

SUMMARY
Removal of introns from pre-mRNAs via splicing provides a versatile means of genetic regulation that is often disrupted in human diseases. To decipher how splicing occurs in real time, we have directly examined with single-molecule sensitivity the kinetics of intron excision from pre-mRNA in the nucleus of living human cells. By using two different RNA labeling methods, MS2 and λN, we show that beta-globin introns are transcribed and excised in 20-30 s. We further show that replacing the weak polypyrimidine (Py) tract in mouse immunoglobulin μ (IgM) pre-mRNA by a U-rich Py decreases the intron lifetime, thus providing direct evidence that splice site strength influences splicing kinetics. We also found that RNA polymerase II transcribes at elongation rates ranging between 3 and 6 kb min-1, and that transcription can be rate limiting for splicing. These results have important implications for mechanistic understanding of co-transcriptional splicing regulation in the live-cell context.
Keywords: pre-mRNA splicing, intron lifetime, live-cell imaging, spinning-disk confocal microscopy
INTRODUCTION
In eukaryotes, extensive modification and alternative processing of the initial products of gene transcription can profoundly affect the diversity and function of the proteins that are generated from a single gene (Nilsen and Graveley, 2010). In addition to providing a versatile means of genetic regulation, removal of introns from pre-mRNAs by splicing is implicated in many human genetic diseases, either as a direct cause, a modifier of disease severity or a determinant of disease susceptibility (Cooper et al., 2009).
Splicing is carried out by the spliceosome, an elaborate macromolecular machine composed of uridine-rich small nuclear RNAs (UsnRNAs) packaged as ribonucleoprotein particles (snRNPs) that in human cells function in conjunction with over 200 distinct non-snRNP auxiliary proteins (Will and Luhrmann, 2011). Spliceosomes build anew on every intron that is synthesized and then disassemble for the next round of splicing (Staley and Guthrie, 1998). Thus, and unlike many other cellular enzymes that contain pre-formed stable active sites, the spliceosome exhibits exceptional compositional and structural dynamics. The dynamic assembly of a single spliceosome has recently been followed in real time in whole-cell extracts (Hoskins et al., 2011). This study revealed that association of spliceosomal subcomplexes with pre-mRNA occurs in an ordered pathway, that every subcomplex binding step is reversible, and that early binding events do not fully commit a pre-mRNA to splicing (Hoskins et al., 2011). This implies that potentially any step during spliceosome formation might be subject to regulation. Spliceosome assembly is indeed highly regulated: depending on the combinatorial effect of proteins that either promote or repress the recognition of the core splicing sequences, splice sites in pre-mRNA can be differentially selected to produce multiple mRNA isoforms through alternative splicing.
In vitro, transcripts generated by RNA polymerase II are spliced within 15-60 minutes (Das et al., 2006). In contrast, electron microscopy analysis of Drosophila embryo genes and Balbiani ring genes in the salivary glands of the dipteran Chironomus tentans revealed that intron excision occurs within 2.5-3 minutes after transcription (Beyer and Osheim, 1988; Wetterberg et al., 2001), thus suggesting that pre-mRNA splicing is much more efficient in the cell nucleus than in nuclear extracts. Moreover, a large body of compelling evidence indicates that pre-mRNA splicing is tightly coupled to transcription and should not, therefore, be studied in isolation from other stages of gene expression (Maniatis and Reed, 2002). In particular, a kinetic model has been proposed whereby the spliceosome requires more time to assemble at certain regulated splice sites; pending on the elongation rate, such sites can be recognized when transcription is slow, but skipped when transcription is fast (de la Mata et al., 2003; Eperon et al., 1988; Nogues et al., 2002). However, no study to date has directly compared the splicing kinetics of pre-mRNAs containing distinct splice sites.
Here, we monitored splicing and intron turnover by combining genomic integration of a single reporter gene in human cells, intron labeling with fluorescent proteins, and spinning-disk confocal microscopy. We have successfully measured the lifetime of single introns in the nucleus of living cells, and we show that different types of introns have distinct splicing kinetics, depending on their relative position in the transcript, size and splice site strength.
RESULTS
An assay to visualize intron removal from pre-mRNA in living cells
As a first model system, we used the well characterized human beta-globin (HBB) gene. The HBB gene has a simple structure composed of two constitutively spliced introns (Figure 1A), and the processes leading to maturation of its primary transcripts have been extensively studied both in vitro and in vivo. In order to make beta-globin pre-mRNA visible in living cells, binding sites for either the coat protein of bacteriophage MS2 or the antiterminator protein N of bacteriophage λ were inserted in each intron (Figure 1A and S1), and the resulting transgenes were stably integrated into the genome of human host cells. Each MS2 binding site consists of a 19 nucleotide RNA stem-loop containing a single base change that significantly enhances MS2 coat protein binding (Lowary and Uhlenbeck, 1987), whereas λN binds to a minimal 15-nt RNA stem-loop termed boxB (Franklin, 1985). A cassette coding for 24 tandemly repeated MS2 stem-loops or 25 boxB repeats was inserted in either the first or the second intron, preserving the consensus 5’ splice donor, lariat branch point and 3’ splice acceptor sites. To avoid lack of control over the copy number and position of the integrated transgenes, we used a strategy that involves the site-specific recombinase Flp (flippase). Thus, all cell lines generated in this study have a single HBB gene integrated at the same site in the genome. Transcription of the HBB gene is driven by the human cytomegalovirus (CMV) promoter, and conditional expression of beta-globin is regulated using a system derived from the tetracycline-resistance operon (TetO) (Figure 1A).
Figure 1. Visualization of beta-globin introns.
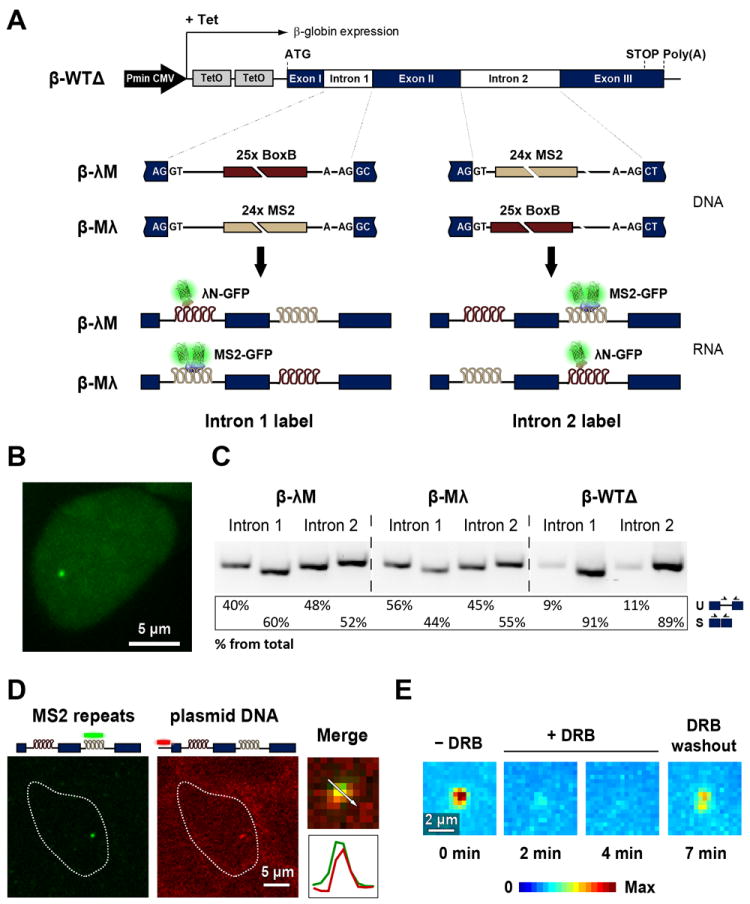
(A) Illustration of the structure of the HBB transgene. Tetracycline-inducible expression is under the control of a minimal human cytomegalovirus promoter (Pmin CMV). Binding sites for MS2 and λN were inserted in either the first or second intron. The interaction of MS2 and λN proteins fused to GFP with the intronic stem loops allows the transcribed pre-mRNAs to be visualized. (B) Representative HEK-293 cell expressing beta-globin transcripts tagged with MS2-binding sites in the second intron. Cells were transiently transfected with a plasmid encoding MS2-GFP and imaged after incubation with tetracycline. (C) RT-PCR analysis of beta-globin RNA uncleaved at the poly(A) site. RNA was extracted from cells that express the beta-globin transcripts tagged in the first or second intron with MS2 and λN fluorescent fusion proteins. PCR primers (arrows) used to detect spliced (S) and unspliced (U) RNA, and results from semiquantitative analysis of PCR product abundance are shown in the diagram. (D) Double RNA and DNA FISH shows introns labeled with MS2 at the transcription site: after transcriptional induction, cells expressing β-λM transcripts were fixed and hybridized with Cy5-labeled probe complementary to the MS2 repeats (RNA FISH, pseudocolored green) and Cy3-labeled probe targeting the vector used for beta-globin gene integration (DNA FISH, pseudocolored red). Double hybridization merged images are shown and enlarged insets depict the transcription site with the corresponding intensity line scans. (E) A spinning disk confocal microscope was used to obtain 4D movies of cells expressing beta-globin transcripts tagged with MS2-GFP in the second intron. Z-stacks of optical sections were obtained every 60 s. Maximum-intensity projections images of fluorescence at the transcription site were generated for each time point and pseudocolored. Images were acquired before (-DRB) and after (+DRB) incubation with 75 μM DRB for 2 and 4 min. DRB was subsequently washed out, and 7 min later the same transcription site was re-imaged. See also Figures S1 and S2 and Table S1.
We engineered three isogenic human embryonic kidney 293 (HEK-293) cell lines that stably express a single copy of the HBB gene. The first cell line expresses a non-tagged version of the HBB gene; in order to have the two introns with approximately the same length we used a HBB variant with a shorter intron 2 (β-WTΔ, Figure S1). The second cell line expresses the HBB gene tagged with λN binding sites in the first intron and MS2 binding sites in the second intron (β-λM, Figure S1). The third cell line expresses the HBB gene tagged with MS2 binding sites in the first intron and λN binding sites in the second intron (β-Mλ, Figure S1).
These cells were transiently transfected with plasmids encoding the green fluorescent protein fused in-frame to either the carboxyl terminus of MS2 coat protein (MS2-GFP) or λN protein (λN-GFP). Both fusion proteins contain a nuclear localization signal that confines the chimera to the nucleus. When the GFP fusion proteins were expressed in the absence of transcriptional activation of the HBB gene by tetracycline, diffuse fluorescence was detected throughout the nucleus and tended to accumulate in nucleoli. Following transcriptional induction, a fluorescent dot was detected in the nucleoplasm (Figure 1B).
To determine whether the insertion of MS2 and λN binding sites in introns interfered with pre-mRNA splicing, we carried out RT-PCR analysis. RNA was isolated from cells expressing wild-type beta-globin (i.e., devoid of MS2 or λN binding sites) and cells co-expressing the intronically tagged beta-globin variants and MS2 and λN fusion proteins. RNA was reversed transcribed using an oligonucleotide that is complementary to a sequence downstream of the beta-globin poly(A) site. The resulting cDNA was then PCR amplified using primers that specifically detect spliced and unspliced beta-globin transcripts (Figure 1C). This RT-PCR experiment detects transcripts that have not yet been cleaved at the poly(A) site and are therefore expected to be close to the gene template. The results indicate that beta-globin pre-mRNA molecules tagged with intronic MS2 and λN fluorescent fusion proteins are already spliced while the transcripts are still uncleaved. However, compared to wild-type pre-mRNAs, transcripts containing tagged introns are less efficiently spliced. This implies that tagged pre-mRNAs take longer to be spliced and/or that some may fail to be spliced.
We next examined the localization of RNAs tagged with MS2 stem loops by fluorescent in situ hybridization (FISH). Cells expressing β-λM transcripts were hybridized with a probe complementary to the MS2 stem loops and a probe complementary to the hygromycin resistance gene in the plasmid used for transfection, which reveals the site of integration of the HBB gene in the host genome (Figure 1D). The resulting nuclear dots co-localized, showing that intronic MS2 stem loops are predominantly detected at the transcription site. Cells were alternatively hybridized with a probe complementary to the MS2 stem loops and a probe complementary to full-length beta-globin RNA (Figure S2). We observed precise co-localization of fluorescent dots, indicating that the majority of intronic MS2 stem loops detected in the nucleus are either part of nascent pre-mRNA molecules or correspond to excised introns located in close proximity to the transcription site. The observation that introns appear restricted to the transcription site is consistent with our previous data indicating that beta-globin pre-mRNA is spliced co-transcriptionally (Custodio et al., 1999; de Almeida et al., 2010). Moreover, we have shown that splicing defective mutant beta-globin pre-mRNAs are retained at the transcription site (Custodio et al., 1999; de Almeida et al., 2010). A similar retention mechanism could explain why we do not detect unspliced tagged RNAs in the nucleoplasm.
Monitoring the dynamics of intron turnover at the transcription site
Time-lapse multi-plane spinning disk confocal microscopy was used to monitor intron dynamics at the site of transcription. An immediate observation in the recorded movies was that the fluorescence intensity was not constant over time but rather showed cycles of fluorescence gain and loss (Movie S1). If the observed increase in fluorescent signal results from binding of MS2- or λN-GFP fusion proteins to newly synthesized MS2 or λN stem loops, the appearance of a fluorescent dot will require ongoing transcription. We therefore treated cells co-expressing β-λM transcripts and MS2-GFP with the reversible inhibitor of transcription 5,6-dichloro-1-β-D-ribobenzimidazole (DRB). Figure 1E shows maximum-intensity z-projection images of the β-globin transcription site in a cell before and after treatment with DRB. After washing out DRB, the same cell was re-imaged revealing re-appearance of the fluorescent signal (Figure 1E). Time-lapse analysis of 30 cells indicated that the fluorescence intensity at the transcription site decreased progressively and the signal was no longer distinguishable from background fluorescence after incubation with DRB for 4-5 min. The signal consistently re-appeared within 2-3 min after washing out DRB. These results support the view that the fluorescent signal at the transcription site results from synthesis of RNA sequences that bind GFP fusion proteins. Since the arrays of RNA motifs that bind MS2 and λN proteins form stem-loop structures, it is estimated that they occupy a volume of less than 250 nm in diameter (assuming a length of 0.3 nm per nucleotide; see also (Grunwald and Singer, 2010)). Thus, they should appear as diffraction-limited objects.
In order to rigorously quantify the fluorescence emitted from a single transcription site, we developed an image analysis application (Figure S3) to track single transcription sites in 3D and quantify their total fluorescence intensity (TFI) over time by Gaussian fitting (Figure 2A-C). As graphically depicted in Figure 2D, the fluorescence intensity shows fluctuations with time, with TFI increasing to a given maximum and returning to background levels. Similar results were observed for the first and second β-globin introns labeled with either MS2 or λN-GFP fusion proteins (Figure S4).
Figure 2. Fluctuations in fluorescence intensity at the transcription site.
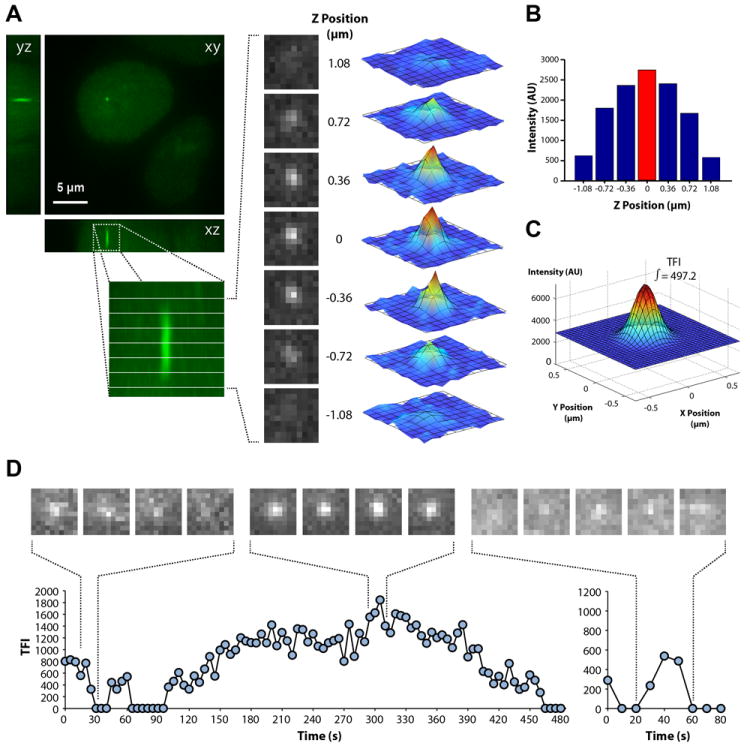
For quantification of total fluorescence intensity (TFI), a z-stack of 8 optical sections spaced by 0.36 μm and centered on the transcription site was recorded at 5 s intervals. (A) The diffraction-limited fluorescent dot is visible in the central planes of the z-stack as a rotationally symmetric point spread function which is longer along the optical z-axis, as expected. Each of the images in these central planes can be fitted by a 2D Gaussian function. (B) At each time point, we tracked the position of the transcription site in 3D and determined the plane corresponding to the highest intensity value. (C) A 2D Gaussian fit was performed at the plane of highest intensity and the result was plotted as an analytical 2D Gaussian function, the integral of which was defined as the TFI for the transcription site. (D) For each time-lapse series, the TFI was plotted over time in line graphs. A sequence of images depicting fluorescence fluctuations in the highest intensity plane is shown on top of each graph. See also Figure S3 and Movie S1.
Assuming that increments in the fluorescence signal result from de novo transcription of intronic MS2 or λN-binding sites, fluorescence loss could reflect either intron excision or release of unspliced RNA from the site of transcription. Although we cannot rule out that some unspliced RNAs are released to the nucleoplasm, the observation that approximately 50% of fluorescently tagged β-globin introns are spliced before cleavage at the poly(A) site (Figure 1C) argue that at least half of the fluorescence loss events are due to splicing.
First and second beta-globin introns have different lifetimes
Fluctuations in total fluorescence intensity values measured at the transcription site were visible in most time-lapse series, with significant cell-to-cell variation in the patterns of fluorescence gain and loss. Although in some time-lapse series the total fluorescence intensity did not drop to background levels, in many movies the phase of fluorescence loss reached background level (Figure 2D). According to the recent finding that in mammalian cells pre-mRNAs are synthesized in bursts (Chubb and Liverpool, 2010), we consider it most likely that a complete disappearance of fluorescence reflects a period of transcriptional silence during which introns were excised from all previously synthesized pre-mRNAs. Analysis of time lapses with fluctuations around background reveals two types of cycles. The first is generally longer than 200 s, reaches high fluorescence intensity values and shows a complex pattern of sub-fluctuations. The second is much shorter (typically less than 50 s) and fluorescence intensity values are lower (Figure 2D). Since high fluorescent intensity indicates accumulation of multiple transcripts, we sought to estimate how many pre-mRNA molecules were present at the transcription site during each cycle of fluorescence gain and loss. For this, we took advantage of spliceostatin A (SSA), a potent splicing inhibitor that induces leakage of unspliced beta-globin pre-mRNAs to the nucleoplasm (Martins et al., 2011). SSA inhibits splicing in vivo and in vitro by targeting the SF3b complex and blocking formation of a catalytic spliceosome subsequent to the recruitment of U2 snRNP to the pre-mRNA (Corrionero et al., 2011; Kaida et al., 2007; Roybal and Jurica, 2010). Live-cell imaging of cells treated with 100 ng ml-1 SSA for 4-8 hours revealed a multitude of diffraction-limited objects diffusing throughout the nucleus, and similar results were observed for introns labeled with either MS2 or λN-GFP fusion proteins (Figure 3A, C and Movie S2). Based on previous studies (Grunwald and Singer, 2010; Larson et al., 2011; Shav-Tal et al., 2004), we reasoned that the diffraction-limited objects that diffuse throughout the nucleus of SSA-treated cells correspond to individual mRNP particles, each containing a single fluorescently labeled intron. In agreement with this view, images of diffusing introns labeled with either MS2 or λN-GFP show single-peaked distributions of fluorescence intensity values (Figure 3B, D), indicating that the population of unspliced RNA primarily consists of individual particles. These observations further demonstrate that if unspliced pre-mRNAs were diffusing away from the transcription site in untreated cells, they would be detected by our visualization system.
Figure 3. Counting the number of introns at the transcription site.
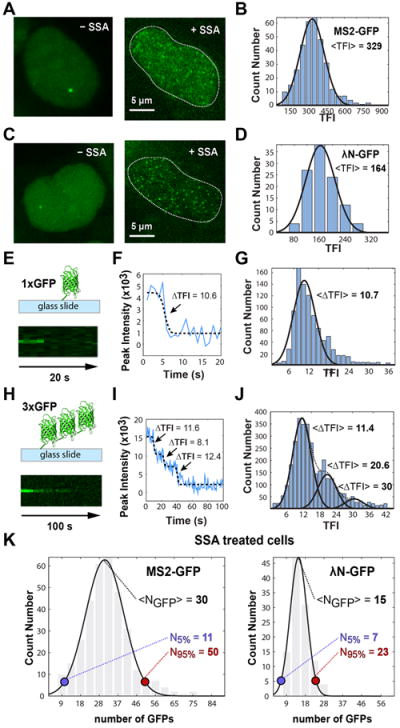
Single optical plane images of cells expressing β-λM transcripts tagged with MS2-GFP (A) and λN-GFP (C) were imaged before and after treatment with 100 ng ml-1 SSA for 4-8 hours. Contrasting to transcription sites that are relatively immobile in the nucleus, unspliced introns in SSA-treated cells diffuse rapidly throughout the nucleoplasm (Movie S2). Distributions of total fluorescence intensity (TFI) measured in SSA-treated cells expressing β-λM transcripts tagged with MS2-GFP (B) and λN-GFP (D). TFI was estimated by 2D Gaussian fitting as described in Figure 2. (E, H) Schematic illustrating GFP molecules adsorbed to glass coverslips (top) and pseudo-line plot (xt plot obtained from xyt timecourse) of photobleaching sequence (bottom). We analyzed single GFP molecules (E) and particles containing three GFP molecules synthesized in tandem (H). A single optical plane was continuously recorded using 1 s exposure per image. (F, I) The intensity profile over time was plotted in a graph and fitted to a sigmoid shaped function for single GFP molecules (black dashed line in F) or sum of sigmoid shaped functions for triple GFP particles (black dashed line in I) to detect bleaching events. The ΔTFI associated with each bleaching event was estimated by 2D Gaussian fitting and normalized for an exposure time of 15 ms for direct comparison purposes (see Methods). Arrows indicate photobleaching of one GFP molecule. (G) Distribution of ΔTFI corresponding to bleaching events for single GFP molecules (n = 1001). Data from a total of 420 images recorded in 22 time series. (J) Distribution of ΔTFI corresponding to bleaching events for triple GFP particles (n = 3358). The first peak corresponds to single bleaching events, the second peak corresponds to two simultaneous bleaching events and the third peak is due to the less frequent situation of bleaching of 3 GFP molecules in the same time point. Data from a total of 931 images recorded in 22 time series. (K) Distribution of GFP molecules bound to individual unspliced pre-mRNA molecules diffusely detected in the nucleoplasm of SSA-treated cells. Data from the histograms depicted in panels B and D were calibrated using 10.7 as the average TFI corresponding to a single GFP molecule (for 15 ms exposure time). N5% and N95% indicate the lower and upper limits for the number of GFPs contained within 2 standard deviations of the average value (about 95% of values). See also Movie S2.
In parallel, we determined the total fluorescence intensity (TFI) emitted by a single GFP molecule. For this, we used either purified single GFP molecules (Figure 3E) or particles containing 3 GFP molecules synthesized in tandem (Figure 3H). Single and triple GFP molecules were adsorbed to the surface of glass coverslips and imaged as isolated diffraction limited fluorescent objects. Bleaching of GFP molecules was induced by continuous imaging while fluorescence intensity was monitored as a function of time (Figure 3E, H). Bleaching of single GFP molecules occurred in a single step (Figure 3F), whereas up to three photobleaching events were detected for the triple GFP particles (Figure 3I), as expected assuming that each GFP molecule in the triple particle undergoes independent stochastic bleaching. The distribution of ΔTFI estimated for single GFP bleaching events is depicted in Figure 3G. The results show that the average ΔTFI associated with bleaching events on single GFP molecules is similar to the most observed average ΔTFI corresponding to single bleaching steps in triple GFP particles (Figure 3J). We used the average ΔTFI corresponding to the amount of light emitted by a single GFP molecule to calibrate the TFI measurements of single introns obtained from SSA-treated cells. The results show that on average each MS2-labeled intron binds 30 GFPs and each λN-labeled intron binds 15 GFPs, with distributions ranging between 11 and 50 (for 2 standard deviations: 95%) for MS2 and between 7 and 23 (95%) for λN (Figure 3K). This is in very good agreement with the maximum number of GFP molecules expected to bind to a single intron labeled with 24 MS2 stem-loops or 25 boxB motifs, taking into account that the MS2 protein binds as a dimer (Valegard et al., 1994) and that the boxB detection system we used is composed of four copies of λN peptide fused to three monomeric GFP molecules (Daigle and Ellenberg, 2007). Thus, our quantitative data are consistent with the view that each particle diffusing in the nucleus of SSA-treated cells contains a single fluorescently labeled intron.
Having determined the TFI value that corresponds to a single fluorescently labeled intron, we next analyzed cycles of fluorescence gain and loss that started and returned to background level and calculated the number of introns present when the TFI reached a maximum. We estimated that the long cycles involve synthesis of up to 10 or more labeled introns, whereas the short cycles contain only 1, 2 or 3 introns (Figure 4A). This suggests that the long cycles result from transcriptional bursts that involve initiation by several consecutive polymerases, leading to superposition of multiple pre-mRNAs at distinct stages of their life cycle. Consistent with this view, during a long cycle we typically observe sub-fluctuations in intensity corresponding to synthesis and disappearance of one or more introns. Such fluctuations are not observed in the short cycles. We therefore conclude that the long fluorescence cycles correspond to the time it takes to transcribe and process an asynchronous population of multiple pre-mRNAs. In contrast, the short cycles reflect the time it takes to transcribe and excise either a single intron or a group of 2-3 synchronous introns. Thus, the duration of a short cycle corresponds to the intron lifetime. We measured the duration of the short cycles and found that the most frequent lifetime value is 20 s for the first intron and 30 s for the second intron (Figure 4B, C). Similar lifetime distributions were observed for introns labeled with either MS2-GFP or λN-GFP (Figure S4), indicating that the observed intron kinetics is independent of the fluorescent labeling technique. These results also argue that the phase of fluorescence loss in the short cycles reflects splicing rather than release of unspliced RNAs from the site of transcription. Indeed, if transcripts were released unspliced, loss of fluorescence for pre-mRNAs labeled on the first intron would only occur after transcription elongation through the second intron and last exon (~2000 nucleotides). Thus, if transcripts were released unspliced, cycles of fluorescence gain and loss for the first intron should be longer than second intron cycles.
Figure 4. The first and second beta-globin introns have distinct lifetimes.
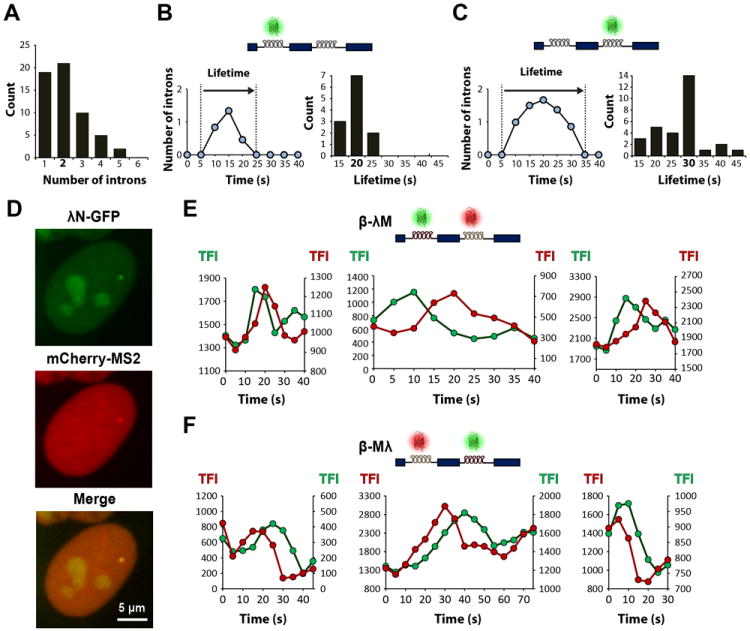
(A) Distribution of the number of introns present at the transcription site when the TFI reaches maximum value, for short cycles starting and returning to background fluorescence. Data from 57 cycles observed in 41 independent time lapses of cells expressing λN-GFP or MS2-GFP labeling either the first or the second intron. (B) Representative graph depicting a cycle of fluorescence gain and loss in a cell expressing beta-globin transcripts tagged in the first intron. TFI was converted into number of introns. The lifetime is defined as the total duration of the cycle. The histogram on the right depicts the distribution of lifetime values (n = 38). (C) Representative graph depicting a cycle of fluorescence gain and loss in a cell expressing beta-globin transcripts tagged in the second intron. The histogram on the right depicts the distribution of lifetime values (n = 75). (D) Simultaneous detection of both beta-globin introns: representative maximum-intensity projection image of a cell expressing the HBB gene tagged with λN binding sites in the first intron and MS2 binding sites in the second intron (β-λM cell line) and co-transfected with λN-GFP (green) and mCherry-MS2 (red). (E) Dual-line plots of TFI over time for cycles observed in β-λM cells, with first intron labeled with λN-GFP (green) and second intron with mCherry-MS2 (red). (F) Dual-line plots of TFI over time for cycles observed in β-Mλ cells, with first intron labeled with mCherry-MS2 (red) and second intron with λN-GFP (green). See also Figure S4.
To further confirm that loss of fluorescence is due to splicing and not to release of unspliced pre-mRNA, we sought to directly visualize the dynamics of both beta-globin introns simultaneously. We reasoned that if transcripts were released unspliced, then fluorescence associated with the first intron should increase before fluorescence associated with the second intron and both fluorescent signals should decrease simultaneously. Cells expressing the HBB gene tagged with λN binding sites in the first intron and MS2 binding sites in the second intron (β-λM) were co-transfected with two plasmids, one that encodes λN protein fused to GFP and another that encodes MS2 protein fused to the red fluorescent protein mCherry (Figure 4D). 3D time-series were captured at 5 s intervals following sequential illumination of the sample with the appropriate lasers and TFI values calculated. We observed that the green fluorescence intensity associated with intron 1 started to increase before the red fluorescence in intron 2, and then decreased while intron 2 red fluorescence was still increasing or at its maximum (Figure 4E). Similar results were observed in cells expressing the β-Mλ construct, with intron 1 labeled red and intron 2 labeled green (Figure 4F). These data strongly suggest that intron 1 is excised while intron 2 is still present in the nascent transcript, arguing against release of unspliced transcripts.
Intron lifetime differs pending on splice site strength and intron size
To study the effect of splice site strength and intron size on splicing kinetics we introduced 24 tandemly repeated MS2 binding sites in mouse immunoglobulin μ (IgM) gene reporters (Figure 5A). A single copy of each transgene was stably integrated into the genome of HEK-293 cells through site-specific DNA recombination. Contrasting to beta-globin pre-mRNA, which contains strong splice sites and is constitutively spliced, mouse IgM is a regulated splicing substrate (Tsurushita et al., 1987; Watakabe et al., 1989). In particular, the intron between exons M1 and M2 contains weak 3’ splice site sequences and requires an enhancer located in exon M2 for efficient splicing (Watakabe et al., 1993). To determine whether presence of a weak or a strong 3’ splice site affects splicing kinetics, we compared wild-type IgM M1-M2 (IgM-weakPy; Figure 5A) and a mutant constructed by replacing the weak 12-nucleotide polypyrimidine (Py) tract of IgM by the U-rich 14-nucleotide Py tract of adenovirus major late promoter pre-mRNA (IgM-strongPy; Figure 5A). To investigate the influence of intron size on splicing kinetics, we extended intron length in the IgM-strong Py construct by inserting fragments derived from the first intron of mouse RNA polymerase II gene before and after the MS2 binding sites (Figure 5A and S5A).
Figure 5. Visualization of the intron located between IgM exons M1 and M2.
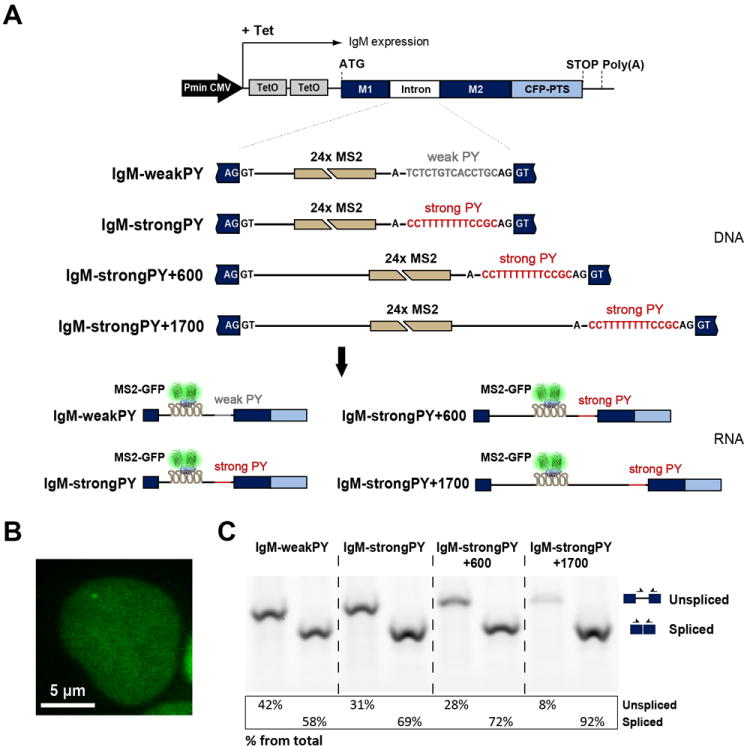
(A) Illustration of the structure of the IgM transgenes. Tetracycline-inducible expression is under the control of a minimal human cytomegalovirus promoter (Pmin CMV). Binding sites for MS2 were inserted as indicated. The interaction of MS2-GFP with the intronic stem loops allows the transcribed pre-mRNAs to be visualized. (B) Representative HEK-293 cell expressing IgM-weakPy transcripts. Cells were transiently transfected with a plasmid encoding MS2-GFP and imaged after incubation with tetracycline. (C) RT-PCR analysis of IgM RNA uncleaved at the poly(A) site. RNA was extracted from the indicated cell lines. PCR primers (arrows) used to detect spliced and unspliced RNA, and results from semiquantitative analysis of PCR product abundance are shown in the diagram. See also Figure S5 and Table S1.
Following transcriptional activation of the IgM reporter genes by tetracycline in cells expressing MS2-GFP, a fluorescent dot was detected in the nucleoplasm corresponding to nascent pre-mRNA (Figure 5B). Because the IgM reporter genes were engineered to encode a cyan fluorescent protein (CFP) fused to a peroxisomal targeting signal (PTS) at the carboxyl terminus, and the CFP-PTS sequence was inserted in frame with spliced M1 and M2 exons, detection of cyan fluorescence in peroxisomes confirmed that pre-mRNAs were correctly spliced and exported to the cytoplasm (Figure S5B).
As previously described for beta-globin RNA, we carried out RT-PCR analysis of IgM RNA using for reverse transcription an oligonucleotide that is complementary to the sequence downstream of the poly(A) site (Figure 5C). This analysis detects uncleaved RNAs, which are presumably still attached to the gene template at the site of transcription. Consistent with the finding that CFP is correctly targeted to peroxisomes, the RT-PCR results confirm that all types of pre-mRNA tested are spliced. The proportion of spliced relative to unspliced RNA differs, however, between constructs, being highest (>90%) for IgM+1700 (Figure 5C). Compared to the other constructs, IgM+1700 contains the MS2 array further apart from both 5’ and 3’ splice sites; this may reduce interference with spliceosome assembly thereby resulting in higher splicing efficiency.
Fluctuations in fluorescence intensity at the site of transcription were quantitatively analyzed as described for beta-globin pre-mRNA. To estimate intron lifetime, we selected discrete cycles of fluorescence starting at background level and involving synthesis of a single transcript (Figure 6A and S6). The results show that IgM introns with weak Py tract have an average lifetime of 43 ± 14 s (n=31). In contrast, introns with strong Py tract have a significantly shorter lifetime: 31 ± 6 s (n=37; p=4.3×10-6). Compared to the short IgM pre-mRNA with strong Py tract, inserting ~600 nucleotides upstream of the MS2 array does not significantly affect, as expected, the duration of the discrete cycles of fluorescence: 31 ± 8 s (n=29). These cycles no longer correspond to the full intron lifetime because the time it takes to synthesize the extra 600 nucleotides before the MS2 array is not detected. Insertion of ~1.000 nucleotides downstream of the MS2 array, on the contrary, results in significantly longer cycles of fluorescence: 49 ± 14 s (n=33; p=9.6×10-9) (Figure 6B, C).
Figure 6. Intron lifetime is influenced by splice site strength and intron size.
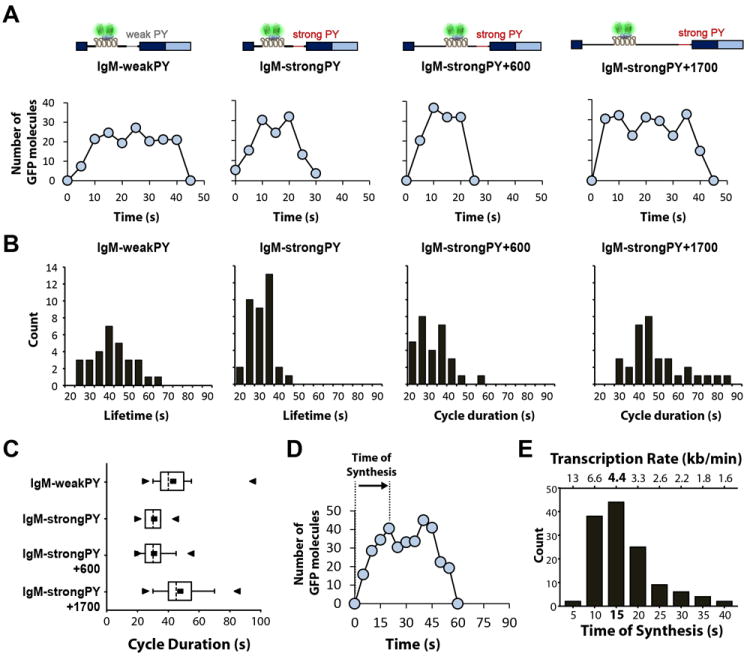
(A) Representative graphs depicting cycles of fluorescence gain and loss involving synthesis of a single transcript (maximum fluorescence intensity up to 40 GFP molecules) in cells expressing the indicated IgM constructs. TFI was converted into number of GFP molecules. (B) Histograms showing the duration of cycles of fluorescence gain and loss involving synthesis of a single transcript. In the case of IgM-weakPY and IgM-strongPY constructs, the cycle duration values correspond to intron lifetime; for IgM-strongPy+600 and IgM-strongPY+1700 constructs, the cycle duration excludes time of synthesis of the ~600 nucleotides transcribed before the MS2 array. (C) Box-and-Whisker plots of lifetime values for the indicated IgM constructs showing the mean (■), median (dotted line), minimum (►), maximum (◄), 25% quartile (box) and 10% quartile (whiskers). (D) Representative graph depicting a cycle of fluorescence gain and loss involving synthesis of a single transcript. The time of synthesis of the MS2 array (1093 nucleotides) is defined as the time elapsed since fluorescence intensity starts to increase above background until it reaches a maximum level. (E) Distribution of the values estimated for time of synthesis and rates of transcription (n = 130). See also Figure S6.
We also determined the time elapsed since the fluorescence intensity starts to increase above background until it peaks (Figure 6D) and we obtained the distribution shown in Figure 6E. From this we conclude that RNA polymerase II transcribes the array of MS2 stem loops (~1,093 nucleotides) at rates ranging predominantly between 3 and 6 kb min-1. Assuming such elongation rates, transcription of the additional 1.000 nucleotides present in the IgM+1700 intron would take 12 to 24 s. Thus, the prolonged fluorescent cycles observed for this construct most likely result from the extra time needed to transcribe the longer intron.
DISCUSSION
We have developed a system that makes it experimentally possible to observe in real time the transcription and turnover of fluorescently labeled introns in living cells with single-molecule resolution.
By using two different RNA labeling methods, MS2 and λN, we show that transcription and excision of beta-globin introns occurs in 20-30 s, and that compared to the second intron, the first intron has a shorter lifetime. The longer lifetime of the second intron may relate to the fact that intron 2 in beta-globin pre-mRNA is a terminal intron, and there is a well-established connection between splicing of terminal introns and 3’ end processing (Niwa et al., 1992). More specifically, co-transcriptional coupling between splicing of beta-globin last intron, 3’ end processing, and transcriptional termination has been proposed to allow for a time lag important for a check on the integrity of the transcript before cleavage and release to the nucleoplasm (Dye and Proudfoot, 1999).
In addition to beta-globin, we further engineered mouse immunoglobulin μ (IgM) gene reporters containing a single intron between exons M1 and M2. When the weak wild type Py tract was replaced by the strong Py of adenovirus major late promoter pre-mRNA, we observed a mean intron lifetime of 31 s. A significantly longer mean intron lifetime (43 s) was measured for the wild type pre-mRNAs containing the weak Py tract, providing for the first time direct evidence that splice site strength influences splicing kinetics.
We found that RNA Polymerase II elongates through the DNA sequence corresponding to 24 MS2 binding sites inserted in an intron at rates between 3 and 6 kb min-1. This is consistent with the previously reported value of 3.8 kb min-1, estimated by quantitative RT-PCR after reversible transcriptional inhibition induced by DRB (Singh and Padgett, 2009). An elongation rate of 4.3 kb min -1 was also inferred from computational models of photobleaching data obtained in living cells using a cassette coding for 24 binding sites for MS2 (Darzacq et al., 2007). In contrast, more recent real-time observations in yeast revealed slower transcription rates in the range of 1.2 to 2.7 kb min-1 (Hocine et al., 2013; Larson et al., 2011). By using global run-on sequencing, elongation rates were reported to vary as much as 4-fold (ranging between 0.37 and 3.57 kb per min) at different genomic loci and in response to different stimuli (Danko et al., 2013).
When we compared the duration of fluorescence cycles for short and long pre-mRNAs with strong Py tract, we obtained mean values of 31 and 48 s, respectively. Based on the estimated elongation rates, we conclude that the extra time required to transcribe the longer intron is sufficient to account for the observed difference. Thus, our results indicate that transcription can be rate limiting for splicing. This finding may explain the slow splicing rates, ranging between 5 and 12 minutes, that have been reported for endogenous long mammalian genes (Kessler et al., 1993) (Singh and Padgett, 2009). It is important to note, however, that splicing may not always occur immediately after transcription of the splice sites. Particularly in the case of alternative splicing, intron excision must be delayed until all sequences involved in the choice are transcribed. Accordingly, some regulated introns have been shown to be excised after release from the transcription site (Vargas et al., 2011) and a more recent global study suggests that a subset of splicing events are completed only after the polymerase transcribes past the poly(A) site but before the mRNA is released (Bhatt et al., 2012).
In summary, our work provides a valuable experimental system that already proved useful for addressing fundamental questions concerning the dynamic control of pre-mRNA splicing in living cells. We anticipate that improved methodologies for single and double labeling of individual pre-mRNA molecules (Wu et al., 2012), and in particular the ability to image endogenous RNA (Lionnet et al., 2011) will contribute further unprecedented real time insight into how specific regulatory sequence motifs and splicing protein factors influence intron dynamics and impinge on alternative splicing decisions.
EXPERIMENTAL PROCEDURES
Cells, transfection and RNA analysis
All methodologies used for cell manipulations are described in detail in the Extended Experimental Procedures.
Spinning-disk confocal microscopy and image analysis
Imaging was performed using a recently described microscopy setup (Boulant et al., 2011; Kural et al., 2012). For image analysis, we developed an application written in MATLAB (Mathworks; Natick, MA) to track single transcription sites in 3D and quantify their total fluorescence intensity (TFI) over time by Gaussian fitting. The software was implemented as an interactive graphical user interface that allows for single time point analysis in a time lapse sequence as well as fully automated analysis of multiple time lapse sequences (batch processing mode), with parameterization of tracking and Gaussian fitting procedures and visualization of fitted data for quality assessment (Figure S3; see also Extended Experimental Procedures).
Supplementary Material
HIGHLIGHTS.
First study on splicing dynamics in living cells with single-molecule sensitivity.
beta-globin introns are transcribed and excised in 20-30 s.
Splice site strength influences splicing kinetics.
Transcription is rate limiting for excision of long introns.
Acknowledgments
We gratefully acknowledge Eric Marino and members of the Kirchhausen laboratory for their help and support. We also thank M. Yoshida, Jens Lykke-Andersen, Niels Gehring, Edouard Bertrand, Jan Ellenberg, Michael Antoniou, and Juan Valcárcel for the gifts of spliceostatin A, MS2, BoxB, MS2-GFP, λN-GFP, β-globin plasmids, and IgM plasmids, respectively. We thank Marisa Cabrita and Sérgio Marinho for excellent technical help and our colleagues José Braga, Noélia Custódio, Sandra Martins and Sérgio de Almeida for insightful discussion. We are very thankful to Stephen C. Harrison for critical reading of the manuscript.
FUNDING
This work was supported by a grant from Fundação para a Ciência e Tecnologia, Portugal (PTDC/SAU-GMG/118180/2010), and the Harvard Medical School-Portugal Program in Translational Research and Information. R.M.M. was supported by a grant from Fundação para a Ciência e Tecnologia, Portugal (SFRH/BPD/66611/2009) and received an EMBO Short Term Fellowship (ASTF272.00-2011). J.R. holds a Ciência 2008 position from the Portuguese Ministry of Science and Technology. E.M. was supported in part by NIH grant National Institutes of Health U54 AI057159, New England Regional Center of Excellence in Biodefense and Emerging Infectious Diseases) and T.K. was supported in part by NIH grant GM 075252.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beyer AL, Osheim YN. Splice site selection, rate of splicing, and alternative splicing on nascent transcripts. Genes & development. 1988;2:754–765. doi: 10.1101/gad.2.6.754. [DOI] [PubMed] [Google Scholar]
- Bhatt DM, Pandya-Jones A, Tong AJ, Barozzi I, Lissner MM, Natoli G, Black DL, Smale ST. Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions. Cell. 2012;150:279–290. doi: 10.1016/j.cell.2012.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulant S, Kural C, Zeeh JC, Ubelmann F, Kirchhausen T. Actin dynamics counteract membrane tension during clathrin-mediated endocytosis. Nature cell biology. 2011;13:1124–1131. doi: 10.1038/ncb2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubb JR, Liverpool TB. Bursts and pulses: insights from single cell studies into transcriptional mechanisms. Current opinion in genetics & development. 2010;20:478–484. doi: 10.1016/j.gde.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrionero A, Minana B, Valcarcel J. Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes & development. 2011;25:445–459. doi: 10.1101/gad.2014311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Custodio N, Carmo-Fonseca M, Geraghty F, Pereira HS, Grosveld F, Antoniou M. Inefficient processing impairs release of RNA from the site of transcription. The EMBO journal. 1999;18:2855–2866. doi: 10.1093/emboj/18.10.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle N, Ellenberg J. LambdaN-GFP: an RNA reporter system for live-cell imaging. Nature methods. 2007;4:633–636. doi: 10.1038/nmeth1065. [DOI] [PubMed] [Google Scholar]
- Danko CG, Hah N, Luo X, Martins AL, Core L, Lis JT, Siepel A, Kraus WL. Signaling Pathways Differentially Affect RNA Polymerase II Initiation, Pausing, and Elongation Rate in Cells. Molecular cell. 2013;50:212–222. doi: 10.1016/j.molcel.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzacq X, Shav-Tal Y, de Turris V, Brody Y, Shenoy SM, Phair RD, Singer RH. In vivo dynamics of RNA polymerase II transcription. Nature structural & molecular biology. 2007;14:796–806. doi: 10.1038/nsmb1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Dufu K, Romney B, Feldt M, Elenko M, Reed R. Functional coupling of RNAP II transcription to spliceosome assembly. Genes & development. 2006;20:1100–1109. doi: 10.1101/gad.1397406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida SF, Garcia-Sacristan A, Custodio N, Carmo-Fonseca M. A link between nuclear RNA surveillance, the human exosome and RNA polymerase II transcriptional termination. Nucleic acids research. 2010;38:8015–8026. doi: 10.1093/nar/gkq703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Mata M, Alonso CR, Kadener S, Fededa JP, Blaustein M, Pelisch F, Cramer P, Bentley D, Kornblihtt AR. A slow RNA polymerase II affects alternative splicing in vivo. Molecular cell. 2003;12:525–532. doi: 10.1016/j.molcel.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Dye MJ, Proudfoot NJ. Terminal exon definition occurs cotranscriptionally and promotes termination of RNA polymerase II. Molecular cell. 1999;3:371–378. doi: 10.1016/s1097-2765(00)80464-5. [DOI] [PubMed] [Google Scholar]
- Eperon LP, Graham IR, Griffiths AD, Eperon IC. Effects of RNA secondary structure on alternative splicing of pre-mRNA: is folding limited to a region behind the transcribing RNA polymerase? Cell. 1988;54:393–401. doi: 10.1016/0092-8674(88)90202-4. [DOI] [PubMed] [Google Scholar]
- Franklin NC. Conservation of genome form but not sequence in the transcription antitermination determinants of bacteriophages lambda, phi 21 and P22. Journal of molecular biology. 1985;181:75–84. doi: 10.1016/0022-2836(85)90325-0. [DOI] [PubMed] [Google Scholar]
- Grunwald D, Singer RH. In vivo imaging of labelled endogenous beta-actin mRNA during nucleocytoplasmic transport. Nature. 2010;467:604–607. doi: 10.1038/nature09438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocine S, Raymond P, Zenklusen D, Chao JA, Singer RH. Single-molecule analysis of gene expression using two-color RNA labeling in live yeast. Nature methods. 2013;10:119–121. doi: 10.1038/nmeth.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins AA, Friedman LJ, Gallagher SS, Crawford DJ, Anderson EG, Wombacher R, Ramirez N, Cornish VW, Gelles J, Moore MJ. Ordered and dynamic assembly of single spliceosomes. Science. 2011;331:1289–1295. doi: 10.1126/science.1198830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, Watanabe H, Kitahara T, Yoshida T, Nakajima H, et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nature chemical biology. 2007;3:576–583. doi: 10.1038/nchembio.2007.18. [DOI] [PubMed] [Google Scholar]
- Kessler O, Jiang Y, Chasin LA. Order of intron removal during splicing of endogenous adenine phosphoribosyltransferase and dihydrofolate reductase pre-mRNA. Molecular and cellular biology. 1993;13:6211–6222. doi: 10.1128/mcb.13.10.6211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kural C, Tacheva-Grigorova SK, Boulant S, Cocucci E, Baust T, Duarte D, Kirchhausen T. Dynamics of intracellular clathrin/AP1- and clathrin/AP3-containing carriers. Cell reports. 2012;2:1111–1119. doi: 10.1016/j.celrep.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson DR, Zenklusen D, Wu B, Chao JA, Singer RH. Real-time observation of transcription initiation and elongation on an endogenous yeast gene. Science. 2011;332:475–478. doi: 10.1126/science.1202142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionnet T, Czaplinski K, Darzacq X, Shav-Tal Y, Wells AL, Chao JA, Park HY, de Turris V, Lopez-Jones M, Singer RH. A transgenic mouse for in vivo detection of endogenous labeled mRNA. Nature methods. 2011;8:165–170. doi: 10.1038/nmeth.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary PT, Uhlenbeck OC. An RNA mutation that increases the affinity of an RNA-protein interaction. Nucleic acids research. 1987;15:10483–10493. doi: 10.1093/nar/15.24.10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T, Reed R. An extensive network of coupling among gene expression machines. Nature. 2002;416:499–506. doi: 10.1038/416499a. [DOI] [PubMed] [Google Scholar]
- Martins SB, Rino J, Carvalho T, Carvalho C, Yoshida M, Klose JM, de Almeida SF, Carmo-Fonseca M. Spliceosome assembly is coupled to RNA polymerase II dynamics at the 3’ end of human genes. Nature structural & molecular biology. 2011;18:1115–1123. doi: 10.1038/nsmb.2124. [DOI] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa M, MacDonald CC, Berget SM. Are vertebrate exons scanned during splice-site selection? Nature. 1992;360:277–280. doi: 10.1038/360277a0. [DOI] [PubMed] [Google Scholar]
- Nogues G, Kadener S, Cramer P, Bentley D, Kornblihtt AR. Transcriptional activators differ in their abilities to control alternative splicing. The Journal of biological chemistry. 2002;277:43110–43114. doi: 10.1074/jbc.M208418200. [DOI] [PubMed] [Google Scholar]
- Roybal GA, Jurica MS. Spliceostatin A inhibits spliceosome assembly subsequent to prespliceosome formation. Nucleic acids research. 2010;38:6664–6672. doi: 10.1093/nar/gkq494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shav-Tal Y, Darzacq X, Shenoy SM, Fusco D, Janicki SM, Spector DL, Singer RH. Dynamics of single mRNPs in nuclei of living cells. Science. 2004;304:1797–1800. doi: 10.1126/science.1099754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Padgett RA. Rates of in situ transcription and splicing in large human genes. Nature structural & molecular biology. 2009;16:1128–1133. doi: 10.1038/nsmb.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley JP, Guthrie C. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell. 1998;92:315–326. doi: 10.1016/s0092-8674(00)80925-3. [DOI] [PubMed] [Google Scholar]
- Tsurushita N, Avdalovic NM, Korn LJ. Regulation of differential processing of mouse immunoglobulin mu heavy-chain mRNA. Nucleic acids research. 1987;15:4603–4615. doi: 10.1093/nar/15.11.4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valegard K, Murray JB, Stockley PG, Stonehouse NJ, Liljas L. Crystal structure of an RNA bacteriophage coat protein-operator complex. Nature. 1994;371:623–626. doi: 10.1038/371623a0. [DOI] [PubMed] [Google Scholar]
- Vargas DY, Shah K, Batish M, Levandoski M, Sinha S, Marras SA, Schedl P, Tyagi S. Single-molecule imaging of transcriptionally coupled and uncoupled splicing. Cell. 2011;147:1054–1065. doi: 10.1016/j.cell.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watakabe A, Inoue K, Sakamoto H, Shimura Y. A secondary structure at the 3’ splice site affects the in vitro splicing reaction of mouse immunoglobulin mu chain pre-mRNAs. Nucleic acids research. 1989;17:8159–8169. doi: 10.1093/nar/17.20.8159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watakabe A, Tanaka K, Shimura Y. The role of exon sequences in splice site selection. Genes & development. 1993;7:407–418. doi: 10.1101/gad.7.3.407. [DOI] [PubMed] [Google Scholar]
- Wetterberg I, Zhao J, Masich S, Wieslander L, Skoglund U. In situ transcription and splicing in the Balbiani ring 3 gene. The EMBO journal. 2001;20:2564–2574. doi: 10.1093/emboj/20.10.2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will CL, Luhrmann R. Spliceosome structure and function. Cold Spring Harbor perspectives in biology. 2011;3 doi: 10.1101/cshperspect.a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Chao JA, Singer RH. Fluorescence fluctuation spectroscopy enables quantitative imaging of single mRNAs in living cells. Biophysical journal. 2012;102:2936–2944. doi: 10.1016/j.bpj.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.