

Abstract
Aims
Chronic administration of cocaine attenuates delta opioid receptor (DOPR) signaling in the striatum and the desensitization is mediated by the indirect actions of cocaine on dopamine D1 receptors (D1R). In addition, DOPR and D1R co-exist in some rat striatal neurons. In the present study, we examined the underlying mechanism of DOPR desensitization by D1R activation.
Main methods
NG 108-15 cells stably expressing HA-rat D1 receptor (HA-D1R) and Chinese hamster ovary (CHO) cells stably expressing both FLAG-mouse DOPR (FLAG-DOPR) and HA-D1R were used as the cell models. Receptor binding, [35S]GTPγS binding, receptor phosphorylation and western blot were conducted to examine DOPR affinity, expression, internalization, downregulation, desensitization, phosphorylation and phosphorylated ERK1/2.
Key findings
Pretreatment with either the DOPR agonist DPDPE or the D1R agonist SKF-82958 for 30 min attenuated DPDPE-stimulated [35S]GTPγS binding to G proteins, demonstrating homologous and heterologous desensitization of the DOPR, respectively. SKF-82958 pretreatment did not affect the level of DOPR or affinity of DOPR antagonist or agonists, nor did it induce phosphorylation, internalization or down-regulation of the DOPR in the CHO-FLAG-DOPR/HA-D1R cells. Pretreatment of cells with inhibitors of PKA, MEK1 and PI3K, but not PKC, attenuated SKF-82958-induced desensitization of the DOPR. The D1R agonist SKF-82958 enhanced phosphorylation of ERK1/2, and pretreatment with inhibitors of MEK1 and PI3K, but not PKA and PKC reduced the effect. These results indicate that activation of ERK1/2 and/or PKA, but not PKC, is involved in D1 receptor-induced heterologous desensitization of the DOPR.
Significance
This study provides possible mechanisms underlying D1R activation-induced DOPR desensitization.
Keywords: Heterologous desensitization, delta opioid receptor, dopamine D1 receptor, PKA, ERK1/2
Introduction
Dopaminergic system interacts with the opioid system, and together they collectively modulate emotional, locomotor, and goal-directed behaviors. These two systems interact at functional, cellular and anatomical levels. In vivo administration of dopaminergic drugs has profound effects on the expression and function of opioid receptors. For example, chronic exposure to cocaine, a non-selective indirect dopaminergic agonist, upregulates mu opioid receptors in brains of rodents and humans (Unterwald et al., 1992; Zubieta et al., 1996). Similar results are found with chronic administration of selective D2 receptor agonist quinpirole (Chen et al., 1993). Kappa opioid receptor binding is also increased after chronic cocaine exposure in rodents and humans (Hurd and Herkenham, 1993; Unterwald et al., 1994). Of note, the brain regions that show the largest increases in opioid receptor levels are those that are innervated by dopaminergic terminals. While repeated cocaine administration increases mu and kappa opioid receptors, neither cocaine (Unterwald et al., 1994; Azaryan et al., 1996) nor selective dopamine receptor agonists (Chen et al., 1993) produce changes in DOPR binding or mRNA levels. Despite stable receptor levels, significant desensitization of DOPR signaling has been demonstrated (Unterwald et al., 1993; Unterwald and Cuntapay, 2000).
Chronic administration of cocaine in rats attenuates DOPR-induced inhibition of adenylyl cyclase activity in striatum (Unterwald et al., 1993), but does not change the number of the DOPR or the levels of four major G proteins (Unterwald et al., 1994; Perrine et al., 2005). DOPR desensitization is mediated by the indirect effects of cocaine on dopamine D1 receptors. Chronic administration of the D1R agonist SKF-82958, but not the D2 receptor agonist quinpirole, attenuates DOPR-induced stimulation of [35S]GTPγS binding, indicative of decreased coupling of DOPR with G proteins (Unterwald and Cuntapay, 2000). On an anatomical level, both D1R and DOPR are found in high levels in the striatum (Mansour et al., 1988; Wamsley et al., 1989) and, more recently, D1R and DOPR have been shown by immunoelectron microscopy to co-exist in individual neurons in a subset of rat dorsolateral striatal and accumbens neurons (Ambrose et al., 2006; Ambrose-Lanci et al., 2008). Thus, it is likely that D1R-mediated desensitization of the DOPR occurs at the cellular level in individual neurons.
The functional consequences of cocaine-induced DOPR desensitization have also been investigated. Genetic deletion of DOPR produces a phenotype of increased anxiety- and depression-like behaviors (Filliol et al., 2000), demonstrating the importance of this receptor in regulating mood and emotional behaviors. Chronic administration of cocaine in rats attenuates DOPR signaling with increasing anxiety- and depression-like behaviors (Perrine et al., 2008). The anxiety- and depression-like responses were reduced by acute administration of the selective DOPR agonist SNC80 (Perrine et al., 2008). In addition, abuse of and withdrawal from cocaine in humans causes psychiatric disorders, including psychosis, depression and anxiety (Kampman et al., 2001), and cocaine-induced anxiety and depression are frequently resistant to treatment with classical anxiolytics and antidepressants suggesting a novel pathology.
Although extensive evidence indicates interactions of these two systems, the molecular mechanisms underlying cocaine or D1R agonist-induced desensitization of the DOPR has not yet been clarified. The complexity and heterogeneity of neurons in the brain preclude the use of brain tissues for such investigations. In this study, we examined the molecular mechanisms underlying the D1R-mediated heterologous desensitization of the DOPR in clonal mouse neuroblastoma × rat glioma hybrid (NG108-15) cells stably transfected with D1R and in CHO cells stably transfected with both the DOPR and D1R.
Materials and methods
Materials
[3H]Diprenorphine (58 Ci/mmol), [3H]SCH23390 (91 Ci/mmol), [32P]orthophosphate (8500–9100 Ci/mmol) and [35S]GTPγS (1000–1200 Ci/mmol) were purchased from PerkinElmer Life Sciences (Boston, MA). SNC-80 ((+)-4-[(aR)-a-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide) was provided by National Institute on Drug Abuse (Bethesda, MD). Rabbit polyclonal antibody against the FLAG epitope, GDP, GTPγS, NaF, Na pyrophosphate and kinase inhibitors [PD98059, wortmannin, GF1092036X and Rp-adenosine 3′,5′-cyclic monophosphorothioate (Rp-cAMP-SH)] were obtained from Sigma-Aldrich (St. Louis, MO). The following compounds or reagents were purchased from indicated companies: Geneticin, Mediatech (Herndon, VA); Lipofectamine 2000, Opti-MEM I reduced serum and hygromycin B, Invitrogen (Carlsbad, CA); Naloxone HCl, (±)-chloro-APB HBr (SKF-82958) and fluphenazine dihydrochloride, Research Biochemicals International (Natick, MA); Calyculin A and anti-pERK1/2 antibody, Cell Signaling Technology (Danvers, MA); DPDPE, Chiron Mimotopes Peptide Systems (San Diego, CA); Pansorbin, Calbiochem (San Diego, CA); Complete protease inhibitor cocktail, Roche Diagnostics (Indianapolis, IN).
Generation of the clonal NG 108-15 cells stably expressing HA-rat D1 dopamine receptor (HA-D1R)
HA-D1R in HA-D1R/pCMV5 (a generous gift from Dr. Marc G. Caron of Duke University) was subcloned into the HindIII and XbaI sites of the mammalian expression vector pcDNA3.1 (invitrogen, with hygromycin resistant gene). HA epitope tag is located 5′ to the initiation codon of the D1R. NG108-15 mouse neuroblastoma × rat glioma hybrid cells endogenously expressing ~0.6 pmol DOPR/mg membrane protein were transiently transfected with 10 μg HA-D1R in pcDNA 3.1 using Lipofectamine 2000 reagent. After two days, cells were cultured in Dulbecco’s modified Eagle’s medium F12 HAM supplemented with 0.5 mg/ml hygromycin B, 10% fetal calf serum, 100 units/ml penicillin, 100 μg/ml streptomycin and HAT media supplement for 2 weeks. Then cells were counted, diluted and cultured in 96-well plates at an average of 1 cell/well. The wells containing single cells were monitored. Clonal cells were cultured for about another 2 weeks and transferred into 24-well plates and grown until confluent. [3H]diprenorphine binding (for the native DOPR) and [3H]SCH23390 binding (for the D1R) were performed on intact cells. The cells expressing both DOPR and D1R were selected for the further study.
Generation of clonal CHO cells stably expressing both FLAG-mouse DOPR (FLAG-mDOPR) and HA-D1R
Clonal CHO cells stably expressing FLAG-mDOPR/pcDNA3 (geneticin resistant) established as described previously (Chen et al., 1995) were transfected with HA-D1R/pcDNA 3.1 (hygromycin resistant) and the clonal CHO cells stably expressing both FLAG-mDOPR/pcDNA3 and HA-D1R/pcDNA 3.1 were generated and selected with 0.5 mg/ml of geneticin and hygromycin B using the similar procedures. FLAG epitope tag is located 5′ to the initiation codon of the DOPR. The selected double clonal CHO-FLAG-DOPR/HA-D1R cells were cultured in Dulbecco’s modified Eagle’s medium F12 HAM supplemented with 0.1 mg/ml geneticin and hygromycin B, 10% fetal calf serum, 100 U/ml penicillin and 100 μg/ml streptomycin in a humidified atmosphere consisting of 5% CO2 and 95% air at 37 °C. Clonal cell lines stably expressing about ~2 pmol/mg protein of each receptor were used to study the molecular mechanism by which dopamine D1R activation results in desensitization of DOPR function.
Saturation and competition binding assays
CHO cells stably expressing FLAG-DOPR/HA-D1R were pretreated with vehicle, DPDPE 10 μM or SKF-82958 10 μM in Dulbecco’s modified Eagle’s F12 HAM medium without serum for 30 min in a humidified 5% CO2 incubator at 37°C. The cells were then collected and washed twice with cold PBS containing calyculin A 10 nM and membranes were prepared as described previously (Xu et al., 1999) in the presence of 10 mM NaF and 10 mM Na pyrophosphate to inhibit phosphatases Saturation binding of [3H]diprenorphine or [3H]SCH23390 to the NG108-15-/HA- D1R or the CHO-FLAG-DOPR/HA-D1R was performed with at least six concentrations of [3H]diprenorphine or [3H]SCH23390 (ranging from 25 pM to 2 nM), and Kd and Bmax values were determined with the Prism program (GraphPad Software Inc., San Diego, CA). Binding was carried out in 50 mM Tris-HCl buffer containing 1 mM EGTA (pH 7.4) at room temperature for 1 h in duplicate in a volume of 1 mL with ~10–20 μg of membrane protein. Naloxone (for DOPR) or fluphenazine (for D1R) (10 μM) was used to define nonspecific binding. Competitive inhibition of [3H]diprenorphine binding by drugs was performed with [3H]diprenorphine at a concentration close to its Kd value (~0.3 nM). Binding data were analyzed and Ki values were determined with GraphPad Prism software.
Effect of SKF-82958 treatment on DOPR-mediated [35S]GTPγS binding
Clonal NG108-15-HA-D1R or CHO-FLAG-DOPR/HA-D1R cells were pre-incubated in Dulbecco’s modified Eagle’s F12 HAM medium without serum for overnight. Clonal cells were pretreated with vehicle, DPDPE 10 μM, or SKF-82958 10 μM for 30 min in a humidified 5% CO2 incubator at 37°C. Clonal cells were then collected and washed three times on ice with ice-cold phosphate-buffered saline (PBS)(pH 7.0) containing calyculin A 10 nM, and membranes were prepared in the presence of 10 mM NaF and 10 mM Na pyrophosphate to inhibit phosphatases. [35S]GTPγS binding was performed as described previously (Zhu et al., 1997). Briefly, membranes (containing 10 μg of proteins) were incubated with 10 μM GDP and ~0.2 nM [35S]GTPγS in the presence or absence of different concentration of DPDPE in a reaction buffer (50 mM HEPES, 100 mM NaCl, 5 mM MgCl2, 1 mM EDTA) in a final volume of 0.5 ml. Nonspecific binding was determined in the presence of 10 μM GTPγS. After 60 min of incubation at 30°C, bound and free [35S]GTPγS were separated by filtration with GF/B filters under reduced pressure and the filter was washed. Radioactivity in filters was determined by liquid scintillation counting. Data were analyzed and EC50 values for potency and Emax values for efficacy were determined with GraphPad Prism software.
For the experiments to determine effects of kinase inhibitors on SKF-82958-induced desensitization of DOPR, CHO-FLAG-DOPR/HA-D1R cells were pretreated with vehicle, the PKA inhibitor Rp-cAMP-SH (10 μM), the PKC inhibitor GF 109203X (10 μM), PI3K inhibitor wortmannin (100 nM) and MEK1 inhibitor PD98059 (10 μM) for 20 min, respectively, and then incubated with vehicle or SKF-82958 (10 μM) for 30 min in the presence of calyculin A (10 nM). [35S]GTPγS binding was performed as described above.
Effect of SKF-82958 treatment on phosphorylation of the DOPR
CHO-FLAG-DOPR/HA-D1R cells were transferred into 60-mm dishes, cultured overnight to confluence and grown in 1 ml/well phosphate-free medium at 37°C for 2 h. [32P]Orthophosphate (0.25 mCi/well) was added and incubated for another 2 h, and medium was aspirated. Cells were incubated with 10 μM DPDPE or 10 μM SKF-82958 for 30 min at 37°C, solubilized for 1 h with solubilization buffer (1% Triton X-100, 50 mM Tris HCl, 150 mM NaCl, 1 mM EDTA, 20 nM calyculin A, and 10% complete protease inhibitor cocktail (pH7.5) and centrifuged at 100,000g for 1 h. Immunoprecipitation of FLAG-DOPR was performed with rabbit anti-FLAG polyclonal antibody followed by Pansorbin. Immunoprecipitated materials were resolved with SDS-PAGE followed by gel drying and autoradiography. Quantitation of receptor phosphorylation was performed with the OptiQuant software program.
Effect of SNC-80 or SKF-82958 treatment on cell surface and total DOPR
Internalized receptors were assessed as we described previously (Li et al., 1999). CHO-FLAG-DOPR/HA-D1R cells were incubated with DOPR agonist SNC-80 (100 nM) or D1 R agonist SKF-82958 (10 μM) for 30 min (for internalization) or 24 h (for downregulation) at 37°C and washed three times on ice with cold PBS. Binding was performed on intact cells with [3H]diprenorphine in Kreb’s buffer solution (130 mM NaCl, 4.8 mM KCl, 1.2 mM KH2PO4, 1.3 mM CaCl2, 1.2 mM MgSO4, 10 mM glucose, and 25 mM HEPES at pH 7.4). Total receptor levels were assessed by binding with 1nM [3H]diprenorphine in the presence or absence of 10 μM naloxone, whereas surface receptors were measured by binding with 1nM [3H]diprenorphine in the presence or absence of 10 μM DPDPE. Binding was performed at 4°C for 3 h. While diprenorphine, which is a hydrophobic ligand, can bind to both cell surface and intracellular receptors, DPDPE, a hydrophilic peptide ligand, binds only to the cell surface receptors. Thus, the difference between the total receptors and the cell surface receptors represents intracellular receptors.
Immunoblot analysis of phosphorylated ERK1/2
CHO-FLAG-DOPR/HA-D1R cells were cultured in 12-well plate in MEM complete medium for 24 h, washed and incubated with OPTI-MEM for 2 h. Kinase Inhibitors (see Fig. 5) were added to the cells and pre-incubated for 20 min followed by stimulation of D1R with SKF-82958 for 30 min. The reaction was stopped by washing the cells with PBS followed by the addition of 200 μl/well SDS loading buffer to lyses cells. Cell lysates were boiled for 5 min and 50 μl each of the samples (20 μg total protein/lane) was loaded to 10 % SDS-PAGE for separation. Immunoblotting was performed with anti-pERK1/2 antibody (1:5000).
Figure 5. Effects of pretreatment of kinase inhibitors on SKF-82958-induced ERK phosphorylation.
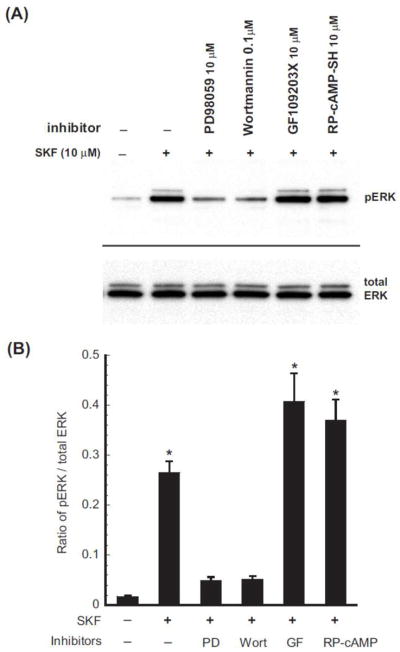
CHO-FLAG-mDOPR/HA-D1R cells were pretreated with the PKC inhibitor GF 109203X), the MEK1 inhibitor PD 98059, the PI3K inhibitor wortmannin or the PKA inhibitor Rp-cAMP-SH at indicated concentrations for 20 min, and then vehicle or 10 μM SKF-82958 for 30 min. Cells were dissolved in 200 μl 4x SDS loading buffer, and 50 μl/lane loaded and resolved by 10% PAGE. Rabbit anti-pERK1/2 was used for ERK phosphorylation. This represents one of the three experiments performed.
Statistical Analysis
For comparison of more than two groups, data were analyzed by one-way analysis of variance (ANOVA) followed by Dunnett’s t test using Prism 3.0 (GraphPad Software, Inc., San Diego, CA). For comparison of two groups, Student’s t test was performed. P < 0.05 was set as the level of significance in all statistical analyses.
RESULTS
Characterization of D1R and DOPR in clonal NG 108-15-HA-D1R cells and CHO-FLAG-DOPR/HA-D1R cells
Receptor expression
Saturation binding of [3H]diprenorphine to the DOPR or [3H]SCH23390 to the D1R in cell membranes was performed to determine binding affinities and receptor levels. Kd and Bmax values of [3H]diprenorphine binding to the endogenous DOPR in NG 108-15-HA-D1R cells were 0.27 ± 0.03 nM and 0.77 ± 0.09 pmol/mg protein, respectively, which are similar to those of the untransfected NG 108-15 cells (Huang et al., 2007). Kd and Bmax values of [3H]SCH23390 binding to the D1R were 1.66 ± 0.58 nM and 180 ± 34 fmol/mg protein, respectively.
Stable expression of the D1R in NG108-15 cells was difficult to maintain; therefore, we established clonal CHO cells stably transfected with the mouse DOPR and the rat D1R (CHO-FLAG-DOPR/HA-D1R cells) for mechanistic studies. Similar saturation binding was performed. Kd and Bmax values of [3H]diprenorphine binding to the DOPR were determined to be 0.40 ± 0.04 nM and 2.78 ± 0.08 pmol/mg protein (n=3), respectively. Kd and Bmax values of [3H]SCH23390 binding to the D1R were calculated to be 0.10± 0.02 nM and 2.17±0.14 pmol/mg protein, respectively. The Kd values are similar to published data (Monsma, Jr. et al., 1990; Zhu et al., 1997).
Receptor signaling
We then carried out functional assays to ensure proper receptor signaling. We found that the DOPR agonist DPDPE dose-dependently increased [35S]GTPγS binding to membranes of NG108-15/HA-D1R cells and CHO-FLAG-DOPR/HA-D1R cells (Fig. 1A and 1B). EC50 values of DPDPE were determined to be 59.9 ± 15.1 nM and 47.0 ± 10.9 nM, respectively (Table 1 and 2). In addition, the D1R agonist SKF-82958 dose-dependently increased cAMP level in NG108-15/HA-D1R cells and CHO-FLAG-DOPR/HA-D1R cells (data not shown).
Figure 1. Effects of DPDPE and SKF-82958 pretreatment of (A) NG108-15-HA-D1R cells and (B) CHO-FLAG-DOPR/HA-D1R cells on DPDPE-stimulated [35S]GTPγS binding.

NG108-15-HA-D1R cells and CHO-FLAG-DOPR/HA-D1R cells were pretreated with vehicle, DPDPE 10 μM or SKF-82958 10 μM for 30 min at 37°C. Cells were then collected and washed and membranes were prepared. [35S]GTPγS binding to cell membranes was measured in the absence and presence of increasing concentrations of DPDPE. Data are expressed as mean ± s.e.m. of three independent experiments performed in duplicate. EC50 and Emax values are shown in Table 1 (NG108-15-HA-D1R cells) and Table 2 (CHO-FLAG-DOPR/HA-D1R cells).
Table 1. Effects of DPDPE and SKF-82958 pretreatment on EC50 and Emax values of DPDPE in stimulating [35S]GTPγS binding to membranes of NG 108-15-HA-D1R cells.
See Fig. 1 legend. Data are expressed as mean ± s.e.m. of three independent experiments performed in duplicate.
| EC50 (nM) | Emax (fmol/mg protein) | |
|---|---|---|
| Control | 20.9 ± 2.5 | 41.1 ± 1.6 |
| DPDPE | 59.9 ± 15.1* | 15.1 ± 0.2** |
| SKF | 53.9 ± 9.1* | 30.8 ± 0.6** |
P<0.05,
P<0.01, compared to the control by one-way ANOVA, followed by Dunnett’s t test.
Table 2. Effects of DPDPE and SKF-82958 pretreatment on Ki values of DOPR agonists in inhibiting [3H]diprenorphine binding and on EC50 and Emax values of DPDPE in stimulating [35S]GTPγS binding to membranes of CHO-FLAG-DOPR/HA-D1R cells.
See Fig. 1 legend. In addition, competitive inhibition of [3H]diprenorphine by DPDPE and SNC-80 binding was conducted and Ki values were determined. Data are expressed as mean ± s.e.m. of three independent experiments performed in duplicate.
| Pretreatment | Ki (nM) | EC50 (nM) | Emax (fmol/mg protein) | |
|---|---|---|---|---|
| DPDPE | SNC-80 | DPDPE | ||
| Control | 8.5 ± 1.2 | 0.83 ± 0.23 | 39.3 ± 6.5 | 48.3 ± 6.8 |
| DPDPE | 21.8 ± 5.1* | 1.11 ± 0.31 | 47.0 ± 10.9 | 14.3 ± 1.1** |
| SKF | 5.6 ± 0.4 | 0.59 ± 0.16 | 60.9 ± 8.9 | 33.4 ± 2.8* |
P<0.05,
P<0.01, compared to the control by one-way ANOVA, followed by Dunnett’s t test.
Taken together, these results indicate that NG108-15/HA-D1R cells or CHO-FLAG-DOPR/HA-D1R cells can be used as cell models to study mechanisms underlying the interactions between DOPR and D1R.
The D1R agonist SKF-82958 induced heterologous desensitization of endogenous DOPR
NG 108-15-HA-D1R cells
NG 108-15-HA-D1R cells were pretreated with vehicle, 10 μM DPDPE, a selective DOPR agonist, or 10 μM SKF-82958 for 30 min. DPDPE-stimulated [35S]GTPγS binding was performed and its EC50 and Emax values were determined (Fig. 1A, Table 1). Basal [35S]GTPγS binding did not differ among the control and the treatment groups (control, 17.8 ± 2.5 fmol/mg protein; DPDPE-treated, 15.2 ± 2.5 fmol/mg protein; SKF-82958-treated, 14.5 ± 1.9 fmol/mg protein, n=3) and was subtracted from stimulated [35S]GTPγS binding data. Compared with the control group, pretreatment of cells with DPDPE or SKF-82958 significantly decreased the Emax value and increased the EC50 value for DPDPE-stimulated [35S]GTPγS binding (P<0.01 and P<0.05, respectively); however, the decrease in Emax was less with SKF-82958 pretreatment than with DPDPE pretreatment. It is important to demonstrate D1R activation-induced heterologous desensitization of the DOPR in NG108-15 cells, a neuron-like cell line.
CHO-FLAG-DOPR/HA-D1R cells
Similar experiments were performed in CHO-FLAG-DOPR/HA-D1R cells. Cells were pretreated with vehicle, 10 μM DPDPE or 10 μM SKF-82958 for 30 min and DPDPE-stimulated [35S]GTPγS binding was performed on membranes. Basal [35S]GTPγS binding did not differ among the control and the treatment groups (control, 39.0 ± 9.4 fmol/mg protein; DPDPE-treated, 41.9 ± 7.6 fmol/mg protein; SKF-82958-treated, 42.6 ± 7.0 fmol/mg protein, n=3) and was subtracted from the stimulated [35S]GTPγS binding data. Pretreatment with DPDPE or SKF-82958 resulted in a reduction in the Emax value of DPDPE by 70.4% or 30.9%, respectively, with no significant change in its EC50 value (Fig. 1B and Table 2). When [35S]GTPγS binding was performed with the nonpeptide DOPR agonist SNC80 at 10 μM following SKF-82958 pretreatment, [35S]GTPγS binding was reduced by 23.2% ± 5.7% (mean ± s.e.m.) (n=3) compared with the control. Therefore, the responses of both the peptide agonist DPDPE and the nonpeptide agonist SNC80 were attenuated.
Taken together, these results indicate that in addition to homologous desensitization, the DOPR undergoes heterologous desensitization following activation of the D1R, in both NG108-15/HA-D1R cells and in CHO-FLAG-DOPR/HA-D1R cells. Incubation with DPDPE or SKF-82958 resulted in a significant increase in EC50 of DPDPE in NG108-15 cells, but not in CHO cells. This is probably due to the presence of spare DOPRs in the clonal CHO cells.
DPDPE, but not SKF-82958, induced phosphorylation of the DOPR
We examined if D1R activation caused phosphorylation of the DOPR. CHO-FLAG-DOPR/HA-D1R cells were treated with 10 μM DPDPE or 10 μM SKF-82958 for 30 min. SKF-82958 did not induce phosphorylation of the DOPR, whereas DPDPE did promote profound DOPR phosphorylation (Fig. 2), indicating that heterologous desensitization of the DOPR induced by D1R agonist does not result from phosphorylation of the DOPR.
Figure 2. DPDPE, but not SKF-82958, induced phosphorylation of the DOPR in CHO-FLAG-mDOPR/HA-D1R cells.

Cells were grown in 1 ml/well phosphate-free medium in 6-well plates at 37°C for 2 h. [32P]Orthophosphate (0.25 mCi/well) was added and incubated for another 2 h, and medium was aspirated. Cells were incubated with vehicle, 10 μM DPDPE or 10 μM SKF-82958 for 30 min at 37°C, solubilized for 1 h with solubilization buffer and centrifuged at 100,000g for 1 h. Immunoprecipitation of FLAG-mDOPR was performed with rabbit anti-FLAG polyclonal antibody followed by Pansorbin. Immunoprecipitated materials were resolved with SDS-PAGE followed by gel drying and autoradiography. This represents one of the three experiments performed. The 54-KDa band is the DOPR.
Pretreatment with SKF-82958 for 30 min did not affect affinity or level of the DOPR
CHO-FLAG-DOPR/HA-D1R cells were pretreated with vehicle, 10 μM DPDPE or 10 μM SKF-82958 for 30 min. Saturation binding of [3H]diprenorphine to the DOPR and [3H]SCH23390 binding to the D1R were performed and Kd and Bmax values were calculated. The data summarized in Table 3 shows that pretreatment with DPDPE or SKF-82958 did not affect Kd and Bmax values of [3H]diprenorphine binding to the DOPR or [3H]SCH23390 binding to the D1R. Since diprenorphine is an antagonist, we also determined if affinities of the agonists SNC-80 and DPDPE to the DOPR were affected using competitive inhibition of [3H]diprenorphine binding. While pretreatment with DPDPE increased the Ki value of DPDPE, but not of SNC-80, pretreatment with SKF-82958 did not affect Ki values of either agonist (Table 2). These results indicate that the observed heterologous desensitization induced by the D1R agonist is not due to changes in affinity or expression level of the DOPR.
Table 3. Effects of DPDPE and SKF-82958 pretreatment on Kd and Bmax values of [3H]diprenorphine binding and [3H]SCH23390 binding to the CHO-FLAG-hDOPR/HA-D1R.
Cells were treated with vehicle, DPDPE 10 μM or SKF-82958 10 μM for 30 min, collected and washed and membranes were prepared. Saturation binding of [3H]diprenorphine to FLAG-hDOR/HA-D1R was performed and Kd and Bmax values were calculated as described in the Method. Data are expressed as mean ± s.e.m. of three independent experiments performed in duplicate.
| [3H]Diprenorphine | [3H]SCH23390 | |||
|---|---|---|---|---|
| Kd (nM) | Bmax (pmol/mg protein) | Kd (nM) | Bmax (fmol/mg protein) | |
| Control | 0.40 ± 0.04 | 2.78 ± 0.08 | 0.10 ± 0.02 | 2.17 ± 0.14 |
| DPDPE | 0.60 ± 0.17 | 2.50 ± 0.10 | 0.08 ± 0.01 | 2.04 ± 0.12 |
| SKF | 0.26 ± 0.05 | 2.65 ± 0.45 | 0.10 ± 0.02 | 2.54 ± 0.88 |
Effects of pretreatment of SNC-80 or SKF-82958 for 30 min or 24 h on DOPR trafficking
CHO-FLAG-DOPR/HA-D1R cells were pretreated with vehicle, the DOPR agonist SNC-80 (100 nM) or the D1R agonist SKF-82958 (10 μM) for 30 min or 24 h. Because of stability issue, the non-peptide selective DOPR agonist SNC-80 was used instead of the peptide agonist DPDPE. Total and cell surface DOPRs were determined by [3H]diprenorphine binding. Pretreatment with SKF-82958 for 30 min or 24 h did not affect total or cell surface DOPRs (Fig. 3A and 3B). In contrast, pretreatment with the DOPR agonist SNC-80 for 30 min significantly reduced cell surface DOPR, without affecting the total DOPR, indicating DOPR internalization (Fig. 3A). In vehicle-treated cells, 76.0 ± 2.6% (n=3) of the total DOPR was present on cell surface.
Figure 3. Effects of SKF-82958 treatment on total and cell surface DOPR in CHO-FLAG-mDOPR/HA-D1R cells.

Cells were pretreated with vehicle, 100 nM SNC-80 or 10 μM SKF-82958 for 30 min (A) or 24 h (B) as described in Methods. Cells were then chilled and washed and [3H]diprenorphine (~1nM) binding was carried out on intact cells. For total and cell-surface receptors, nonspecific binding was defined as the binding in the presence of 10 μM naloxone and 10 μM DPDPE, respectively. Data were normalized against vehicle-treated cells (100%). The data represent the mean ± s.e.m. of three independent experiments performed in duplicate.
Pretreatment with SNC-80 for 24 h reduced the total DOPR to 19 ± 1.4% of the control, indicating DOPR downregulation (Fig. 3B). These results demonstrate that the heterologous desensitization induced by the D1R agonist is not related to internalization and downregulation of DOPR.
Multiple downstream signaling pathways underlying the D1 receptor-induced heterologous desensitization of DOPR
Heterologous desensitization of seven-transmembrane receptors (7TMRs) often involves kinase activity. Activation of the D1R has been shown to activate PKA, PKC and extracellular signal-regulated kinase 1/2 (ERK1/2) (Flores-Hernandez et al., 2000; Chao et al., 2002a; Chao et al., 2002b; Zhang et al., 2004; Mangiavacchi and Wolf, 2004; Chen et al., 2004b; Hopf et al., 2005; Rex et al., 2010; Rankin and Sibley, 2010). To examine if these kinases are involved in D1R-induced desensitization of the DOPR, CHO-FLAG-DOPR/HA-D1R cells were pretreated for 20 min with vehicle, the PKA inhibitor Rp-cAMP-SH (10 μM), the PKC inhibitor GF 109203X (10 μM), the PI3K inhibitor wortmannin (100 nM) or the MEK1 inhibitor PD98059 (10 μM), and then incubated with vehicle or SKF-82958 (10 μM) for 30 min in the presence of the phosphatase inhibitor calyculin A (10 nM). The concentration of each inhibitor has been reported in the literature to produce significant kinase inhibition (Toullec et al., 1991; Horvath and DeLeo, 2009; Machida et al., 2011; Allen et al., 2011). Membranes were prepared and DPDPE-induced [35S]GTPγS binding was performed and EC50 and Emax values were determined. Basal [35S]GTPγS binding did not differ among the control groups and all the treatment groups and was subtracted from the agonist-induced [35S]GTPγS binding. The PKA inhibitor Rp-cAMP-SH, the PI3K inhibitor wortmannin and MEK1 inhibitor PD98059 significantly inhibited D1R activation-induced desensitization of the DOPR (Table 4 and Fig. 4A, 4B, 4D and 4E). In contrast, the PKC inhibitor GF 109203X did not affect the cross desensitization (Table 4 and Fig. 4A and 4C). These results demonstrate that PKA, PI3K and ERK1/2, but not PKC, signaling pathways play important roles in the heterologous desensitization of DOPR by D1R activation.
Table 4. Effects of pretreatment with kinase inhibitors on SKF-82958-induced desensitization of DOPR in CHO-FLAG-hDOPR/HA-D1R cells.
EC50 and Emax values of DPDPE in stimulating [35S]GTPγS binding to membranes of CHO-FLAG-hDOPR/HA-D1R cells pretreated with or without the inhibitors were calculated. Each value represents the mean ± s.e.m. of three to four independent experiments performed in duplicate.
| EC50 (nM) | Emax (fmol/mg protein) | ||
|---|---|---|---|
| Control | vehicle | 20.3 ± 2.1 | 59.6 ± 5.9 |
| SKF | 35.3 ± 10.7 | 31.6 ± 3.6* | |
|
| |||
| PKC inhibitor GF 109203X | vehicle | 20.3 ± 6.5 | 57.2 ± 3.8 |
| SKF | 16.7 ± 4.1 | 34.3 ± 4.7* | |
|
| |||
| MEK1 inhibitor PD 98059 | vehicle | 22.6 ± 6.0 | 57.9 ± 4.4 |
| SKF | 22.2 ± 5.1 | 60.5 ± 4.2 | |
|
| |||
| PI3K inhibitor wortmannin | vehicle | 17.1 ± 2.9 | 60.5 ± 4.7 |
| SKF | 21.3 ± 8.8 | 47.5 ± 3.2 | |
|
| |||
| PKA inhibitor Rp-cAMP-SH | vehicle | 20.3 ± 2.1 | 66.8 ± 7.8 |
| SKF | 17.5 ± 4.8 | 56.9 ± 3.1 | |
P<0.05, compared to the vehicle control by two tailed t test.
Figure 4. Effects of pretreatment of kinase inhibitors on SKF-82958-induced heterologous desensitization of DOPR.

CHO-FLAG-mDOPR/HA-D1R cells were pretreated with the PKC inhibitor GF109203X (10 μM), the MEK1 inhibitor PD 98059 (10 μM), the PI3K inhibitor wortmannin (100 nM) or the PKA inhibitor Rp-cAMP-SH (10 μM) for 20 min, and then vehicle or 10 μM SKF-82958 and 10 nM calyculin A for 30 min. Membranes were prepared. [35S]GTPγS binding to cell membranes was measured in the presence of increasing concentrations of DPDPE. Each point represents the mean ± s.e.m. of three to four independent experiments performed in duplicate. EC50 and Emax values are shown in Table 4.
We next investigated whether the involvements of PKA, PI3K and ERK1/2 were through ERK1/2 phosphorylation. As shown as Figure 5, treatment with 10 μM SKF-82958 for 30 min produced a marked increase in ERK1/2 phosphorylation compared with the control. In addition, pretreatment of 10 μM PD98059 (MEK1 inhibitor) and 0.1 μM wortmannin (PI3K inhibitor), but not 10 μM GF109203 (PKC inhibitor) and 10 μM Rp-cAMP-SH (PKA inhibitor), significantly reduced the increase, indicating that MEK1 and PI3K, but not PKC and PKA, pathways were involved in ERK1/2 activation by D1R. Therefore, inhibition of cross desensitization by MEK1 and PI3K inhibitors, but not PKA inhibitor, is likely due to inhibition of ERK1/2 phosphorylation. Taken together, these data indicate that ERK1/2 and PKA downstream signaling pathways underlie the D1 receptor-induced heterologous desensitization of DOPR.
Discussion
The present study demonstrates heterologous desensitization of the DOPR by activation of the D1R in both NG 108-15/HA-D1R cells and CHO/FLAG-DOPR/HA-D1R cells. This heterologous desensitization of the DOPR appears to be associated with D1R-activated ERK1/2 and PKA signaling pathways, but does not involve changes in ligand binding affinity, phosphorylation, internalization or downregulation of the DOPR. These results provide some mechanistic insights for our previous findings that chronic administration of cocaine or a D1R agonist in the rat attenuates DOPR signaling in the rostral caudate putamen and nucleus accumbens (Unterwald et al., 1993; Unterwald and Cuntapay, 2000). In addition, these observations provide evidence at the cellular level for possible functional consequence of the co-expression of D1R and DOPR in neurons in the dorsolateral striatum and nucleus accumbens (Ambrose et al., 2006; Ambrose-Lanci et al., 2008).
7TMRs undergo desensitization following prolonged or repeated activation. There are two types of desensitization, homologous and heterologous. In homologous desensitization, only the activated receptor is desensitized, whereas in heterologous desensitization, activation of a 7TMR leads to desensitization of another receptor. The mechanisms underlying homologous desensitization of 7TMRs are similar among receptors [for a review, see (Gainetdinov et al., 2004)]. In contrast, the mechanisms underlying heterologous desensitization are more diverse and involve multiple mechanisms, including phosphorylation and internalization of the 7TMRs, changes in G proteins and depletion of downstream second messengers (Hosey, 1999; Gainetdinov et al., 2004)
PKA-dependent and ERK-dependent pathways are involved in the heterologous desensitization of DOPR by D1R
PKA pathway
Dopamine D1-like receptors (D1R and D5R) are coupled to Gs/olf to activate adenylyl cyclase, increase intracellular cAMP and enhance the activity of PKA. The PKA inhibitor Rp-cAMP-SH blocked desensitization of the DOPR by D1R activation, indicating that PKA signaling pathway is involved in the reduced coupling of the DOPR and Gi/o following activation of D1R. D1R/Gs/cAMP/PKA pathway has been shown to regulate the activity of other receptors, for example, AMPA and GABAA receptors (Flores-Hernandez et al., 2000; Chao et al., 2002a; Chao et al., 2002b; Mangiavacchi and Wolf, 2004)
ERK1/2 signaling
MAP kinases play important roles in the transduction of a wide variety of extracellular signals. Activation of D1R was found to activate ERK1/2, but not p38 MAP kinase and c-Jun N-terminal kinases, in SK-N-MC neuroblastoma cells (Zhang et al., 2004; Chen et al., 2004b). We found that the D1R agonist enhanced phosphorylation of ERK1/2, and inhibitors of MEK1 and PI3K reduced the increase in pERK1/2, indicating that the D1R agonist activates ERK1/2 in a MEK1- and PI3K-dependent fashion. The finding that pretreatment with the PI3K inhibitor wortmannin or the MEK1 inhibitor PD98059 reduced the heterologous desensitization of DOPR by the D1R agonist SKF-82958 supports the notion that PI3K and MEK1 are upstream elements of ERK1/2 and involved in the heterologous desensitization of DOPR by D1R.
Since either the PKA or MEK inhibitor totally abolished D1R activation-induced DOPR desensitization and PKA inhibitor did not affect ERK1/2 phosphorylation, we believe that these two pathways leading to DOPR desensitization are independent of each other.
How PKA and ERK1/2 activation by D1R activation leads to DOPR desensitization remains to be determined. Since we observed DOPR desensitization following only 30 min activation of the D1R, it is not likely due to changes at the transcription or translational level. DOPR agonist-induced [35S]GTPγS binding was the functional endpoint; therefore, changes should occur at the receptor or G proteins. However, we found that D1R activation did not enhance DOPR phosphorylation. Phosphorylation and down-regulation of G proteins have been demonstrated to underline heterologous desensitization of GPCRs (for example, (Yatomi et al., 1992; Green et al., 1992; Seasholtz et al., 1997). We observed that D1R activation did not affect phosphorylation or levels of Gαi2 and Gαi3 (data not shown). The lack of alterations in G proteins is consistent with our previous observations that chronic administration of cocaine did not change the levels of four G proteins α subunits (Gi1, Gs, Golf and Go) in the rat striatum (Perrine et al., 2005). In addition, as [35S]GTPγS is not hydrolyzable, the involvement of RGS proteins seems unlikely.
Role of receptor phosphorylation
Our finding that the D1R agonist SKF-82958 did not increase phosphorylation of the DOPR indicates that DOPR phosphorylation is not necessary for heterologous desensitization of DOPR by D1R activation. Heterologous desensitization of 7TMRs may or may not involve receptor phosphorylation [for example, (Grimm et al., 1998; Bundey and Nahorski, 2001; Chen et al., 2004a)].
Role of receptor affinity and trafficking
Our findings that pretreatment of D1R agonist did not change ligand affinity to DOPR (Tables 2 and 3) or induce internalization and down-regulation (Fig. 3) demonstrate that heterologous desensitization of DOPR by D1R activation is not attributable to changes of DOPR affinity or trafficking. Our present results are in accord with our previous in vivo findings (Unterwald et al., 1994) that acute or chronic cocaine treatment does not affect [3H]deltorphin or [3H]DPDPE binding to the DOPR in the striatum or nucleus accumbens (Unterwald et al., 1994; Turchan et al., 1999). Receptor internalization and downregulation have been shown to be associated with (Shapira et al., 2003) or not involved in (Grimm et al., 1998; Szabo et al., 2003) heterologous desensitization of 7TMRs.
Here we found that pretreatment with a D1R agonist for 30 min induced ERK1/2- and PKA-mediated heterologous desensitization of the DOPR, without causing DOPR internalization or down-regulation. Ambrose-Lanci et al. (Ambrose-Lanci et al., 2008) reported that chronic cocaine administration (14 days) followed by 48-h withdrawal resulted in an increase in the percentage of DOPR localized intracellularly in both core and shell of the nucleus accumbens in male rats with no change in DOPR protein expression, indicating DOPR internalization. Perhaps the longer-term activation of D1R, indirectly by cocaine, causes DOPR internalization. Alternatively, activation of D1 receptor by chronic cocaine administration may desensitize the DOPR and lead to a compensatory increase in presynaptic opioid peptides and upon withdrawal the opioid peptides cause DOPR internalization. These results underscore complex interactions between the dopamine receptor and DOPR in NAc.
Conclusion
In summary, these data demonstrate the functional interaction of D1R and DOPR; activation of D1R produced heterologous desensitization of DOPR in two cell lines, similar to the previous findings in neurons. Activation of PKA and ERK1/2 pathways by D1R activation is involved in heterologous desensitization of the DOPR. These data provide important insights into the molecular interactions of dopamine and opioid receptors and shed lights on the underlying mechanism of altered anxiety and mood phenotypes following chronic cocaine exposure.
Acknowledgments
We would like to acknowledge the contribution of Dr. Shane Perrine to this study. This work was supported by funds provided by the NIH grants DA018326 (to EMU), DA17302 (to LLC) and P30 DA13429.
Funding source
This work was supported by funds provided by the NIH grants DA018326 (to EMU), DA17302 (to LLC) and P30 DA13429.
Abbreviations
- CHO cells
Chinese hamster ovary cells
- CHO-FLAG-mDOPR
clonal CHO cells stably transfected with FLAG-mouse δ opioid receptor cDNA in pcDNA3 vector
- DPDPE
[D-Pen2,D-Pen5]-Enkephalin
- CHO-FLAG-DOPR/HA-D1R cells
clonal CHO cells stably transfected with FLAG-DOPR cDNA in pcDNA3 vector and HA-D1R cDNA in pcDNA3.1 vector
- NG108-15-HA-D1R cells
clonal NG108-15 cells stably transfected with HA-D1R cDNA in pcDNA3.1 vector
- Rp-cAMP-SH
Rp-adenosine 3′,5′-cyclic monophosphorothioate
- SNC-80
(+)-4-[(aR)-a-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide
Footnotes
Conflict of interest statement
Authors have no conflicts to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen CE, Worsley MA, King AE, Boissonade FM. Fos expression induced by activation of NMDA and neurokinin-1 receptors in the trigeminal subnucleus caudalis in vitro: role of protein kinases. Brain Res. 2011;1368:19–27. doi: 10.1016/j.brainres.2010.10.072. [DOI] [PubMed] [Google Scholar]
- Ambrose LM, Gallagher SM, Unterwald EM, Van Bockstaele EJ. Dopamine-D1 and delta-opioid receptors co-exist in rat striatal neurons. Neurosci Lett. 2006;399:191–6. doi: 10.1016/j.neulet.2006.02.027. [DOI] [PubMed] [Google Scholar]
- Ambrose-Lanci LM, Peiris NB, Unterwald EM, Van Bockstaele EJ. Cocaine withdrawal-induced trafficking of delta-opioid receptors in rat nucleus accumbens. Brain Res. 2008;1210:92–102. doi: 10.1016/j.brainres.2008.02.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azaryan AV, Coughlin LJ, Buzas B, Clock BJ, Cox BM. Effect of chronic cocaine treatment on mu- and delta-opioid receptor mRNA levels in dopaminergically innervated brain regions. J Neurochem. 1996;66:443–8. doi: 10.1046/j.1471-4159.1996.66020443.x. [DOI] [PubMed] [Google Scholar]
- Bundey RA, Nahorski SR. Homologous and heterologous uncoupling of muscarinic M(3) and alpha(1B) adrenoceptors to Galpha(q/11) in SH-SY5Y human neuroblastoma cells. Br J Pharmacol. 2001;134:257–64. doi: 10.1038/sj.bjp.0704229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao SZ, Ariano MA, Peterson DA, Wolf ME. D1 dopamine receptor stimulation increases GluR1 surface expression in nucleus accumbens neurons. J Neurochem. 2002a;83:704–12. doi: 10.1046/j.1471-4159.2002.01164.x. [DOI] [PubMed] [Google Scholar]
- Chao SZ, Lu W, Lee HK, Huganir RL, Wolf ME. D(1) dopamine receptor stimulation increases GluR1 phosphorylation in postnatal nucleus accumbens cultures. J Neurochem. 2002b;81:984–92. doi: 10.1046/j.1471-4159.2002.00877.x. [DOI] [PubMed] [Google Scholar]
- Chen C, Li J, Bot G, Szabo I, Rogers TJ, Liu-Chen L-Y. Heterodimerization and cross-desensitization between the mu-opioid receptor and the chemokine CCR5 receptor. Eur J Pharmacol. 2004a;483:175–86. doi: 10.1016/j.ejphar.2003.10.033. [DOI] [PubMed] [Google Scholar]
- Chen C, Xue JC, Zhu J, Chen YW, Kunapuli S, de Riel JK, Yu L, Liu-Chen L-Y. Characterization of irreversible binding of beta-funaltrexamine to the cloned rat mu opioid receptor. J Biol Chem. 1995;270:17866–70. doi: 10.1074/jbc.270.30.17866. [DOI] [PubMed] [Google Scholar]
- Chen J, Rusnak M, Luedtke RR, Sidhu A. D1 dopamine receptor mediates dopamine-induced cytotoxicity via the ERK signal cascade. J Biol Chem. 2004b;279:39317–30. doi: 10.1074/jbc.M403891200. [DOI] [PubMed] [Google Scholar]
- Chen JF, Aloyo VJ, Weiss B. Continuous treatment with the D2 dopamine receptor agonist quinpirole decreases D2 dopamine receptors, D2 dopamine receptor messenger RNA and proenkephalin messenger RNA, and increases mu opioid receptors in mouse striatum. Neuroscience. 1993;54:669–80. doi: 10.1016/0306-4522(93)90238-b. [DOI] [PubMed] [Google Scholar]
- Filliol D, Ghozland S, Chluba J, Martin M, Matthes HW, Simonin F, Befort K, Gaveriaux-Ruff C, Dierich A, LeMeur M, Valverde O, Maldonado R, Kieffer BL. Mice deficient for delta- and mu-opioid receptors exhibit opposing alterations of emotional responses. Nat Genet. 2000;25:195–200. doi: 10.1038/76061. [DOI] [PubMed] [Google Scholar]
- Flores-Hernandez J, Hernandez S, Snyder GL, Yan Z, Fienberg AA, Moss SJ, Greengard P, Surmeier DJ. D(1) dopamine receptor activation reduces GABA(A) receptor currents in neostriatal neurons through a PKA/DARPP-32/PP1 signaling cascade. J Neurophysiol. 2000;83:2996–3004. doi: 10.1152/jn.2000.83.5.2996. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107–44. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- Green A, Milligan G, Dobias SB. Gi down-regulation as a mechanism for heterologous desensitization in adipocytes. J Biol Chem. 1992;267:3223–9. [PubMed] [Google Scholar]
- Grimm MC, Ben-Baruch A, Taub DD, Howard OM, Resau JH, Wang JM, Ali H, Richardson R, Snyderman R, Oppenheim JJ. Opiates transdeactivate chemokine receptors: delta and mu opiate receptor-mediated heterologous desensitization. J Exp Med. 1998;188:317–25. doi: 10.1084/jem.188.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf FW, Mailliard WS, Gonzalez GF, Diamond I, Bonci A. Atypical protein kinase C is a novel mediator of dopamine-enhanced firing in nucleus accumbens neurons. J Neurosci. 2005;25:985–9. doi: 10.1523/JNEUROSCI.3099-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath RJ, DeLeo JA. Morphine enhances microglial migration through modulation of P2X4 receptor signaling. J Neurosci. 2009;29:998–1005. doi: 10.1523/JNEUROSCI.4595-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosey MM. What molecular events underlie heterologous desensitization? Focus on “receptor phosphorylation does not mediate cross talk between muscarinic M(3) and bradykinin B(2) receptors” [comment] [Review] [24 refs] Am J Physiol. 1999;277:C856–C858. doi: 10.1152/ajpcell.1999.277.5.C856. [DOI] [PubMed] [Google Scholar]
- Huang P, Xu W, Yoon SI, Chen C, Chong PL, Liu-Chen LY. Cholesterol reduction by methyl-beta-cyclodextrin attenuates the delta opioid receptor-mediated signaling in neuronal cells but enhances it in non-neuronal cells. Biochem Pharmacol. 2007;73:534–49. doi: 10.1016/j.bcp.2006.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd YL, Herkenham M. Molecular alterations in the neostriatum of human cocaine addicts. Synapse. 1993;13:357–69. doi: 10.1002/syn.890130408. [DOI] [PubMed] [Google Scholar]
- Kampman KM, Volpicelli JR, Mulvaney F, Alterman AI, Cornish J, Gariti P, Cnaan A, Poole S, Muller E, Acosta T, Luce D, O’Brien C. Effectiveness of propranolol for cocaine dependence treatment may depend on cocaine withdrawal symptom severity. Drug Alcohol Depend. 2001;63:69–78. doi: 10.1016/s0376-8716(00)00193-9. [DOI] [PubMed] [Google Scholar]
- Li J-G, Luo LY, Krupnick JG, Benovic JL, Liu-Chen L-Y. U50, 488H-induced internalization of the human kappa opioid receptor involves a beta-arrestin- and dynamin-dependent mechanism. Kappa receptor internalization is not required for mitogen-activated protein kinase activation. J Biol Chem. 1999;274:12087–94. doi: 10.1074/jbc.274.17.12087. [DOI] [PubMed] [Google Scholar]
- Machida T, Ohta M, Onoguchi A, Iizuka K, Sakai M, Minami M, Hirafuji M. 5-Hydroxytryptaime induces cyclooxygenase-2 in rat vascular smooth muscle cells: Mechanisms involving Src, PKC and MAPK activation. Eur J Pharmacol. 2011;656:19–26. doi: 10.1016/j.ejphar.2010.12.044. [DOI] [PubMed] [Google Scholar]
- Mangiavacchi S, Wolf ME. D1 dopamine receptor stimulation increases the rate of AMPA receptor insertion onto the surface of cultured nucleus accumbens neurons through a pathway dependent on protein kinase A. J Neurochem. 2004;88:1261–71. doi: 10.1046/j.1471-4159.2003.02248.x. [DOI] [PubMed] [Google Scholar]
- Mansour A, Khachaturian H, Lewis ME, Akil H, Watson SJ. Anatomy of CNS opioid receptors. Trends Neurosci. 1988;11:308–14. doi: 10.1016/0166-2236(88)90093-8. [DOI] [PubMed] [Google Scholar]
- Monsma FJ, Jr, Mahan LC, McVittie LD, Gerfen CR, Sibley DR. Molecular cloning and expression of a D1 dopamine receptor linked to adenylyl cyclase activation. Proc Natl Acad Sci U S A. 1990;87:6723–7. doi: 10.1073/pnas.87.17.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrine SA, Schroeder JA, Unterwald EM. Behavioral sensitization to binge-pattern cocaine administration is not associated with changes in protein levels of four major G-proteins. Brain Res Mol Brain Res. 2005;133:224–32. doi: 10.1016/j.molbrainres.2004.10.025. [DOI] [PubMed] [Google Scholar]
- Perrine SA, Sheikh IS, Nwaneshiudu CA, Schroeder JA, Unterwald EM. Withdrawal from chronic administration of cocaine decreases delta opioid receptor signaling and increases anxiety- and depression-like behaviors in the rat. Neuropharmacol. 2008;54:355–64. doi: 10.1016/j.neuropharm.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin ML, Sibley DR. Constitutive phosphorylation by protein kinase C regulates D1 dopamine receptor signaling. J Neurochem. 2010;115:1655–67. doi: 10.1111/j.1471-4159.2010.07074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex EB, Rankin ML, Yang Y, Lu Q, Gerfen CR, Jose PA, Sibley DR. Identification of RanBP 9/10 as interacting partners for protein kinase C (PKC) gamma/delta and the D1 dopamine receptor: regulation of PKC-mediated receptor phosphorylation. Mol Pharmacol. 2010;78:69–80. doi: 10.1124/mol.110.063727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seasholtz TM, Gurdal H, Wang HY, Cai G, Johnson MD, Friedman E. Heterologous desensitization of the rat tail artery contraction and inositol phosphate accumulation after in vitro exposure to phenylephrine is mediated by decreased levels of Galphaq and Galphai. J Pharmacol Exp Ther. 1997;283:925–31. [PubMed] [Google Scholar]
- Shapira M, Gafni M, Sarne Y. Long-term interactions between opioid and cannabinoid agonists at the cellular level: cross-desensitization and downregulation. Brain Res. 2003;960:190–200. doi: 10.1016/s0006-8993(02)03842-8. [DOI] [PubMed] [Google Scholar]
- Szabo I, Wetzel MA, Zhang N, Steele AD, Kaminsky DE, Chen C, Liu-Chen L-Y, Bednar F, Henderson EE, Howard OM, Oppenheim JJ, Rogers TJ. Selective inactivation of CCR5 and decreased infectivity of R5 HIV-1 strains mediated by opioid-induced heterologous desensitization. J Leukoc Biol. 2003;74:1074–82. doi: 10.1189/jlb.0203067. [DOI] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–81. [PubMed] [Google Scholar]
- Turchan J, Przewlocka B, Toth G, Lason W, Borsodi A, Przewlocki R. The effect of repeated administration of morphine, cocaine and ethanol on mu and delta opioid receptor density in the nucleus accumbens and striatum of the rat. Neuroscience. 1999;91:971–7. doi: 10.1016/s0306-4522(98)00637-x. [DOI] [PubMed] [Google Scholar]
- Unterwald EM, Cox BM, Kreek MJ, Cote TE, Izenwasser S. Chronic repeated cocaine administration alters basal and opioid-regulated adenylyl cyclase activity. Synapse. 1993;15:33–8. doi: 10.1002/syn.890150104. [DOI] [PubMed] [Google Scholar]
- Unterwald EM, Cuntapay M. Dopamine-opioid interactions in the rat striatum: a modulatory role for dopamine D1 receptors in delta opioid receptor-mediated signal transduction. Neuropharmacol. 2000;39:372–81. doi: 10.1016/s0028-3908(99)00154-9. [DOI] [PubMed] [Google Scholar]
- Unterwald EM, Horne-King J, Kreek MJ. Chronic cocaine alters brain mu opioid receptors. Brain Res. 1992;584:314–8. doi: 10.1016/0006-8993(92)90912-s. [DOI] [PubMed] [Google Scholar]
- Unterwald EM, Rubenfeld JM, Kreek MJ. Repeated cocaine administration upregulates kappa and mu, but not delta, opioid receptors. Neuroreport. 1994;5:1613–6. doi: 10.1097/00001756-199408150-00018. [DOI] [PubMed] [Google Scholar]
- Wamsley JK, Gehlert DR, Filloux FM, Dawson TM. Comparison of the distribution of D-1 and D-2 dopamine receptors in the rat brain. J Chem Neuroanat. 1989;2:119–37. [PubMed] [Google Scholar]
- Xu W, Ozdener F, Li J-G, Chen C, de Riel JK, Weinstein H, Liu-Chen L-Y. Functional role of the spatial proximity of Asp114(2.50) in TMH 2 and Asn332(7. 49) in TMH 7 of the mu opioid receptor. FEBS Lett. 1999;447:318–24. doi: 10.1016/s0014-5793(99)00316-6. [DOI] [PubMed] [Google Scholar]
- Yatomi Y, Arata Y, Tada S, Kume S, Ui M. Phosphorylation of the inhibitory guanine-nucleotide-binding protein as a possible mechanism of inhibition by protein kinase C of agonist-induced Ca2+ mobilization in human platelet. Eur J Biochem. 1992;205:1003–9. doi: 10.1111/j.1432-1033.1992.tb16867.x. [DOI] [PubMed] [Google Scholar]
- Zhang L, Lou D, Jiao H, Zhang D, Wang X, Xia Y, Zhang J, Xu M. Cocaine-induced intracellular signaling and gene expression are oppositely regulated by the dopamine D1 and D3 receptors. J Neurosci. 2004;24:3344–54. doi: 10.1523/JNEUROSCI.0060-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Luo L-Y, Chen C, Liu-Chen L-Y. Activation of the cloned human κ opioid receptor by agonists enhances [35S]GTPγS binding to membranes: Determination of potencies and efficacies of ligands. J Pharmacol Exp Ther. 1997;282:676–84. [PubMed] [Google Scholar]
- Zubieta JK, Gorelick DA, Stauffer R, Ravert HT, Dannals RF, Frost JJ. Increased mu opioid receptor binding detected by PET in cocaine-dependent men is associated with cocaine craving. Nat Med. 1996;2:1225–9. doi: 10.1038/nm1196-1225. [DOI] [PubMed] [Google Scholar]