

Abstract
Synthetic biology may be viewed as an effort to establish, formalize, and develop an engineering discipline in the context of biological systems. The ability to tune the properties of individual components is central to the process of system design in all fields of engineering, and synthetic biology is no exception. A large and growing number of approaches have been developed for tuning the responses of cellular systems, and here we address specifically the issue of tuning the rate of response of a system: given a system where an input affects the rate of change of an output, how can the shape of the response curve be altered experimentally? This affects a system’s dynamics as well as its steady-state properties, both of which are critical in the design of systems in synthetic biology, particularly those with multiple components. We begin by reviewing a mathematical formulation that captures a broad class of biological response curves and use this to define a standard set of varieties of tuning: vertical shifting, horizontal scaling, and the like. We then survey the experimental literature, classifying the results into our defined categories, and organizing them by regulatory level: transcriptional, post-transcriptional, and post-translational.
Keywords: synthetic biology, tuning, tunable, response curves, biological rates
Synthetic biology includes a concerted effort to formalize an engineering discipline suitable for the design and implementation of novel biological systems.1−8 Analogies to well-established fields such as mechanical or electrical engineering are often drawn, but it has also been noted9 that biology presents a number of particular challenges for engineering applications: the biological environment is noisy, our understanding of cellular dynamics is imperfect, and our tools for creating and manipulating biological systems are limited and still under active development. Engineering in a cell is currently, and perhaps in some ways fundamentally, more difficult than engineering in steel or silicon.
One advantage offered by the advanced state of development in other branches of engineering is the ability to tune the way individual components respond to their inputs. Let us introduce the generic idea of a process: a system that accepts an input (mechanical force, electrical current, or biomolecular concentration) and responds dynamically by changing its output (bending, current flow, or concentration of another biomolecule) at some predictable rate. We will refer to the relationship between the input to a process and the rate at which the process changes its output as a response curve: a mapping from input levels to output rates of change. Mechanical and electrical engineering projects have an extensive ability to tune these response curves: a girder can flex at a desired rate or resonate at a desired frequency; a circuit element can slew its current or voltage output rapidly or gradually. This tunability allows engineers the powerful abilities to design individual components with desired behaviors and to integrate multiple components by ensuring that inputs and outputs match across different processes.
Synthetic biology will require these same tuning capabilities, for the same reasons: if we are to build complex systems in biology, we must be able to tune both the internal dynamics of individual systems and to match the output/input levels of connected systems. A growing library of experimental work has demonstrated the ability to tune biological response curves, and here we will review a number of approaches that have been implemented in vivo in a variety of biological contexts. After an introduction to the mathematical description of response curves, we will group our discussion into sections on transcriptional, post-transcriptional, and post-translational levels of regulation. It is a positive sign for the future progress of synthetic biology that there are now so many publications on this topic, but it also means that we cannot claim that this review is exhaustive.
Response Curves: Models and Mechanisms
We begin by establishing a mathematical notation to be used throughout the remainder of the review. Because tuning can take many forms, we want our description to be as broadly applicable as possible. We confine ourselves to population-averaged quantities, without addressing the range of single-cell distributions that can generate a given population-level response. Average rates of response in a biochemical system can often be represented by curves that rise steadily from a minimum rate to asymptotically approach some maximum rate as the input is increased (for activating inputs), or fall steadily from a maximum to a minimum rate (for repressing inputs) (see Figure 1A). Nonlinear, monotonically increasing curves of this general type can describe Michaelis-Menten kinetics for enzyme-catalyzed reactions, the rate of transcription from a promoter as a function of an activating or repressing transcription factor protein, and a variety of other examples. The frequent appearance of these saturating response curves in biology arises because many in vivo biochemical reactions are rate-limited by the concentration of some conserved macromolecule (such as DNA or an enzyme). The specific shapes of these curves are governed by the details of individual systems, and parameter changes lead to a range of alterations (see Figures 1B–G).
Figure 1.

Basic ways in which to transform the shape of sigmoidal response curves. Dark curves are reference curves; light curves are altered curves. Also shown is a Hill function representation for each of the curves; parameters responsible for each of the transformations are bolded. Note that these transformations are not linearly independent. In order to affect only the leakage level in panel E, k′ and k must be tuned in opposite directions such that their sum remains constant. Experimental methods for achieving these transformations are discussed in the main text.
We will focus on biological processes whose response curves can be described (or well approximated) by a single first order differential equation of the form
| 1 |
where x and y are the input and output of the process, respectively, and f(x) defines the response curve. Strictly speaking, such equations arise only from elementary chemical reactions or from multistep reaction systems where a strong separation of time-scales yields a single rate-limiting step. In many situations, however, it is possible to approximate more complex systems with simplified first-order systems, often informed by empirical observations of the system in question; this approach finds common use, and we will adopt it here.
The Hill function10,11 provides a semiempirical approach capable of capturing the class of response curves of interest. The function describes the average fraction of binding sites (of some biomacromolecule, say) occupied by an input ligand, as a function of unbound ligand concentration, x:
| 2 |
with the parameters K and n described below. It has a sigmoidal shape, ranging between 0 and 1 as x increases; the approach to 1 represents saturation, where binding sites are nearly fully occupied at all times.
The K parameter (the Hill constant) is related to the dissociation constant between the ligand and the macromolecule: it is equal to the ligand concentration for which half of all the possible binding sites become occupied. It therefore also serves as a rough indicator of the level of ligand concentrations needed to induce saturation (x ≫ K).
In some cases, if a macromolecule is already bound by a ligand, the binding affinity of subsequent ligands to that macromolecule becomes enhanced or reduced; this is known as cooperative binding, quantified in the Hill function by n (the Hill coefficient). n = 1 indicates a noncooperative reaction; n > 1 indicates cooperativity, where affinity increases in the presence of previously bound ligands; and 0 < n < 1 indicates negative cooperativity, where affinity is reduced. The larger the value of n, the steeper the slope of the Hill function.
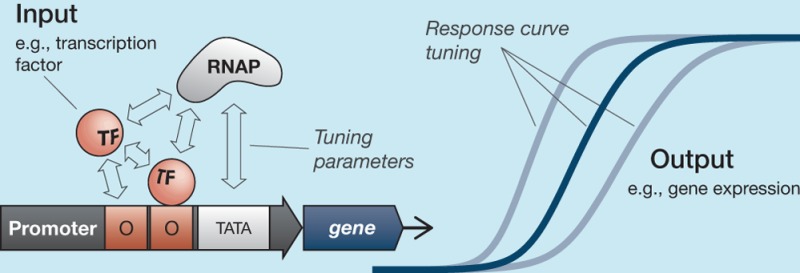
Synthetic biologists often take advantage of time-scale separations or leverage longer time-scales of interest to model multistep processes as single-step events using an empirically parametrized Hill function. The modeling of gene expression that is transcriptionally or translationally activated or repressed by an input signal (see Figure 2) commonly follows this practice, since the shape of the Hill function has been shown to agree well with experimental evidence.12 In such cases, the rate of change of protein concentration may be described by combining basal (unregulated) gene production with a Hill function term used to describe the up- or down-regulation of gene expression by a regulatory species. For activation, the expression rate increases by an additional amount proportional to θ (where x is the concentration of the regulatory species), such that the rate of new protein production may be described by
 |
3 |
where y is the concentration of the protein being expressed, k′ is the basal rate of production, k is the maximum additional production rate arising from up-regulation, and the bracketed term is an increasing sigmoidal Hill function. Repression, on the other hand, may be modeled by replacing the regulated production term with k(1 – θ) such that
 |
4 |
Here, k′ + k is the basal (unregulated) expression rate, k′ accounts for the fact that complete repression may not be possible, and the bracketed term is a decreasing sigmoidal Hill function.
Figure 2.

Simplified view of the biochemical mechanisms behind regulated transcription and their relation to tuning the rate response curve for gene expression. The input and output signals are the molecular concentrations of a transcription factor protein (TF) and an expressed protein, respectively. RNAP = RNA polymerase, O = operator site (TF binding sequence), TATA = RNAP binding sequence (TATA box in eukaryotes and archaea, −10 and −35 consensus sequences in bacteria), RBS = ribosome binding site. (Top) Transcriptional activation where the promoter-bound TFs promote the recruitment of RNAPs, increasing the probability per unit time that a RNAP will bind. Tuning parameters are in reference to eq 3. (Bottom) Transcriptional repression via steric inhibition, wherein one or more TFs physically block RNAP binding to the promoter or impede its progress along the template DNA strand (the latter case is illustrated here). Tuning parameters are in reference to eq 4. In general, mechanisms for both activation and repression vary31 and can involve more complex actions including altering DNA secondary structure and recruiting additional coregulator proteins; in eukaryotes, RNAP binding is mediated by a suite of accessory proteins.
This type of description greatly abstracts the realities of biological processes: in addition to combining multistep processes such as gene expression into a single step, it neglects fluctuations, assuming instead that a population-averaged view will be sufficient for at least any initial design work. In cases where these realities cannot be neglected, these simplifications will need to be reconsidered.
Rates of change tend to be difficult to measure experimentally. Consequently, reports of steady-state input-output (or “dose-response”) functions are seen in literature far more often than the rate response curves we describe above. Fortunately, it is possible to interconvert the two if the ancillary processes contributing to steady state are well-known. Consider a biochemical process described by eq 1, where the output y represents the concentration of a protein whose production rate is given by the response curve f(x). The total intracellular protein concentration, ytot, depends both on the protein’s rate of production and its rate of removal; in the simplest case, this can be represented by a linear degradation term in the rate equation for ytot:
| 5 |
where kd is a first-order rate constant. Setting eq 5 to zero, we can easily extract the steady-state relationship for total intracellular y as a function of constant-valued x: ytot,ss = f(xss)/kd (the ss subscript denotes steady-state). This is simply the original process’s rate function scaled by kd, implying that both the production rate and steady-state response curves share the same characteristic shape governed by the same biological parameters. More generally, the degradation rate can be nonlinear, but it remains possible to extract the f(x) rate response curve.
There are several motivations for tuning response curves in synthetic biology design work. When creating an analog control system,13−21 the nature of the response curve is critical in determining the feedback properties of the controller. More generally, the intersections and slopes of response curves determine the locations and stability properties of steady states, and controlling steady-state positions is required for virtually any multicomponent engineered system; this includes digital logic systems, where steady states determine the values and degree of separation of the digital ON and OFF states.22−26
Types of Tuning
Let us examine how each of the transformation types depicted in Figure 1 relates in principle to the Hill function description and to underlying biomolecular interactions. To make the discussion more concrete, we will continue to use transcriptionally regulated gene expression (Figure 2) as a running example; any tuning mechanisms mentioned in this context will be further expanded upon in the next section. Recall that for this particular process, the input and output signals are molecular concentrations of the transcription factor (TF) protein and the expressed protein, respectively.
Vertical Scaling
In Figure 1B, the response curves are scaled vertically, amounting to multiplication of the function f(x) (i.e., equal scaling of k′ and k in our Hill function representation). Most directly, this is done by creating multiple replicates of the entire process. For the gene expression example, this is analogous to changing the promoter-gene copy number. Alternatively, altering translational efficiency by modifying the RBS strength or through codon optimization would also vertically scale the response curve.
Vertical Shifting
Figure 1C shows the response curve shifting vertically, corresponding to a change in k′ alone. This could be achieved by introducing or tuning a constitutive source of y output (i.e., one that is not regulated by the input signal x). In the gene expression example, such an output source would amount to gene transcription from a constitutive promoter, supplementing transcription from the x-regulated promoter.
Vertical Extension
Figure 1D shows a transformation type that we have termed vertical extension, where the curve is scaled vertically but with the low end level fixed. Such a transformation would result from changing k alone. If gene expression were up-regulated by an activating TF, presumably one whose binding helped to recruit RNA polymerase (RNAP), then this could be achieved by tuning the activation potency of each bound TF protein, i.e., changing the probability per unit time that an RNAP will bind per bound TF. For down-regulated expression, we are not aware of any single-step method of transforming the response curve in this manner without also affecting the final leakage level; such tuning would likely require combining other transformations. For example, in the special case where k′ = 0, vertical extension becomes identical to vertical scaling, and a supplementary vertical shift could be used to adjust the baseline level to a nonzero value.
Leakage
We refer to the low-end level of the response curves as the output leakage level. It represents the portion of the process that is always activated in the up-regulating case, and the portion that cannot be repressed in the down-regulating case. Tuning this level without affecting the high-end saturation level (Figure 1E) is equivalent to tuning both k′ and k, while keeping their sum k′ + k constant. For down-regulated gene expression, this would amount to varying the repression strength of each bound TF protein. In the up-regulated case, we are not aware of a direct method to produce such tuning; achieving the effect would likely require combining other transformations.
Horizontal Scaling
Horizontal scaling, illustrated in Figure 1F, results from tuning of the Hill constant K (increasing K scales the curve to the right), which is related to the effective binding affinity of the input signal to the process. For transcriptionally regulated gene expression, this corresponds to tuning the binding affinity of the TF to the promoter.
Steepness
Changes in curve steepness are shown in Figure 1G and result from tuning the Hill coefficient n (increasing n leading to increasing steepness). Having a steep or switch-like steady-state response curve is often referred to as having ultrasensitivity in a biochemical process.27−30 Biochemically, changing steepness requires adjusting the effective binding cooperativity. For transcriptionally regulated gene expression, this implies the cooperative binding of multiple TFs to the same promoter (or another biochemical process that mimics this effect).
Dynamic Range
Often times, published experimental results report only values at the extremes of a biological response curve (uninduced and fully saturated induction) or in some instances just the ratios of the saturated levels in the form of “fold increases”. In such cases, the precise nature of the tuning can be ambiguous. Where possible, we speculate on plausible tuning effects for the full response curve; however, if this is not possible, we simply refer to the observed tuning as a change in the response curve’s “dynamic range”, which in reality can be achieved in many ways, particularly through vertical scaling, vertical extension, leakage tuning, or combinations thereof.
Note that all of the above descriptions assume that the output is not subject to biological limitations beyond those imposed by the input signal itself, but if this is not the case, it could change the nature of the apparent tuning. As an example, the output of an activated process could hit an absolute maximum rate for the cell, perhaps because of limitations in the availability of a substrate (e.g., nucleotides or amino acids) or a facilitating enzyme shared among other processes (e.g., polymerases or ribosomes). In this case, changes that would normally result in vertical shifting could manifest instead as leakage, as the high end of the curve ran into the upper limit of attainable rates while the low end continued to shift up and down. We assume in what follows that such global saturation is not at work in the systems discussed, but the possibility should be recognized.
Biological Options for Tuning
We now survey some specific examples of response curve tuning from the experimental literature, grouped by the biological levels at which they operate: at the transcriptional level, through post-transcriptional effects, or at the post-translational level. All input and output values will refer to molecular concentrations and rates of concentration change, respectively, unless otherwise noted. Note that while the curves in Figure 1 provide a useful framework for discussing types of tuning, experimental results are of course rarely so clean. Beyond the inevitable experimental noise, it is often the case that secondary biochemical effects lead to secondary tuning effects.
Transcription
The stability of DNA, the wide array of molecular biology techniques available for its manipulation, and the generally modular structure of an operon have combined to make transcriptional regulation a natural first target in the development of synthetic biology. Methods for controlling and tuning gene expression via transcription act predominantly through mutations to operons and transcription factors (TFs); most often, process inputs in this section will be TF concentrations.
Gene Copy Number and Location
Consider an operon consisting of a gene under the control of a TF-regulated promoter. The expression response curve for the gene could be vertically shifted upward by introducing additional copies of the gene under the control of a constitutive (unregulated) promoter, as this would contribute a flat baseline expression rate. On the other hand, increasing the copy number of the full operon would act as a multiplier for the rate at which mRNA is produced and therefore translated, leading to vertical scaling of the original expression response curve. This can be accomplished by inserting multiple repeats of an operon into the genome34 or by carrying the operon on plasmids, where copy number is variable. Plasmid copy number is typically controlled by changing the plasmid’s origin of replication, although Chen et al.(35) showed that the copy number of widely used 2-μm-based plasmids inserted into yeast can be increased by decreasing the output and stability of a selective marker gene produced by that plasmid. A library of plasmids combining these two effects exhibited up to a 3-fold increase of a constitutively expressed reporter gene from the same plasmid, indicating an increase in copy number.
A recent study by Block et al.(32) has demonstrated that the proximity of the output protein operon or the TF expressing operon to the origin of replication of a bacterial chromosome can vertically scale or affect the leakage level, respectively, of the corresponding expression response curve; see Figure 3A. In a related study, Bikard et al.(36) shuffled the order of genes in a polycistronic operon encoding tryptophan production to increase the dynamic range between saturated expression levels as high as 11-fold over the native arrangement. These efforts demonstrate that tuning is possible solely by controlling genetic context.
Figure 3.

Examples of experimental tuning curves for transcriptional regulation. (A) Vertical scaling (left) and leakage tuning (right) on log-scale plots, both achieved by varying the position of a promoter relative to a bacterial genome’s origin of replication.32 The gray and green curves represent operons nearest and furthest from the origin of replication, respectively. Image used by permission of Oxford University Press. (B) Tuning of steepness and leakage, by varying the position of operator sites relative to the TATA box.33 Copyright 2007 National Academy of Sciences of the United States of America.
Promoter Modifications
Regulating gene expression by controlling the promoter region of an operon dominated early work in genetic control and remains a primary technique today. Promoters are modular genetic units that often function across entire kingdoms, and the wide range of well-known native and synthetically designed promoters across a variety of species offers choices for expression ranges; the Registry of Standard Biological Parts (http://partsregistry.org) provides a convenient catalog of available and pretested options. Promoters are increasingly being characterized under similar genetic conditions for comparisons of strength.37−39
While earlier studies developed our understanding of the nucleotide architecture of promoters and the correlated mechanisms by which promoters function (reviewed in refs (40) and (41)), many recent investigations have focused on the tunable nature of this relationship. Particular targets for tuning have been the binding strengths of communal proteins that make up the general transcriptional machinery (e.g., RNAP and sigma factors), which we focus on first, and the binding strengths and activities of TFs, which we cover in the subsequent section on operator site modification.
The binding sites for the RNAPs, the TATA box for eukaryotes, and the −10 and −35 hexameric upstream regions in prokaryotes, are a set of consensus sequences that vary across the kingdoms in which they are found. An example of the sequence dependence of the binding strength was demonstrated by Eandwar et al.,42 who showed that mutations of the binding region of the T7 promoter, targeted by the T7 RNAP (originally from T7 bacteriophage but used widely as a heterologous RNAP in bacteria and eukaryotes), reduced the binding affinity 2- to 3-fold. In principle, this would horizontally scale outward the T7 RNAP vs expression response curve and vertically scale downward any TF vs expression response curve. The precision with which this is possible has been aided by more recent efforts to create libraries of TATA box43,44 and hexameric sequence45,46 mutations.
Since the discovery of the consensus sequences, mutations in the surrounding regions have also been known to influence the output of gene expression,47,48 with vertical scaling of the expression response curve over as much as 3–4 orders of magnitude.49
Upstream sequence (UP-element) interactions with the C-terminal domains of the RNAP have also become a target for controlling gene expression.50 A recent study by Rhodius et al.(51) determined the upstream contributions to promoter strength using a library of 60 mutated promoters. This library included mutations distal to the −35 hexamer, as far as −65 bp upstream. Different mutations of the UP-elements led to vertical scaling, achieving 2-fold increases and 4-fold decreases in gene expression. These findings were then modeled along with mutations in the core promoter regions to include all the DNA elements that contribute to promoter strength. In a study of over 2,800 constructs in yeast, Gertz et al.(52) assembled a library of enhancers with random combinations of operator sites upstream from a promoter, offering a finely tuned range of basal expression.
Operator Site Modification
Modifying the sequence, number, or position of operator sites within a promoter are common tuning techniques in synthetic biology. Since sequence modification will likely affect TF-promoter binding affinity, horizontal scaling of the TF vs expression response curve can be expected. Adding multiple copies of an operator site will permit multiple TFs to bind to a single promoter and may therefore increase maximal activation or repression levels, leading to upward vertical extension or less leakage, respectively. It should also result in outward horizontal scaling, since the number of potential TF binding locations is greater, thereby requiring a higher TF concentration to reach binding saturation. Furthermore, if TF binding to adjacent operator sites is cooperative, then response curve steepness would also be affected. Finally, we would expect operator site location to affect the ability of a TF to recruit or hinder the binding of RNAPs, therefore leading to changes in vertical extension or leakage, respectively. In practice, however, tuning results are rarely so straightforward. Moving operator site position, for example, could lead to significant changes in the secondary structure of the DNA, TF binding notwithstanding, and therefore lead to additional vertical scaling effects.
Murphy et al.(33) (and an early study by Heins et al.(53) that did not report full response curves) used operator site modification in S. cerevisiae, varying the number of operator sites binding the TF repressor TetR, and their proximity to the TATA box, to obtain a variety of expression response curves with differences in vertical scaling, steepness, and most prominently, leakage levels (ranging from approximately 0.2% to 35% of the unrepressed output). A sampling of these observed curves is shown in Figure 3B. The experimental response was measured as a function of the chemical inducer anhydrotetracycline (aTc), which acts to reduce the binding of constitutively expressed TetR to the operator site(s), and the curves thus show activation as a function of increasing aTc; if measurements were taken while varying the concentration of TetR directly, we would expect a decreasing response curve representing repression.
With regards to TF effectiveness, many operator sites show optimal proximities from the RNAP binding sites.54,55 For repressors, the mechanism by which the TF prevents the binding of the polymerase to the DNA likely determines the influence of operator position. A recent study by Garcia et al.(55) has suggested that the mechanism of repression by the Lac repressor is different for operator positions centered at −60 vs +11, resulting in differences in leakage.
Libraries of operator site and TF mutants are also increasing in number. These typically target the binding affinities of the operator-TF pairs and are similar in their goals and methods to promoter libraries.56 Milk et al.(57) combined a library of mutations in the Lac repressor at three residues known to interact with the operator, with a library of symmetric mutations in the Lac operator at bases 5 to 7, to produce a range of repression options spanning a 35-fold difference in leakage. Maity et al.(58) found that single-nucleotide changes to lac O1, the primary operator of the E. coli TF repressor LacI, led to changes of up to 6- and 12-fold in repressed and nonrepressed expression levels, respectively, indicating a combination of tuning types at work.
Promoter Escape
While the tuning approaches discussed thus far focus on regulating transcription initiation (RNAP binding), progress has also been made concerning the regulation of promoter escape: the ability of the transcriptional complex to dissociate itself from the promoter and allow elongation of the full transcript, the failure of which leads to abortive transcripts. The 20-nucleotide sequence directly downstream of the transcription start site can have a dramatic influence on the efficiency of promoter escape. Kammerer et al.(59) showed that the bacteriophage T5 N25 promoter and its derivative, the N25 antipromoter, exhibit very different rates of promoter escape (roughly 1.7 and 0.6 min–1, respectively) and ratios of abortive to productive transcripts (40 and 300, respectively), despite differing only in the initial portion of their transcribed sequences (+3 to +20). Chander et al.(60) demonstrated finer tuning using individual mutations to the 20-nucleotide sequence. These changes should in principle introduce a vertical scaling of the expression response curve.
Using a library of 43 variants and a highly abortive promoter, Hsu et al.(61) demonstrated a 25-fold range of promoter escape efficiency in vitro, resulting in an mRNA increase ranging from 5% to 150% above the native level, and vertical scaling of the rate of gene expression. Manipulation of promoter escape efficiency has since been demonstrated in vivo in E. coli,62 suggesting a key gene expression tuning approach for operons where promoter escape is rate limiting.
Modular Transcription Factor Domains
Typical eukaryotic TFs have a modular structure comprising two to three domains:31 a DNA binding domain (DBD), a trans-activating or trans-silencing domain (TAD, TSD), and an optional signal-sensing domain (SSD) that affects TF activity primarily by modulating the DBD binding affinity for its cognate DNA operator sequence in a signal-dependent manner. Modularity is conferred by the fact that these domains typically function independently, allowing for the creation of chimeric TFs through domain mixing63,64 with tuning implications that vary with the domains involved (see below).
Signal-Sensor Domains
Signal-sensor domains (SSDs) that respond to exogenous stimuli (e.g., small molecules, light, etc.) permit externally inducible control over effective TF-promoter binding affinity. In principle, this leads to horizontal shifting of the expression response curve that corresponds to TF concentration as the process input (and not the exogenous signal). To date, a wide variety of eukaryotic TFs have been created by co-opting inducible DNA binding proteins from bacteria (see ref (65) for many examples) and inserting their cognate operator sites into minimal promoters.66
trans-Activator and trans-Silencer Domains
For an activating TF, functionality can also be adjusted by varying the type and/or number of trans-activator domains (TADs). Since TADs recruit transcriptional machinery through direct binding interactions with coactivator proteins, varying these domains changes the activation potential of each individual TF, thereby in principle achieving vertical extension (adjusting the high-end saturation limit of the activation response curve). Although the potent Herpes simplex VP16 TAD67,68 is most commonly used, graded regulation has been demonstrated by fusing tandem repeats of VP16-derived minimal domains (e.g., the quad-repeating VP64 TAD) and other TADs such as human NF-κB-derived p65 and human-derived E2F4.69−71 If cognate operator sites are cloned downstream of a constitutive promoter, a DBD alone can function as a transcriptional repressor through steric RNAP hindrance, although the repression potential can often be increased by fusing a trans-silencer domain (TSD) such as yeast-derived Ume6 or human kox1-derived KRAB.72,73 TSDs typically recruit corepressor and subsequently histone proteins that alter DNA accessibility. In principle, the use and variation of TSDs would vary the leakage level in the expression response curve.
TAL-Effectors and Zinc Fingers
While importing heterologous TFs into a system of interest has been a productive strategy, there are limits to both how many such TFs are currently available and the degree of orthogonality achievable between them. These limitations have inspired the creation of synthetic TFs, constructed by fusing together TADs with protein domains engineered to bind particular DNA sequences with high specificity. These synthetic TFs have introduced the ability to activate gene expression in eukaryotes without the need for either native or heterologous promoter-TF pairs.
Transcription activator-like effectors (TALEs), first discovered in the Xanthomonus genus, have recently become an important focus of the synthetic TF field. TALEs comprise tandem repeats of small 33–35 amino acid domains, each of which recognizes and binds a single nucleotide. Covalent linkage of these domains into engineered arrays allows for the highly specific recognition and targeting of longer, user-specified nucleotide sequences.74 In a recent study targeting regions in a DNase I hypersensitive site in human HEK293T cells, Maeder et al.(75) created TALEs with varying numbers of domain repeats, allowing them to incrementally tune the dynamic range of expression between 5.3- to 114-fold. In addition, fusion to two distinct TADs, p65 and VP64, were compared. The VP64 construct yielded consistently higher expression, which we speculate is a result of vertically extending the response curve.
In another study, Perez-Pinera et al.(76) engineered several TALEs (using the VP64 TAD) to target various upstream regions within four endogenous gene promoters (distributed within 600 bp of the transcription start site) in human HEK293T cells. They observed modest transcriptional activation when using individual TALEs, but considerable synergistic activation effects for three of the four genes when expressing certain combinations of TALEs, with increases in mRNA abundance spanning a striking 4 orders of magnitude. By systematically varying these combinations, output gene expression levels were tunable over a 500-fold range (here TALEs were constitutively expressed from a common promoter, and therefore full TALE vs expression response curves were not reported since TALE concentrations were not titrated).
Similar in concept to TALEs, eukaryotic zinc fingers (ZFs) are small (∼30 amino acid) modular domains that bind to 3 bp DNA regions with engineerable sequence specificity and can be linked into multifinger arrays that recognize longer sequences.77 In recent work, Khalil et al.(78) used the OPEN platform79 to construct an artificial library of specific and orthogonal ZF array-promoter pairs, in particular, three-finger arrays that bind to cognate 9-bp operators. They then fused the arrays to a minimal VP16 TAD to create a library of synthetic TFs (ZF-TFs) that activated expression with dynamic ranges ranging from 1.3- to 6-fold and then showed that key TF properties could be rationally and independently adjusted to further tune transcriptional output. First, they multimerized ZF-binding operator sites in order to recruit greater numbers of ZF-TFs. For promoters harboring one, two, and eight tandem operators, corresponding increases in maximal expression were observed (indicating a possible increase in vertical extension, although full response curves were not presented); interestingly, a less-obvious decrease in leakage was also seen. They then performed structure-guided mutation of the ZF array backbone (i.e., outside the DNA recognition helices) to decrease its DNA-binding affinity, presumably implying outward horizontal scaling of the expression response curve. Decreased expression levels were indeed seen as the number of mutated residues increased from one to four. Finally, they tested configurations in which two different ZF-TFs could dimerize via the addition of modular PDZ protein–protein interaction domains from metazoan cells. In this case, expression from a single promoter containing the two corresponding operator sites was shown to be synergistic in nature when both ZF-TF types were present, with increased vertical extension due to the interaction.
In a recent study, Lohmueller et al.(80) fused various leucine zipper (LZ) homodimerization domains to ZF-TFs and found that these added domains improved activation and repression up to 2.5- and 7.5-fold, respectively, using a human c-Jun LZ (dimerization kd = 448 μM) and up to 10-fold and 8-fold, respectively, using a stronger homodimerizing yeast GCN4 LZ (kd = 8 nM). This corresponds to vertical extension tuning in the activating case and leakage tuning in the repressive case.
Orthogonal RNA Polymerases
RNAPs that are orthogonal to native promoters offer an alternative to activating TFs. Orthogonality permits varied concentrations within a cell without compromising native cell function and therefore varied rates of transcription exclusively from cognate (RNAP-specific) promoters. Temme et al.(81) developed a set of four orthogonal variants of the heterologous T7 RNAP along with cognate polymerase-specific promoters for use in E. coli. Similar to swapping TF-promoter pairs, these T7 RNAP variants could be interchanged to alter the expression response curve, albeit coarsely and unsystematically (published results show varying output levels at saturating T7 RNAP concentrations, corresponding to varying vertical scalings or extensions, but omit full response curves); some finer tuning, however, was achieved via mutagenesis to a 5-bp strength-determining region of the promoter.
Nuclear Localization and Export Sequences
In eukaryotic cells, genetic material and transcriptional machinery is contained in the nucleus, segregated from translational and metabolic machinery in the cytoplasm. The bidirectional translocation of TFs through the nuclear envelope (via nuclear pore complexes) facilitates another layer of transcriptional regulation, as transfer rates vary among different proteins and in some cases for the same protein depending on its state of post-translational modification (e.g., phosphorylation state). While macromolecules smaller than ∼40 kDa can passively diffuse through these pores, most proteins with intranuclear function undergo active but selective transport mediated by nuclear import and export receptors that recognize and bind to certain short amino acid nuclear localization sequences (NLSs) and nuclear export sequences (NESs), respectively.82,83 Sequence variation generates different binding affinities, which correlate to protein import and export rates and therefore nuclear concentration.84 By fusing different NLSs and (in some cases) NESs to the termini of proteins, researchers have shown the ability to adjust the ratio of in vivo steady-state nuclear to cytoplasmic accumulation over a wide range in different eukaryotic cells including yeast.84−86 Furthermore, variants of so-called classical NLSs have been generated with quantified binding affinities to Importin (the import receptor protein) ranging over several orders of magnitude.84,87 Within our Hill function representation of gene expression, increasing the ratio of nuclear to cytoplasmic TF by some factor would be equivalent to decreasing the Hill constant, K, by the same factor (where the input x remains representative of total nuclear-plus-cytoplasmic TF concentration), thereby horizontally scaling the expression response curve.
Nuclesome-Disfavoring Sequences
Recently, the use of nuclesome-disfavoring sequences has emerged as a promising tuning technique in eukaryotes. TF-promoter binding is regulated by nucleosomes, segments of DNA wrapped around histone proteins that are the fundamental repeating unit of chromatin structure, that restrict access to potential operator sites. In a recent study, Raveh-Sadka et al.(88) tuned transcription activation in yeast by targeting local nucleosome organization, accomplished by the insertion of poly(dA:dT) tracts (homopolymeric stretches of deoxyadenosine nucleotides, highly prevalent in natural eukaryotic promoters and known to disfavor nucleosome formation) into a specific promoter. Rational fine-tuning was demonstrated by systematically varying the length, composition (i.e., purity), and relative distance from the activating TF operator site of the inserted poly(dA:dT) tract. Manipulating these tracts affects TF access to its cognate operator site, which in principle results in modulation of the average TF to DNA binding affinity and, therefore, horizontal scaling of the expression response curve. There are two standout benefits of this technique: First, it offers much finer control over gene expression than possible even with singular point mutations to the TF operator site. Second, it works around the problem of limited orthogonal TFs; for example, poly(dA:dT) tracts can generate a multiplicity of responses from different promoters using the same common TF.
Post-Transcription
Expression control at the translational level represents a promising alternative to the control of transcription initiation. mRNA transcripts are often targeted by RNA-based regulators on the basis of Watson-Crick base pairing. This has enabled researchers to design and tune novel regulators using model-based techniques, permitting a systematic engineering approach not yet available for the protein effectors used in transcriptional control. In this section, we present methods to tune constitutive translation rates (modifying ribosome binding sites (RBSs), codons, and mRNA degradation rates) and translation rates that are inducible by noncoding RNA or other small molecule effectors (riboregulators, aptamers, and RNA interference (RNAi)). For a general review of natural RNA-based regulatory devices, see ref (89); the engineering and current diversity of synthetic devices are reviewed in refs (90) and (91), respectively.
Ribosome Binding Site Modifications
Modifying the RBS on an mRNA transcript alters the efficiency of translation initiation, thereby in principle vertically scaling the overall expression response curve. Currently, the RBS is one of the most attractive options for tuning because its strength can be, in large part, forward-engineered using model-based design. Citing the previous work of others as their foundation, Salis et al.(92) developed a predictive method for designing synthetic RBSs for any gene of interest based on statistical thermodynamic modeling. Experimental validation of over 100 predictions in E. coli showed the method’s predictive accuracy to be within a factor of 2.3 over an impressive 5 orders of magnitude of translational efficiency.
Orthogonal Ribosomes
In addition to modifying the RBS, the ribosome itself can be modified such that it becomes orthogonal to wild-type translation, recognizing instead synthetic RBSs.93−95 By modifying internal segments of the orthogonal ribosome’s rRNA, horizontal scaling of translation can be achieved.96 Through a combination of computational design and experimental measurement, Chubiz and Rao97 demonstrated that orthogonal ribosomes could display apparent vertical scaling by varying the sequence of the 16S rRNA. They further demonstrated tuning of dose-responses to inducers of either rRNA or cognate orthogonal ribosome mRNA, achieving vertical scaling and steepness tuning.97
Start Codon Modification
While AUG is the most commonly used start codon in most species, translation can also be initiated from alternative start codons that differ in efficacy and differ between species. In E. coli, for example, varying start codon usage can vertically scale an expression response curve, both up and down relative to the standard AUG start codon.98 Similar possibilities exist in eukaryotes wherein start condons seem to be especially sensitive to genetic context. For example, the presence and length of flanking polyU or polyA sequences can induce translation initiation from yeast non-AUG codons such as UUG, ACU, and ACG that otherwise exhibit almost no translational activity.99 Context sensitivity is also a property of prokaryote start codons, where vertical scaling can arise from flanking polyA/U sequences, nearby stem-loops, and variations in the proximity and strength of the RBS.98,100
Codon Optimization
In synonymous codon optimization, the triplet RNA sequences coding for amino acids are replaced with alternative triplets coding for the same residue. Organisms display kinetic preference for certain codons sequences, and codon replacements can significantly affect the efficiency of translation elongation, thereby altering translation rates and total gene expression levels.101
Welch et al.(102) devised a partial least-squares based model to correlate synonymous codon choices for a particular gene with its observed expression level in E. coli. (Interestingly, the codon choices that maximized expression were not necessarily those most commonly found in native E. coli transcripts.) Combining this model with a genetic algorithm allowed for the generation of synthetic transcript sequences that produced precalculated expression levels. In principle, codon optimization allows for vertical scaling of a gene expression response curve, under the assumption that codon mutations do not interact with other control elements.
Riboregulators
A riboregulator is an RNA sequence that responds to the Watson-Crick (sense-antisense) base pairing of a signaling nucleic acid molecule, commonly for the purpose of regulating translation. Isaacs et al.(103) introduced a short DNA sequence complementary to and directly upstream from the RBS, such that the 5′ UTR of resulting mRNA transcripts, referred to as cis-repressed mRNA (crRNA), folded naturally to form a stem-loop structure that sequestered the RBS and inhibited translation initiation with extremely low leakage levels (down to 2% in E. coli). Activation was then achieved by independently transcribing noncoding RNA, referred to as trans-activating RNA (taRNA), designed to target and hybridize to the crRNA, unfold the stem-loop structure, and expose the RBS. Tweaking sequence complementarities provided limited coarse-tuning of the taRNA vs expression rate response curve: alterations to the stem of the crRNA stem-loop structure resulted in modest variations in leakage levels, while taRNA truncation and alterations to the taRNA-crRNA hybridization sequence influenced activation levels, possibly at least in part a result of horizontal shifting of the response curve, since variations to taRNA-crRNA binding affinity were observed.
Practically speaking, the easiest way to obtain differing responses using riboregulators is to simply choose from a set of precharacterized heterogeneous riboregulators; if these riboregulators are functionally orthogonal, they can be used simultaneously and effectively within the same cell. Isaacs et al.(103) produced two orthogonal crRNA-trRNA riboregulator pairs for E. coli that exhibited 8- and 19-fold repression-to-activation dynamic ranges, and this set was later expanded by Callura et al.(104) to include two additional orthogonal pairs with ∼70- and ∼200-fold dynamic ranges. Recently, Mutalik et al.(105) used a model-guided design approach, involving hybridization free energy calculations and data clustering algorithms, to forward engineer new families of five and six mutually orthogonal trans-repressed riboregulators for E. coli with consistent and predictable leakage levels. In the process, riboregulators with leakage levels ranging from 10% to 95% of the nonrepressed expression level were isolated.
Aptamers
A post-transcriptional aptamer is a sequence of nucleotides designed, typically through an in vitro selection process, to bind strongly and specifically to a ligand of interest.106 Such sequences are playing an increasingly prominent role in post-transcriptional control devices: by serving as allosteric sites built into mRNA transcripts, they enable coupling between exogenous ligand concentrations and translation rates.107 Aptamers are normally sourced synthetically: given a particular ligand of interest, the space of possible nucleotide sequences is methodically searched via a directed-evolution procedure known as SELEX (Systematic Evolution of Ligands by EXponential enrichment).108−111
By coupling aptamer technology to trans-acting RNA molecules, Bayer and Smolke112 were able to engineer several ligand-controlled riboregulator systems, called antiswitches due to the fact that a ligand-induced conformational change exposes an antisense sequence (otherwise sequestered by a proximate complementary sequence) that binds a target mRNA transcript and blocks its translation. Tuning was demonstrated in S. cerevisiae by varying the conformational equilibrium of the trans-acting RNA itself: lengthening the sequestering sequence or introducing mismatches between it and the antisense domain increased or decreased, respectively, the amount of effector ligand required to repress target mRNA repression. This resulted in horizontal scaling and changes to the vertical extension of the ligand vs expression response curve; the latter reflecting changes to the fraction of the trans-acting RNA molecules with their antisense domains exposed in the absence of ligands.
Carothers et al.(113) created a set of tunable RNA-based devices capable of delivering a wide range of gene expression outputs. One set of RNA structures took the form of ribozymes: RNA structures able to catalyze reactions. These ribozymes catalyzed 5′ UTR cleavage in target mRNA, leading to increased mRNA half-life and thus to greater gene expression. A second set of RNA structures, classified as aptazymes, exhibited similar UTR-cleavage activity but were also augmented with aptamer sequences allowing for ligand-sensitive cleavage rates. A sophisticated modeling approach was used to guide the design process, combining a biochemical kinetic model with RNA folding simulations. Twenty-eight distinct RNA systems were constructed and characterized experimentally, with widely varying gene expression levels suggesting ligand vs expression response curves with various vertical scalings. Importantly, the model was successful at predicting the observed experimental results, and analysis of the model offered direct guidance in the design process by identifying important steps in the biochemical kinetics and predicting sequence mutations likely to affect those steps. This highlights the strong potential for tuning ribozyme/aptazyme devices through systematic, model-guided design.
Transcriptional Attenuators
In antisense RNA-mediated transcriptional attenuation, the binding of an antisense RNA to an “attenuator sequence” in the 5′ UTR of a nascent mRNA transcript causes it to fold into a configuration that exposes an intrinsic transcriptional terminator hairpin, resulting in premature transcription termination.114 Lucks et al.(115) designed three such antisense RNA sequences to function orthogonally in E. coli. In a method mirroring the repetition of operator sites in a promoter, series insertion of an additional identical attenuator sequence into the 5′ UTR of the target transcript steepened and reduced the leakage level of the antisense vs expression response curve in a manner agreeing remarkably well with the multiplication of single attenuator Hill functions (when normalized to 1); see Figure 4A. This suggests that attenuators in series function independently, as in the case for engineered tandem ribozyme devices.116
Figure 4.

Examples of experimental tuning curves for post-transcriptional regulation. (A) Dose-response curves for IPTG-inducible pT181 transcriptional attenuators. Curves are shown for wild-type (left) and mutant (right) attenuators designed to function orthogonally. Repression curves for a single repeat of the attenuator (circles) and for two attenuators in tandem (squares) are shown; addition of the second attenuator leads to a decrease in leakage and an increase in steepness. Image from ref (115). Copyright 2011 National Academy of Sciences of the United States of America. (B) Tuning of horizontal scaling and leakage of shRNA switches via modulation of the 3′ length (left) and 5′ length (right) of the region of complementarity between the competing strand and the shRNA stem sequence of a hairpin transcript. On each plot, results are shown for multiple sequence lengths. Image from ref (117). Reprinted by permission from Macmillan Publishers Ltd: Molecular Systems Biology, Beisel, et al., 4, 224, copyright 2008.
mRNA Degradation Control
The previously mentioned post-transcriptional strategies affect translation efficiency per mRNA transcript. An alternative approach to tuning translational rates involves controlling the transcript concentrations themselves; this would lead to vertical scaling of the gene expression response. To this end, researchers have developed targeted methods for manipulating the rate of mRNA degradation.
Early work examined factors involved in controlling mRNA stability, most notably the effect of hairpin secondary structures in the 5′ UTR of the transcript (reviewed in ref (118)). By introducing rationally designed hairpins into E. coli mRNA, Carrier and Keasling119 were able to influence half-lives over an order-of-magnitude range. More recently, Babiskin and Smolke120 developed an RNA device in S. cerevisiae enabling aptamer-mediated transcript cleavage. This was done by inserting into the 3′ UTR of the transcript of interest a hairpin-shaped formation amenable to cleavage by the ribonuclease Rnt1p and containing an aptamer sequence that leads to inhibited cleavage activity when ligand-bound; in the absence of ligand, Rnt1p cleavage proceeds normally and the transcript, with its polyA tail removed, is quickly degraded.
Babiskin and Smolke120 employed three different strategies to tune the monotonically increasing ligand vs gene expression response curve. Changes to a key region of the hairpin sequence controlling cleavage efficiency yielded various combinations of vertical extension and leakage tuning, typically with only slight horizontal scaling. Changes to another key region of the hairpin controlling ribonuclease binding affinity and containing the aptamer sequence resulted in more notable horizontal scaling, as was expected; in addition, changes in vertical extension and leakage levels were again observed, likely resulting from nucleotide modifications to the hairpin stem that were required for variable aptamer integration. Finally, positioning multiple hairpin copies (up to three) within the 3′ UTR resulted in outward horizontal scaling and reduced leakage levels due to the increase in potential cleavage targets for Rnt1p. In particular, the expression response curve’s Hill constant, K, increased nearly additively with hairpin copy number, while the leakage level decrease was approximately multiplicative (47%, 20%, and 10% for one to three copies).
RNA Interference
RNA interference is the process whereby small double stranded RNAs (dsRNAs) down-regulate protein expression via either steric hindrance of the ribosome or induced endonuclease cleavage of the target mRNA. Both of these processes are mediated by the RNA-induced silencing complex (RISC), which comprises a number of interacting proteins, and the single strand from the dsRNA complementary to the target site.
The development of RNA interference as a therapeutic tool to silence gene expression has spurred the search for novel and improved pharmaceutical properties for medicinal purposes (reviewed by Rettig and Behlke121). The potential to introduce synthetic small interfering RNAs (siRNAs) into cells and tissues has led to a wide-ranging examination of siRNA properties, particularly their stability under hostile conditions (such as in blood) and their silencing strength. The search has largely involved screening based on sequence and target sites122,123 and on chemical modifications.124−126 Efforts have typically focused on identifying the strongest and most robustly silencing siRNAs, but for tuning purposes the range of characteristics across an entire library is of interest: choosing siRNAs with varying binding affinities potentially allows one to achieve horizontal scaling of an siRNA vs gene expression response curve, while choosing those with varying silencing strengths has the potential to influence leakage. Thus far, a strong focus on finding the strongest silencers has meant that other library candidates have rarely been characterized; collecting data on the full range of silencing strengths obtained from a library would provide valuable tuning information for future applications.
One potential method of delivering siRNAs to human cells is through the bloodstream, in which case their serum stability is critical. Hong et al.(127) demonstrated that a large proportion of native siRNAs are serum stable and that RNA duplexes in serum are cleaved preferentially at two sequence-dependent dinucleotide sites, which can be avoided during design in order to improve stability. For siRNAs containing these dinucleotide sites, even single modifications to the sugar backbone within the sites were sufficient to significantly increase stability. Varying siRNA serum stability led to horizontal scaling of the silencing response to a given dose of siRNA.
A study by Patel et al.(128) compared the potency across sets of standard siRNA constructs used to target various sites on single genes essential for cell growth. Although the general trend was that longer, chemically modified siRNAs yielded more effective silencing, the specific target sequence also played a substantial role, leading the authors to suggest that the length, chemical modification state, and target site sequence should all be considered as factors in an siRNA’s level of silencing. Carrying out short-term growth assays showed horizontal scaling over an order of magnitude of siRNA concentrations.
Using an in vivo method for controlling siRNA concentration, Beisel et al.(117) described the use of small hairpin RNA (shRNA)-based switches that respond to chemical induction through an aptameric distal loop embedded in the hairpin sequence. The hairpin exists in equilibrium between two conformations, one that is efficiently processed by the RNAi machinery and another that is not. In the latter, the aptamer is exposed and binding of a ligand stabilizes this conformation, effectively preventing the hairpin from forming its active siRNA state. Thus, higher intracellular ligand concentrations reduce silencing of the shRNA’s target protein. Aptamers responsive to hypoxanthine, tetracycline, and theophylline ligands were successfully tested in human HEK293T cells. Guided by a semiempirical thermodynamic model, the lengths of the complementary RNA regions in the aptamer-stabilized shRNA conformations were varied, yielding systematic changes to the expression response curve, primarily in horizontal scaling and leakage; see Figure 4B.
In a follow-up study, Beisel et al.(129) inserted RNA aptamers into the basal stems of shRNAs, rather than the distal loops. In this system, the shRNA sequence itself was inserted into the 3′ UTR of a fluorescent reporter, allowing for direct monitoring of shRNA levels. Interestingly, the physical size of a mismatched basal bulge was found to correlate directly with the degree of repression of the shRNA against its target, suggesting that it provides a steric cue for processing of the hairpin. This allowed for leakage tuning through modulation of the basal bulge size, though the choice of size was limited by the requirement of efficient aptamer-ligand binding. Vertical scaling was achieved by adding more copies of the shRNA in tandem onto the 3′ UTR of the reporter gene; up to four copies were added, separated by spacer sequences. Each added copy reduced both uninduced and fully induced expression levels, consistent with a vertical scaling effect. Different spacer sequence lengths were also tested, separating two copies of the shRNA in the 3′ UTR. Increasing the spacer length was found to decrease leakage without appreciably affecting the maximal activity level in this case.
Tunable Intergenic Regions
Pfleger et al.(130) describe a method for tuning the relative expression of multiple genes within an operon using tunable intergenic regions (TIGRs): intergene nucleotide sequences containing control elements that include mRNA secondary structures, RNase cleavage sites, and RBS sequestering sequences. TIGRs are designed for placement between two genes in a polycistronic operon so that upon transcription, the RNase cleavage site is cut and two distinct transcripts emerge, each containing a residual portion of the TIGR sequence (at the 3′ and 5′ ends, respectively) that modulates transcript stability and translational efficiency. Moreover, large secondary structures in the TIGR can lead to premature transcription termination, heavily affecting the transcriptional efficiency of the second gene in the operon. By assembling and screening large libraries of TIGRs, Pfleger et al.(130) demonstrated that TIGRs could vary the relative expression levels of two bicistronic genes over a 100-fold range (offering in principle vertical scaling of an expression response curve). Furthermore, they simultaneously tuned the expression of three genes within an operon encoding a heterologous mevalonate biosynthetic pathway in E. coli in order to optimize its output flux.
Protein-Based Systems
Another recent development is the use of protein-based systems to implement control at the post-transcriptional level. Stapleton et al.(131) described such a system, built around the regulatory protein L7Ae, that enabled tunable translational repression. Provided that the recognition sequence for L7Ae (or a variant thereof) is present upstream of the coding region of interest, the L7Ae protein will sterically block translation of the downstream sequences. The repression is entirely translational and does not affect the expression of other cistrons translated from internal ribosome entry sites (IRESs), nor is it expected to appreciably modulate the degradation rate of the transcript. In vivo tunability was achieved via an informed trial-and-error process: the wild-type L7Ae binding sequence was mutated with the intent of reducing the repressive strength of the interaction to varying degrees compared to the wild-type interaction. The authors achieved considerable horizontal scaling, but the range of experimental results did not make it possible to determine if they also obtained vertical scaling; basal expression remained largely unchanged across all trials. One apparent advantage of protein-based translational control over RNAi-based strategies is that the protein-based systems may require relatively less ancillary machinery thereby reducing the risk of saturating dynamics, whereas in the case of RNAi, it has been observed that multiple targets or multiple shRNAs in the same cellular system occasionally saturate the RNA-induced silencing complex responsible for transcript regulation.132
Post-Translation
Biological devices that respond by producing new proteins are forced to operate on long time-scales (minutes to hours or even days) by the delays inherent in the processes of transcription and translation. Post-translational systems, involving protein-protein interactions, can respond much more quickly (fractions of a second to minutes) to changing inputs. Unfortunately, this increased speed currently comes at the cost of significantly increased difficulty in tuning protein function.5,133,134 Transcriptional initiation and much of post-transcriptional processing are highly modular and accessible through well-established molecular biology protocols, allowing designers to freely substitute promoters, coding regions, and untranslated regions while keeping basic functionality largely unchanged. Proteins, by contrast, function through chemical interactions that are strongly dependent on their physical structure, and the complexity of this structure-function relationship makes rational protein design challenging. These issues mean that there are as yet fewer examples of tuning available in the post-translational space than in the previous two levels of regulation.
Dose-Responsive Enzymatic Catalysis
Consider a process describing an enzyme-catalyzed biochemical reaction, where the process input is the amount of available substrate, and the output response is the reaction rate. Such a response curve is often described by well-known Michaelis-Menten kinetics wherein the reaction rate varies from zero to some saturating value Vmax = kcatE (for effective enzyme concentration E) as the substrate concentration increases (see Figure 5A). In our Hill-function notation, such a process has k′ = 0, k = Vmax, and n = 1. (We offer this function as an example of an enzyme-catalyzed response curve, but please note that the assumptions underlying the Michaelis-Menten equation will not apply to all post-translational systems. Signaling cascades in particular will tend to violate the assumption that the catalyzing enzyme is present in much lower concentrations than a target substrate.)
Figure 5.

Tuning of enzymatic reactions. (A) Michaelis-Menten kinetics for an enzyme-catalyzed reaction: the rate of the reaction x→y varies with the input concentration of the substrate, x, and with the enzymatic catalyst’s effective concentration and catalytic efficiency: Vmax = kcatE. (B) The relationship between Vmax and the input ligand can be characterized (and itself tuned): each marked point, i–iv, on the Vmax vs ligand concentration curve yields a different response curve shown in panel A. (C) A protein switch, in which the protein of interest (POI) contains autoinhibitory domains that bind and inactivate the enzyme’s catalytic activity, reducing the enzyme’s effective concentration. The presence of a competitively binding ligand can relieve the inhibition and restore catalytic activity.
Many naturally occurring enzymes change their catalytic activity as a function of the binding of some intra- or extracellular ligand. This results in a change in the effective value of Vmax, thereby vertical scaling the Michaelis-Menten response curve (see the relationship between Figures 5A and B). (In this special case with k′ = 0, this also represents vertical extension.) Therefore, this can be seen as a form of response curve tuning, where the tuning strength is dependent on ligand concentration.
Protein Switches
Designers needing to control processes whose enzyme catalysts do not natively respond to any ligand may benefit from reengineering of the enzyme itself. Protein switches are enzymes engineered to have inducible ON and OFF states in terms of their catalytic activity: the binding of a ligand flips individual proteins between their active and inactive conformations. This permits the tuning situation described above, where effective Vmax is tuned through ligand concentration. In many such systems, the ligand vs Vmax dose-response curve is also easily tunable. Additional coverage of protein switches may be found in several recent reviews.135−138
Most strategies for generating protein switches involve fusing or inserting modular domains into the protein of interest such that they disrupt or facilitate, either sterically or conformationally, the activity of the target protein. Examples of inputs for this type of regulation are small molecules,139 light,140−143 ions,144 and redox conditions.145 One important approach is to construct an autoinhibitory pair of domains that dimerize and inhibit protein activity when no competitive ligand is present. In the presence of the ligand, the dimerization is disrupted, allowing the protein to become active (Figure 5C); we describe two such switches, below.
Dueber et al.(27) controlled the activity of the actin polymerizing protein N-WASP by fusion of both an SH3 protein domain and an SH3-binding peptide, such that in the absence of competing (nonfused) SH3-binding peptides, N-WASP was autoinhibited and rendered inactive. The dose-response between free SH3-binding peptides and active N-WASP was showed to be tunable by controlling modular components such as the number of SH3 domains and SH3-binding peptides fused to each N-WASP, as well as their binding affinities. The primary result of doing so was steepness modulation, as shown in Figure 6A; however, steepness was difficult to manipulate independent of secondary changes to horizontal scaling, vertical shifting, and vertical scaling. The autoinhibitory paradigm was also expanded to include PDZ and GBD interaction domains, which were subsequently assembled together to regulate N-WASP under the control of a three-input AND gate operating on fast time-scales.
Figure 6.

Examples of experimental tuning curves for post-translational regulation. (A) Steepness tuning by varying the number and binding affinity of an autoinhibitory domain in protein switches.27 Reprinted by permission from Macmillan Publishers Ltd.: Nature Biotechnology, Dueber et al., 25, 660–662, copyright 2007. (B) Steepness tuning and horizontal scaling by varying the binding affinity of a decoy domain in a peptide system.30
Lu et al.(30) reported results on a peptide system in which an autoinhibitory PDZ domain was fused to a binding domain for SH3 peptides. In the absence of SH3, the PDZ formed a loop structure, while binding of SH3 to its domain prevented this autoinhibition, placing the peptide into a loop-free conformation. The conformation of the system was determined through fluorescence measurements, with the looped state defined as inactive and the loop-free state defined as active. Decoy sites, able to bind SH3 without affecting the activation state of the system, were then added to the peptide and shown to have an extensive ability to tune the relationship between the peptide’s activation state and the level of SH3 present. Use of a single autoinhibitory PDZ pair resulted in a noncooperative dose-response to free SH3, while varying the numbers and binding affinities of the decoy domains led to a variety of dose-responses to SH3, implementing horizontal scaling and steepness tuning (Figure 6B).
Structure Rescue
Structure rescue is another promising strategy for tuning enzymatic activity. Allosteric control is engineered into an enzyme by structurally weakening the enzyme through mutation to the point that its enzymatic activity is abolished, then identifying a small molecule able to bind to the mutant protein and restore its original structure. Average enzymatic activity therefore becomes tunable through the concentration of the small molecule, providing inducible vertical scaling of the Michaelis-Menten curve via control over Vmax. Deckert et al.(146) successfully restored β-glycosidase activity in a W33G mutant by rescue with indole, adopting the approach that the small molecule inducer should be exactly complementary to the residue(s) missing in the mutant protein, to achieve structure rescue. They focused on buried and tightly packed tryptophan residues that, when mutated to glycine and supplemented with indole, yielded a protein structure highly similar to the original. The best mutant was able to fully restore original activity levels in the presence of sufficient indole and thus was exogenously tunable from effectively zero activity to its wild-type level.
Protein Half-Life Modulation
We have focused mainly on rates of production of biochemical species, but of course one can also treat degradation as its own process with a response curve (typically Michaelis-Menten) to be tuned. To this end, a number of techniques have been developed. One method involves tagging proteins with a short amino acid recognition sequence to induce degradation by an alternative set of proteolytic machinery. The ssrA tag, for example, induces degradation catalyzed by the ClpXP protease system,147 and control of some combination of the tag sequence variation, the ClpXP protease level, and the level of SspB (an optional adaptor protein) has been shown to effectively tune both Vmax (vertical scaling) and the Michaelis constant K (horizontal scaling).148−151
Tunable degradation has also been achieved by adding modular domains to proteins that promote proteolytic degradation in the presence (or absence) of a bound ligand. Most such ligands are small molecules, including Shield ligands,152,153 hydrophobic libraries for Halo-tags,154 auxin,155 and trimethoprim (TMP).156
Scaffolding
Proteins, DNA, and RNA have all been used to construct “scaffolds” that co-localize multiple interacting molecules by assembling them into a single physical complex, enhancing interaction rates. In simple cases, this would serve to tune the effective Vmax of a biochemical reaction, providing vertical scaling for the reaction process.
Dueber et al.(157) built synthetic protein scaffolds bearing modular SH3, PDZ, and GBD interaction domains that spatially recruit three metabolic enzymes tagged with cognate peptide ligands in order to enhance the production of mevalonate and glucaric acid. Varying the number of interaction domains fused to the scaffold allowed for optimization of the stoichiometry between the recruited enzymes, resulting in a 77-fold improvement in mevalonate production, as well as a 3-fold improvement in glucaric acid production (despite already high yields). Note that in this study, the relationship between the scaffold concentration and total product production was nonmonotonic, rising to a peak at intermediate scaffold concentrations. Although the variation of the interaction domain stoichiometry resulted in prominent changes to peak height and location, these response curves are not captured by the sigmoidal response curves we have focused on in this review.
DNA and RNA molecules can also be used as scaffolds. For example, enzymes can be fused to zinc fingers or other programmable DNA-binding domains158−160 and RNA aptamers can be designed to bind enzymatic partners.161
Beyond Rate Tuning
Our final sections address topics that diverge from the issue of tuning production rates but do bear on the broader problem of systematically creating biological devices: the use of network structure effects to tune net steady-state response curves and the creation of consistent, modular systems.
Network Extension
Extending or restructuring the internal signaling network of a process can introduce complex and significant transient dynamics that cannot be accurately described by eq 1. For example, relatively simple feedforward or feedback connections can give rise to delayed responses, oscillation, or temporal adaptation;162 this implies time-dependent rate response curves. Although important in many design scenarios, such behavior is beyond the scope of this review. In this section, we address only rate response curves for which this behavior is insignificant, focusing our attention on how network extension has been used to tune a process’s steady-state response curve.
In principle, network extension is a strategy limited only by the availability of orthogonal parts that can be tuned into similar ranges of responsiveness; in terms of a steady-state response, the size of a process’s internal network structure is irrelevant. Here, however, we review a few examples where network extension is mainly confined to the addition of a single component or network node.
Linear Cascades: Sensitivity Tuners
A simple example of network extension is provided in the work of a University of Cambridge team, who constructed and characterized a set of 15 transcription-based “sensitivity tuners” in E. coli for the 2009 iGEM competition.163 Each tuner is essentially a phage-derived activating TF-promoter pair designed to be inserted, in series, between the input and output signals of a linear transcriptional cascade in order to modify the shape of its rate response curve. Each tuner was characterized in an otherwise consistent construct described by x → A → y, where → denotes promoter up-regulation, and x, A, and y represent chemical inducer, phage activator, and reporter protein levels, respectively (see Figure 7A). Describing the rate response curve as in eq 3, they found that, across the set of 15 tuners, K varied by an order of magnitude, k′ was fairly consistent, k depended to a large extent on the tuner’s activator type (and less on the promoter choice), and n shifted between values of around 2.25 to 4 (experimental resolution was too small to characterize n with much confidence), leading to horizontal scaling, vertical extension, and steepness tuning, respectively. Another early study by Hooshangi et al.,164 investigating synthetic transcriptional cascades comprising one, two, and three repression stages in E. coli (see Figure 7B), demonstrated that as cascade length increases the overall steady-state response curve steepens (increasing n).
Figure 7.

Schematics describing the general biochemical network designs reviewed in the Network Extension section. (A) A two-promoter construct containing an interchangeable sensitivity tuner, which comprises the gene for a transcriptional activator protein and its cognate promoter. Our schematic has been drawn based on the description provided in ref (163). (B) From ref (164). Transcriptional cascade comprising one, two, and three repression stages. Hooshangi et al., Proc. Natl. Acad. Sci., 102, 3581–3586. Copyright 2005 National Academy of Sciences, U.S.A. (C) From ref (28). An internal feedback loop is engineered into the yeast mating MAPK pathway via the downstream expression of a pathway modulator protein that binds to the Ste5 scaffold protein through a leucine zipper interaction. Feedback polarity is determined by the choice of either a positive or negative modulator, which up- or down-regulates the MAPK cascade, respectively; feedback gain is tuned by varying promoter strength or varying the leucine zipper domains to affect binding affinity. From Bashor, et al., Science, 2008, 319, 1539. Adapted with permission from AAAS. (D) From ref (165). Two coupled positive feedback loops are engineered into a two-component signaling system via downstream expression of both the transmembrane receptor and intracellular TF proteins. Adapted by permission from Macmillan Publishers Ltd.: Molecular Systems Biology, Palani et al., 7, 7, copyright 2011. (E) From ref (166). Two parallel pathways constitutively repress the expression of an output protein. One pathway uses a TF repressor to repress transcription, while the other pathway represses translation using an shRNA to induce RNAi-mediated mRNA degradation. In the bottom schematic, a single input signal down-regulates TF and shRNA production thereby up-regulating expression of the output protein. Adapted from Cell 130, Deans et al. A tunable genetic switch based on RNAi and repressor proteins for regulating gene expression in mammalian cells, 363–372. Copyright 2007, with permission from Elsevier.
Feedback Loops: Engineered Scaffold Interactions
Bashor et al.(28) engineered feedback into the yeast mating MAP kinase pathway, a post-translational signal transduction cascade mediated by the Ste5 scaffold protein, which co-localizes signaling molecules by assembling them into a single physical complex, thereby promoting correct pathway connectivity. An internal feedback loop was generated by expressing a pathway modulator protein as a downstream product of the signaling pathway and recruiting it back to the upstream pathway via an artificial binding site on Ste5 created by fusing cognate leucine zipper interaction (heterodimerization) modules to the scaffold and modulator proteins (see Figure 7C).
The system was then tuned by adjusting the polarity and strength of the feedback signal: the former by using either a positive or negative modulator protein and the latter by changing the strength of either the leucine zipper interaction (using zipper pairs with varying Kd) or the promoter controlling modulator expression (both to the same effect). Positive feedback was shown to vertically scale (upward) and steepen (from n∼0.12 to 2.42) the overall network’s steady-state response curve while negative feedback resulted in vertical scaling (downward). Furthermore, by having the feedback positive modulator displace a constitutively expressed negative modulator, response curve steepness was further increased to n∼2.84.
Feedback Loops: Bistability
Through the addition of a pair of coupled positive feedback loops, Palani and Sarkar165 were able to engineer bistability into a two-component signaling system (a transmembrane receptor that signals an intracellular TF via phosphorylation). The switching between stable fixed points allowed for steady-state response curves with very high steepness; in some cases, apparent Hill coefficients of n > 20 were achieved. In this network, shown in Figure 7D, extracellular ligand binding to the transmembrane receptor activates the intracellular TF; the activated TF then activates receptor expression (the first positive feedback loop) as well as its own expression (the second positive feedback loop). Experimental implementation in S. cerevisiae used the transmembrane receptor CRE1 from A. thaliana to bind the cytokinin ligand isopentenyl adenine (IP), and the yeast TF SKN7 as the intracellular TF. A variety of responses curves to IP induction were obtained by varying the number of SKN7 operator site repeats in the corresponding promoters, thus effectively varying the positive feedback strength. Across five variants of the network, steepness tuning with Hill coefficients ranging from n∼2 to 20 was observed accompanied by changes in vertical extension.
Parallel Pathways: Reducing Leakage
In cases where tighter repression of gene expression is desired, synthetic biologists have had success employing multiple repression mechanisms in parallel. This has typically involved supplementing TF repression with a post-transcriptional mechanism such as RNAi-mediated mRNA degradation. Deans et al.,166 for example, used short-hairpin RNAs (shRNAs) to induce RNAi mediated degradation and controlled both shRNA and TF repressor production using the same input signal, as shown in Figure 7E. Since both the TF and the shRNA are constitutively produced in the absence of the input signal, the presence of the shRNA pathway reduces steady-state expression leakage. Rinauldo et al.(167) and Xie et al.(25) employed similar techniques using small-interfering RNA (siRNA) and microRNA (miRNA), respectively.
Consistency and Modularity
Sophisticated tuning of one component in a biological system is of little use if the component’s behavior is easily disrupted when its cellular context is changed. It is an ongoing challenge in synthetic biology to create systems that are consistent (able to display the same behavior repeatedly, and in the face of global background variations such as differing cell strains) and modular (maintaining their behavior when linked with other engineered systems).3,18,168−172
A variety of phenomena wherein one transcriptional process suppresses a second transcriptional process are collected under the label of transcriptional interference.173 This includes physically proximate promoters competing directly for RNAP access, RNAPs initiating transcription from one promoter colliding with other RNAPs or blocking them from transcribing from a downstream promoter, and post-transcriptional interactions such as RNAP inactivation or RNA interference leading one transcriptional process to reduce the effective transcriptional or translational rates of another.
Insulated Promoter-Gene Cassettes
Miller et al.(174) created a set of promoter-reporter cassettes in which they placed a transcriptional termination sequence from E. coli upstream of the promoter regions. This made use of the established ability of such termination sequences to reduce transcription initiated from promoters other than the target.175 Using one or two copies of the termination sequence, they reduced such external transcription by 94% and 97% respectively. In Davis et al.,45 transcriptional interference was minimized by providing insulated cassettes: promoters flanked by controlled sequences both upstream and downstream of the transcriptional initiation site (−105 to +55), thereby providing the promoter a consistent functional neighborhood. Inserting a 24-nucleotide sequence known to activate transcription in some promoters showed a wide range of resulting changes in activity in promoters lacking the flanking insulating sequences, but the promoter outputs were very consistent when the insulating sequences were included.
Promoter Position Relative to ORI
As discussed in the transcriptional regulation section, Block et al.(32) studied the effect of promoter location relative to the genomic origin of replication (ORI) on gene expression levels, finding that promoters closer to the ORI expressed at higher levels. This suggests a mechanism for tuning response curves, but also provides a cautionary note in terms of consistency: if a promoter-gene pair is inserted into the genome at random, its behavior may vary significantly with distance from the ORI. Consistent performance will require control of that positioning, though Block et al.(32) found that expression levels were robust to two other forms of alteration: the orientation of the genes and the distance between genes and their TFs. Similar considerations also apply to promoters on plasmid vectors: promoter position relative to the plasmid ORI will also cause variation in effective copy number of the promoter and thus alter gene expression levels. Transcription-replication interference was observed by Mirkin and Mirkin176 who found that promoters could, if oriented such that they transcribed genes in the opposite direction to that in which DNA replication proceeded, cause significant interference with plasmid replication. Such interference would lower the plasmid copy number and reduce gene expression levels.
CRISPR Editing
An alternative method of ensuring transcriptional consistency focuses on removing unwanted elements from the mRNA transcript itself, after transcription but before translation. Qi et al.(177) made use of the bacterial clustered regularly interspaced short palindromic repeat (CRISPR) system to protect the expression of target genes from upstream effects. Screening a randomly generated library of 30-nucleotide 5′ UTRs upstream of the RBS and gene coding sequence of a fluorescent reporter gene, they measured gene expression of the reporter in E. coli, and observed a wide range of expression levels across the library, suggesting variable levels of transcriptional interference induced by the UTRs. Inserting a CRISPR cleavage site targeted by the Csy4 protein reduced the observed relative standard deviations of reporter expression by nearly 3-fold in the presence of the Csy4 protein, suggesting that the designed system acts to insulate the mRNA from upstream effects; by cleaving at the inserted cleavage site, Csy4 produces processed mRNA strands that are more independent of their upstream genetic context and thus yield more consistent protein levels when translated.
Ribozyme-Based Insulator Parts
A study by Lou et al.(178) used sequences designed for ribozyme cleavage as a mechanism for buffering transcripts from their upstream context. The mechanisms employed in this and the work by Qi et al.(177) are conceptually similar, both based on cleaving away the 5′ UTRs to prevent their having an impact on gene expression levels.179 Lou et al.(178) constructed a logical NOT gate circuit based on transcriptional regulation, and tested several different promoters to drive expression of the first gene in the circuit. They observed that the NOT gate’s transfer function, in this case, the mapping between this promoter’s activity and the downstream expression level of the circuit’s output protein, varied substantially as a function of the promoter chosen. Screening a library of prospective insulator parts identified the cleavage sequence RiboJ as the best insulator, and placing the RiboJ sequence between the circuit driving promoter and its gene served to effectively eliminate the transfer function discrepancies. Transfer functions collapsed quite strikingly onto a single curve independent of the promoter being used, illustrating the insulating effect of removing inconsistent untranslated regions from the start of the resulting mRNA strands.
Noise
A pervasive issue working against consistent behavior in biological systems is the noise inherent in biochemical reactions operating in regimes where fluctuations are not averaged away. The topic of noise in biological systems is beyond the scope of this review but has been extensively reviewed elsewhere.180−184
Discussion
Clearly there are many challenges to be addressed before tuning in synthetic biology achieves the status it enjoys in traditional engineering disciplines. Noise effects, while increasingly well understood theoretically, can be significant or design-breaking in experimental implementations. Coupling multiple systems in a biological context often has unexpected effects, despite design efforts to achieve true modularity. Biology inherently operates in a more interconnected context than mechanical or electronic systems, making perfect functional compartmentalization correspondingly more challenging and raising the question of how difficult it will be to combine multiple types of tuning to affect a target process. Biology also comes with substantial context-dependence, making it difficult to craft portable designs that can be implemented across organisms or indeed across genomic variants of a single species, without significant risk of their operation being disrupted by the local context.
For all of these challenges, synthetic biology is on a promising path, with the library of parts, methods, and approaches growing rapidly, year by year; the already extensive list of tuning options available to designers in synthetic biology seems certain to continue to grow. A combination of awareness of tuning applications and improved experimental methods may lead experimentalists to report full tuning curves more consistently and to characterize their systems in terms of rates of change in addition to steady-state values. Our hope is that evaluating the tuning potential of a new method or system will come to be a standard part of the process of characterizing it and that libraries of components will be augmented with collections of tuning methods, increasing the range and sophistication of solutions available for synthetic biology design problems.
Acknowledgments
Funding has been provided by the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Ontario Research Fund (ORF).
The authors declare no competing financial interest.
References
- Andrianantoandro E.; Basu S.; Karig D. K.; Weiss R. (2006) Synthetic biology: new engineering rules for an emerging discipline. Mol. Syst. Biol. 2, 2006–0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber D.; Fussenegger M. (2007) Mammalian synthetic biology: Engineering of sophisticated gene networks. J. Biotechnol. 130, 329–345. [DOI] [PubMed] [Google Scholar]
- Lu T. K.; Khalil A. S.; Collins J. J. (2009) Next-generation synthetic gene networks. Nat. Biotechnol. 27, 1139–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherji S.; van Oudenaarden A. (2009) Synthetic biology: understanding biological design from synthetic circuits. Nat. Rev. Genet. 10, 859–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel C.; Yus E.; Serrano L. (2010) Engineering signal transduction pathways. Cell 140, 33–47. [DOI] [PubMed] [Google Scholar]
- Ruder W. C.; Lu T.; Collins J. J. (2011) Synthetic biology moving into the clinic. Science 333, 1248–1252. [DOI] [PubMed] [Google Scholar]
- Voigt C. A., Ed. (2011) Synthetic Biology Part A: Methods for Part/Device Characterization and Chassis Engineering, Academic Press Inc., San Diego, Methods in Enzymology, Vol. 497. [Google Scholar]
- Voigt C. A., Ed. (2011) Synthetic Biology Part B: Computer Aided Design and DNA Assembly, Academic Press Inc., San Diego, Methods in Enzymology, Vol. 498. [Google Scholar]
- Kwok R. (2010) Five hard truths for synthetic biology. Nature 463, 288–290. [DOI] [PubMed] [Google Scholar]
- Hill A. V. (1910) The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves. J. Physiol. 40(Suppl), 4–7. [Google Scholar]
- Weiss J. N. (1997) The Hill equation revisited: uses and misuses. FASEB J. 11, 835–841. [PubMed] [Google Scholar]
- Ptashne M. (1986) A Genetic Switch: Phage Lambda and Higher Organisms, Cell Press and Blackwell Scientific Publications, Cambridge, MA. [Google Scholar]
- Ingalls B. P., Yi T.-M., Iglesias P. A. (2006) Using control theory to study biology, in System Modeling in Cellular Biology (Szallasi Z., Periwal V., and Jorg S., Eds.), pp 243–267, MIT Press, Cambridge, MA. [Google Scholar]
- Kærn M., Weiss R. (2006) Synthetic gene regulatory systems, in System Modeling in Cellular Biology (Szallasi Z., Periwal V., and Jorg S., Eds.), pp 269–295, MIT Press, Cambridge, MA. [Google Scholar]
- Iglesias P. A., Ingalls B. P. (2009) Control Theory and Systems Biology, MIT Press, Cambridge, MA. [Google Scholar]
- Ang J.; Bagh S.; Ingalls B. P.; McMillen D. R. (2010) Considerations for using integral feedback control to construct a perfectly adapting synthetic gene network. J. Theor. Biol. 266, 723–738. [DOI] [PubMed] [Google Scholar]
- Sarpeshkar R. (2010) Ultra Low Power Bioelectronics, Chapter 24, pp 753–786, Cambridge University Press, Cambridge, MA. [Google Scholar]
- Randall A.; Guye P.; Gupta S.; Duportet X.; Weiss R. (2011) Design and connection of robust genetic circuits. Methods Enzymol. 497, 159–186. [DOI] [PubMed] [Google Scholar]
- Drengstig T.; Jolma I. W.; Ni X. Y.; Thorsen K.; Xu X. M.; Ruoff P. (2012) A basic set of homeostatic controller motifs. Biophys. J. 103, 2000–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slusarczyk A. L.; Lin A.; Weiss R. (2012) Foundations for the design and implementation of synthetic genetic circuits. Nat. Rev. Genet. 13, 406–420. [DOI] [PubMed] [Google Scholar]
- Ang J.; McMillen D. R. (2013) Physical constraints on biological integral control design for homeostasis and sensory adaptation. Biophys. J. 104, 505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer B. P.; Fischer C.; Fussenegger M. (2004) BioLogic gates enable logical transcription control in mammalian cells. Biotechnol. Bioeng. 87, 478–484. [DOI] [PubMed] [Google Scholar]
- Anderson J. C.; Voigt C. A.; Arkin A. P. (2007) Environmental signal integration by a modular AND gate. Mol. Syst. Biol. 3, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagh S.; Mandal M.; McMillen D. R. (2010) Minimal genetic device with multiple tunable functions. Phys. Rev. E 82, 021911. [DOI] [PubMed] [Google Scholar]
- Xie Z.; Liu S. J.; Bleris L.; Benenson Y. (2010) Logic integration of mRNA signals by an RNAi-based molecular computer. Nucleic Acids Res. 38, 2692–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z.; Wroblewska L.; Prochazka L.; Weiss R.; Benenson Y. (2011) Multi-input RNAi-based logic circuit for identification of specific cancer cells. Science 333, 1307–1311. [DOI] [PubMed] [Google Scholar]
- Dueber J. E.; Mirsky E. A.; Lim W. A. (2007) Engineering synthetic signaling proteins with ultrasensitive input/output control. Nat. Biotechnol. 25, 660–662. [DOI] [PubMed] [Google Scholar]
- Bashor C. J.; Helman N. C.; Yan S. D.; Lim W. A. (2008) Using engineered scaffold interactions to reshape MAP kinase pathway signaling dynamics. Science 319, 1539–1543. [DOI] [PubMed] [Google Scholar]
- O’Shaughnessy E. C.; Palani S.; Collins J. J.; Sarkar C. A. (2011) Tunable signal processing in synthetic MAP kinase cascades. Cell 144, 119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M. S.; Mauser J. F.; Prehoda K. E. (2012) Ultrasensitive synthetic protein regulatory networks using mixed decoys. ACS Synth. Biol. 1, 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchman D. S. (1997) Transcription factors: An overview. Int. J. Biochem. Cell Biol. 29, 1305–1312. [DOI] [PubMed] [Google Scholar]
- Block D. H. S.; Hussein R.; Liang L. W.; Lim H. N. (2012) Regulatory consequences of gene translocation in bacteria. Nucleic Acids Res. 40, 8979–8992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy K. F.; Balazsi G.; Collins J. J. (2007) Combinatorial promoter design for engineering noisy gene expression. Proc. Natl. Acad. Sci. U.S.A. 104, 12726–12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volfson D.; Marciniak J.; Blake W. J.; Ostroff N.; Tsimring L. S.; Hasty J. (2006) Origins of extrinsic variability in eukaryotic gene expression. Nature 439, 861–864. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Partow S.; Scalcinati G.; Siewers V.; Nielsen J. (2012) Enhancing the copy number of episomal plasmids in Saccharomyces cerevisiae for improved protein production. FEMS Yeast Res. 12, 598–607. [DOI] [PubMed] [Google Scholar]
- Bikard D.; Julié-Galau S.; Cambray G.; Mazel D. (2010) The synthetic integron: an in vivo genetic shuffling device. Nucleic Acids Res. 38, e153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly J. R.; Rubin A. J.; Davis J. H.; Ajo-Franklin C. M.; Cumbers J.; Czar M. J.; de Mora K.; Glieberman A. L.; Monie D. D.; Endy D. (2009) Measuring the activity of BioBrick promoters using an in vivo reference standard. J. Biol. Eng. 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partow S.; Siewers V.; Bjørn S.; Nielsen J.; Maury J. (2010) Characterization of different promoters for designing a new expression vector in Saccharomyces cerevisiae. Yeast 27, 955–964. [DOI] [PubMed] [Google Scholar]
- Qin J. Y.; Zhang L.; Clift K. L.; Hulur I.; Xiang A. P.; Ren B.-Z.; Lahn B. T. (2010) Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PLoS One 5, e10611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D. J.; Minchin S. D.; Busby S. J. W. (2012) Activating transcription in bacteria. Annu. Rev. Microbiol. 66, 125–52. [DOI] [PubMed] [Google Scholar]
- Shandilya J.; Roberts S. G. E. (2012) The transcription cycle in eukaryotes: From productive initiation to RNA polymerase II recycling. Biochim. Biophys. Acta 1819, 391–400. [DOI] [PubMed] [Google Scholar]
- Bandwar R. P.; Jia Y. P.; Stano N. M.; Patel S. S. (2002) Kinetic and thermodynamic basis of promoter strength: Multiple steps of transcription initiation by T7 RNA polymerase are modulated by the promoter sequence. Biochemistry 41, 3586–3595. [DOI] [PubMed] [Google Scholar]
- Blake W. J.; Balázsi G.; Kohanski M. A.; Isaacs F. J.; Murphy K. F.; Kuang Y.; Cantor C. R.; Walt D. R.; Collins J. J. (2006) Phenotypic consequences of promoter-mediated transcriptional noise. Mol. Cell 24, 853–865. [DOI] [PubMed] [Google Scholar]
- Murphy K. F.; Adams R. M.; Wang X.; Balazsi G.; Collins J. J. (2010) Tuning and controlling gene expression noise in synthetic gene networks. Nucleic Acids Res. 38, 2712–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J. H.; Rubin A. J.; Sauer R. T. (2011) Design, construction and characterization of a set of insulated bacterial promoters. Nucleic Acids Res. 39, 1131–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster R. C.; Jones D. L.; Phillips R. (2012) Tuning promoter strength through RNA polymerase binding site design in Escherichia coli. PLoS Comput. Biol. 8, e1002811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solem C.; Jensen P. R. (2002) Modulation of gene expression made easy. Appl. Environ. Microbiol. 68, 2397–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis T.; Wang X.; Collins J. J. (2009) Diversity-based, model-guided construction of synthetic gene networks with predicted functions. Nat. Biotechnol. 27, 465–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mey M.; Maertens J.; Lequeux G. J.; Soetaert W. K.; Vandamme E. J. (2007) Construction and model-based analysis of a promoter library for E. coli: an indispensable tool for metabolic engineering. BMC Biotechnol. 7, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross W.; Gosink K. K.; Salomon J.; Igarashi K.; Zou C.; Ishihama A.; Severinov K.; Gourse R. L. (1993) A third recognition element in bacterial promoters: DNA binding by the α subunit of RNA polymerase. Science 262, 1407–1413. [DOI] [PubMed] [Google Scholar]
- Rhodius V. A.; Mutalik V. K.; Gross C. A. (2012) Predicting the strength of UP-elements and full-length E. coli σE promoters. Nucleic Acids Res. 40, 2907–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz J.; Siggia E. D.; Cohen B. A. (2009) Analysis of combinatorial cis-regulation in synthetic and genomic promoters. Nature 457, 215–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heins L.; Frohberg C.; Gatz C. (1992) The Tnl0-encoded Tet repressor blocks early but not late steps of assembly of the RNA polymerase II initiation complex in vivo. Mol. Genet. Genomics 232, 328–331. [DOI] [PubMed] [Google Scholar]
- Berens C.; Hillen W. (2003) Gene regulation by tetracyclines: Constraints of resistance regulation in bacteria shape TetR for application in eukaryotes. Eur. J. Biochem. 270, 3109–3121. [DOI] [PubMed] [Google Scholar]
- Garcia H. G.; Sanchez A.; Boedicker J. Q.; Osborne M.; Gelles J.; Kondev J.; Phillips R. (2012) Operator sequence alters gene expression independently of transcription factor occupancy in bacteria. Cell Rep. 2, 150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokobayashi Y.; Weiss R.; Arnold F. H. (2002) Directed evolution of a genetic circuit. Proc. Natl. Acad. Sci. U.S.A. 99, 16587–16591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milk L.; Daber R.; Lewis M. (2010) Functional rules for lac repressor-operator associations and implications for protein-DNA interactions. Protein Sci. 19, 1162–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maity T. S.; Jha R. K.; Strauss C. E.; Dunbar J. (2012) Exploring the sequence-function relationship in transcriptional regulation by the lac O1 operator. FEBS Lett. 279, 2534–2543. [DOI] [PubMed] [Google Scholar]
- Kammerer W.; Deuschle U.; Gentz R.; Bujard H. (1986) Functional dissection of Escherichia coli promoters: information in the transcribed region is involved in late steps of the overall process. EMBO J. 5, 2995–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chander M.; Austin K. M.; Aye-Han N.-N.; Sircar P.; Hsu L. M. (2007) An alternate mechanism of abortive release marked by the formation of very long abortive transcripts. Biochemistry 46, 12687–12699. [DOI] [PubMed] [Google Scholar]
- Hsu L. M.; Cobb I. M.; Ozmore J. R.; Khoo M.; Nahm G.; Xia L.; Bao Y.; Ahn C. (2006) Initial transcribed sequence mutations specifically after promoter escape properties. Biochemistry 45, 8841–8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman S. R.; Ebright R. H.; Nickels B. E. (2009) Direct detection of abortive RNA transcripts in vivo. Science 324, 927–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptashne M., Gann A. (2001) Genes and Signals, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Mapp A. K.; Ansari A. Z. (2007) A TAD further: Exogenous control of gene activation. ACS Chem. Biol. 2, 62–75. [DOI] [PubMed] [Google Scholar]
- Ramos J. L.; Martinez-Bueno M.; Molina-Henares A. J.; Teran W.; Watanabe K.; Zhang X. D.; Gallegos M. T.; Brennan R.; Tobes R. (2005) The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 69, 326–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson M.; Weber W.; Fussenegger M. (2011) De novo design and construction of an inducible gene expression system in mammalian cells. Methods Enzymol. 497, 239–253. [DOI] [PubMed] [Google Scholar]
- Triezenberg S. J.; Kingsbury R. C.; McKnight S. L. (1988) Functional dissection of VP16, the trans-activator of herpes simplex virus immediate early gene expression. Genes Dev. 2, 718–729. [DOI] [PubMed] [Google Scholar]
- Sadowski I.; Ma J.; Triezenberg S.; Ptashne M. (1988) GAL4-VP16 is an unusually potent transcriptional activator. Nature 335, 563–564. [DOI] [PubMed] [Google Scholar]
- Emami K. H.; Carey M. (1992) A synergistic increase in the potency of a multimerized VP16 transcriptional activation domain. EMBO J. 11, 5005–5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron U.; Gossen M.; Bujard H. (1997) Tetracycline-controlled transcription in eukaryotes: novel transactivators with graded transactivation potential. Nucleic Acids Res. 25, 2723–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber W.; Kramer B. P.; Fux C.; Keller B.; Fussenegger M. (2002) Novel promoter/transactivator configurations for macrolide- and streptogramin-responsive transgene expression in mammalian cells. J. Gene Med. 4, 676–686. [DOI] [PubMed] [Google Scholar]
- Park K. S.; Lee D. K.; Lee H.; Lee Y.; Jang Y. S.; Kim Y. H.; Yang H. Y.; Lee S. I.; Seol W.; Kim J. S. (2003) Phenotypic alteration of eukaryotic cells using randomized libraries of artificial transcription factors. Nat. Biotechnol. 21, 1208–1214. [DOI] [PubMed] [Google Scholar]
- Fussenegger M.; Morris R. P.; Fux C.; Rimann M.; von Stockar B.; Thompson C. J.; Bailey J. E. (2000) Streptogramin-based gene regulation systems for mammalian cells. Nat. Biotechnol. 18, 1203–1208. [DOI] [PubMed] [Google Scholar]
- Mak A. N.-S. A.; Bradley P. P.; Bogdanove A. J. A.; Stoddard B. L. B. (2013) TAL effectors: function, structure, engineering and applications. Curr. Opin. Struct. Biol. 23, 93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder M. L.; Linder S. J.; Reyon D.; Angstman J. F.; Fu Y.; Sander J. D.; Joung J. K. (2013) Robust, synergistic regulation of human gene expression using TALE activators. Nat. Methods 10, 243–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pinera P.; Ousterout D. G.; Brunger J. M.; Farin A. M.; Glass K. A.; Guilak F.; Crawford G. E.; Hartemink A. J.; Gersbach C. A. (2013) Synergistic and tunable human gene activation by combinations of synthetic transcription factors. Nat. Methods 10, 239–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klug A. (2010) The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu. Rev. Biochem. 79, 213–231. [DOI] [PubMed] [Google Scholar]
- Khalil A. S.; Lu T. K.; Bashor C. J.; Ramirez C. L.; Pyenson N. C.; Joung J. K.; Collins J. J. (2012) A synthetic biology framework for programming eukaryotic transcription functions. Cell 150, 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder M. L.; et al. (2008) Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol. Cell 31, 294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmueller J. J.; Armel T. Z.; Silver P. A. (2012) A tunable zinc finger-based framework for Boolean logic computation in mammalian cells. Nucleic Acids Res. 40, 5180–5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temme K.; Zhao D. H.; Voigt C. A. (2012) Refactoring the nitrogen fixation gene cluster from Klebsiella oxytoca. Proc. Natl. Acad. Sci. U.S.A. 109, 7085–7090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange A.; Mills R. E.; Lange C. J.; Stewart M.; Devine S. E.; Corbett A. H. (2007) Classical nuclear localization signals: Definition, function, and interaction with Importin α. J. Biol. Chem. 282, 5101–5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marfori M.; Mynott A.; Ellis J. J.; Mehdi A. M.; Saunders N. F.; Curmi P. M.; Forwood J. K.; Boden M.; Kobe B. (2011) Molecular basis for specificity of nuclear import and prediction of nuclear localization. Biochim. Biophys. Acta 1813, 1562–77. [DOI] [PubMed] [Google Scholar]
- Hodel A. E.; Harreman M. T.; Pulliam K. F.; Harben M. E.; Holmes J. S.; Hodel M. R.; Berland K. M.; Corbett A. H. (2006) Nuclear localization signal receptor affinity correlates with in vivo localization in Saccharomyces cerevisiae. J. Biol. Chem. 281, 23545–23556. [DOI] [PubMed] [Google Scholar]
- Luo M.; Pang C. W. M.; Gerken A. E.; Brock T. G. (2004) Multiple nuclear localization sequences allow modulation of 5-lipoxygenase nuclear import. Traffic 5, 847–854. [DOI] [PubMed] [Google Scholar]
- Kakar M.; Davis J. R.; Kem S. E.; Lim C. S. (2007) Optimizing the protein switch: Altering nuclear import and export signals, and ligand binding domaine. J. Controlled Release 120, 220–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodel M. R.; Corbett A. H.; Hodel A. E. (2001) Dissection of a nuclear localization signal. J. Biol. Chem. 276, 1317–1325. [DOI] [PubMed] [Google Scholar]
- Raveh-Sadka T.; Levo M.; Shabi U.; Shany B.; Keren L.; Lotan-Pompan M.; Zeevi D.; Sharon E.; Weinberger A.; Segal E. (2012) Manipulating nucleosome disfavoring sequences allows fine-tune regulation of gene expression in yeast. Nat. Genet. 44, 743–750. [DOI] [PubMed] [Google Scholar]
- Serganov A.; Nudler E. (2013) A decade of riboswitches. Cell 152, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J. C.; Bloom R. J.; Smolke C. D. (2011) Engineering biological systems with synthetic RNA molecules. Mol. Cell 43, 915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A. L.; Wolf J. J.; Smolke C. D. (2012) Synthetic RNA switches as a tool for temporal and spatial control over gene expression. Curr. Opin. Biotechnol. 23, 679–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salis H. M.; Mirsky E. A.; Voigt C. A. (2009) Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 27, 946–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rackham O.; Chin J. W. (2005) Cellular logic with orthogonal ribosomes. J. Am. Chem. Soc. 127, 17584–17585. [DOI] [PubMed] [Google Scholar]
- Rackham O.; Chin J. W. (2005) A network of orthogonal ribosome·mRNA pairs. Nat. Chem. Biol. 1, 159–166. [DOI] [PubMed] [Google Scholar]
- Rackham O.; Wang K.; Chin J. W. (2006) Functional epitopes at the ribosome subunit interface. Nat. Chem. Biol. 2, 254–258. [DOI] [PubMed] [Google Scholar]
- An W.; Chin J. W. (2009) Synthesis of orthogonal transcription-translation networks. Proc. Natl. Acad. Sci. U.S.A. 106, 8477–8482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubiz L. M.; Rao C. V. (2008) Computational design of orthogonal ribosomes. Nucleic Acids Res. 36, 4038–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterman I. A. I.; Evfratov S. A. S.; Sergiev P. V. P.; Dontsova O. A. O. (2013) Comparison of mRNA features affecting translation initiation and reinitiation. Nucleic Acids Res. 41, 474–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.-J.; Lin G.; Chang K.-J.; Yeh L.-S.; Wang C.-C. (2008) Translational efficiency of a non-AUG initiation codon is significantly affected by its sequence context in yeast. J. Biol. Chem. 283, 3173–3180. [DOI] [PubMed] [Google Scholar]
- Egbert R. G.; Klavins E. (2012) Fine-tuning gene networks using simple sequence repeats. Proc. Natl. Acad. Sci. U.S.A. 109, 16817–16822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin J. B.; Kudla G. (2011) Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 12, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch M.; Govindarajan S.; Ness J. E.; Villalobos A.; Gurney A.; Minshull J.; Gustafsson C. (2009) Design parameters to control synthetic gene expression in Escherichia coli. PLoS One 4, e7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs F. J.; Dwyer D. J.; Ding C. M.; Pervouchine D. D.; Cantor C. R.; Collins J. J. (2004) Engineered riboregulators enable post-transcriptional control of gene expression. Nat. Biotechnol. 22, 841–847. [DOI] [PubMed] [Google Scholar]
- Callura J. M.; Cantor C. R.; Collins J. J. (2012) Genetic switchboard for synthetic biology applications. Proc. Natl. Acad. Sci. U.S.A. 109, 5850–5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutalik V. K.; Qi L.; Guimaraes J. C.; Lucks J. B.; Arkin A. P. (2012) Rationally designed families of orthogonal RNA regulators of translation. Nat. Chem. Biol. 8, 447–454. [DOI] [PubMed] [Google Scholar]
- Hermann T.; Patel D. J. (2000) Adaptive recognition by nucleic acid aptamers. Science 287, 820–825. [DOI] [PubMed] [Google Scholar]
- Weigand J. E.; Suess B. (2009) Aptamers and riboswitches: perspectives in biotechnology. Appl. Microbiol. Biotechnol. 85, 229–236. [DOI] [PubMed] [Google Scholar]
- Ellington A. D.; Szostak J. W. (1990) In vitro selection of RNA molecules that bind specific ligands. Nature 346, 818–822. [DOI] [PubMed] [Google Scholar]
- Tuerk C.; Gold L. (1990) Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249, 505–510. [DOI] [PubMed] [Google Scholar]
- Stoltenburg R.; Reinemann C.; Strehlitz B. (2007) SELEX—A (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng. 24, 381–403. [DOI] [PubMed] [Google Scholar]
- Gopinath S. C. B. (2007) Methods developed for SELEX. Anal. Bioanal. Chem. 387, 171–182. [DOI] [PubMed] [Google Scholar]
- Bayer T. S.; Smolke C. D. (2005) Programmable ligand-controlled riboregulators of eukaryotic gene expression. Nat. Biotechnol. 23, 337–343. [DOI] [PubMed] [Google Scholar]
- Carothers J. M.; Goler J. A.; Juminaga D.; Keasling J. D. (2011) Model-driven engineering of RNA devices to quantitatively program gene expression. Science 334, 1716–1719. [DOI] [PubMed] [Google Scholar]
- Brantl S.; Wagner E. G. H. (2002) An antisense RNA-mediated transcriptional attenuation mechanism functions in Escherichia coli. J. Bacteriol. 184, 2740–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucks J. B.; Qi L.; Mutalik V. K.; Wang D.; Arkin A. P. (2011) Versatile RNA-sensing transcriptional regulators for engineering genetic networks. Proc. Natl. Acad. Sci. U.S.A. 108, 8617–8622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Win M. N.; Smolke C. D. (2008) Higher-order cellular information processing with synthetic RNA devices. Science 322, 456–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisel C. L.; Bayer T. S.; Hoff K. G.; Smolke C. D. (2008) Model-guided design of ligand-regulated RNAi for programmable control of gene expression. Mol. Syst. Biol. 4, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrier T. A.; Keasling J. D. (1997) Controlling messenger RNA stability in bacteria: strategies for engineering gene expression. Biotechnol. Prog. 13, 699–708. [DOI] [PubMed] [Google Scholar]
- Carrier T. A.; Keasling J. D. (1999) Library of synthetic 5′ secondary structures to manipulate mRNA stability in Escherichia coli. Biotechnol. Prog. 15, 58–64. [DOI] [PubMed] [Google Scholar]
- Babiskin A. H.; Smolke C. D. (2011) Engineering ligand-responsive RNA controllers in yeast through the assembly of RNase III tuning modules. Nucleic Acids Res. 39, 5299–5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig G. R.; Behlke M. A. (2012) Progress towards in vivo use of siRNAs-II. Mol. Ther. 20, 483–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strapps W. R.; Pickering V.; Muiru G. T.; Rice J.; Orsborn S.; Polisky B. A.; Sachs A.; Bartz S. R. (2010) The siRNA sequence and guide strand overhangs are determinants of in vivo duration of silencing. Nucleic Acids Res. 38, 4788–4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinev V. V. (2012) Design and quality control of short interfering RNA. Mol. Biol. 46, 739–754. [PubMed] [Google Scholar]
- Choung S.; Kim Y. J.; Kim S.; Park H. O.; Choi Y. C. (2006) Chemical modification of siRNAs to improve serum stability without loss of efficacy. Biochem. Biophys. Res. Commun. 342, 919–927. [DOI] [PubMed] [Google Scholar]
- Gaglione M.; Potenza N.; Fabio G. D.; Romanucci V.; Mosca N.; Russo A.; Nevellino E.; Cosconati S.; Messere A. (2013) Tuning RNA interference by enhancing siRNA/PAZ recognition. ACS Med. Chem. Lett. 4, 75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Q.; Dallas A.; Ilves H.; Shorenstein J.; Behlke M. A.; Johnston B. H. (2010) Effects of chemical modification on the potency, serum stability, and immunostimulatory properties of short shRNAs. RNA 16, 118–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J.; Huang Y.; Li J.; Yi F.; Zheng J.; Huang H.; Wei N.; Shan Y.; An M.; Zhang H.; Ji J.; Zhang P.; Xi Z.; Du Q.; Liang Z. (2010) Comprehensive analysis of sequence-specific stability of siRNA. FASEB J. 24, 4844–4855. [DOI] [PubMed] [Google Scholar]
- Patel R.; t’Wallant N. C.; Herbert M. H.; White D.; Murison J. G.; Reid G. (2009) The potency of siRNA-mediated growth inhibition following silencing of essential genes is dependent on siRNA design and varies with target sequence. Oligonucleotides 19, 317–328. [DOI] [PubMed] [Google Scholar]
- Beisel C. L.; Chen Y. Y.; Culler S. J.; Hoff K. G.; Smolke C. D. (2011) Design of small molecule-responsive microRNAs based on structural requirements for Drosha processing. Nucleic Acids Res. 39, 2981–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleger B. F.; Pitera D. J.; Smolke C. D.; Keasling J. D. (2006) Combinatorial engineering of intergenic regions in operons tunes expression of multiple genes. Nat. Biotechnol. 24, 1027–1032. [DOI] [PubMed] [Google Scholar]
- Stapleton J. A.; Endo K.; Fujita Y.; Hayashi K.; Takinoue M.; Saito H.; Inoue T. (2012) Feedback control of protein expression in mammalian cells by tunable synthetic translational inhibition. ACS Synth. Biol. 1, 83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcintyre G. J.; Arndt A. J.; Gillespie K. M.; Mak W. M.; Fanning G. C. (2011) A comparison of multiple shRNA expression methods for combinatorial RNAi. Genet. Vaccines Therapy 9, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünberg R.; Serrano L. (2010) Strategies for protein synthetic biology. Nucleic Acids Res. 38, 2663–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim W. A. (2010) Designing customized cell signalling circuits. Nat. Rev. Mol. Cell Biol. 11, 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golynskiy M. V.; Koay M. S.; Vinkenborg J. L.; Merkx M. (2011) Engineering protein switches: sensors, regulators, and spare parts for biology and biotechnology. ChemBioChem 12, 353–361. [DOI] [PubMed] [Google Scholar]
- Stratton M. M.; Loh S. N. (2011) Converting a protein into a switch for biosensing and functional regulation. Protein Sci. 20, 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha J.-H.; Loh S. N. (2012) Protein conformational switches: from nature to design. Chemistry 18, 7984–7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson E. J.; Tabor J. J. (2012) Post-translational tools expand the scope of synthetic biology. Curr. Opin. Chem. Biol. 16, 1–7. [DOI] [PubMed] [Google Scholar]
- Kim S. B.; Otani Y.; Umezawa Y.; Tao H. (2007) Bioluminescent indicator for determining protein-protein interactions using intramolecular complementation of split click beetle luciferase. Anal. Chem. 79, 4820–4826. [DOI] [PubMed] [Google Scholar]
- Yazawa M.; Sadaghiani A. M.; Hsueh B.; Dolmetsch R. E. (2009) Induction of protein-protein interactions in live cells using light. Nat. Biotechnol. 27, 941–945. [DOI] [PubMed] [Google Scholar]
- Kennedy M. J.; Hughes R. M.; Peteya L. A.; Schwartz J. W.; Ehlers M. D.; Tucker C. L. (2010) Rapid blue-lightĐmediated induction of protein interactions in living cells. Nat. Methods 7, 973–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggsbee C. W.; Deiters A. (2010) Recent advances in the photochemical control of protein function. Trends Biotechnol. 28, 468–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland D.; Lin Y.; Wagner E.; Hope C. M.; Zayner J.; Antoniou C.; Sosnick T. R.; Weiss E. L.; Glotzer M. (2012) TULIPs: tunable, light-controlled interacting protein tags for cell biology. Nat. Methods 9, 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills E.; Truong K. (2011) Ca2+-mediated synthetic biosystems offer protein design versatility, signal specificity, and pathway rewiring. Chem. Biol. 18, 1611–1619. [DOI] [PubMed] [Google Scholar]
- Peng Q.; Kong N.; Wang H.-C. E.; Li H. (2012) Designing redox potential-controlled protein switches based on mutually exclusive proteins. Protein Sci. 21, 1222–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckert K.; Budiardjo S. J.; Brunner L. C.; Lovell S.; Karanicolas J. (2012) Designing allosteric control into enzymes by chemical rescue of structure. J. Am. Chem. Soc. 134, 10055–10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman S.; Roche E.; Zhou Y. N.; Sauer R. T. (1998) The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev. 12, 1338–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersch G. L.; Baker T. A.; Sauer R. T. (2004) SspB delivery of substrates for ClpXP proteolysis probed by the design of improved degradation tags. Proc. Natl. Acad. Sci. U.S.A. 101, 12136–12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz M. B.; Leibler S. (2000) A synthetic oscillatory network of transcriptional regulators. Nature 403, 335–338. [DOI] [PubMed] [Google Scholar]
- Grilly C.; Strickler J.; Pang W. L.; Bennett M. R.; Hasty J. (2007) A synthetic gene network for tuning protein degradation in Saccharomyces cerevisiae. Mol. Syst. Biol. 3, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong W. W.; Tsai T. Y.; Liao J. C. (2007) Single-cell zeroth-order protein degradation enhances the robustness of synthetic oscillator. Mol. Syst. Biol. 3, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaszynski L. A.; Chen L.-c.; Maynard-Smith L. A.; Ooi A. G. L.; Wandless T. J. (2006) A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 126, 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonger K. M.; Chen L.-c.; Liu C. W.; Wandless T. J. (2011) Small-molecule displacement of a cryptic degron causes conditional protein degradation. Nat. Chem. Biol. 7, 531–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neklesa T. K.; Tae H. S.; Schneekloth A. R.; Stullberg M. J.; Corson T. W.; Sundberg T. B.; Raina K.; Holley S. A.; Crews C. M. (2011) Small-molecule hydrophobic tagging-induced degradation of HaloTag fusion proteins. Nat. Chem. Biol. 7, 538–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K.; Fukagawa T.; Takisawa H.; Kakimoto T.; Kanemaki M. (2009) An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods 6, 917–922. [DOI] [PubMed] [Google Scholar]
- Iwamoto M.; Björklund T.; Lundberg C.; Kirik D.; Wandless T. J. (2010) A general chemical method to regulate protein stability in the mammalian central nervous system. Chem. Biol. 17, 981–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dueber J. E.; Wu G. C.; Malmirchegini G. R.; Moon T. S.; Petzold C. J.; Ullal A. V.; Prather K. L. J.; Keasling J. D. (2009) Synthetic protein scaffolds provide modular control over metabolic flux. Nat. Biotechnol. 27, 753–759. [DOI] [PubMed] [Google Scholar]
- Conrado R. J.; et al. (2012) DNA-guided assembly of biosynthetic pathways promotes improved catalytic efficiency. Nucleic Acids Res. 40, 1879–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H.; Jung S.-C.; Bui L. M.; Kang K. H.; Song J.-J.; Kim S. C. (2013) Improved production of l-threonine in Escherichia coli by use of a DNA scaffold system. Appl. Environ. Microbiol. 79, 774–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmel F. C. (2012) DNA-based assembly lines and nanofactories. Curr. Opin. Biotechnol. 23, 516–521. [DOI] [PubMed] [Google Scholar]
- Delebecque C. J.; Lindner A. B.; Silver P. A.; Aldaye F. A. (2011) Organization of intracellular reactions with rationally designed RNA assemblies. Science 333, 470–474. [DOI] [PubMed] [Google Scholar]
- Alon U. (2007) An Introduction to Systems Biology: Design Principles of Biological Circuits, Mathematical and Computational Biology Series, Chapman & Hall, Boca Raton, FL. [Google Scholar]
- University of Cambridge (2009), International Genetically Engineered Machine (iGEM). http://2009.igem.org/Team:Cambridge.
- Hooshangi S.; Thiberge S.; Weiss R. (2005) Ultrasensitivity and noise propagation in a synthetic transcriptional cascade. Proc. Natl. Acad. Sci. U.S.A. 102, 3581–3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palani S.; Sarkar C. A. (2011) Synthetic conversion of a graded receptor signal into a tunable, reversible switch. Mol. Syst. Biol. 7, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans T. L.; Cantor C. R.; Collins J. J. (2007) A tunable genetic switch based on RNAi and repressor proteins for regulating gene expression in mammalian cells. Cell 130, 363–372. [DOI] [PubMed] [Google Scholar]
- Rinaudo K.; Bleris L.; Maddamsetti R.; Subramanian S.; Weiss R.; Benenson Y. (2007) A universal RNAi-based logic evaluator that operates in mammalian cells. Nat. Biotechnol. 25, 795–801. [DOI] [PubMed] [Google Scholar]
- Del Vecchio D.; Ninfa A. J.; Sontag E. D. (2008) Modular cell biology: retroactivity and insulation. Mol. Syst. Biol. 4, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agapakis C. M.; Silver P. A. (2009) Synthetic biology: exploring and exploiting genetic modularity through the design of novel biological networks. Mol. Biosyst. 5, 704–713. [DOI] [PubMed] [Google Scholar]
- Purnick P. E. M.; Weiss R. (2009) The second wave of synthetic biology: from modules to systems. Nat. Rev. Mol. Cell Biol. 10, 410–422. [DOI] [PubMed] [Google Scholar]
- Del Vecchio D. (2012) Modularity in signaling systems. Phys. Biol. 9, 045008. [DOI] [PubMed] [Google Scholar]
- Kittleson J. T.; Wu G. C.; Anderson J. C. (2012) Successes and failures in modular genetic engineering. Curr. Opin. Chem. Biol. 16, 329–336. [DOI] [PubMed] [Google Scholar]
- Shearwin K. E.; Callen B. P.; Egan J. B. (2005) Transcription interference—a crash course. Trends Genet. 21, 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller W. G.; Leveau J. H. J.; Lindow S. E. (2000) Improved gfp and inaZ broad-host-range promoter-probe vectors. Mol. Plant-Microbe Interact. 13, 1243–1250. [DOI] [PubMed] [Google Scholar]
- Simons R. W.; Houman F.; Kleckner N. (1987) Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene 53, 85–96. [DOI] [PubMed] [Google Scholar]
- Mirkin E. V.; Mirkin S. M. (2005) Mechanisms of transcription-replication collisions in bacteria. Mol. Cell. Biol. 25, 888–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L.; Haurwitz R. E.; Shao W.; Doudna J. A.; Arkin A. P. (2012) RNA processing enables predictable programming of gene expression. Nat. Biotechnol. 30, 1002–1006. [DOI] [PubMed] [Google Scholar]
- Lou C.; Stanton B.; Chen Y.-J.; Munsky B.; Voigt C. A. (2012) Ribozyme-based insulator parts buffer synthetic circuits from genetic context. Nat. Biotechnol. 30, 1137–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashor C. J.; Collins J. J. (2012) Insulating gene circuits from context by RNA processing. Nat. Biotechnol. 30, 1061–1062. [DOI] [PubMed] [Google Scholar]
- Paulsson J. (2004) Summing up the noise in gene networks. Nature 427, 415–418. [DOI] [PubMed] [Google Scholar]
- Kærn M.; Elston T. C.; Blake W. J.; Collins J. J. (2005) Stochasticity in gene expression: from theories to phenotypes. Nat. Rev. Genet. 6, 451–464. [DOI] [PubMed] [Google Scholar]
- Swain P. S.; Longtin A. (2006) Noise in genetic and neural systems. Chaos 16, 026101. [DOI] [PubMed] [Google Scholar]
- Maheshri N.; O’Shea E. K. (2007) Living with noisy genes: How cells function reliably with inherent variability in gene expression. Annu. Rev. Biophys. 36, 413–434. [DOI] [PubMed] [Google Scholar]
- Balazsi G.; van Oudenaarden A.; Collins J. J. (2011) Cellular decision making and biological noise: from microbes to mammals. Cell 144, 910–925. [DOI] [PMC free article] [PubMed] [Google Scholar]