

Abstract
Recent evidence established that the cell envelope of Mycobacterium tuberculosis, the bacillus causing tuberculosis (TB), is coated by an α-glucan-containing capsule that has been implicated in persistence in a mouse infection model. As one of three known metabolic routes to α-glucan in mycobacteria, the cytoplasmic GlgE-pathway converts trehalose to α(1 → 4),α(1 → 6)-linked glucan in 4 steps. Whether individual reaction steps, catalyzed by trehalose synthase TreS, maltokinase Pep2, and glycosyltransferases GlgE and GlgB, occur independently or in a coordinated fashion is not known. Here, we report the crystal structure of M. tuberculosis TreS, and show by small-angle X-ray scattering and analytical ultracentrifugation that TreS forms tetramers in solution. Together with Pep2, TreS forms a hetero-octameric complex, and we demonstrate that complex formation markedly accelerates maltokinase activity of Pep2. Thus, complex formation may act as part of a regulatory mechanism of the GlgE pathway, which overall must avoid accumulation of toxic pathway intermediates, such as maltose-1-phosphate, and optimize the use of scarce nutrients.
Introduction
The cell envelope of Mycobacterium tuberculosis, the organism causing tuberculosis (TB), remains a central focus of TB research. It contributes critically to virulence and survival in the host, yet our understanding of how its complex architecture is assembled is fragmentary. Pathways of cell wall synthesis continue to be scrutinized for potential novel therapeutic targets, which are urgently needed to help ease the burden of the global TB epidemic and the threat posed by rising antibiotic resistance.1−3
Evidence from cryo-electron microscopy has recently confirmed that mycobacteria are coated by a capsular layer.4 The capsular layer is composed of α-glucan (80–90%), arabinomannan (10–20%), and proteins of the ESX-1 secretion system,4 and could offer novel serological biomarkers for TB infection.5 The dominant component of the capsular layer, α-glucan, is a polymer composed of α(1 → 4)-linked glucose units with α(1 → 6)-branching. To date, three metabolic pathways in mycobacteria are known to synthesize α-glucan (at least as an intermediate) including the classical GlgC–GlgA pathway, the Rv3032 pathway, and the GlgE pathway. These pathways share common nodes or are linked by synthetic lethal interactions.6,7 Although the α-glucan capsule is shed readily in detergent-containing liquid cultures, a knockout of GlgA compromised persistence of tubercle bacilli in a mouse infection model, suggesting that persistence requires an intact capsule.8 However, the extent to which the three pathways contribute to the synthesis of capsular α-glucan remains to be established. In the recently discovered 4-step GlgE pathway7,9 (Figure 1), the starting material is trehalose (α,α-1,1-diglucose), which is isomerized to maltose (α,α-1,4-diglucose), in a step catalyzed by M. tuberculosis trehalose synthase, TreS, followed by phosphorylation of maltose to maltose-1-phosphate, catalyzed by maltokinase Pep2. The phospho-activated disaccharide is then incorporated into the growing α-(1 → 4)-glucan chain by glycosyltransferase GlgE, while glycosyltransferase GlgB mediates α(1 → 6)-branching of the chain.7,10 Mycobacteria can generate trehalose, a carbohydrate store and stress protectant, from cytosolic glycogen by the TreX–TreY–TreZ pathway or from glucose-1-phosphate via GalU and OtsA–OtsB.6 In addition, trehalose can be recycled between the cell wall and the cytoplasm by an ABC transporter system that is essential for virulence.11 Such metabolic networks ensure that M. tuberculosis makes optimal use of scarce carbohydrates, a major limitation of its ecological niche.
Figure 1.

Diagram of the GlgE-pathway of mycobacterial α-glucan synthesis. Conversion of trehalose to maltose-1-phosphate proceeds through isomerization (TreS) and phosphorylation (Pep2) of the disaccharide (generated with ChemBioDraw Ultra).
Classical and reverse genetics have shown that both glgE and glgB are essential genes, while simultaneous inactivation of the treS gene recovered viability. In contrast, the ΔtreS deletion on its own had no effect on growth.7 This phenotypic pattern was interpreted as the consequence of self-poisoning by the toxic pathway intermediate maltose-1-phosphate, as the latter is produced (by TreS and Pep2), but not processed further. Limitations of nutrient supply and synthesis of toxic pathway intermediates suggest that α-glucan synthesis is subject to regulatory control. To date we do not know what mechanisms regulate α-glucan synthesis nor whether enzymes of the GlgE pathway work independently or in a coordinated fashion.
Here, we report structural, biophysical, and biochemical evidence demonstrating that formation of a large multicatalytic complex of TreS and Pep2 affects enzymatic activity of the latter, suggesting that complex formation may be part of a regulatory mechanism of the GlgE pathway.
Results and Discussion
X-ray Crystal Structure of TreS
The structure of M. tuberculosis TreS was solved by molecular replacement to a resolution of 2.6 Å (Figure 2A,B). The asymmetric unit of the crystal lattice contains two copies of TreS (chains A and B), which were refined using noncrystallographic symmetry restraints. As a result, the two copies of TreS superimpose closely with a root-mean-square deviation (rmsd) of 0.6 Å for 537 paired carbon-α (Cα) positions. The electron density covers residues 12 to 586 (of 601 amino acids total), whereby the uncleaved N-terminal affinity-tag, as well as residues 1–11 and residues 587–601 are disordered. In addition, density is poor or absent for residues 426–429 in chain A and residues 353–381 in chain B.
Figure 2.
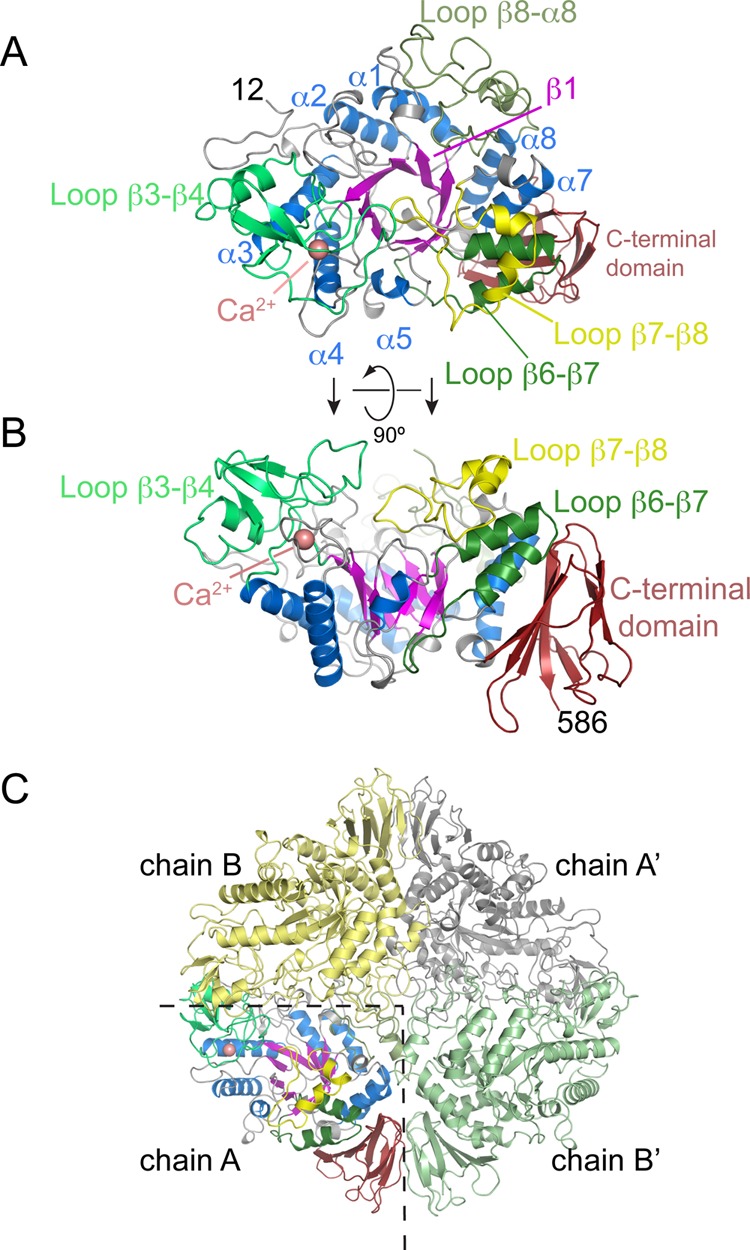
Overall fold of M. tuberculosis trehalose synthase TreS and its tetrameric assembly in the crystal. (A) Top view of the structure, with a (β/α)8-barrel fold (blue helices, magenta strands) as the conserved core and an antiparallel β-sandwich domain at the C-terminus (dark red). Selected loops connecting successive β-strands in the (β/α)8-fold are highlighted. (B) Orthogonal view of panel A. (C) Quaternary structure of TreS, containing 2 copies of each of chain A and B. Primes denote copies linked by crystallographic symmetry.
By sequence similarity, TreS belongs to family GH13 of glycoside hydrolases (www.cazy.org).12 The prototypic structures of this large enzyme family comprise two conserved domains: a catalytic domain with a (β/α)8 barrel-like fold (known as the triosephosphate isomerase or TIM barrel fold), and a C-terminal β-sandwich domain (Figures 2A,B and S1A, Supporting Information). The closest structural neighbor according to distance matrix alignment (DALI13) is Neisseria polysaccharea amylosucrase (PDB entry 1ZS2,14 27% sequence identity), aligning with an rmsd of 2.7 Å over 492 aligned Cα positions (Figure 3A). The core TIM barrel is decorated with extended loops, for some of which specific functional roles are discernible (Figure S1A, Supporting Information). For instance, the β3-β4 loop contains a calcium binding site, close to the active site but of unknown mechanistic significance (Figures 2A,B and S1B, Supporting Information). The β7−β8 loop folds over the central β-barrel in chain A but is disordered in chain B for residues 353–381, while the β6−β7 loop includes a helix-turn-helix motif that replaces the canonical helix α6 of the TIM-barrel fold.
Figure 3.
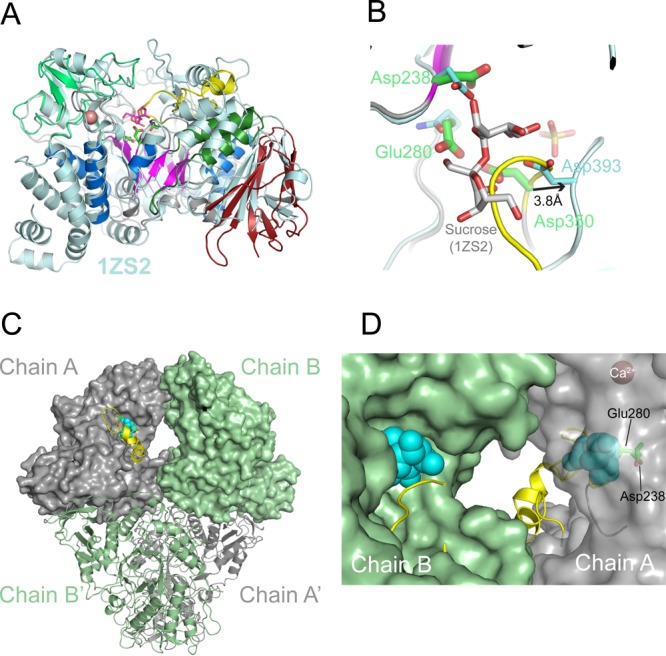
Structural homology and active site geometry of TreS. (A) Superposition of the TreS monomer with its closest structural neighbor, sucrose-bound structure of Neisseria polysaccharea amylosucrase (PDB entry 1ZS2(14)). Sticks in magenta indicate the amylosucrase-bound sucrose, and sticks in green indicate the TreS catalytic residues (Asp238, Glu280, and Asp350), with Ca2+ site as a sphere in salmon. (B) Position of sucrose (sticks in gray) in the active site of TreS (chain A), based on the structural superposition in panel A. The side chains of the catalytic triad of TreS (green) and amylosucrase (cyan) are shown. The β7−β8 loop (yellow) overlaps with the putative substrate binding site. (C) Molecular surfaces of chains A and B in the TreS tetramer. Spheres in light blue represent sucrose according to the superposition in panel A, while the β7−β8 loop is shown as a ribbon (yellow). (D) Close-up view of the cavity between the active sites of chains B and A (view of panel C rotated by 180° about the vertical axis). The surface of chain A is left transparent to reveal the location of the catalytic residues and of the (modeled) substrate.
The Ca2+ ion is octahedrally coordinated by Oδ1 of Asp208 and the carbonyl oxygen of Leu243 at the apexes and Asp140 and Glu245, a water molecule and the carbonyl oxygen of Tyr242 in the central plane of the octahedron (Figure S1B, Supporting Information). The identity of this site as calcium is supported by strong anomalous density (7.4σ above background, calculated with diffraction data recorded at 1.77 Å; Figure S1B, Supporting Information) and the overlap with a corresponding Ca2+ site in a structural neighbor (α-amylase SusG of Bacteroides thetaiotaomicrometer, 3K8K).15
Analysis of packing interfaces (PISA,16www.ebi.ac.uk/msd-srv/prot_int/pistart.html) suggested that TreS may form tetramers in solution (2TreSA + 2TreSB, Figure 2C), with an extensive buried solvent-accessible surface of ∼11 000 Å2. When testing self-assembly by analytical ultracentrifugation, a tetrameric species was found (see below). Intersubunit contacts are primarily mediated by the C-terminal β-sandwich domain, the Ca2+-binding β3−β4 loop, and at the center of the tetramer, by the β8−α8 loop (Figure 2C).
Differential Ordering of β7−β8 Loop in the Active Site
A recent mechanistic study has identified the catalytic residues of TreS in the closely related (83% identity) M. smegmatis species,17 which correspond to Asp238 (nucleophile), Glu280 (general acid), and Asp350 (unknown function) in M. tuberculosis. These residues are situated at the top (C-terminal) rim of the β-barrel (Figure 3A). By soaking and cocrystallization, we sought unsuccessfully to generate substrate/product-bound complexes of TreS. The structure contains several bound sulfate ions, including one in each of the active site of chains A and B (Figure S1C, Supporting Information). High sulfate concentrations were critical to grow crystals of TreS and to maintain crystal stability during soaking, promoting sulfate binding in the active site (Figure S1C, Supporting Information), which may interfere with substrate binding.
Superimposing chain A of TreS with sucrose-bound structures of amylosucrase (PDB entry 1ZS2(14)) or trehalulose synthase (2PWE18) shows that the β7−β8 loop, containing the catalytic Asp350, overlaps with the position of the substrate (Figure 3B). Indeed, in sucrose-bound amylosucrase, the β7−β8 loop is in a markedly different conformation, with the aspartic acid corresponding to Asp350 (Asp393) shifted away by 3.8 Å from the substrate-binding site and forming H-bonds with the glucosyl moiety of the sugar. In chain B of TreS, density for the β7−β8 loop (at a 1σ contour level) is invisible for residues 353–381, despite the bound active site sulfate, suggesting inherent flexibility of this loop.
The crystal tetramer of TreS possesses two 2-fold rotational symmetry axes, mapping subunit A onto subunit A′ and B onto B′ (by crystallographic symmetry), and chain A onto chain B (A′ onto B′, by the noncrystallographic 2-fold rotation) (Figures 2C and 3C). The active sites of each noncrystallographic pair of subunits (A–B and A′–B′) are connected by an internal cavity or tunnel, and the substrate binding sites are separated by about 35 Å. In a state where the β7−β8-loop is disordered (as is the case for chain B), the active site is wide open to solvent (Figure 3D). Yet even when this loop ordered the active site remains solvent-accessible.
TreS-catalyzed isomerization of trehalose to maltose proceeds through a double displacement mechanism, which involves a covalently bound glucosyl-enzyme intermediate.17 Such mechanism raises the question whether, between glycosylation and deglycosylation of TreS, the leaving group (the noncovalently bound glucose molecule) diffuses out of the active sit or is retained, reorients, and reattacks the covalently bound half of the disaccharide. Recent evidence using 13C-labeled glucose in addition to unlabeled disaccharide indicated that no labeled glucose was incorporated into the product, strongly suggesting that isomerization occurs without release of the leaving glucose molecule to solvent.17 Hydrolysis of trehalose (and maltose) to glucose occurs as a side reaction (see below), suggesting that retention of the leaving glucose molecule is imperfect. Nonetheless, the ordered vs disordered state of the β7−β8 loop in chains A and B, respectively, may be linked to enabling retention of the hydrolyzed substrate in the active site.
In the conformation of chain A, the β7−β8 loop overlaps with the substrate binding site (Figure 3B), but relatively minor structural adjustments of this loop suffice to make space for a disaccharide. It is conceivable that the loop acts as a clamp, helping to hinder diffusion of the leaving glucose out of the active site. Of the three catalytic carboxylic acids (Asp238, Glu280, and Asp350),17 Asp350 is located in the β7−β8 loop. Furthermore, in this loop, residues 350 to 366 are strictly conserved in mycobacteria, and only two conservative substitutions (Ser to Thr and Asp to Ser) occur in TreS of Corynebacterium glutamicum, a nonpathogenic surrogate organism. Sequence conservation could provide conserved, specificity-determining contacts with the noncovalently bound glucose. At the same time, conformational flexibility could provide plasticity required to facilitate the reorientation of the cleaved glucose moiety, together favoring reorientation of the leaving glucose over release from the active site. Very recently a crystal structure M. smegmatis TreS was determined, with structural features consistent to our findings.19
Solution X-ray Scattering of TreS
Crystal packing interfaces had suggested that TreS assembles as a tetramer in solution. We wondered whether this apparent tetramer (the “crystal tetramer”) described how TreS assembles in solution and probed self-assembly of TreS by small-angle X-ray scattering (SAXS).
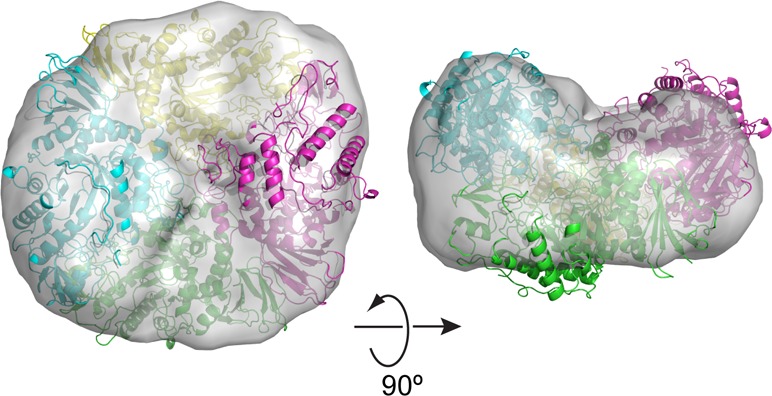
Guinier plot analysis (PRIMUS20) confirmed that TreS does not aggregate in the solution state, with a radius of gyration of Rg = 4.71 nm (Figure S2A, Supporting Information). The distance distribution function is approximately bell-shaped, indicative of a globular protein (Figure S2B, Supporting Information). We next calculated theoretical scattering curves based on the crystal structure of TreS using CRYSOL,21 assuming monomeric, dimeric, and tetrameric configurations. Pronounced deviations from the experimental data are evident at low scattering angles for calculated curves assuming a monomer or dimer. In contrast, the tetrameric configuration was able to reproduce the features of the measured scattering curve at low scattering angles (Figure 4A), although systematic deviations are still apparent at s ≥ 0.1 nm–1. The discrepancy indicates that the arrangement of the subunits in the solution tetramer may differ from that in the crystal tetramer, likely reflecting the absence of packing constraints imposed in the lattice. Nevertheless, a bead model calculated solely on the basis of the scattering curve (DAMMIF22 and DAMAVER23) envelops the crystal tetramer and shows internal features compatible with the tetrameric structure (Figure 4B).
Figure 4.
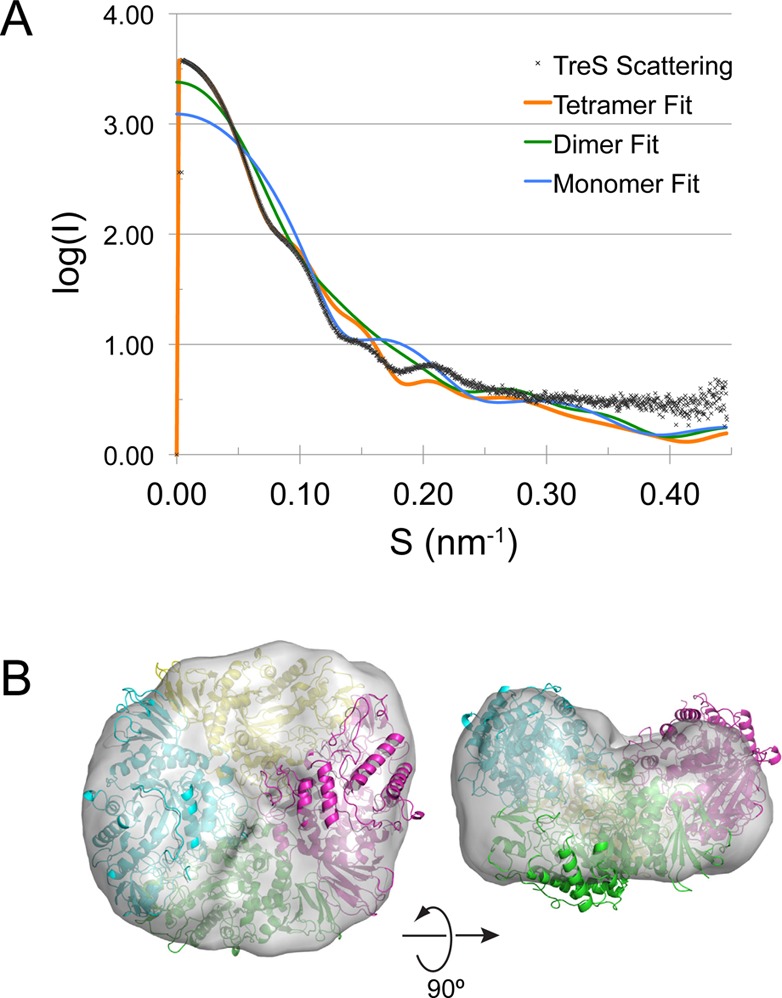
Solution small-angle X-ray scattering analysis of TreS. (A) Scattering curves for TreS calculated on the basis of the TreS structure in monomeric, dimeric, and tetrameric configuration are superimposed over the measured scattering curve. (B) Envelope of the TreS tetramer determined by ab initio calculation from the solution scattering data and superimposed over the crystal tetramer of TreS.
Thus, the solution scattering data indicate that the TreS tetramer observed in the crystal lattice describes the assembly in the solution state, although the SAXS-derived molecular envelope, derived without assuming internal symmetry, suggests that the solution tetramer is not strictly symmetrical.
TreS and Pep2 Form a Noncovalent Complex
In the genome of M. tuberculosis, TreS and Pep2 are encoded by separate genes (Rv0126 and Rv0127, respectively), but they appear as a gene fusion in a considerable number of bacterial species.6 We, therefore, investigated whether TreS and Pep2 form a noncovalent complex. First, we examined elution of TreS and Pep2 from a size exclusion resin. Calibration of the resin using bovine serum albumin (BSA) showed a dominant monomer peak at 137 mL (66.5 kDa) and a weaker dimer peak at 116 mL (133 kDa), consistent with published data.24 Trehalose synthase (monomer mass 72 kDa) on its own eluted with a dominant peak at 107 mL (Figure S3A, Supporting Information), while M. tuberculosis Pep2 (monomer mass 52 kDa) showed a continuous size distribution with three distinct peaks at 108, 114, and 149 mL. Compared to the BSA standard, these peaks could represent tetrameric, trimeric, and monomeric assembly states of Pep2, respectively. Analyzing the elution of a mixture of TreS with Pep2 (nominal molar ratio of 1:2) resulted in a dominant peak at 98 mL, over a background of a size distribution that resembled that of Pep2 alone.
Analyzing the elution fractions by denaturing polyacrylamide gel electrophoresis (SDS-PAGE; Figure S3B, Supporting Information) demonstrates that the peak at 98 mL includes TreS and Pep2 in approximately equal parts (by visual inspection of band intensity), whereas on its own, Pep2 starts eluting from the column only at 105 mL, with the peak fractions eluting between 110 and 115 mL. Thus, TreS and Pep2 coelute and do so earlier than on their own, strongly suggesting complex formation.
Stoichiometry of the TreS:Pep2 Complex
Next, we analyzed the sedimentation behavior of TreS and Pep2 by analytical ultracentrifugation. In the absence of Pep2, TreS sediments at a sedimentation coefficient of 11S, with a much smaller maximum at 16S (Figure 5A). Fitting a single frictional coefficient, these two peaks correspond to molecular masses of 260 000 and 470 000, respectively, indicating that TreS exists predominantly as a tetramer in solution (monomer mass of TreS is 72 kDa), with only a weak tendency of forming higher oligomers. The fact that two peaks are seen is indicative that the exchange between the tetramer and octamer is slow on the time scale of sedimentation (ca. 10–5 s–1). No evidence of a TreS monomer or dimer was found in this velocity experiment or the size exclusion profile. When analyzed on its own, Pep2 displays maxima at sedimentation coefficients of 3.8S and 8.9S, corresponding to molecular masses of 54 000 and 188 000, respectively. Compared to the calculated mass of recombinant Pep2 (52 kDa), the 3.8S peaks represents a Pep2 monomer, while the 8.9S peak could represent a mixture of Pep2 trimers and tetramers, which were both apparent in the size exclusion profile of Pep2.
Figure 5.
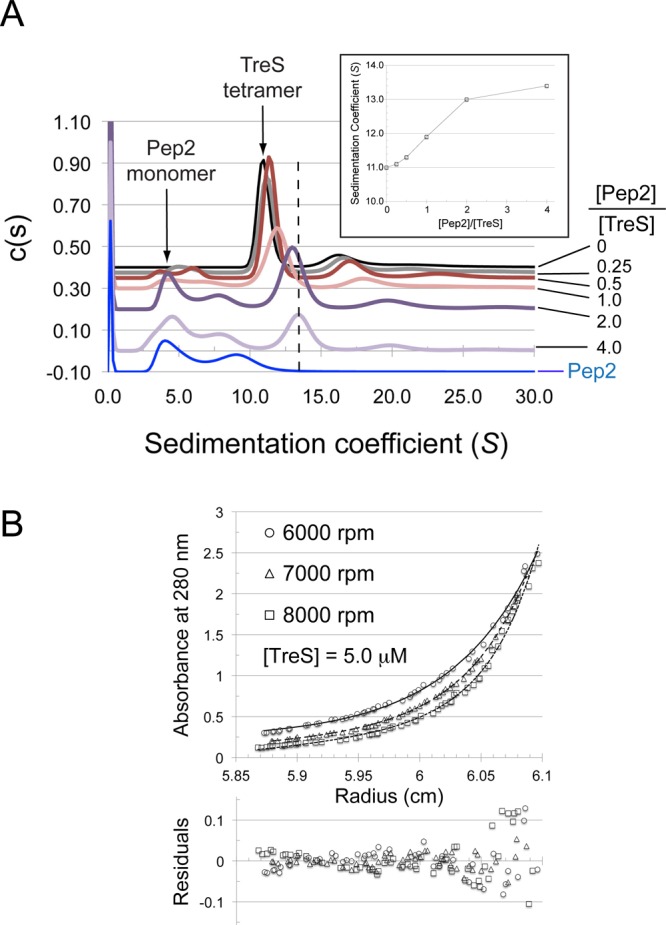
Sedimentation behavior of TreS in the absence and presence of Pep2. (A) Sedimentation velocity analysis of TreS, Pep2, and molar mixtures of the two as indicated on the right of the panel (absolute concentration of TreS at 4.5 μM). With the exception of the trace in blue (Pep2 alone, 9 μM), c(s) traces have been spaced along the vertical axis according to the molar ratio of [Pep2]/[TreS]. Arrows indicate the positions of the Pep2 monomer and TreS tetramer, respectively. The dashed line highlights the shift of the TreS tetramer peak in response to the addition of Pep2. (Inset) Sedimentation coefficient of the TreS tetramer peak as a function of the concentration ratio [Pep2]/[TreS]. (B) Sedimentation equilibrium analysis of the TreS:Pep2 complex. Fit of a noninteracting species model consisting of the TreS:Pep2 complex and free Pep2 (fixed at 52 kDa). TreS and Pep2 are at a concentration of 5 and 10 μM, respectively. Rotation speeds are indicated. Equilibrium was allowed to establish over a 24 h period at each speed, and several absorbance scans were recorded per rotation speed, the last of each was used in the data analysis.
Adding Pep2 to TreS in molar ratios of 0.25:1, 0.5:1, 1:1, 2:1, and 4:1 (with respect to monomers), distinctly altered the c(s) distribution profile. The peak at 11S, representing the TreS tetramer, shifted continually to about 13.1S until the molar ratio was 2:1, and changed only slowly thereafter (Figure 5A + inset). Within in the limits of the resolution of the c(s) distribution, the profiles indicated a continuous shift of the original 11S peak, rather than this peak disappearing and a new peak (at 13.1S) appearing instead. Thus, the c(s) distributions suggest that Pep2 gradually associates with the tetramer of TreS in a fast exchange on the time scale of sedimentation. The shifted TreS peak (13.1S) at a 4:1 ratio of Pep2:TreS corresponds to 345 kDa, fitted with a single frictional ratio. This mass would be compatible with four TreS plus two Pep2 subunits. However, fitting only a single frictional ratio, which varied between 1.11 and 1.48 (Figure S4A, Supporting Information), for a complex mixture of species, significant over- or underestimates of actual masses are possible. Therefore, we performed an analysis of the TreS:Pep2 complex by analytical ultracentrifugation in sedimentation equilibrium mode. A mixture of Pep2:TreS at nominal molar ratio of 2:1 was analyzed at 3 different concentrations and 3 different rotation speeds (Figures 5B, S4B, and S4C, Supporting Information).
Assuming that only a single species (the TreS:Pep2 complex) is present and fitting its molecular weight yielded an experimental mass of 452 861 Da [±4670 Da] (global fit over data from three speeds and three sample concentrations), but still led to systematic deviations in the residual plots. In contrast, a model of 2 noninteracting species in which Pep2 (present at a 2:1 molar ratio to TreS) was assumed to remain monomeric when not part of the TreS:Pep2 complex, led to a fitted mass of 506 101 Da [±9975 Da] (global fit) for the complex, with residuals showing no systematic deviation (Figure s5B, S4B, and S4C, Supporting Information). Thus, the fitted mass of the TreS:Pep2 complex is most compatible with a complex of TreS tetramer bound to four Pep2 subunits (calculated mass 491 000).
Pep2 Activity Varies in the Presence of TreS in a Dose-Dependent Fashion
Initially, we probed activities of recombinant trehalose synthase and maltokinase in an end-point assay, monitoring reaction products by thin layer chromatography.7 Incubation of TreS with trehalose or maltose interconverted the substrates (Figure S5A, Supporting Information), as reported previously for M. smegmatis TreS.25 At high enzyme concentrations, we also observed the hydrolysis of the disaccharide to glucose, as reported for M. smegmatis TreS.17,25 Incubating trehalose with both TreS and Pep2 produces maltose-1-phosphate (Figure 6A, lanes 11–13). Phosphorylation of maltose is ATP-dependent (Figure 6A, lane 8), consistent with previous evidence,26 but Pep2 does not phosphorylate trehalose under the assay conditions (Figure 6A, lane 6). Likewise, TreS does not produce a phosphorylated sugar when incubated with either trehalose or maltose (Figure 6A, lanes 4 and 5). Thus, the 2-step conversion from trehalose to maltose-1-phosphate occurs only when both enzymes, TreS and Pep2, are present.
Figure 6.

Activity of TreS and Pep2 and evidence for TreS affecting Pep2 activity. (A) Thin layer chromatography analysis of reaction products demonstrating Pep2-catalyzed conversion of maltose to maltose-1-phosphate and conversion of trehalose to maltose-1-phosphate when both TreS and Pep2 are present. (B,C) Michaelis–Menten kinetics of M. tuberculosis Pep2 activity, monitored by an enzyme-coupled assay (see Methods). Experiments were done in triplicate and Michaelis–Menten parameters (Table 2) determined using the GraphPad Prism software. (D) Hydrolysis activity of M. smegmatis TreS (0.4 μM) in the absence and presence of M. smegmatis Pep2-K145A. Molar ratios of Pep2-K145A:TreS are indicated, and data points for [TreS] = 0.8 μM (Figure S6C, Supporting Information) are included for comparison. Data points were fitted to a linear function (vi = k[Tre]).
We then asked whether complex formation between TreS and Pep2 affects the catalytic activity of the constituent enzymes. To this end, we designed a continuous enzyme assay that quantified Pep2-catalyzed phosphorylation of maltose by coupling ATP-to-ADP conversion to oxidation of NADH, monitoring the latter fluorimetrically. Varying maltose at a fixed initial ATP concentration or varying ATP at a fixed initial maltose concentration resulted in Michaelis–Menten type kinetics in both cases (Figures 6B,C). Fixing ATP at 0.5 mM and increasing the concentration of TreS (from nil to a molar ratio of 4 TreS to 1 Pep2) tripled Vmax in a nearly linear fashion (Figures 6C and S5B, Supporting Information; Table 2). In contrast, KM increased moderately from the TreS-free to the TreS:Pep2 complex state, after which any change remained within the experimental error (Figure S5C, Supporting Information; Table 2). Varying ATP at a fixed maltose concentration (20 mM) mirrored this pattern in that Vmax even quadrupled, while the KM for ATP increased when TreS was added, but remained invariant within error for [TreS]/[Pep2] ratios ≥ 1 (Figures 6C, S5B, and S5C, Supporting Information; Table 2). This effect was protein-specific: adding bovine serum albumin (BSA) to Pep2 at the same molar ratios resulted in changes of activity that were nonsystematic and distinctly smaller in magnitude (±30%; Figure S5D, Supporting Information), underscoring that the pronounced and dose-dependent increase of Vmax of Pep2 was specific for TreS, likely a consequence of complex formation.
Table 2. Michaelis–Menten Parameters of Pep2-Catalyzed Phosphorylation of Maltose.
| [TreS]/[Pep2] | 0 | 1:1 | 2:1 | 4:1 |
|---|---|---|---|---|
| Maltose ([ATP] = 0.5 mM) | ||||
| Vmax (μmol min–1 mg–1) | 707 ± 50 | 1164 ± 148 | 1625 ± 399 | 2170 ± 360 |
| KM (mM) | 0.17 ± 0.04 | 0.341 ± 0.12 | 0.540 ± 0.31 | 0.457 ± 0.19 |
| kcat (sec–1) | 613.6 | 1010.4 | 1410.6 | 1883.7 |
| kcat/KM (sec–1 M–1 106) | 3.71 | 2.96 | 2.61 | 4.12 |
| ATP ([Mal] = 20 mM) | ||||
| Vmax (μmol min–1 mg–1) | 1185 ± 70 | 1904 ± 76 | 3416 ± 146 | 4164 ± 221 |
| KM (mM) | 0.031 ± 0.005 | 0.050 ± 0.005 | 0.058 ± 0.006 | 0.049 ± 0.007 |
| kcat (sec–1) | 1028.6 | 1652.8 | 2965.3 | 3614.6 |
| kcat/KM (sec–1 M–1 106) | 33.7 | 33.1 | 51.5 | 73.2 |
In order to test whether complex formation also affected the activity of TreS, we monitored M. smegmatis TreS-catalyzed conversion of trehalose to glucose in an enzyme-coupled continuous assay (see Methods). Since the coupling reactions involved conversion of ATP to ADP, it was necessary to rule out ATP depletion by Pep2-catalyzed conversion of maltose to maltose-1-phosphate. We designed three point mutants of M. smegmatis Pep2 (K145A, Q309A, and D321A) aiming to abrogate ATP binding. Two of these mutants (K145A and D321A) showed activity levels that were indistinguishable from the baseline of the assay (Figure S6A, Supporting Information). We chose the Pep2-K145A mutant for further experimentation, verifying by size exclusion chromatography that this mutant still formed a complex with TreS (Figure S6B, Supporting Information). Because of trace contaminations of glucose in the substrate, we were limited to probing activity at trehalose concentrations of less than half the KM (∼85 mM).17 We, therefore, compared the slopes Δvi/Δ[Tre] of the vi vs [Tre] plot at different ratios of [Pep2-K145A]/[TreS] and calibrated the resulting slopes against activity measurements at different TreS concentrations (without Pep2 present), thus mimicking an increase or decrease in TreS hydrolysis activity (Figure S6C, Supporting Information). We observed a systematic increase of TreS-catalyzed hydrolysis as Pep2-K145A was added up to a molar ratio [Pep2-K145A]/[TreS] of 4. However, the change was small, exceeding only slightly the unsystematic variation observed when adding BSA (Figure S6D, Supporting Information) and remaining distincly below the increase of activity observed when doubling the TreS concentration (Figures 6D and S6C, Supporting Information). We conclude that complex formation increases Pep2 activity markedly, but affects TreS activity to a far lesser extent.
Evidence for TreS:Pep2 Complex Formation in Vivo
In order to probe whether TreS and Pep2 form a complex in vivo, we overexpressed and purified His6-tagged M. smegmatis Pep2 from cell extracts of Mycobacterium smegmatis mc2155 using Ni-NTA and ion exchange chromatography. To detect endogenous TreS, we analyzed column fractions for their ability to convert trehalose to maltose-1-phosphate, which requires the presence of both TreS and Pep2. As the purification involved two successive chromatography steps, in which the Ni-NTA matrix selected for the His-tagged Pep2, observation of TreS activity indicated complex formation. Coomassie-staining failed to visualize endogenous TreS enzyme on the SDS gel of the untreated column fractions (Figure 7A, main panel), but thin-layer chromatography showed fraction-specific conversion of trehalose to maltose-1-phosphate (Figure 7B). Concentrating the maltose-1-phosphate producing fractions 20-fold revealed two bands that line up with TreS and Pep2 controls, respectively (Figure 7A, inset). Taken together these results provide evidence that TreS and Pep2 form a complex in the cellular context.
Figure 7.
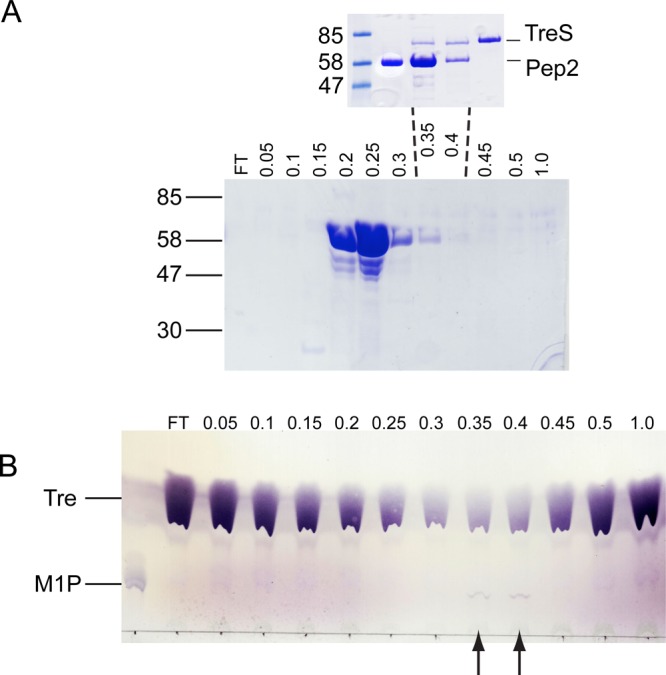
Evidence for formation of the TreS:Pep2 complex in M. smegmatis mc2155. (A) His6-tagged Pep2 was purified from M. smegmatis extracts by Ni-NTA and ion exchange chromatography, and anion exchange column fractions were analyzed by Coomassie-stained SDS-PAGE, with NaCl concentrations (in M, across the top), and molecular weight standards (in kDa, on the left) as indicated. FT = flow through. (A, inset) Coomassie-stained SDS-PAGE of ion exchange fractions eluted at 0.35 and 0.4 M NaCl after ∼20-fold concentration, flanked by TreS- and Pep2 controls. (B) Thin-layer chromatogram of reactions mixtures (100 μL) containing 100 mM trehalose (Tre) and 20 μL of fractions of the anion exchange column fractions shown in panel A. The position of maltose-1-phosphate (M1P) and NaCl concentrations (in M) are indicated. Arrows highlight the two fractions producing maltose-1-phosphate.
Conclusions and Final Remarks
To our knowledge, complex formation between TreS and Pep2 in mycobacteria has not been reported previously but is perhaps not surprising given the treS-pep2 gene fusion in a considerable number of bacterial species.6 Yet the size of this complex (a hetero-octamer of ∼0.5 × 106 Da) is unexpected. A survey of structural neighbors of TreS (according to analyses of crystal packing interfaces) suggests that monomeric and dimeric biological assemblies prevail by far. Our data indicate that the TreS-Pep2 hetero-octamer is built on the platform of a TreS tetramer. On the basis of size exclusion chromatography, a hexameric assembly has previously been reported for M. smegmatis TreS.25 However, size exclusion, in a strict sense, sizes on hydrodynamic radii rather than mass; hence, accurate mass estimates are difficult to obtain. In contrast, our analytical ultracentrifugation and SAXS data clearly indicate a tetramer as the preferred assembly state of TreS in solution. The shift of the dominant TreS tetramer peak to higher S-values in the c(s) profile on adding Pep2 indicates that Pep2 subunits bind to the TreS tetramer, as opposed to the TreS tetramer dissociating and reassembling into a TreS:Pep2 octameric complex. The complex appears to be in equilibrium between the octameric state and a state of free Pep2 and free TreS tetramer, as including free Pep2 in the model removed systematic deviations between data and fit. Excess of one or the other partner appears to be required to reach a saturated complex, which is also consistent with the influence of TreS on Pep2 activity.
Complex formation with TreS significantly upregulates the activity of Pep2, an effect that is not explained by nonspecific protein–protein interactions, as the BSA control demonstrates. While activity of TreS also increases in response to complex formation, the effect is subtle by comparison. In the absence of a structure for Pep2 (or the Pep2:TreS complex), the mechanistic basis of the 3-fold (maltose) or 4-fold (ATP) increase of Vmax is not obvious, but complex formation might drive Pep2 into a conformation that favors catalysis by reducing the sampling of conformational states that are compatible with substrate binding but do not allow turnover.
A key requirement during α-glucan synthesis is to avoid accumulation of toxic pathway intermediates. Stress by increased levels of maltose-1-phosphate appears to result in inhibition of respiration and the induction of the SOS regulon, which controls the DNA damage response.7 Complex formation would be an effective strategy to direct the flow of pathway intermediates through consecutive catalytic steps. The open active site of the TreS tetramer and the tunnel between the active sites within the TreS tetramer would offer several mechanistic options for docking of partner enzymes and the efficient transfer of pathway intermediates. Thus, our study provides a first hint to the intriguing possibility of the GlgE pathway being organized as a multicatalytic machinery that controls the flow of pathway intermediates by complexation between pathway enzymes.
Methods
Recombinant Proteins
The genes of M. tuberculosis trehalose synthase treS (Rv0126) and pep2 (Rv0127) were amplified by polymerase chain reaction (PCR, primers in Table S1, Supporting Information) from M. tuberculosis H37Rv genomic DNA. The PCR products were ligated into NdeI and HindIII sites of plasmid pET28a (Novagen). E. coli BL21 (DE3) cells were heat-transformed with plasmids, encoding either TreS or Pep2, and cultured on agar plates (LB/kanamycin 25 μg/mL). A single colony was used to inoculate 10 mL of LB broth, 1% (w/v) glucose, and kanamycin (25 μg/mL), followed by incubation overnight (37 °C). The liquid culture was centrifuged and resuspended in fresh Terrific Broth media (TB27) with kanamycin (50 μg/mL), and incubated at 37 °C (200 rpm). At OD600 = 0.4, the cultures cooled to 16 °C for 3 h, and protein expression was induced using 1 mM IPTG (TreS) or 0.1 mM IPTG (Pep2), followed by further incubation (21 h, 16 °C, 200 rpm). Cells were harvested (7000g, 10 min), washed with phosphate buffered saline (PBS), resuspended in 30 mL lysis buffer (25 mM HEPES-NaOH pH 7.6, 1 M NaCl, 20% (v/v) glycerol), and frozen at −80 °C until further use.
Protease inhibitor cocktail (Roche), 1 mM PMSF, 10 mM MgCl2, and 10 μg/mL DNase I were added to the resuspended cell pellet, which was passed 4 times through a French Press (Thermo Spectronic FA-078). The cleared lysate (27,000g, 30 min, 4 °C), was diluted 4-fold with buffer A (25 mM HEPES-NaOH pH 7.6, 1 M NaCl, 10% (v/v) glycerol), filtered (0.45 μm pore size), and loaded on a pre-equilibrated Ni-NTA column (5 mL, GE Healthcare). The column was washed with buffer A (5 column volumes), and buffer A + 20 mM, 40 mM, and 60 mM imidazole, respectively. The protein was eluted with 500 mM imidazole in buffer A, and fractions were analyzed by SDS-PAGE. The eluate was diluted 20-fold with buffer B (20 mM Bis-Tris pH 6.5), filtered (0.45 μm), and applied on a HiTrap Q-column (1 mL, GE Healthcare Life Sciences) pre-equilibrated with 20 mM Bis-Tris pH 6.5 and 50 mM NaCl. The column was washed with buffer B supplemented with NaCl (50 to 500 mM, steps of 50 mM). Fractions were analyzed by SDS-PAGE and pooled, followed by concentration in Amicon Ultra-4 centrifugal filter units, then loaded on a HiPrep Sephacryl 26/60 S-200HR column (GE Healthcare). Fractions containing protein were concentrated as described before.
Crystallization and Structure Determination
Crystals of TreS were grown by vapor diffusion in 96-well plates, using a Mosquito liquid handling system (TTP Labtech) to set up crystallization drops containing 100 nL of TreS (80 mg mL-1) + 100 nL of reservoir solution. Sizable crystals grew over a reservoir of 0.1 M sodium citrate pH 5.6, 0.5 M (NH4)2SO4, and 1 M Li2SO4. Crystals were soaked in mother liquor supplemented with either 15% (v/v) glycerol or 100 mM maltose or both, and frozen in liquid nitrogen. Diffraction data were recorded from a single crystal on beamline I04 (Diamond Light Source, Table 1). Data were processed and scaled (XDS and XSCALE28) and the structure phased by molecular replacement (PHASER29) using an ensemble of aligned search models (PDB entries 2ZE0,301UOK,312PWE,181WZA32), truncating nonconserved side chains (CHAINSAW33). Using 2-fold noncrystallographic symmetry averaging of the MR-phased map led to interpretable density, and the model was completed through iterative rounds of building and refinement (COOT34 and REFMAC535).
Table 1. X-ray Diffraction Data and Refinement Statistics.
| X-ray diffraction data | |
|---|---|
| beamline | Diamond I04 |
| wavelength (Å) | 0.9795 |
| space group | P3212 |
| cell parameters a,b,c (Å) | 161.6, 161.6, 139.1 |
| molecules per asymmetric unit | 2 |
| resolution (Å) | 29.2–2.60 |
| high resolution shell (Å) | 2.74–2.60 |
| Rmergea (%) | 6.0 (66.8) |
| total observations | 298633 |
| unique reflections | 63561 |
| I/σ(I)a | 16.1 (1.7) |
| completenessa (%) | 99.6 (99.6) |
| multiplicitya | 4.7 (4.7) |
| refinement | |
|---|---|
| resolution range (Å) | 29.16–2.60 |
| unique reflections | 60392 |
| Rcryst (%) | 21.3 |
| Rfree (%) | 24.0 |
| No. of non-hydrogen atoms | 8845 |
| protein | 8737 |
| solvent | 108 |
| rmsd bonds (Å) | 0.006 |
| rmds angles (deg) | 1.0 |
| B-factors | |
| wilson (Å2) | 70.6 |
| average (Å2) | 69.2 |
| protein (Å2) | 69.1 |
| solvent (Å2) | 74.1 |
| rmsd B-factors | 1.3 |
| ramachandran plotb | |
| favored (%) | 96.9 |
| allowed (%) | 3.1 |
| disallowed (number) | 0 |
| score, percentilec | 1.65, 99th |
Numbers in parentheses refer to the high resolution shell.
Ramachandran analysis of backbone dihedral angles was done using Molprobity (molprobity.biochem.duke.edu).
The Molprobity score provides, on a scale of X-ray resolution, an overall assessment of the quality of the protein geometry relative to a set of reference structures; the 100th percentile is among the best, the 0th percentile among the worst structures at comparable resolution (N = 6237; 2.6 Å ± 0.25 Å).
Small Angle X-ray Scattering
Solution scattering data of TreS (concentration range 8.1 to 2.3 mg mL–1) were recorded on beamline BM29 at ESRF, Grenoble. The protein was buffered in 20 mM Bis-Tris pH 6.5, 150 mM NaCl, and the sample cell equilibrated at 20 °C. Exposures comprised 10 frames exposed for 2 s each in flow mode, which were merged. The detector images were integrated and reduced to 1-dimensional scattering curves, and buffer contributions to scattering were subtracted using the beamline software BsxCuBE. Scattering curves were displayed using the program PRIMUS.20 All SAXS data analyses were performed using programs of the ATSAS suite version 2.5 (www.embl-hamburg.de/biosaxs/software.html).
Size Exclusion Chromatography
The elution of TreS and Pep2, a 2:1 molar mixture of Pep2:TreS from a Sephacryl S-200HR resin (320 mL column volume), was monitored by UV absorbance at 280 nm with a flow rate of 0.5 mL/min, and fractions (5 mL) were analyzed by SDS-PAGE. Proteins were in 20 mM Bis-Tris pH 6.5, 150 mM NaCl.
Analytical Ultracentrifugation
Sedimentation velocity and equilibrium experiments used a Beckman Optima XL-A analytical ultracentrifuge equipped with absorbance optics. Protein samples were dialyzed into 20 mM Bis-Tris pH 6.5, 150 mM NaCl, and, for the velocity experiments, loaded into cells with 2-channel Epon centerpieces and quartz windows. A total of 120 absorbance scans (280 nm) were recorded (25 000 rpm, 20 °C) for each sample, representing the full extent of sedimentation of the sample. Data analysis was performed using SEDFIT, fitting a single frictional ratio (Figure S4A, Supporting Information).36 The sedimentation equilibrium experiment (96 h total duration) was conducted at a rotor temperature of 4 °C. Samples buffered as above were loaded into 6-channel Epon centerpieces with quartz windows, and data were recorded at 6000, 7000, and 8000 rpm, respectively, at three different sample concentrations (see captions of Figures 5 and S4, Supporting Information). At each rotation speed, the sample was allowed to reach equilibrium during a 24 h period. The data analysis was performed using SEDPHAT.37 Parameters for solvent density and viscosity and for the partial specific volume (v̅p) of the proteins were calculated using SEDNTERP (sednterp.unh.edu).
Overexpression of Pep2 in M. smegmatis
DNA primers (Table S1, Supporting Information) were obtained from MWG, and DNA of the M. smegmatis pep2 gene was amplified by PCR. The purified PCR fragment (Qiagen) was ligated into the pSD26 plasmid (BamH1 and EcoRV), which was transformed into M. smegmatis mc2155 cells by electroporation. Cells were selected on LB/hygB+ (50 μg/mL) agar plates, and a single colony was used to inoculate a 3 mL culture (LB/hygB+ at 50 μg/mL + 0.05% Tween-80) and grown for 2 d at 37 °C. The culture was propagated (1% v/v inoculation) into 50 mL (2 d growth) and 1 L, grown to midlog phase, induced with 0.2% w/v acetamide, and incubated overnight (16 °C). Cells were harvested (7000g, 10 min, 4 °C), resuspended in 25 mM Hepes-NaOH, pH 7.5, 300 mM NaCl, 10% v/v glycerol, 1 mM β-mercaptoethanol, 10 mM MgSO4, DNase, and lysozyme, and lysed by sonication (12 cycles of 60 s on, 90 s off at maximum amplitude). The lysate was cleared (27 000g, 30 min, 4 °C), the supernatant filtered (0.45 μm) and loaded on a pre-equilibrated Ni-NTA HiTrap HP column (GE Life Science, 5 mL) and washed with buffer A (25 mM Hepes-NaOH, pH 7.5, 300 mM NaCl, 10% v/v/glycerol) adding imidazole in steps of 20, 40, 60, 80, 100, 150, 200, 250, 300, and 500 mM (5 column volumes per step). Protein containing fractions were analyzed by SDS-PAGE, pooled, and diluted 6-fold into buffer B (20 mM Bis-Tris pH 6.0, 10% glycerol) prior to loading on an anion exchange column (HiTrap QHP, 1 mL, GE Life Science). Protein was eluted with a stepwise NaCl gradient (Figure 7A).
Generation of Inactive Pep2 Mutants
A structural model for Pep2 was generated using the fold prediction server Phyre2 (http://www.sbg.bio.ic.ac.uk/phyre2)38 based on the sequence of M. smegmatis Pep2. Comparison with ATP-bound structural homologues suggested three residues (K145, Q309, and D321) as critical for Mg2+ and/or ATP binding. Individual point mutations to alanine were introduced into the pep2 gene of M. smegmatis using the Quikchange approach (Stratagene, primers in Table S1, Supporting Information). The His6-tagged proteins were expressed and purified as described for wild-type Pep2. Activity of the mutants was tested with the assay described below (Figure S6A, Supporting Information).
End Point Assays Probing Trehalose Synthase and Maltokinase Activity
Reaction mixtures probing TreS activity were in a final volume of 100 μL, containing 50 mM HEPES-KOH buffer (pH 7.5), 100 mM maltose or trehalose, and TreS. Reaction mixtures probing maltokinase activity (100 μL) consisted of 50 mM HEPES-KOH buffer, pH 7.5, 10 mM MgCl2, 5 mM ATP, and 560 nM Pep2 and were initiated by the addition of 100 mM maltose. All reactions were at 37 °C for 1 h and stopped by adding an equal volume of ice-cold acetone, followed by centrifugation (20 000g, 4 °C, 10 min). The supernatant was transferred and dried by speedvac (Hetovac). To discriminate trehalose from maltose, the dried reaction product was dissolved in 200 μL water and 2 μL of the solution applied on silica gel concentrating zone HPTLC plate (Merck, Darmstadt, Germany) and developed in the solvent system butanol/pyridine/water (7:3:1 v/v/v). In order to detect phosphorylated reaction products (Figures 6A and 7B), maltose-1-phosphate was separated from maltose using aluminum backed silica gel 60 F254 TLC plates (Merck, Darmstadt, Germany), using the solvent system butanol/ethanol/water (5:3:2 v/v/v), developing the plate three times in the same solvent system. Conversion of trehalose to maltose-1-phosphate was probed by incubating both TreS (140 nM) and Pep2 (0, 35, 70, and 140 nM) in a reaction mixture (50 mM HEPES-KOH buffer, pH 7.5, 10 mM MgCl2, 5 mM ATP) and initiating turnover with 100 mM trehalose as substrate. To visualize the products, the dried TLC plates were sprayed with α-naphthol sulfuric acid stain, followed by mild charring with a heat gun.
Continuous Assay
In order to monitor phosphorylation of maltose, conversion of ATP to ADP was enzymatically coupled to oxidation of NADH (via pyruvate kinase and lactate dehydrogenase), and the latter monitored fluorimetrically (excitation 340 nm and emission 450 nm). Fluorescence units were converted to concentrations of ADP by an internal calibration curve for NADH. The assays were performed in triplicate in 96-well plates, using a BMG PHERAstar FS microtiter plate reader and MARS and GraphPad Prism software to record and analyze data, respectively.
Reaction mixtures contained 50 mM HEPES-NaOH (pH 7.5), 50 mM NaCl, 10 mM MgCl2, and Pep2 at 0.2 μM (when varying ATP) or 1.2 μM (when varying maltose), adding TreS to final molar ratios [TreS]/[Pep2] of 0, 1, 2, and 4. The coupling reagents were present at these initial concentrations: 4 mM phosphoenolpyruvate, 2 units of pyruvate kinase, 2 units of lactate dehydrogenase, and 0.1 mM NADH.
Enzymatic activity of TreS was quantified by monitoring conversion of trehalose to glucose (the hydrolysis side reaction),17 coupling formation of glucose to oxidation of NADH (monitored as above) via hexokinase, pyruvate kinase, and lactate dehydrogenase. The reaction mixtures contained 50 mM HEPES-NaOH (pH 7.5), 50 mM NaCl, 1 mM MgCl2, 1.2 μM ATP, 4 mM phosphoenolpyruvate, and 0.1 mM NADH in addition to 0.5 units of hexokinase and 2 units each of pyruvate kinase and lactate dehydrogenase. Reactions were initiated by TreS (0.4 μM final concentration) or the TreS:Pep2 complex (TreS at 0.4 μM) at varying molar ratios of Pep2 to TreS for trehalose concentrations from 0 to 25 mM. Above 40 mM trehalose, instant NADH oxidation became noticeable in the absence of TreS, suggesting that the trehalose batch used contained trace amounts of glucose. Given a KM of ∼85 mM for trehalose,17 the glucose contamination precluded the determination of the Michaelis parameters of the reaction. Instead, slopes Δvi/Δ[Tre] of the vi vs [Tre] plot were compared to assess changes in enzymatic activity in response to the addition of Pep2. To calibrate changes of enzyme activity, we tested hydrolysis activity at TreS concentrations between 0.02 and 1.2 μM (Figure S6C, Supporting Information) as well as in the presence of BSA at variable molar ratios [BSA]/[TreS], with TreS at 0.4 μM (Figure S6D, Supporting Information).
Acknowledgments
G.S.B. acknowledges support from a Personal Research Chair from J. Bardrick, and a Royal Society Wolfson Research Merit Award. D.J.S. is partially supported by the Science and Technology Facilities Council (U.K.). This work used a crystal imaging system obtained through Birmingham Science City Translational Medicine Clinical Research and Infrastructure Trials Platform. We thank Diamond Light Source and ESRF for access to synchrotron beamlines and their staff for support during experiments. M. smegmatis ΔglgE was a gift from R. Kalscheuer, and competent cells of M. smegmatis mc2155 were prepared by V. Nataraj. We are grateful to R. Parslow for supporting the AUC experiments, V. Bowruth and N. Veerapen for help with the TLC experiments, J. B. Jackson and A. L. Lovering for critical comments on the manuscript, and T. Dafforn for useful discussions throughout the project. This work is supported by funding from the Wellcome Trust (084923/B/08/Z).
Supporting Information Available
Structure factors and coordinates for the crystal structure of M. tuberculosis TreS are deposited in the PDB under accession code 4LXF. Supplementary Figures S1–S6 and Supplementary Table S1. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Brennan P. J.; Crick D. C. (2007) The cell-wall core of Mycobacterium tuberculosis in the context of drug discovery. Curr. Top. Med. Chem. 7, 475–488. [DOI] [PubMed] [Google Scholar]
- Favrot L.; Ronning D. R. (2012) Targeting the mycobacterial envelope for tuberculosis drug development. Expert Rev. Anti-Infect. Ther. 10, 1023–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2012) Global Tuberculosis Report 2012.
- Sani M.; Houben E. N.; Geurtsen J.; Pierson J.; de Punder K.; van Zon M.; Wever B.; Piersma S. R.; Jimenez C. R.; Daffe M.; Appelmelk B. J.; Bitter W.; van der Wel N.; Peters P. J. (2010) Direct visualization by cryo-EM of the mycobacterial capsular layer: a labile structure containing ESX-1-secreted proteins. PLoS Pathog 6, e1000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X.; Prados-Rosales R.; Jenny-Avital E. R.; Sosa K.; Casadevall A.; Achkar J. M. (2012) Comparative evaluation of profiles of antibodies to mycobacterial capsular polysaccharides in tuberculosis patients and controls stratified by HIV status. Clin Vaccine Immunol 19, 198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra G.; Chater K. F.; Bornemann S. (2011) Unexpected and widespread connections between bacterial glycogen and trehalose metabolism. Microbiology 157, 1565–1572. [DOI] [PubMed] [Google Scholar]
- Kalscheuer R.; Syson K.; Veeraraghavan U.; Weinrick B.; Biermann K. E.; Liu Z.; Sacchettini J. C.; Besra G.; Bornemann S.; Jacobs W. R. J. (2010) Self-poisoning of Mycobacterium tuberculosis by targeting GlgE in an alpha-glucan pathway. Nat. Chem. Biol. 6, 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambou T.; Dinadayala P.; Stadthagen G.; Barilone N.; Bordat Y.; Constant P.; Levillain F.; Neyrolles O.; Gicquel B.; Lemassu A.; Daffe M.; Jackson M. (2008) Capsular glucan and intracellular glycogen of Mycobacterium tuberculosis: biosynthesis and impact on the persistence in mice. Mol. Microbiol. 70, 762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbein A. D.; Pastuszak I.; Tackett A. J.; Wilson T.; Pan Y. T. (2010) Last step in the conversion of trehalose to glycogen: a mycobacterial enzyme that transfers maltose from maltose 1-phosphate to glycogen. J. Biol. Chem. 285, 9803–9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal K.; Kumar S.; Sharma S.; Garg S. K.; Alam M. S.; Xu H. E.; Agrawal P.; Swaminathan K. (2010) Crystal structure of full-length Mycobacterium tuberculosis H37Rv glycogen branching enzyme: insights of N-terminal beta-sandwich in substrate specificity and enzymatic activity. J. Biol. Chem. 285, 20897–20903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalscheuer R.; Weinrick B.; Veeraraghavan U.; Besra G. S.; Jacobs W. R. J. (2010) Trehalose-recycling ABC transporter LpqY-SugA-SugB-SugC is essential for virulence of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 107, 21761–21766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel B. L.; Coutinho P. M.; Rancurel C.; Bernard T.; Lombard V.; Henrissat B. (2009) The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 37, D233–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L.; Rosenstrom P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skov L. K.; Mirza O.; Sprogoe D.; van der Veen B. A.; Remaud-Simeon M.; Albenne C.; Monsan P.; Gajhede M. (2006) Crystal structure of the Glu328Gln mutant of Meisseria polysaccharea amylosucrase in complex with sucrose and maltoheptaose. Biocatal. Biotransform. 24, 99–105. [Google Scholar]
- Koropatkin N. M.; Smith T. J. (2010) SusG: a unique cell-membrane-associated alpha-amylase from a prominent human gut symbiont targets complex starch molecules. Structure 18, 200–215. [DOI] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Pan Y. T.; He S.; Lam M.; Brayer G. D.; Elbein A. D.; Withers S. G. (2011) Mechanistic analysis of trehalose synthase from Mycobacterium smegmatis. J. Biol. Chem. 286, 35601–35609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravaud S.; Robert X.; Watzlawick H.; Haser R.; Mattes R.; Aghajari N. (2007) Trehalulose synthase native and carbohydrate complexed structures provide insights into sucrose isomerization. J. Biol. Chem. 282, 28126–28136. [DOI] [PubMed] [Google Scholar]
- Caner S.; Nguyen N.; Aguda A.; Zhang R.; Pan Y. T.; Withers S. G.; Brayer G. D. (2013) The structure of the Mycobacterium smegmatis trehalose synthase (TreS) reveals an unusual active site configuration and acarbose binding mode. Glycobiology 23, 1075–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev P. V.; Volkov V. V.; Sokolova A. V.; Koch M. H. J.; Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282. [Google Scholar]
- Svergun D. I.; Barberato C.; Koch M. H. J. (1995) CRYSOL: a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773. [Google Scholar]
- Franke D.; Svergun D. I. (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov V. V.; Svergun D. I. (2003) Uniqueness of ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk J. A.; Smit J. A. (2000) Size-exclusion chromatography–multiangle laser light scattering analysis of beta-lactoglobulin and bovine serum albumin in aqueous solution with added salt. J. Chromatogr., A 867, 105–112. [DOI] [PubMed] [Google Scholar]
- Pan Y. T.; Koroth Edavana V.; Jourdian W. J.; Edmondson R.; Carroll J. D.; Pastuszak I.; Elbein A. D. (2004) Trehalose synthase of Mycobacterium smegmatis: purification, cloning, expression, and properties of the enzyme. Eur. J. Biochem. 271, 4259–4269. [DOI] [PubMed] [Google Scholar]
- Mendes V.; Maranha A.; Lamosa P.; da Costa M. S.; Empadinhas N. (2010) Biochemical characterization of the maltokinase from Mycobacterium bovis BCG. BMC Biochem 11, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E. F., and Maniatis T. (1987) Molecular Cloning: A Laboratory Manual.
- Kabsch W. (2010) XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai T.; Hung V. S.; Morinaka K.; Kobayashi T.; Ito S. (2008) Crystal structure of GH13 alpha-glucosidase GSJ from one of the deepest sea bacteria. Proteins 73, 126–133. [DOI] [PubMed] [Google Scholar]
- Shirai T.; Hung V. S.; Morinaka K.; Kobayashi T.; Ito S. (1997) The refined crystal structure of Bacillus cereus oligo-1,6-glucosidase at 2.0 A resolution: structural characterization of proline-substitution sites for protein thermostabilization. J. Mol. Biol. 269, 142–153. [DOI] [PubMed] [Google Scholar]
- Sivakumar N.; Li N.; Tang J. W.; Patel B. K.; Swaminathan K. (2006) Crystal structure of AmyA lacks acidic surface and provide insights into protein stability at poly-extreme condition. FEBS Lett. 580, 2646–2652. [DOI] [PubMed] [Google Scholar]
- (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vistica J.; Dam J.; Balbo A.; Yikilmaz E.; Mariuzza R. A.; Rouault T. A.; Schuck P. (2004) Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal. Biochem. 326, 234–256. [DOI] [PubMed] [Google Scholar]
- Kelley L. A.; Sternberg M. J. (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.