

Abstract
AIM
To review recent developments in the understanding, diagnosis, and treatment of Sturge–Weber syndrome (SWS).
METHOD
Members of the Brain Vascular Malformation Consortium Sturge–Weber National Workgroup contributed their expertise, to review the literature, and present promising directions for research.
RESULTS
The increasing number of reports dealing with SWS over the last decade reflects progress in the diagnosis and understanding of the neurological involvement. The proliferation of centers and advocacy groups to care for patients with SWS and to stimulate research has aided the development of new insights into the clinical manifestations and the pathophysiology of neurological progression, and the development of novel hypotheses to direct future research. Many key questions remain, but the tools and networks to answer them are being developed.
INTERPRETATION
This review summarizes important new knowledge and presents new research directions that are likely to provide further insights, earlier diagnosis, improved treatments, and possibly, prevention of this syndrome.
A new effort funded in the United States by the National Institutes of Health (NIH), the National Institute of Neurological Disorders and Stroke, and the NIH Office of Rare Diseases Research has been initiated to facilitate novel, coordinated research among centers on the problem of Sturge–Weber syndrome (SWS). This initiative, the Brain Vascular Malformation Consortium, assembles patients, advocates, and basic and clinical researchers to establish a national database, develop new biomarkers, and identify the cause of SWS. The etiology of SWS is unknown, and no true animal model exists. Early presymptomatic diagnosis is limited by the low sensitivity of neuroimaging in young infants and accurate prognostication is constrained by the clinical variability of SWS. Treatment remains controversial.
Recent research has provided new insights into the cause of SWS and has pointed to promising directions for diagnosis and treatment. We review these new insights, describe the novel approaches being developed, and outline research goals to improve the understanding, diagnosis, and treatment of SWS.
ADVANCES AND QUESTIONS IN THE DEFINITION, NEUROLOGICAL SYMPTOMS, AND PRESENTATION OF SWS
SWS (also known as encephalofacial angiomatosis) is a sporadic congenital condition marked by capillary malformations of the skin (port-wine birthmark, also known as port-wine stain/nevus) in the V1 distribution of the face (forehead and/or eyelid). This may occur alone or in combination with the V2 and V3 distributions, by cerebral venous malformations (leptomeningeal angiomatosis), and by glaucoma with ocular capillary venous vascular malformations.1,2
Of all patients who present with a facial port-wine nevus, only 8 to 20% develop neurological symptoms, so the presence of a port-wine nevus is not sufficient to diagnose SWS.3,4 If the port-wine nevus includes the ophthalmic division of the trigeminal nerve, particularly the eyelid, the likelihood of SWS increases.3 When there is a unilateral facial nevus, the leptomeningeal angiomatosis tends to be ipsilateral. Conversely, rarely, a patient will have radiological features of SWS but no facial involvement.5,6 These individuals may be less likely to have occipital lobe brain involvement (and more likely to have only frontal/parietal involvement). A bilateral port-wine nevus is associated with a higher risk of SWS brain involvement.1 There are many questions about the spectrum of SWS, such as which factors have the greatest prognostic value and how the spectrum correlates with etiology.
Occasionally, children with SWS also have extensive cutaneous capillary malformations, limb hypertrophy, and vascular and lymphatic malformations that collectively are consistent with Klippel–Trénaunay syndrome; these children have been classified as having Klippel–Trénaunay–Weber syndrome. Whether there is truly an overlap between these two rare syndromes or whether they are distinct entities is unknown.7-9
Epilepsy, cognitive impairment, disorders of behaviour, including attention deficit disorders, headache, spastic hemiparesis, and visual field defects are common.10,11 Transient ischemic attacks and strokes also occur.12 The tremendous variations in the extent of neurological involvement and in the severity of symptoms present great challenges in caring for patients with SWS and in counseling their families. The neurological features can vary in severity and may evolve over time. Pascual-Castroviejo and colleagues noted in their series of 55 patients with SWS with long-term follow-up that the intracranial involvement frequently progresses during early childhood but not later.13 The site and extent of brain involvement influence the neurological manifestations. Leptomeningeal angiomatosis typically involves the parietal and occipital lobes but may involve the entire hemisphere.14 Bilateral cerebral involvement is uncommon.15-17 Experimental blood flow studies using the 133Xe single-photon emission computed tomography (SPECT) technique have shown decreased flow in the involved cerebral region, and one blood flow study showed that a seizure can cause a significant reduction of flow in remote, uninvolved cerebral regions, in effect a vascular steal phenomenon.1
Epilepsy develops in 75 to 80% of average patients with SWS; up to 93% of those who have bilateral cerebral involvement will develop epilepsy.15,18 A questionnaire survey of participants in the Sturge-Weber Foundation (SWF) registry found that seizures manifest in the first year of life in about 75% of those who develop epilepsy.19 Typical seizures are partial (either simple or complex partial), but infantile spasms and myoclonic seizures may occur.20-22 There is limited evidence that seizures in young children may occur in clusters separated by seizure-free periods.23 Some reports suggest that early-onset seizures correlate with more severe epilepsy, more cognitive impairment, and greater hemiparesis, but there is little longitudinal evidence to support this contention.14,23 Prospectively gathered natural history data are critical to advancing our knowledge of prognostic factors, developing effective biomarkers, and guiding the development of new treatments.
Cognition may vary from normal to developmental delay and from mild learning disabilities to severe intellectual disability. In another survey of patients with SWS from the SWF registry, the need for special education varied widely, but increased greatly when epilepsy was an associated diagnosis.10 Better treatments are needed in this area to improve long-term outcomes. A major goal must be to diagnose brain involvement earlier and to administer neuroprotective measures before irreversible brain damage can occur. Factors associated with developmental delay include bilateral cerebral lesions, the extent of cerebral atrophy, the presence of intractable epilepsy, and the presence of multiple types of epileptic seizures. Bilateral cerebral involvement is associated with normal cognition in only 10% of patients with SWS.15
Behavior problems also occur in patients with SWS. In a survey that compared patients with SWS with their siblings, the patients had more problems in a range of behavioral domains, including poorer social skills, more mood problems, and poorer compliance.24 Lower cognitive function, epilepsy, and greater frequency of seizures are associated with more psychological problems. In one case series, the presence of hemiparesis was associated with poorer adaptive functioning.25 Therefore, hemiparesis in a child with SWS may signal the need for neuropsychological assessment to guide treatment and educational needs.
Headaches in patients with SWS have been reported to occur commonly. The evidence primarily comes from two patient surveys (including one from the SWF registry) as well as isolated case reports.26,27 In the first study, 14% of the SWF registry responded to a mail survey regarding the frequency and type of headache. Of the respondents, 28% reported symptoms consistent with migraine, which is more frequent than the 17% of females and 5% of males in the general population who have migraine.26 In a subsequent anonymous online survey of individuals with SWS performed by SWF, 74 of 104 respondents reported migraine headaches.28 In many older patients, migraines are more an impairment than seizures; this issue deserves further study (see ‘Updates in the treatment of Sturge–Weber syndrome’ below). Since selection bias is possible in self-reported surveys, the frequencies of headache derived from these studies should be considered to be preliminary.
Based on the location of the leptomeningeal angiomatosis, one would not be surprised that spastic hemiparesis and visual field defects occur in patients with SWS. What is noteworthy is that no prevalence data for these two neurological complications have been reported to date.
An intriguing and perplexing problem is the occurrence of stroke-like episodes marked by transient hemiparesis or visual field deficits. These episodes may be difficult to distinguish from postictal Todd’s paresis, as is illustrated by two recently reported cases.29 However, the stroke-like episodes in SWS are frequently more prolonged than a postictal paresis and may last days, weeks, or months, or become permanent. Several small case series suggest that SWS patients may have regional cerebral hypoperfusion where areas are at risk for sustained ischemia. In one brain magnetic resonance imaging (MRI) study, apparent diffusion coefficients were elevated in the white matter of 15 participants with SWS compared with an unaffected comparison group.30 One single-patient case report described concurrent transient hemiparesis and abnormal magnetic resonance brain diffusion in the corresponding contralateral hemisphere.31 These stroke-like episodes are commonly triggered by minor head injury in toddlers with SWS.32 No data address the prevalence of these episodes, and estimates of their frequency vary widely. In one series, 119 stroke-like episodes occurred in 20 children over 2 years; the episodes were less frequent in those patients treated with aspirin.17 In another series of 55 patients, no participant reported stroke-like episodes, but it was unclear whether they were questioned about this symptom.13 From our experience, patients or family members often do not understand the term ‘stroke-like episode’ unless it is explained to them. This topic needs a functional, agreed-upon definition of ‘a stroke-like episode’ if research to understand these episodes is to progress.
Endocrine disorders are a newly recognized aspect of SWS that deserves mention. Germain-Lee and colleagues, in collaboration with SWF, determined that patients with SWS have a greatly increased risk of growth hormone deficiency.33 In addition, Comi et al. reported the occurrence of central hypothyroidism in patients with SWS, which may relate to anticonvulsant use and is important to diagnose and treat.8 The exact nature of the interaction between the SWS vascular malformation and endocrine disorders is not well understood and should be the focus of future study.
DIAGNOSIS OF SWS BRAIN INVOLVEMENT
The diagnosis of SWS is straightforward in an individual with a port-wine nevus, glaucoma, clinical evidence of cerebral involvement, and neuroimaging confirmation. Diagnosis is far more difficult, however, in a neonate who has a facial port-wine birthmark and who is neurologically asymptomatic. Computed tomography (CT) and contrast-enhanced brain MRI may not detect abnormalities in a newborn infant. Scanning may need to be repeated at ages 1 to 2 years in uncertain cases.34 Epileptic seizures may not appear until some time in the first 3 years of life and occasionally even later.19 Several imaging approaches have been reported, including the use of post-contrast fluid-attenuated inversion recovery (FLAIR) and susceptibility-weighted imaging (SWI) MRI, but the reports were based on small numbers of cases so that selectivity and sensitivity testing of these imaging methods has yet to be performed.35,36 In older symptomatic children, the typical MRI findings include leptomeningeal enhancement, dilated deep draining vessels (venous malformation), and a glomus malformation (enlarged, enhancing choroid plexus). The ‘Neuroimaging and pathogenesis’ section below presents more details.
NEUROIMAGING AND PATHOGENESIS
Neuroimaging, particularly MRI, is used extensively to establish the diagnosis of SWS and to evaluate the extent and severity of intracranial involvement. Advanced structural and functional neuroimaging techniques contribute valuable knowledge toward increasing understanding about the pathophysiology of disease progression.
The conventional criterion standard for the radiological diagnosis of SWS is a T1-weighted brain MRI with gadolinium contrast. This technique delineates the leptomeningeal angioma and can demonstrate an enlarged choroid plexus and dilated transmedullary as well as subependymal veins. SWI MRI is a recently developed, contrast-free MRI technique that is superior to conventional gadolinium-enhanced MRI for showing fine details of deep transmedullary and periventricular veins. Case reports suggest these dilated veins may occur in children with SWS before conventional MRI shows definite structural changes.37,38 SWI detects with great sensitivity the calcified cortex in SWS (Fig. 1). Together, SWI scans and T1-weighted, gadolinium-enhanced scans provide complementary information regarding brain abnormalities in SWS. When possible, SWI should be incorporated into clinical brain MRI protocols when scanning patients with suspected SWS.39 Perfusion-weighted imaging generates maps of cerebral blood volume and blood flow that can show decreased perfusion in areas with pial enhancement and, in some cases, in adjacent areas.40,41 In SWS the hypoperfused parenchyma can correlate with motor deficits and disability scores.41 A recent study also demonstrated that decreased perfusion in the affected hemisphere is associated with frequent seizures, long duration of epilepsy, and brain atrophy.42 These preliminary perfusion-weighted imaging results suggest that low perfusion areas are detrimental as they could contribute to both seizures and neurological deficit in SWS. MR spectroscopy has identified abnormal metabolism in regions that macroscopically appear normal. Recently, in a cohort of 16 children with unilateral SWS, MR spectroscopy demonstrated abnormal neuronal and cell membrane marker levels in frontal lobes that appeared normal on conventional MRI.43 In those children, tissue concentrations of N-acetyl-aspartate and choline were significantly decreased in the frontal lobes, spectroscopic changes that have been associated with early-onset seizures and poor motor functions.43
Figure 1.
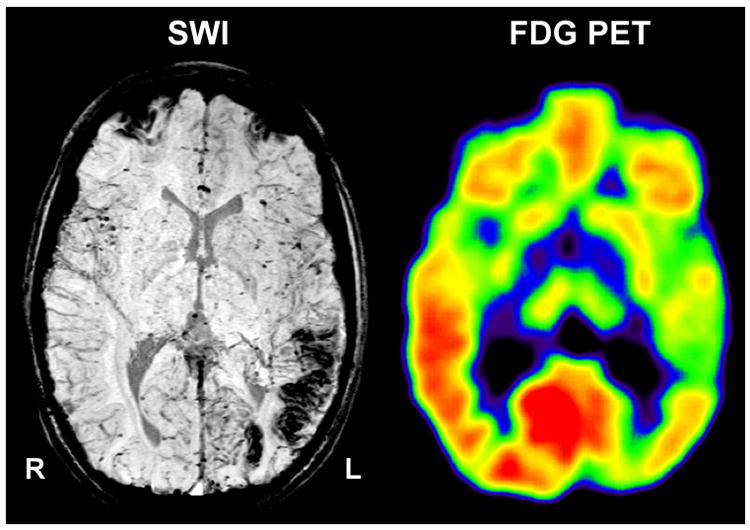
Co-registered susceptibility weighted imaging (SWI) and 2-deoxy-2[18F]fluoro-D-glucose positron emission tomography (FDG-PET) in a child with Sturge–Weber syndrome and left hemispheric involvement. Native SWI shows calcified areas in the left occipital and posterior temporal cortex. Hypometabolism on PET extends into the anterior temporal cortex.
Recent MRI studies have combined diffusion-weighted imaging and MRI volumetry to show clinically significant subcortical abnormalities in patients with SWS. Increased apparent diffusion coefficients in normal-appearing hemispheric white matter and the pons suggest that subcortical abnormalities may contribute to neurocognitive deficits.30 Hemispheric white matter atrophy independently predicts cognitive dysfunction in children with unilateral SWS.44 Thalamic diffusion abnormalities have been strongly correlated with IQ measures.45 Diffusion tensor imaging can provide further information by allowing the study of white matter tract integrity and enabling the detection of early changes in specific cerebral pathways (such as the corticospinal tract) prior to the onset of neurological symptoms.46 In one case, transient abnormal diffusion was identified concurrent with repeated complex partial seizures.31 Such diffusion changes may indicate focal brain ischemia and/or seizure-induced abnormalities, typically after prolonged or frequent seizures; since seizure control may result in resolution of seizure-induced MRI abnormalities, serial MRI is important to differentiate peri-ictal diffusion from permanent brain ischemia. Positron emission tomography (PET) of brain glucose metabolism can track the extent and severity of cortical dysfunction, which often extends beyond the structural brain abnormalities detected by CT and MRI (Fig. 1).16,47 On longitudinal 18F-deoxyglucose (FDG) PET studies in children with SWS, major metabolic progression typically occurs before 4 years of age and is often related to severe seizures.48 Paradoxical preservation of IQ was observed in children after an early, severe unilateral hemispheric metabolic progression (Fig. 2A). This preservation suggested that early unilateral progression, destroying one hemisphere during the early disease course, may prompt the opposite hemisphere to take over critical cognitive functions.49 Whether such cognitive preservations are long-lasting or are followed by a long-term decline in some cases (as reported in at least one patient following early hemispherectomy) remains to be determined.50 Increased cortical metabolism contralateral to severely hypometabolic cortex may be an imaging marker of such reorganization (Fig. 2B).51 Multimodal imaging studies (comparing MRI and PET images) show that, when structurally intact cortex shows mild hypometabolism, cognitive function is often impaired. This observation raises the possibility that mild cortical dysfunction does not prompt adequate reorganization in other brain regions.47,49 If this is true, perhaps surgical elimination of hypometabolic epileptic cortex may facilitate cognitive improvement even when seizures are not intractable and incapacitating. Advanced multimodal imaging that quantifies abnormal brain structure and function should assess young children with SWS. This multimodal imaging approach may identify those young children who are at greater risk for major neurocognitive deterioration and, therefore, may be candidates for aggressive medical and surgical treatment. As an illustration of this point, a recent study combining diffusion MRI with FDG PET in 20 children with unilateral SWS examined thalamic glucose metabolism.45 When compared with unaffected controls and when correlated with the degree of cortical hypometabolism, children with SWS had significantly higher thalamic glucose metabolic and diffusion asymmetry. The thalamic metabolic asymmetries correlated positively with the degree of cortical hypometabolism, and were strong, independent predictors of poorer cognitive function.
Figure 2.
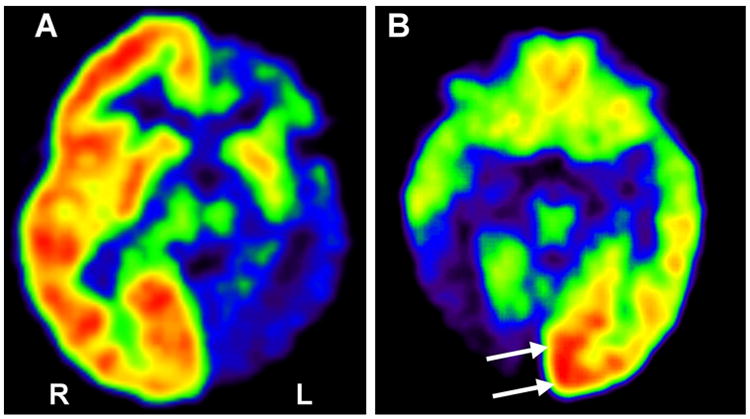
2-deoxy-2[18F]fluoro-D-glucose positron emission tomography (FDG PET)) images of two children with Sturge–Weber syndrome (SWS) and unilateral brain involvement. (a) Severe, extensive hypometabolism in the left hemisphere occurred before 3 years of age; this child had relatively well-preserved cognitive functions (full scale IQ 79, with good verbal skills), suggesting effective reorganization; his seizures became controlled with medication. (b) Increased glucose metabolism (arrows) in the left occipital lobe of a 4-year old child with SWS and severe right occipital damage.
In summary, newer MRI sequences such as SWI have demonstrated more of the structural abnormalities present in patients with SWS and hold promise for increasing our ability to make the early diagnosis of brain involvement in some patients. SWI, along with contrast-enhanced conventional MRI sequences, is strongly recommended for the initial evaluation of infants with suspected SWS. Both diffusion and perfusion MRI are now also available in most centers and can be helpful to assess the extent of brain involvement. Functional neuroimaging with FDG PET should have an increasing role in presurgical planning and prognosis. In addition, it is likely that novel forms of functional neuroimaging will provide us with new insights into the evolution and progression of neurological impairments and perhaps aid selection of optimal treatment for a given patient.
UPDATES IN THE TREATMENT OF SWS
The treatment of SWS brain involvement largely aims at the control of the seizures, headaches and stroke-like episodes. Most treatment outcome data come from surveys, a few small clinical trials, and case series. Important treatment controversies exist, perhaps the most difficult one being the timing of and indications for epilepsy surgery. Given the rarity of SWS, multicentered collaboration will be needed to answer the many important treatment questions.
Glaucoma can present in the newborn infant, so it is important to evaluate neonates for glaucoma and to monitor infants closely for increased intraocular pressure. Topiramate should be used with caution as it can cause acute bilateral closed angle glaucoma.52 This side effect of topiramate is rarely seen in children so it is not an absolute contraindication to use in children, but other anticonvulsants may be better choices.
Stroke-like episodes in children with SWS are common. These episodes are marked by transient neurological deficits that frequently last from several days to weeks. The episodes may occur in the context of a cluster of seizures, may precede a migraine or onset of seizures, or less frequently may occur without apparent seizures or headache. When stroke-like episodes occur after a seizure, they can be distinguished from a Todd paresis by their persistence beyond 24 hours after the seizure. Case reports and clinical experience indicate that stroke-like episodes are commonly triggered by minor head injury in toddlers and young children with SWS.32 Evaluation for suspected episodes includes imaging to exclude brain hemorrhage, which is exceedingly rare, and electroencephalogram (EEG), which may demonstrate ictal activity or slowing. Neuropsychological screening before and after an episode may be used to determine whether the patient fully regains all cognitive and functional skills. Imaging studies during stroke-like episodes may demonstrate a diffusion abnormality without large vessel thrombosis on cerebral angiography.31 Such a finding suggests that the basis of these stroke-like episodes is microvascular stasis.
Whether the prevention of stroke-like episodes improves outcome is unknown. A few studies have examined the use of low-dose aspirin as prophylaxis. Maria et al. reported a cohort in which low-dose aspirin decreased stroke-like episodes.53 Udani et al. reported six children whose seizures greatly decreased after starting low-dose aspirin; one patient’s seizures resurged when the aspirin was temporarily stopped and were effectively suppressed when the aspirin was resumed.54 Aspirin does present risks, as illustrated by a single case of a subgaleal hemorrhage after mild head trauma in a child with SWS on low-dose aspirin.55 What is unanswered by these limited reports is whether low-dose aspirin improves long-term cognitive function and overall quality of life. A hypothetical question is whether it is better to allow the stroke-like episodes to occur at an early age in the hopes that they will ‘burn out’ and then allow plasticity to compensate for any potential ischemic change. Therefore, data on the long-term impact and risks of low-dose aspirin are needed. In addition, there are other approaches to the prevention of stroke-like episodes that have a rational basis but have not been rigorously studied, including providing an annual flu vaccination, excellent hydration during illness, and treatment of fevers.
Migraines often start at an early age in SWS and may precede or follow seizures. A survey of patients with SWS with migraine showed that a subset were safely and effectively treated with triptans.28 The same survey indicated that prophylactic medications are probably underutilized in these patients. Medications such as topiramate, valproic acid, and gabapentin should be considered for migraine prophylaxis in these patients since these medications can effectively prevent seizures and migraines. Equally important is educating the patient about the importance of sufficient, regular sleep, meals, hydration, and exercise in the prevention and control of migraines.
Epilepsy is a frequent complication of SWS. While the control of seizures in some patients may be straightforward, others will have frequent or prolonged seizures. SPECT studies and clinical experience strongly suggest that prolonged seizures in patients with SWS can increase or decrease cerebral perfusion, which may increase the risk of stroke and further brain injury.56 There is some evidence that seizures lead to neurocognitive deterioration over time. Pascual-Castroviejo et al. reported that in 55 participants with SWS followed over 40 years neurocognitive deterioration occurred over time. They noted that levetiracetam provided the best seizure control of the anticonvulsants they tried in this group.13 A comparison of presymptomatic treatment of infants with SWS with phenobarbital with those who were treated after presenting with seizures suggested a better cognitive outcome in the presymptomatically treated group; however, this group had less severe brain involvement and age at seizure onset in the two groups was not mentioned.57 Given this limited evidence, some have advocated aggressive seizure control in the hope of preventing progressive neurological deterioration and improving outcome.58 Aggressive control includes initiation of a chronic antiepileptic drug after the first seizure, adjusting the medication dose in infants and young children every few months for weight gain, and medications to abort acute seizures. Aggressive treatment should be pursued early in infants and young children with SWS in whom the risk of neurocognitive deterioration is greatest. If seizures are not controlled with two or three major antiepileptic drugs used at their effective maximum dose then other treatments should be promptly pursued.
The Atkins diet variation of the ketogenic diet was recently reported to be safe and effective in reducing seizure frequency in five children with SWS who had medically refractory seizures occurring at least monthly.59
The surgical management of medically intractable epilepsy in SWS has been examined in a small number of reports. Surgery for SWS, particularly hemispherectomy, has been performed for the control of medically intractable seizures in unilateral brain involvement.60 Two recent series have demonstrated convincingly that hemispherectomy is more effective in achieving long-term seizure control than focal surgery.61,62 Hemispherectomy, however, results in significant hemiparesis with a hemiparetic gait, little use of the affected hand, and a homonymous hemianopsia. The decision to proceed with hemispherectomy is considerably easier when these deficits are already present and permanent. Therefore, in patients with more focal brain involvement and medically uncontrolled seizures, focal resections should be considered first because of the more limited postoperative morbidity. There is universal agreement that epilepsy surgery is indicated for medically refractory seizures, but the ideal timing can be difficult to establish. Epilepsy surgery in the first months of life must be pursued with caution. An infant may have undiagnosed bilateral brain SWS malformations and in that case should not have surgery. Other patients may experience clusters of seizures separated by many months or years of seizure freedom so that the determination of intractability may require extended observation.23 Nevertheless, a patient with SWS who has epilepsy refractory to at least two seizure medications should be considered for surgery.
Proponents of early surgery argue that early intervention will prevent brain injury and improve outcome, but the evidence is controversial.62,63 Hoffman et al. in 1979 and Ogunmekan et al. in 1989 reported two small series with very promising outcomes.64,65 The latter reported 10 children who underwent hemispherectomy between 1975 and 1987 for medically refractory seizures, extensive hemispheric involvement, and progressive hemiparesis. In eight children who had surgery before 13 months of age, IQ 3 to 12 years later ranged from 88 to 99. They reported that when the surgery was performed early the hemiparesis resolved ‘almost completely’. The two children who had hemispherectomies at ages 8 and 9 years were already severely impaired. Their IQs were 55 and 40 at follow-up; whether this reflected further post-surgical decline was not clear. Shunts were required in three of the eight children who underwent early hemispherectomy. Bourgeois et al. reported a retrospective post-surgical follow-up cohort of 27 children with SWS who were evaluated at between 9 and 17 years 3 months after surgery. Of these, eight had hemispherectomies: three were functional and five were anatomic. The authors concluded that the age when surgery was performed was an important predictor of developmental outcome. Patients who had improved developmental function had had surgery at a mean age of 4 years 5 months (SD 4y 9mo) while those who did not improve had surgery at mean age of 9 years 8 months (SD 4ymo). The limitation with this observation was that four of the patients who ‘did not improve’ had normal development at their last follow-up. If one excludes these four patients then the mean age of those who did not improve was 6 years 3 months and not significantly different from that of those who did improve. If one examines in detail the outcomes of the four cases who had surgery before age 1 year (three hemispherectomies and one partial resection for temporo-parieto-occipital SWS malformation), one had severe developmental delay, two had moderate developmental delay and one had mild developmental delays (based on DQ or IQ) at follow-up, which contrasts with the report of Ogunmekan. In the series of Pascual-Castroviejo et al., early surgery reportedly controlled seizures in two patients, but ‘other neurological problems such as hemiparesis and intellectual deficits showed a less satisfactory response’; these were not characterized further.13 Therefore, the reported outcomes of epilepsy surgery have varied. This is an area where further careful study is needed, particularly since the surgical intervention has evolved. Functional hemispherectomies have replaced anatomic hemispherectomies since they are less likely to require postoperative shunts. A multicenter collaborative study is needed to compare outcomes between early surgery and aggressive medical management. The information gained regarding the optimal timing of epilepsy surgery, patient selection, and optimal medical management will have a major impact upon the treatment of these difficult cases.
In summary, there has been progress in the treatment of the SWS comorbidities of glaucoma, stroke-like episodes, migraine, and epilepsy. There is an urgent need for studies that compare the outcomes of contemporary hemispherectomies with the older anatomic hemispherectomies, and a critical comparison between surgery and aggressive management with the newer anticonvulsants and low-dose aspirin. Three important goals for the future are to: (1) improve education and outreach so the diagnosis of SWS brain involvement is considered at birth in every infant with a facial port-wine birthmark in the V1 distribution; (2) refine presymptomatic screening for brain involvement in young infants suspected to have SWS; and (3) develop better neuroprotective strategies to improve outcome. A small number of studies to date suggest that if treatment can delay the onset of symptoms then this may be enough to improve long-term outcome.1,23 In the future, a better understanding of the pathogenesis of SWS will probably provide new avenues for preventative or neuroprotective treatment.
PATHOGENESIS OF SWS
The somatic mutation model
SWS is a sporadically occurring condition and is noted with equal frequency in males and females.66 The localized abnormalities of blood vessel development and function already described suggest that a disruption of normal development may occur during the first trimester of pregnancy. The abnormal blood vessels are usually localized to a single region on one side of the body. This localization pattern suggests the intriguing hypothesis that somatic mosaicism plays a key role in the pathogenesis of SWS. Somatic mosaicism could explain the genetic basis of several syndromes characterized by the following characteristics: (almost always) sporadic occurrence, distribution of lesions in a scattered or asymmetrical pattern (often not crossing the midline), variable extent of involvement, lack of diffuse involvement of entire organs, and equal sex ratio.67 Intriguingly, the clinical involvement of both the skin and nervous system in SWS has many of these features.
Somatic mosaicism (somatic mutation) is extremely common, occurring in several diseases including leukemias and other cancers as well as immune deficiencies, and occurring in other neurocutaneous disorders with mosaic phenotypes.68-70 Rudolf Happle first proposed the hypothesis that mutations that form autosomal lethal genes could survive in a mosaic state. Briefly, for a normal individual, most cells in the body are likely to harbor at least one somatic mutation, and every gene is likely to undergo somatic mutation repeatedly.71 Happle later refined his hypothesis to include the possibility of rare familial clustering of a trait in which a lethal (recessive) mutation could be maintained in the heterozygous state through mosaicism. During embryonic development of individuals carrying such a mutation, loss of the wild-type allele owing to various chromosomal events (e.g. nondisjunction leading to chromosome loss, mitotic cross-over, or deletion) could eventually lose the wild-type allele (loss of heterozygosity) and unmask the mutant allele with the resulting phenotypic changes. This latter refinement of Happle’s hypothesis was termed ‘paradominant inheritance’.72 Paradominant inheritance is conceptually similar to Knudson’s two-hit mutation theory that explains the basis of inherited retinoblastoma because of an inactivated tumor suppressor gene.73 The difference between Happle’s paradominant inheritance and Knudson’s two-hit mutation theory is that paradominant inheritance applies to somatic mutations occurring early in development that predispose to congenital mutations, whereas the two-hit mutation theory applies to mutations in tumor suppressor genes that occur later in life and predispose to tumorigenesis.
Whether a lethal recessive mutation segregates in families and is uncovered by somatic loss of heterozygosity or whether a somatic point mutation occurs de novo during development, a SWS gene is likely to be essential for survival because of its critical importance for developmental angiogenesis. The effects of a mutant allele are incompatible with normal development from conception. This fact would preclude germline transmission of the SWS mutation. Although chromosomal abnormalities have been associated with SWS, no consistent genomic aberration points to the existence or location of an underlying germline mutation.74,75
A report of typical bilateral SWS occurring in only one member of monozygotic twins supports the notion that a genetic basis for SWS is likely to be due to a somatic rather than germline mutation.76 Consistent with this observation, a study by Huq et al. provided evidence for a possible somatic mutation in affected tissues, but the putative mutation was not identified.77
Somatic mosaicism in SWS?
In the somatic mosaicism scenario, the extent of the SWS phenotype would depend on when the somatic mutation occurred during development. A somatic mutation might occur in a cell derived from the primordial vascular plexus. During the first trimester the primitive vascular plexus invades the adjacent fetal brain, skin, and eye in this region; a somatic mutation would prevent the normal maturation of these vessels. The exact moment in development when the SWS mutation occurs could determine the extent of involvement of the nervous and vascular systems. For example, at 4 to 5 weeks of gestation, the visual cortex is juxtaposed to the optic vesicle and the upper part of the embryonic face. If an SWS gene controls or modulates early angiogenesis, angiogenesis biomarkers might correlate with disease severity. If so, such biomarkers might assist the early diagnosis of SWS in young infants with developing port-wine birthmarks.
The search for an underlying somatic mutation in SWS
The nature of one or more genes that could mutate to cause SWS is speculative. One candidate category is the genes involved in vascular innervation. This notion is based on the observation of Smoller and Rosen that port-wine stains have a significantly lower density of nerves associated with the aberrant blood vessels.78 This observation, which has been validated by others, suggests the hypothesis that defective innervation that impairs modulation of vascular flow is the primary cause of the port-wine stain.79,80 The extent of the vascular anomalies may depend on when during development this defective innervation begins so that early developmental events predispose to some of the neurological problems associated with SWS. Two candidate pathways come to mind that could produce defective vascular innervation; both pathways have connections to axonal guidance and vascular growth. The semaphorins/neuropilin ligand receptor family and the ephrin/ephrin receptor family are both implicated in axonal guidance and angiogenesis. The many genes involved in these complex signaling networks would make compelling candidates for the search for somatic mutations in SWS. Unfortunately, the list of potential candidate genes is too large to test the hypothesis by re-sequencing using conventional molecular genetics technology. Adding to these two potential candidate families are an equally large list of other candidate genes arising from other compelling hypotheses (reviewed in Comi) and the inability to test these hypotheses is evident.79 When the signposts point to nearly everything, the correct road to success is obscured.
Over 20 years have passed since Happle first proposed his somatic mosaic hypothesis for the cause of SWS. This hypothesis has been validated for multiple neurocutaneous syndromes, such as tuberous sclerosis (MIM #191100) and neurofibromatosis (MIM #162200), in which the causative genes have been identified. Those syndromes for which causative genes have been identified share one common characteristic – a much higher frequency of familial segregation that in most cases follows a traditional Mendelian pattern of inheritance. Within the affected families, genetic linkage analysis first identified the approximate genomic position of the causative gene, which effectively reduced the ‘genome space’ needed to search for the gene location. Furthermore, a causative germline mutation that is present in all tissues is easier to identify than a somatic mutation, which might be present only in a particular type of cell in affected tissue, and then possibly only in a fraction of the cells.81 In stark contrast, the identity of the SWS gene has remained elusive. Even with rare examples of more than one affected individual in a family, SWS does not exhibit clear Mendelian segregation, thus precluding the genetic linkage shortcut to gene identification mentioned above.
One recently developed set of tools that should enable a genome-wide, unbiased survey for somatic mutations in affected SWS tissues is the high-throughput, genome-wide technologies for DNA sequencing and genotyping. The comparison of normal and affected tissue from individuals with SWS could enable the identification of somatic point mutations in a critical gene or uncover regions of loss of heterozygosity that unmask the effects of a recessive-lethal mutation. These genome-wide studies are in progress. The identification of the elusive SWS gene will shed new light on the pathogenic pathways involved and will provide new clues to direct research into the prevention and treatment of the disorder.
BIOMARKERS IN SWS: GOALS, DEVELOPMENT, AND FUTURE DIRECTIONS
The development of safe, accurate and minimally invasive biomarkers and tools to monitor clinical status in individuals with SWS has the potential to provide better informed clinical management. Such biomarkers might also provide the opportunity to monitor therapeutic efficacy over time and to distinguish between progressive and stable vascular lesions. Biomarker development is an important goal because of the current difficulties in making an early diagnosis of SWS brain involvement and because of the tremendous variability in clinical progression and symptoms that hinders attempts at prognostication and treatment. The availability of reliable biomarkers is a critical step toward the pursuit of clinical trials. The biomarkers discussed below are not yet recommended as standard clinical tests for the diagnosis of evaluation of SWS, but all show promise in this area and are under active development.
Quantitative EEG
A valid, reliable biomarker of neurological function in SWS is essential because of two aspects of the natural history. First, infants who have a facial port-wine birthmark at birth have an unpredictable extent of neurological involvement. Second, children who have established SWS brain involvement can have widely variable neurological symptoms that wax and wane. It will be enormously helpful to be able to determine which infants with a port-wine stain have SWS (in order to institute prophylactic therapies) and which infants do not (to reassure parents). MRI and CT can define the severity and extent of brain lesions in older children with SWS, but these imaging tools can be falsely negative at birth (see the ‘Neuroimaging and pathogenesis’ section above).2 The repeated use of these imaging tools is constrained in young infants because of the potential for side effects from radiation exposure and intravenous contrast, and the risks that surround sedation.
EEG offers a non-invasive method to assess brain function that can be repeated safely and as often as necessary. Abnormalities of the EEG, specifically decreased voltage amplitudes over an affected hemisphere, were first described in the 1970s.82 The sensitivity and specificity of the clinical EEG is limited because physicians’ interpretations are based upon a qualitative interpretation of the tracing and can vary widely, particularly with a rare entity such as SWS.83 The EEG can be quantified to reduce this variability in interpretation. Mathematical signal processing of the EEG has been used for years in assessing brain ischemia.84 In preliminary studies, quantitative EEG (qEEG) methods were used that originally were developed for the monitoring of carotid endarterectomy85 to evaluate patients with SWS. This study was able to show that qEEG correlated with clinical symptom scores and MRI angioma/atrophy severity scores.86 Eight infants with port-wine birthmark were examined with qEEG: four had SWS brain involvement (a positive MRI with or without neurological symptoms) and four did not (at least one negative MRI and clinically asymptomatic at least to age 2 years). A qEEG threshold was then developed that discriminated between the two groups, and then validated in a second cohort of nine infants (four with neurological involvement and five without). The qEEG threshold correctly identified 100% of the infants with SWS.87 A limitation of these studies is that only three infants with SWS in both cohorts total were asymptomatic at the time of testing but later developed symptoms. Current studies are investigating additional children to establish the sensitivity and specificity of this technique.
Since individuals with known SWS may have acute or sub-acute decreases in neurological function due to worsening seizures, stroke-like episodes or migraines, it is also helpful to have a tool that can predict neurological deterioration to enable the selection of epileptic seizure treatments, cerebral ischemia prophylaxis such as aspirin, and other future treatments. Ongoing studies with qEEG are also investigating whether this tool can aid in prognostication and selection of treatment regimens.
Transcranial Doppler
Transcranial Doppler ultrasound (TCD) is a non-invasive vascular blood flow measuring technique clinically utilized as a screening tool for determining which children with sickle cell disease are at highest risk of stroke.88,89 Studies are ongoing to determine whether TCD will prove to be similarly useful for SWS. While TCD may not be suitable for identifying neurological involvement, it can measure cerebral flow velocity non-invasively and without the use of radionuclides. In one study, TCD was combined with video/digital EEG to measure inter-ictal and ictal middle cerebral artery velocity (MCAV) in a small cohort of patients with SWS with epilepsy.90 MCAV was lower in the more severely affected hemisphere compared with the contralateral side. During seizures, MCAV increased in the affected hemisphere, but flow in the contralateral hemisphere increased four-to six-fold in comparison. Eight children with SWS were studied with TCD in a small pilot study.91 On the side affected by venous angioma, they had decreased arterial flow velocity and increased pulsatility index in the middle and posterior cerebral arteries; these measurements suggested a high resistance pattern of blood flow.91 Since the pulsatility index measures the resistance to blood flow within a vessel, the higher the pulsatility index, the higher the downstream resistance to flow in the blood vessels.92 One possibility is that this TCD pattern may reflect venous stasis within the venous angioma, which may contribute to chronic hypoperfusion of brain tissue. Alternatively, this TCD blood flow pattern could mean the presence of decreased arterial perfusion secondary to cortical vessel hypoplasia resulting from cortical atrophy on the involved side. The occurrence of this TCD pattern in young children with SWS brain involvement before the onset of symptoms or developmental issues gives credence to the venous stasis hypothesis. In addition, descriptions of cerebral angiography in children with SWS has revealed thrombosis and obstruction of cerebral veins as well as a relative lack of superficial cortical veins.93 Furthermore, magnetic resonance perfusion-weighted imaging in children with SWS has suggested that impaired venous drainage contributes to the cerebral hypoperfusion.42 Comparing TCD data from children with SWS to measurements made in healthy children suggests that children with SWS have a significantly more asymmetric blood flow.91,94,95
Therefore, in children with SWS, TCD may provide a non-invasive tool to determine the severity of blood flow abnormalities at baseline and monitor progressive changes over time. Additional studies need to be carried out to ascertain test–retest reliability of TCD, to determine the precise TCD measures that correlate with functional outcome, and to determine whether changes in asymmetry in TCD velocities over time correlate with alterations in neurological status. It would be also useful to correlate TCD measurements with perfusion imaging, SPECT imaging, and PET imaging results; this would enable us to determine how the TCD measurements relate to these other functional imaging modalities in SWS. In summary, TCD abnormalities have been measured in SWS, and TCD could be developed as a biomarker and future clinical tool reflecting impaired cerebral blood flow and stroke risk.
Urine angiogenesis factors
The development of vascular biomarkers has been reported by Marler and et al. who undertook the non-invasive profiling of vascularization in patients with a variety of vascular disorders including infantile hemangioma, lymphatic and capillary-lymphaticovenous malformations, extensive and unremitting capillary malformations and arteriovenous malformations, and other vascular neoplasms.96 These authors demonstrated that matrix metalloproteinases are elevated in the urine of children with these vascular anomalies and that increases in the levels of these urinary matrix metalloproteinases parallels the extent and activity of the vascular anomalies.96 Comi and Moses are currently pursuing a similar approach to validate the use of urine vascular biomarkers in patients with SWS. The continued identification and validation of biomarkers for SWS will be an important complement to current and future therapeutic approaches for the treatment of this disease.96
THE ROLE OF ADVOCACY GROUPS AND PARENT ORGANIZATIONS
SWF was founded in 1987 during an emergence of patient advocacy groups for rare diseases. Patient advocacy organizations improve patient care and access to care, increase the pace of discovery by encouraging collaboration, mold public policy, and serve as patient advocates for better health insurance and reimbursement. SWF has become an effective advocate for global awareness and serves to guide strategic research investigations. Since 1987, SWF’s toll-free telephone line, compassionate staff, website, and international conferences have given patients the resources to feel connected and no longer alone in dealing with their condition. The Internet provides patients even faster access to SWS education and the ability to network with other patients internationally through online discussion forums. The use of diverse social media enables people to access news conveniently on the latest education, advocacy efforts, and research advances.
SWF acts as a liaison with the researchers at the NIH, private institutions, and the US Food and Drug Administration (FDA) in order to increase the pace of discovery. Networking with umbrella organizations expands their ability to assist patients and healthcare providers. For instance, the Coalition of Skin Diseases (CSD), a group of 19 skin disease advocacy groups of which SWF is a member, awarded in excess of $16.5 million dollars in research funding in a 15-year period. It will be beneficial to collaborate with these umbrella organizations as funding for research becomes scarcer. SWF has a strategic research plan and has been partnering with scientists worldwide to support their investigations. For example, SWF’s research seed grants have investigated the impact on quality of life, growth hormone abnormalities, angiogenesis, and other comorbidities in SWS. SWF is also funding research involving animal models and investigating the use of drugs clinically approved for other disorders.
Another model many rare disease groups are adopting is the establishment of Centers of Excellence around the US and abroad. The SWF currently refers patients to and works with 11 SWF Centers around the country. This local and regional access to care makes it emotionally and financially easier for patients to find knowledgeable healthcare providers and to participate in scientific research studies. Currently SWF is collaborating with the Beckman Laser Institute to conduct an FDA-approved investigational new drug study on the use of combined pulsed dye laser and oral sirolimus to treat port-wine stain birthmarks. The involvement of SWF has also significantly aided investigators’ efforts to obtain a grant from the NIH’s Rare Diseases Clinical Research Network (RDCRN) in order to investigate the somatic mutation hypothesis, develop urine biomarkers, and establish a de-identified online patient registry. SWF had a key role in generating a ‘first-generation’ patient registry which has served as a foundation for a second-generation registry (http://rarediseasesnetwork.epi.usf.edu/BVMC/index.htm), funded by the NIH. Looking to the future, SWF is updating its strategic research plan and is fostering SWS awareness with biotechnology companies who have potential drug candidates for therapeutic intervention. As SWF expands its collaborative clinical and scientific research endeavours, it is expected that new discoveries will continue to emerge in SWS.
Ultimately, the patients and their families are flourishing in spite of a rare disease that can be devastating, frustrating, and isolating, in part because the SWF is a community of hope. Other advocacy groups that serve this population include the Vascular Birthmark Foundation, the Sturge–Weber Syndrome Community and Birthmarks.com in the USA, and SWF United Kingdom, SWF Germany, and SWF Canada. However, more can and should be done. With patient participation and more scientific investigations and collaboration, patients with SWS can look forward to a brighter future with new discoveries and therapeutic options.
Conclusion
Momentum is gathering for significant research advances that will answer longstanding fundamental questions such as What causes SWS? What is the optimal way to screen for and diagnose presymptomatic brain involvement? How do we develop information to guide treatment? What is the best way to monitor response to treatment? How do we improve outcome and patient quality of life? These challenging questions require the kind of multidisciplinary and multicentered efforts that are now possible and that promise to provide future breakthroughs in the management of patients with SWS.
What this paper adds.
Assembles a comprehensive range of experts to present a novel view of this complex problem.
Describes biomarkers being developed to characterize SWS brain involvement.
Summarizes the latest treatment and comorbidity studies in patients with SWS.
Acknowledgments
The following individuals are contributing members of the Brain Vascular Malformation Consortium Sturge–Weber National Workgroup: Warren D. Lo, Departments of Pediatrics and Neurology, The Ohio State University and Nationwide Children’s Hospital, Columbus, OH; Douglas A. Marchuk, Duke University; Karen L. Ball, The Sturge-Weber Foundation; Jonathan Pevsner, Hugo Moser Kennedy Krieger Research Institute; Marsha A. Moses, Harvard Medical Center; Csaba Juhász, Pediatric Neurology/PET Center, Children’s Hospital of Michigan, Detroit, MI, Harry T. Chugani, Children’s Hospital of Michigan; Joshua B. Ewen, Kennedy Krieger Institute; Lori C. Jordan; Vanderbilt University; and Anne M. Comi, Hugo Moser Kennedy Krieger Research Institute.
The Brain Vascular Malformation Consortium is a part of the NIH Rare Diseases Clinical Research Network (RDCRN). Funding and/or programmatic support for this project has been provided by BVMC #6203 from the National Institute for Neurological Disorders and Stroke and the NIH Office of Rare Diseases Research (ORDR).
We would like to thank Kira Lanier for her assistance in editing and preparing this manuscript for submission.
ABBREVIATIONS
- FDA
Food and Drug Administration
- MCAV
Middle cerebral artery velocity
- NIH
National Institutes of Health
- PET
Positron emission tomography
- qEEG
Quantitative electroencaphalogram
- SPECT
Single-photon emission computed tomography
- SWF
The Sturge-Weber Foundation
- SWI
Susceptibility-weighted imaging
- SWS
Sturge–Weber syndrome
- TCD
Transcranial Doppler ultrasound
Footnotes
ONLINE MATERIAL/SUPPORTING INFORMATION The references for this article may be found in the online version.
References
- 1.Riela AR, Stump DA, Roach ES, McLean WT, Jr, Garcia JC. Regional cerebral blood flow characteristics of the Sturge-Weber syndrome. Pediatr Neurol. 1985;1:85–90. doi: 10.1016/0887-8994(85)90042-6. [DOI] [PubMed] [Google Scholar]
- 2.Comi AM. Advances in Sturge-Weber syndrome. Curr Opin Neurol. 2006;19:124–8. doi: 10.1097/01.wco.0000218226.27937.57. [DOI] [PubMed] [Google Scholar]
- 3.Tallman B, Tan OT, Morelli JG, et al. Location of port-wine stains and the likelihood of ophthalmic and/or central nervous system complications. Pediatrics. 1991;87:323–7. [PubMed] [Google Scholar]
- 4.Enjolras O, Riche MC, Merland JJ. Facial port-wine stains and Sturge-Weber syndrome. Pediatrics. 1985;76:48–51. [PubMed] [Google Scholar]
- 5.Crosley CJ, Binet EF. Sturge-Weber Syndrome: presentation as a focal seizure disorder without nevus flammeus. Clin Pediatr (Phila) 1978;17:606–9. doi: 10.1177/000992287801700801. [DOI] [PubMed] [Google Scholar]
- 6.Taly AB, Nagaraja D, Das S, Shankar SK, Pratibha NG. Sturge-Weber-Dimitri disease without facial nevus. Neurology. 1987;37:1063–4. doi: 10.1212/wnl.37.6.1063. [DOI] [PubMed] [Google Scholar]
- 7.Furukawa T, Igata A, Toyokura Y, Ikeda S. Sturge-Weber and Klippel-Trenaunay syndrome with nevus of ota and ito. Arch Dermatol. 1970;102:640–5. [PubMed] [Google Scholar]
- 8.Comi AM, Bellamkonda S, Ferenc LM, Cohen BA, Germain-Lee EL. Central hypothyroidism and Sturge-Weber syndrome. Pediatr Neurol. 2008;39:58–62. doi: 10.1016/j.pediatrneurol.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 9.Bonse G. Roentgen findings in a phacomatosis (Sturge-Weber combined with Klippel-Trenaunay) Fortschr Geb Rontgenstr. 1951;74:727–9. [PubMed] [Google Scholar]
- 10.Sujansky E, Conradi S. Outcome of Sturge-Weber syndrome in 52 adults. Am J Med Genet. 1995;57:35–45. doi: 10.1002/ajmg.1320570110. [DOI] [PubMed] [Google Scholar]
- 11.Bodensteiner J, Roach ES. Sturge-Weber syndrome: introduction and overview. In: Bodensteiner J, Roach ES, editors. Sturge-Weber Syndrome. Mt Freedom, NJ: Sturge-Weber Foundation; 1999. pp. 1–10. [Google Scholar]
- 12.Garcia JC, Roach ES, McLean WT. Recurrent thrombotic deterioration in the Sturge-Weber syndrome. Childs Brain. 1981;8:427–33. doi: 10.1159/000120011. [DOI] [PubMed] [Google Scholar]
- 13.Pascual-Castroviejo I, Pascual-Pascual SI, Velazquez-Fragua R, Viano J. Sturge-Weber syndrome: study of 55 patients. Can J Neurol Sci. 2008;35:301–7. doi: 10.1017/s0317167100008878. [DOI] [PubMed] [Google Scholar]
- 14.Thomas-Sohl KA, Vaslow DF, Maria BL. Sturge-Weber syndrome: a review. Pediatr Neurol. 2004;30:303–10. doi: 10.1016/j.pediatrneurol.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 15.Bebin EM, Gomez MR. Prognosis in Sturge-Weber disease: comparison of unihemispheric and bihemispheric involvement. J Child Neurol. 1988;3:181–4. doi: 10.1177/088307388800300306. [DOI] [PubMed] [Google Scholar]
- 16.Chugani HT, Mazziotta JC, Phelps ME. Sturge-Weber syndrome: a study of cerebral glucose utilization with positron emission tomography. J Pediatr. 1989;114:244–53. doi: 10.1016/s0022-3476(89)80790-5. [DOI] [PubMed] [Google Scholar]
- 17.Maria BL, Neufeld JA, Rosainz LC, et al. High prevalence of bihemispheric structural and functional defects in Sturge-Weber syndrome. J Child Neurol. 1998;13:595–605. doi: 10.1177/088307389801301203. [DOI] [PubMed] [Google Scholar]
- 18.Oakes WJ. The natural history of patients with the Sturge-Weber syndrome. Pediatr Neurosurg. 1992;18:287–90. doi: 10.1159/000120677. [DOI] [PubMed] [Google Scholar]
- 19.Sujansky E, Conradi S. Sturge-Weber syndrome: age of onset of seizures and glaucoma and the prognosis for affected children. J Child Neurol. 1995;10:49–58. doi: 10.1177/088307389501000113. [DOI] [PubMed] [Google Scholar]
- 20.Miyama S, Goto T. Leptomeningeal angiomatosis with infantile spasms. Pediatr Neurol. 2004;31:353–6. doi: 10.1016/j.pediatrneurol.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 21.Barbagallo M, Ruggieri M, Incorpora G, et al. Infantile spasms in the setting of Sturge-Weber syndrome. Childs Nerv Syst. 2009;25:111–8. doi: 10.1007/s00381-008-0705-6. [DOI] [PubMed] [Google Scholar]
- 22.Petit F, Auvin S, Lamblin MD, Vallee L. Myoclonic astatic seizures in a child with Sturge-Weber syndrome. Rev Neurol (Paris) 2008;164:953–6. doi: 10.1016/j.neurol.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 23.Kossoff EH, Ferenc L, Comi AM. An infantile-onset, severe, yet sporadic seizure pattern is common in Sturge-Weber syndrome. Epilepsia. 2009;50:2154–7. doi: 10.1111/j.1528-1167.2009.02072.x. [DOI] [PubMed] [Google Scholar]
- 24.Chapieski L, Friedman A, Lachar D. Psychological functioning in children and adolescents with Sturge-Weber syndrome. J Child Neurol. 2000;15:660–5. doi: 10.1177/088307380001501004. [DOI] [PubMed] [Google Scholar]
- 25.Reesman J, Gray R, Suskauer SJ, et al. Hemiparesis is a clinical correlate of general adaptive dysfunction in children and adolescents with Sturge-Weber syndrome. J Child Neurol. 2009;24:701–8. doi: 10.1177/0883073808329529. [DOI] [PubMed] [Google Scholar]
- 26.Klapper J. Headache in Sturge-Weber syndrome. Headache. 1994;34:521–2. doi: 10.1111/j.1526-4610.1994.hed3409521.x. [DOI] [PubMed] [Google Scholar]
- 27.Kossoff EH, Hatfield LA, Ball KL, Comi AM. Comorbidity of epilepsy and headache in patients with sturge-weber syndrome. J Child Neurol. 2005;20:678–82. doi: 10.1177/08830738050200080901. [DOI] [PubMed] [Google Scholar]
- 28.Kossoff EH, Balasta M, Hatfield LM, Lehmann CU, Comi AM. Self-reported treatment patterns in patients with Sturge-Weber syndrome and migraines. J Child Neurol. 2007;22:720–6. doi: 10.1177/0883073807304008. [DOI] [PubMed] [Google Scholar]
- 29.Jansen FE, van der Worp HB, van Huffelen A, van Nieuwenhuizen O. Sturge-Weber syndrome and paroxysmal hemiparesis: epilepsy or ischaemia? Dev Med Child Neurol. 2004;46:783–6. [PubMed] [Google Scholar]
- 30.Arulrajah S, Ertan G, Comi M, Tekes A, Lin DL, Huisman TA. MRI with diffusion-weighted imaging in children and young adults with simultaneous supra- and infratentorial manifestations of Sturge-Weber syndrome. J Neuroradiol. 2010;37:51–9. doi: 10.1016/j.neurad.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 31.Kumar KR, Hon K, Schultz D, Agzarian MJ, Jones DN, Thyagarajan D. Transient changes on brain magnetic resonance imaging in a patient with sturge-weber syndrome presenting with hemiparesis. Neurologist. 2009;15:351–4. doi: 10.1097/NRL.0b013e3181940244. [DOI] [PubMed] [Google Scholar]
- 32.Zolkipli Z, Aylett S, Rankin PM, Neville BG. Transient exacerbation of hemiplegia following minor head trauma in Sturge-Weber syndrome. Dev Med Child Neurol. 2007;49:697–9. doi: 10.1111/j.1469-8749.2007.00697.x. [DOI] [PubMed] [Google Scholar]
- 33.Miller RS, Ball KL, Comi AM, Germain-Lee EL. Growth hormone deficiency in Sturge-Weber syndrome. Arch Dis Child. 2006;91:340–1. doi: 10.1136/adc.2005.082578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Comi AM. Update on Sturge-Weber syndrome: diagnosis, treatment, quantitative measures, and controversies. Lymphat Res Biol. 2007;5:257–64. doi: 10.1089/lrb.2007.1016. [DOI] [PubMed] [Google Scholar]
- 35.Juhasz C, Chugani HT. An almost missed leptomeningeal angioma in Sturge-Weber syndrome. Neurology. 2007;68:243. doi: 10.1212/01.wnl.0000242581.43024.0a. [DOI] [PubMed] [Google Scholar]
- 36.Griffiths PD, Coley SC, Romanowski CA, Hodgson T, Wilkinson ID. Contrast-enhanced fluid-attenuated inversion recovery imaging for leptomeningeal disease in children. AJNR Am J Neuroradiol. 2003;24:719–23. [PMC free article] [PubMed] [Google Scholar]
- 37.Mentzel HJ, Dieckmann A, Fitzek C, Brandl U, Reichenbach JR, Kaiser WA. Early diagnosis of cerebral involvement in Sturge-Weber syndrome using high-resolution BOLD MR venography. Pediatr Radiol. 2005;35:85–90. doi: 10.1007/s00247-004-1333-2. [DOI] [PubMed] [Google Scholar]
- 38.Sehgal V, Delproposto Z, Haacke EM, et al. Clinical applications of neuroimaging with susceptibility-weighted imaging. J Magn Reson Imaging. 2005;22:439–50. doi: 10.1002/jmri.20404. [DOI] [PubMed] [Google Scholar]
- 39.Hu J, Yu Y, Juhasz C, et al. MR susceptibility weighted imaging (SWI) complements conventional contrast enhanced T1 weighted MRI in characterizing brain abnormalities of Sturge-Weber Syndrome. J Magn Reson Imaging. 2008;28:300–7. doi: 10.1002/jmri.21435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Evans AL, Widjaja E, Connolly DJ, Griffiths PD. Cerebral perfusion abnormalities in children with Sturge-Weber syndrome shown by dynamic contrast bolus magnetic resonance perfusion imaging. Pediatrics. 2006;117:2119–25. doi: 10.1542/peds.2005-1815. [DOI] [PubMed] [Google Scholar]
- 41.Lin DD, Barker PB, Hatfield LA, Comi AM. Dynamic MR perfusion and proton MR spectroscopic imaging in Sturge-Weber syndrome: correlation with neurological symptoms. J Magn Reson Imaging. 2006;24:274–81. doi: 10.1002/jmri.20627. [DOI] [PubMed] [Google Scholar]
- 42.Miao Y, Juhász C, Wu J, et al. Clinical correlates of white matter blood flow perfusion changes in Sturge-Weber syndrome: a dynamic MR perfusion-weighted imaging study. AJNR Am J Neuroradiol. 2011;32:1280–5. doi: 10.3174/ajnr.A2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Batista CE, Chugani HT, Hu J, et al. Magnetic resonance spectroscopic imaging detects abnormalities in normal-appearing frontal lobe of patients with Sturge-Weber syndrome. J Neuroimaging. 2008;18:306–13. doi: 10.1111/j.1552-6569.2007.00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Juhasz C, Lai C, Behen ME, et al. White matter volume as a major predictor of cognitive function in Sturge-Weber syndrome. Arch Neurol. 2007;64:1169–74. doi: 10.1001/archneur.64.8.1169. [DOI] [PubMed] [Google Scholar]
- 45.Alkonyi B, Chugani HT, Behen M, et al. The role of the thalamus in neuro-cognitive dysfunction in early unilateral hemispheric injury: a multimodality imaging study of children with Sturge-Weber syndrome. Eur J Paediatr Neurol. 2010;14:425–33. doi: 10.1016/j.ejpn.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sivaswamy L, Rajamani K, Juhasz C, Maqbool M, Makki M, Chugani HT. The corticospinal tract in Sturge-Weber syndrome: a diffusion tensor tractography study. Brain Dev. 2008;30:447–53. doi: 10.1016/j.braindev.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Juhasz C, Haacke EM, Hu J, et al. Multimodality imaging of cortical and white matter abnormalities in Sturge-Weber syndrome. AJNR Am J Neuroradiol. 2007;28:900–6. [PMC free article] [PubMed] [Google Scholar]
- 48.Juhasz C, Batista CE, Chugani DC, Muzik O, Chugani HT. Evolution of cortical metabolic abnormalities and their clinical correlates in Sturge-Weber syndrome. Eur J Paediatr Neurol. 2007;11:277–84. doi: 10.1016/j.ejpn.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee JS, Asano E, Muzik O, et al. Sturge-Weber syndrome: correlation between clinical course and FDG PET findings. Neurology. 2001;57:189–95. doi: 10.1212/wnl.57.2.189. [DOI] [PubMed] [Google Scholar]
- 50.Andrade DM, McAndrews MP, Hamani C, Poublanc J, Angel M, Wennberg R. Seizure recurrence 29 years after hemispherectomy for Sturge Weber syndrome. Can J Neurol Sci. 2011;37:141–4. doi: 10.1017/s0317167100009835. [DOI] [PubMed] [Google Scholar]
- 51.Batista CE, Juhasz C, Muzik O, Chugani DC, Chugani HT. Increased visual cortex glucose metabolism contralateral to angioma in children with Sturge-Weber syndrome. Dev Med Child Neurol. 2007;49:567–73. doi: 10.1111/j.1469-8749.2007.00567.x. [DOI] [PubMed] [Google Scholar]
- 52.Fraunfelder FW, Fraunfelder FT, Keates EU. Topiramate-associated acute, bilateral, secondary angle-closure glaucoma. Ophthalmology. 2004;111:109–11. doi: 10.1016/j.ophtha.2003.04.004. [DOI] [PubMed] [Google Scholar]
- 53.Maria BL, Neufeld JA, Rosainz LC, et al. Central nervous system structure and function in Sturge-Weber syndrome: evidence of neurologic and radiologic progression. J Child Neurol. 1998;13:606–18. doi: 10.1177/088307389801301204. [DOI] [PubMed] [Google Scholar]
- 54.Udani V, Pujar S, Munot P, Maheshwari S, Mehta N. Natural history and magnetic resonance imaging follow-up in 9 Sturge-Weber syndrome patients and clinical correlation. J Child Neurol. 2007;22:479–83. doi: 10.1177/0883073807300526. [DOI] [PubMed] [Google Scholar]
- 55.Greco F, Fiumara A, Sorge G, Pavone L. Subgaleal hematoma in a child with Sturge-Weber syndrome: to prevent stroke-like episodes, is treatment with aspirin advisable? Childs Nerv Syst. 2008;24:1479–81. doi: 10.1007/s00381-008-0662-0. [DOI] [PubMed] [Google Scholar]
- 56.Namer IJ, Battaglia F, Hirsch E, Constantinesco A, Marescaux C. Subtraction ictal SPECT co-registered to MRI (SISCOM) in Sturge-Weber syndrome. Clin Nucl Med. 2005;30:39–40. doi: 10.1097/00003072-200501000-00014. [DOI] [PubMed] [Google Scholar]
- 57.Ville D, Enjolras O, Chiron C, Dulac O. Prophylactic antiepileptic treatment in Sturge-Weber disease. Seizure. 2002;11:145–50. doi: 10.1053/seiz.2001.0629. [DOI] [PubMed] [Google Scholar]
- 58.Comi AM. Sturge-Weber syndrome and epilepsy: an argument for aggressive seizure management in these patients. Expert Rev Neurother. 2007;7:951–6. doi: 10.1586/14737175.7.8.951. [DOI] [PubMed] [Google Scholar]
- 59.Kossoff EH, Borsage JL, Comi AM. A pilot study of the modified Atkins diet for Sturge-Weber syndrome. Epilepsy Res. 2010;92:240–3. doi: 10.1016/j.eplepsyres.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 60.Schropp C, Sorensen N, Krauss J. Early periinsular hemispherotomy in children with Sturge-Weber syndrome and intractable epilepsy – outcome in eight patients. Neuropediatrics. 2006;37:26–31. doi: 10.1055/s-2006-923945. [DOI] [PubMed] [Google Scholar]
- 61.Bourgeois M, Crimmins DW, de Oliveira RS, et al. Surgical treatment of epilepsy in Sturge-Weber syndrome in children. J Neurosurg. 2007;106(1 Suppl):20–8. doi: 10.3171/ped.2007.106.1.20. [DOI] [PubMed] [Google Scholar]
- 62.Arzimanoglou AA, Andermann F, Aicardi J, et al. Sturge-Weber syndrome: indications and results of surgery in 20 patients. Neurology. 2000;55:1472–9. doi: 10.1212/wnl.55.10.1472. [DOI] [PubMed] [Google Scholar]
- 63.Kossoff EH, Buck C, Freeman JM. Outcomes of 32 hemispherectomies for Sturge-Weber syndrome worldwide. Neurology. 2002;59:1735–8. doi: 10.1212/01.wnl.0000035639.54567.5c. [DOI] [PubMed] [Google Scholar]
- 64.Hoffman HJ, Hendrick EB, Dennis M, Armstrong D. Hemispherectomy for Sturge-Weber syndrome. Childs Brain. 1979;5:233–48. doi: 10.1159/000119821. [DOI] [PubMed] [Google Scholar]
- 65.Ogunmekan AO, Hwang PA, Hoffman HJ. Sturge-Weber-Dimitri disease: role of hemispherectomy in prognosis. Can J Neurol Sci. 1989;16:78–80. doi: 10.1017/s0317167100028559. [DOI] [PubMed] [Google Scholar]
- 66.Comi AM. Pathophysiology of Sturge-Weber syndrome. J Child Neurol. 2003;18:509–16. doi: 10.1177/08830738030180080701. [DOI] [PubMed] [Google Scholar]
- 67.Happle R. Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol. 1987;16:899–906. doi: 10.1016/s0190-9622(87)80249-9. [DOI] [PubMed] [Google Scholar]
- 68.Greenman C, Stephens P, Smith R, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–8. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stuart D, Sellers WR. Linking somatic genetic alterations in cancer to therapeutics. Curr Opin Cell Biol. 2009;21:304–10. doi: 10.1016/j.ceb.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 70.Wada T, Candotti F. Somatic mosaicism in primary immune deficiencies. Curr Opin Allergy Clin Immunol. 2008;8:510–4. doi: 10.1097/ACI.0b013e328314b651. [DOI] [PubMed] [Google Scholar]
- 71.Frank SA. Evolution in health and medicine Sackler colloquium: Somatic evolutionary genomics: mutations during development cause highly variable genetic mosaicism with risk of cancer and neurodegeneration. Proc Natl Acad Sci U S A. 2010;107(Suppl 1):1725–30. doi: 10.1073/pnas.0909343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kuster W, Happle R. Neurocutaneous disorders in children. Curr Opin Pediatr. 1993;5:436–40. doi: 10.1097/00008480-199308000-00011. [DOI] [PubMed] [Google Scholar]
- 73.Knudson AG. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–3. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gutierrez AC, Salamanca F, Lisker R, Segovia A. Supernumerary bisatellited chromosome in a family ascertained through a patient with Sturge-Weber syndrome. Ann Genet. 1975;18:45–9. [PubMed] [Google Scholar]
- 75.Habedank M, Kampe G. Familial translocation t(3p-;21q+) associated with both Down’s and Sturge-Weber’s syndrome in unbalanced state. Humangenetik. 1975;29:207–16. doi: 10.1007/BF00297625. [DOI] [PubMed] [Google Scholar]
- 76.Pedailles S, Martin N, Launay V, et al. Sturge-Weber-Krabbe syndrome A severe form in a monozygote female twin. Ann Dermatol Venereol. 1993;120:379–82. [PubMed] [Google Scholar]
- 77.Huq AH, Chugani DC, Hukku B, Serajee FJ. Evidence of somatic mosaicism in Sturge-Weber syndrome. Neurology. 2002;59:780–2. doi: 10.1212/wnl.59.5.780. [DOI] [PubMed] [Google Scholar]
- 78.Smoller BR, Rosen S. Port-wine stains. A disease of altered neural modulation of blood vessels? Arch Dermatol. 1986;122:177–9. doi: 10.1001/archderm.122.2.177. [DOI] [PubMed] [Google Scholar]
- 79.Rydh M, Malm M, Jernbeck J, Dalsgaard CJ. Ectatic blood vessels in port-wine stains lack innervation: possible role in pathogenesis. Plast Reconstr Surg. 1991;87:419–22. doi: 10.1097/00006534-199103000-00003. [DOI] [PubMed] [Google Scholar]
- 80.Cotterill JA, Lanigan SW. Erosive pustular dermatosis of the leg – a definition. Br J Dermatol. 1990;123:548. doi: 10.1111/j.1365-2133.1990.tb01465.x. [DOI] [PubMed] [Google Scholar]
- 81.Devilee P, Cleton-Jansen AM, Cornelisee CJ. Ever since Knudson. Trends Genet. 2001;17:569–73. doi: 10.1016/s0168-9525(01)02416-7. [DOI] [PubMed] [Google Scholar]
- 82.Brenner RP, Sharbrough FW. Electroencephalographic evaluation in Sturge-Weber syndrome. Neurology. 1976;26:629–32. doi: 10.1212/wnl.26.7.629. [DOI] [PubMed] [Google Scholar]
- 83.Gilbert DL, Sethuraman G, Kotagal U, Buncher CR. Meta-analysis of EEG test performance shows wide variation among studies. Neurology. 2003;60:564–70. doi: 10.1212/01.wnl.0000044152.79316.27. [DOI] [PubMed] [Google Scholar]
- 84.Pfurtscheller G, Jonkman EJ, Lopes da Silva FH. Brain Ischemia: Quantitative EEG and Imaging Techniques. Amsterdam, New York: Elsevier; 1984. Osterreichische Akademie der Wissenschaften. [Google Scholar]
- 85.van Putten MJ, Peters JM, Mulder SM, de Haas JA, Bruijninckx CM, Tavy DL. A brain symmetry index (BSI) for online EEG monitoring in carotid endarterectomy. Clin Neurophysiol. 2004;115:1189–94. doi: 10.1016/j.clinph.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 86.Hatfield LA, Crone NE, Kossoff EH, et al. Quantitative EEG asymmetry correlates with clinical severity in unilateral Sturge-Weber syndrome. Epilepsia. 2007;48:191–5. doi: 10.1111/j.1528-1167.2006.00630.x. [DOI] [PubMed] [Google Scholar]
- 87.Ewen JB, Kossoff EH, Crone NE, et al. Use of quantitative EEG in infants with port-wine birthmark to assess for Sturge-Weber brain involvement. Clin Neurophysiol. 2009;120:1433–40. doi: 10.1016/j.clinph.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Goldstein LB, Adams R, Alberts MJ, et al. Primary prevention of ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council: cosponsored by the Atherosclerotic Peripheral Vascular Disease Interdisciplinary Working Group; Cardiovascular Nursing Council; Clinical Cardiology Council; Nutrition, Physical Activity, and Metabolism Council; and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation. 2006;113:e873–e923. doi: 10.1161/01.STR.0000223048.70103.F1. [DOI] [PubMed] [Google Scholar]
- 89.Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998;339:5–11. doi: 10.1056/NEJM199807023390102. [DOI] [PubMed] [Google Scholar]
- 90.Aylett SE, Neville BG, Cross JH, Boyd S, Chong WK, Kirkham FJ. Sturge-Weber syndrome: cerebral haemodynamics during seizure activity. Dev Med Child Neurol. 1999;41:480–5. [PubMed] [Google Scholar]
- 91.Jordan LC, Wityk RJ, Dowling MM, DeJong MR, Comi AM. Transcranial Doppler ultrasound in children with Sturge-Weber syndrome. J Child Neurol. 2008;23:137–43. doi: 10.1177/0883073807307079. [DOI] [PubMed] [Google Scholar]
- 92.Bragoni M, Feldmann E. Transcranial Doppler indices of intracranial hemodynamics. In: Tegeler CH, Babikian VL, Gomez CR, editors. Neurosonology. St Louis, London: Mosby; 1996. pp. 129–39. [Google Scholar]
- 93.Poser CM, Taveras JM. Cerebral angiography in encephalo-trigeminal angiomatosis. Radiology. 1957;68:327–36. doi: 10.1148/68.3.327. [DOI] [PubMed] [Google Scholar]
- 94.Brouwers PJ, Vriens EM, Musbach M, Wieneke GH, van Huffelen AC. Transcranial pulsed Doppler measurements of blood flow velocity in the middle cerebral artery: reference values at rest and during hyperventilation in healthy children and adolescents in relation to age and sex. Ultrasound Med Biol. 1990;16:1–8. doi: 10.1016/0301-5629(90)90079-r. [DOI] [PubMed] [Google Scholar]
- 95.Schoning M, Niemann G, Hartig B. Transcranial color duplex sonography of basal cerebral arteries: reference data of flow velocities from childhood to adulthood. Neuropediatrics. 1996;27:249–55. doi: 10.1055/s-2007-973773. [DOI] [PubMed] [Google Scholar]
- 96.Marler JJ, Fishman SJ, Kilroy SM, et al. Increased expression of urinary matrix metalloproteinases parallels the extent and activity of vascular anomalies. Pediatrics. 2005;116:38–45. doi: 10.1542/peds.2004-1518. [DOI] [PubMed] [Google Scholar]