et al. KATP channels regulate mitogenically induced proliferation in primary rat hepatocytes and human liver cell lines. Implications for liver growth control and potential therapeutic targeting. J Biol Chem. 2000;275:26050–26057. doi: 10.1074/jbc.M001576200. [DOI] [PubMed] [Google Scholar]
- 23.Nunez M, Medina V, Cricco G, et al. Glibenclamide inhibits cell growth by inducing G0/G1 arrest in the human breast cancer cell line MDA-MB-231. BMC Pharmacol Toxicol. 2013;14:6. doi: 10.1186/2050-6511-14-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Payen L, Delugin L, Courtois A, et al. The sulphonylurea glibenclamide inhibits multidrug resistance protein (MRP1) activity in human lung cancer cells. Br J Pharmacol. 2001;132:778–784. doi: 10.1038/sj.bjp.0703863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qian X, Li J, Ding J, et al. Glibenclamide exerts an antitumor activity through reactive oxygen species-c-jun NH2-terminal kinase pathway in human gastric cancer cell line MGC-803. Biochem Pharmacol. 2008;76:1705–1715. doi: 10.1016/j.bcp.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 26.Sheppard DN, Robinson KA. Mechanism of glibenclamide inhibition of cystic fibrosis transmembrane conductance regulator Cl- channels expressed in a murine cell line. J Physiol. 1997;503:333–346. doi: 10.1111/j.1469-7793.1997.333bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stieger B, Fattinger K, Madon J, et al. Drug- and estrogen-induced cholestasis through inhibition of the hepatocellular bile salt export pump (Bsep) of rat liver. Gastroenterology. 2000;118:422–430. doi: 10.1016/s0016-5085(00)70224-1. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki Y, Inoue T, Murai M, et al. Depolarization potentiates TRAIL-induced apoptosis in human melanoma cells: Role for ATP-sensitive K+ channels and endoplasmic reticulum stress. Int J Oncol. 2012;41:465–475. doi: 10.3892/ijo.2012.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H, Zhang Y, Cao L, et al. HERG K+ channel, a regulator of tumor cell apoptosis and proliferation. Cancer Res. 2002;62:4843–4848. [PubMed] [Google Scholar]
- 30.Wondergem R, Cregan M, Strickler L, et al. Membrane potassium channels and human bladder tumor cells: II. Growth properties. J Membr Biol. 1998;161:257–262. doi: 10.1007/s002329900332. [DOI] [PubMed] [Google Scholar]
- 31.Woodfork KA, Wonderlin WF, Peterson VA, et al. Inhibition of ATP-sensitive potassium channels causes reversible cell-cycle arrest of human breast cancer cells in tissue culture. J Cell Physiol. 1995;162:163–171. doi: 10.1002/jcp.1041620202. [DOI] [PubMed] [Google Scholar]
- 32.Yasukagawa T, Niwa Y, Simizu S, et al. Suppression of cellular invasion by glybenclamide through inhibited secretion of platelet-derived growth factor in ovarian clear cell carcinoma ES-2 cells. FEBS Lett. 2012;586:1504–1509. doi: 10.1016/j.febslet.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 33.Zhanping W, Xiaoyu P, Na C, et al. Voltage-gated K+ channels are associated with cell proliferation and cell cycle of ovarian cancer cell. Gynecol Oncol. 2007;104:455–460. doi: 10.1016/j.ygyno.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 34.Zhou Q, Kwan HY, Chan HC, et al. Blockage of voltage-gated K+ channels inhibits adhesion and proliferation of hepatocarcinoma cells. Int J Mol Med. 2003;11:261–266. [PubMed] [Google Scholar]
- 35.Pasello G, Urso L, Silic-Benussi M, et al. International Mesothelioma Interest Group. Glibenclamide sensitizes malignant pleural mesothelioma cells and primary cultures to TRAIL-mediated apoptosis. International Conference of the International Mesothelioma Interest Group 2012 Abstract Book; September 11–14, 2012; Boston, MA. 2012. [Google Scholar]
- 36.Mizuno CS, Chittiboyina AG, Kurtz TW, et al. Type 2 diabetes and oral antihyperglycemic drugs. Curr Med Chem. 2008;15:61–74. doi: 10.2174/092986708783330656. [DOI] [PubMed] [Google Scholar]
- 37.Ashcroft FM. AT. P-sensitive potassium channelopathies: Focus on insulin secretion. J Clin Invest. 2005;115:2047–2058. doi: 10.1172/JCI25495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abdul M, Hoosein N. Voltage-gated potassium ion channels in colon cancer. Oncol Rep. 2002;9:961–964. [PubMed] [Google Scholar]
- 39.Sliwinska A, Blasiak J, Drzewoski J. Effect of gliclazide on DNA damage in human peripheral blood lymphocytes and insulinoma mouse cells. Chem Biol Interact. 2006;162:259–267. doi: 10.1016/j.cbi.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 40.O'Brien RC, Luo M, Balazs N, et al. In vitro and in vivo antioxidant properties of gliclazide. J Diabetes Complications. 2000;14:201–206. doi: 10.1016/s1056-8727(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 41.Sliwinska A, Blasiak J, Kasznicki J, et al. In vitro effect of gliclazide on DNA damage and repair in patients with type 2 diabetes mellitus (T2DM) Chem Biol Interact. 2008;173:159–165. doi: 10.1016/j.cbi.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 42.Pan HZ, Chang D, Feng LG, et al. Oxidative damage to DNA and its relationship with diabetic complications. Biomed Environ Sci. 2007;20:160–163. [PubMed] [Google Scholar]
- 43.Sliwinska A, Sliwinski T, Kasznicki J, et al. Effect of gliclazide on nucleotide excision repair (NER) and non-homologous DNA end joining (NHEJ) in normal and cancer cells. J Physiol Pharmacol. 2010;61:347–353. [PubMed] [Google Scholar]
- 44.Sawada F, Inoguchi T, Tsubouchi H, et al. Differential effect of sulfonylureas on production of reactive oxygen species and apoptosis in cultured pancreatic beta-cell line, MIN6. Metabolism. 2008;57:1038–1045. doi: 10.1016/j.metabol.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 45.Noda Y, Mori A, Cossins E, et al. Gliclazide scavenges hydroxyl and superoxide radicals: An electron spin resonance study. Metabolism. 2000;49:14–16. doi: 10.1016/s0026-0495(00)80079-7. [DOI] [PubMed] [Google Scholar]
- 46.Gier B, Krippeit-Drews P, Sheiko T, et al. Suppression of KATP channel activity protects murine pancreatic beta cells against oxidative stress. J Clin Invest. 2009;119:3246–3256. doi: 10.1172/JCI38817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Corgnali M, Piconi L, Ihnat M, et al. Evaluation of gliclazide ability to attenuate the hyperglycaemic ‘memory’ induced by high glucose in isolated human endothelial cells. Diabetes Metab Res Rev. 2008;24:301–309. doi: 10.1002/dmrr.804. [DOI] [PubMed] [Google Scholar]
- 48.Kimoto K, Suzuki K, Kizaki T, et al. Gliclazide protects pancreatic beta-cells from damage by hydrogen peroxide. Biochem Biophys Res Commun. 2003;303:112–119. doi: 10.1016/s0006-291x(03)00310-3. [DOI] [PubMed] [Google Scholar]
- 49.Sliwinska A, Rogalska A, Szwed M, et al. Gliclazide may have an antiapoptotic effect related to its antioxidant properties in human normal and cancer cells. Mol Biol Rep. 2012;39:5253–5267. doi: 10.1007/s11033-011-1323-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taylor CW, Alberts DS, Ketcham MA, et al. Clinical pharmacology of a novel diarylsulfonylurea anticancer agent. J Clin Oncol. 1989;7:1733–1740. doi: 10.1200/JCO.1989.7.11.1733. [DOI] [PubMed] [Google Scholar]
- 51.Grindey GB, Boder GB, Grossman CS, et al. Further development of diarylsulfonylureas as novel anticancer drugs. Proc Am Assoc Cancer Res. 1987;28:309. [Google Scholar]
- 52.Mohamadi F, Spees MM, Grindey GB. Sulfonylureas: A new class of cancer chemotherapeutic agents. J Med Chem. 1992;35:3012–3016. doi: 10.1021/jm00094a013. [DOI] [PubMed] [Google Scholar]
- 53.Houghton PJ, Houghton JA. Antitumor diarylsulfonylureas: Novel agents with unfulfilled promise. Invest New Drugs. 1996;14:271–280. doi: 10.1007/BF00194530. [DOI] [PubMed] [Google Scholar]
- 54.Taylor CW, Alberts DS, Peng YM, et al. Antitumor activity and clinical pharmacology of sulofenur in ovarian cancer. J Natl Cancer Inst. 1992;84:1798–1802. doi: 10.1093/jnci/84.23.1798. [DOI] [PubMed] [Google Scholar]
- 55.Pratt CB, Bowman LC, Marina N, et al. A phase I study of sulofenur in refractory pediatric malignant solid tumors. Invest New Drugs. 1995;13:63–66. doi: 10.1007/BF02614222. [DOI] [PubMed] [Google Scholar]
- 56.Grindey GB. Current status of cancer drug development: Failure or limited success? Cancer Cells. 1990;2:163–171. [PubMed] [Google Scholar]
- 57.Howbert JJ, Grossman CS, Crowell TA, et al. Novel agents effective against solid tumors: The diarylsulfonylureas. Synthesis, activities, and analysis of quantitative structure-activity relationships J Med Chem. 1990;33:2393–2407. doi: 10.1021/jm00171a013. [DOI] [PubMed] [Google Scholar]
- 58.Houghton PJ, Bailey FC, Houghton JA, et al. Evidence for mitochondrial localization of N-(4-methylphenylsulfonyl)-N′-(4-chlorophenyl)urea in human colon adenocarcinoma cells. Cancer Res. 1990;50:664–668. [PubMed] [Google Scholar]
- 59.Thakar JH, Chapin C, Berg RH, et al. Effect of antitumor diarylsulfonylureas on in vivo and in vitro mitochondrial structure and functions. Cancer Res. 1991;51:6286–6291. [PubMed] [Google Scholar]
- 60.Neubauer BL, Merriman RL, Best KL, et al. Inhibition of PAIII rat prostatic adenocarcinoma growth and metastasis by a new diarylsulfonylurea antitumor agent, LY181984. J Urol. 1992;147:500–504. doi: 10.1016/s0022-5347(17)37288-9. [DOI] [PubMed] [Google Scholar]
- 61.Diab SG, Hilsenbeck SG, Izbicka E, et al. Significant activity of a novel cytotoxic agent, LY295501, against a wide range of tumors in the human tumor cloning system. Anticancer Drugs. 1999;10:303–307. doi: 10.1097/00001813-199903000-00009. [DOI] [PubMed] [Google Scholar]
- 62.Forouzesh B, Takimoto CH, Goetz A, et al. A phase I and pharmacokinetic study of ILX-295501, an oral diarylsulfonylurea, on a weekly for 3 weeks every 4-week schedule in patients with advanced solid malignancies. Clin Cancer Res. 2003;9:5540–5549. [PubMed] [Google Scholar]
- 63.Schultz RM, Andis SL, Toth JE, et al. Effect of albumin on antitumor activity of diarylsulfonylureas. Anticancer Res. 1993;13:1939–1943. [PubMed] [Google Scholar]
- 64.Hwang HS, Moon EY, Seong SK, et al. Characterization of the anticancer activity of DW2282, a new anticancer agent. Anticancer Res. 1999;19:5087–5093. [PubMed] [Google Scholar]
- 65.Lee CW, Hong DH, Han SB, et al. A novel stereo-selective sulfonylurea, 1-[1-(4-aminobenzoyl)-2,3-dihydro-1H-indol-6-sulfonyl]-4-phenyl-imidazolidin-2-on e, has antitumor efficacy in in vitro and in vivo tumor models. Biochem Pharmacol. 2002;64:473–480. doi: 10.1016/s0006-2952(02)01105-x. [DOI] [PubMed] [Google Scholar]
- 66.Piao W, Yoo J, Lee DK, et al. Induction of G(2)/M phase arrest and apoptosis by a new synthetic anti-cancer agent, DW2282, in promyelocytic leukemia (HL-60) cells. Biochem Pharmacol. 2001;62:1439–1447. doi: 10.1016/s0006-2952(01)00796-1. [DOI] [PubMed] [Google Scholar]
- 67.Kim S, Park JH, Koo SY, et al. Novel diarylsulfonylurea derivatives as potent antimitotic agents. Bioorg Med Chem Lett. 2004;14:6075–6078. doi: 10.1016/j.bmcl.2004.09.069. [DOI] [PubMed] [Google Scholar]
- 68.Jee SH, Ohrr H, Sull JW, et al. Fasting serum glucose level and cancer risk in Korean men and women. JAMA. 2005;293:194–202. doi: 10.1001/jama.293.2.194. [DOI] [PubMed] [Google Scholar]
- 69.Yeh HC, Platz EA, Wang NY, et al. A prospective study of the associations between treated diabetes and cancer outcomes. Diabetes Care. 2012;35:113–118. doi: 10.2337/dc11-0255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee MY, Lin KD, Hsiao PJ, et al. The association of diabetes mellitus with liver, colon, lung, and prostate cancer is independent of hypertension, hyperlipidemia, and gout in Taiwanese patients. Metabolism. 2012;61:242–249. doi: 10.1016/j.metabol.2011.06.020. [DOI] [PubMed] [Google Scholar]
- 71.Lo SF, Chang SN, Muo CH, et al. Modest increase in risk of specific types of cancer types in type 2 diabetes mellitus patients. Int J Cancer. 2013;132:182–188. doi: 10.1002/ijc.27597. [DOI] [PubMed] [Google Scholar]
- 72.Hense HW, Kajuter H, Wellmann J, et al. Cancer incidence in type 2 diabetes patients - first results from a feasibility study of the D2C cohort. Diabetol Metab Syndr. 2011;3:15. doi: 10.1186/1758-5996-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Geraldine N, Marc A, Carla T, et al. Relation between diabetes, metformin treatment and the occurrence of malignancies in a Belgian primary care setting. Diabetes Res Clin Pract. 2012;97:331–336. doi: 10.1016/j.diabres.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 74.Zhang PH, Chen ZW, Lv D, et al. Increased risk of cancer in patients with type 2 diabetes mellitus: A retrospective cohort study in China. BMC Public Health. 2012;12:567. doi: 10.1186/1471-2458-12-567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Noto H, Osame K, Sasazuki T, et al. Substantially increased risk of cancer in patients with diabetes mellitus: A systematic review and meta-analysis of epidemiologic evidence in Japan. J Diabetes Complications. 2010;24:345–353. doi: 10.1016/j.jdiacomp.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 76.Habib SL, Rojna M. Diabetes and risk of cancer. ISRN Oncol. 2013 doi: 10.1155/2013/583786. 583786, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Soranna D, Scotti L, Zambon A, et al. Cancer risk associated with use of metformin and sulfonylurea in type 2 diabetes: A meta-analysis. The Oncologist. 2012;17:813–822. doi: 10.1634/theoncologist.2011-0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bosco JL, Antonsen S, Sorensen HT, et al. Metformin and incident breast cancer among diabetic women: A population-based case-control study in Denmark. Cancer Epidemiol Biomarkers Prev. 2011;20:101–111. doi: 10.1158/1055-9965.EPI-10-0817. [DOI] [PubMed] [Google Scholar]
- 79.Bodmer M, Meier C, Krahenbuhl S, et al. Long-term metformin use is associated with decreased risk of breast cancer. Diabetes Care. 2010;33:1304–1308. doi: 10.2337/dc09-1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chlebowski RT, McTiernan A, Wactawski-Wende J, et al. Diabetes, metformin, and breast cancer in postmenopausal women. J Clin Oncol. 2012;30:2844–2852. doi: 10.1200/JCO.2011.39.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Boyle P, Boniol M, Koechlin A, et al. Diabetes and breast cancer risk: A meta-analysis. Br J Cancer. 2012;107:1608–1617. doi: 10.1038/bjc.2012.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Goodwin PJ, Thompson AM, Stambolic V. Diabetes, metformin, and breast cancer: Lilac time? J Clin Oncol. 2012;30:2812–2814. doi: 10.1200/JCO.2012.42.3319. [DOI] [PubMed] [Google Scholar]
- 83.Jiralerspong S, Palla SL, Giordano SH, et al. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol. 2009;27:3297–3302. doi: 10.1200/JCO.2009.19.6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bonanni B, Puntoni M, Cazzaniga M, et al. Dual effect of metformin on breast cancer proliferation in a randomized presurgical trial. J Clin Oncol. 2012;30:2593–2600. doi: 10.1200/JCO.2011.39.3769. [DOI] [PubMed] [Google Scholar]
- 85.Goodwin PJ, Ligibel JA, Stambolic V. Metformin in breast cancer: Time for action. J Clin Oncol. 2009;27:3271–3273. doi: 10.1200/JCO.2009.22.1630. [DOI] [PubMed] [Google Scholar]
- 86.Schumacker PT. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell. 2006;10:175–176. doi: 10.1016/j.ccr.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 87.Wang J, Yi J. Cancer cell killing via ROS: To increase or decrease, that is the question. Cancer Biol Ther. 2008;7:1875–1884. doi: 10.4161/cbt.7.12.7067. [DOI] [PubMed] [Google Scholar]
- 88.Kudin AP, Debska-Vielhaber G, Kunz WS. Characterization of superoxide production sites in isolated rat brain and skeletal muscle mitochondria. Biomed Pharmacother. 2005;59:163–168. doi: 10.1016/j.biopha.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 89.Szewczyk A, Kajma A, Malinska D, et al. Pharmacology of mitochondrial potassium channels: Dark side of the field. FEBS Lett. 2010;584:2063–2069. doi: 10.1016/j.febslet.2010.02.048. [DOI] [PubMed] [Google Scholar]