

Abstract
Background
Impaired gut barrier function and acute lung injury (ALI) are significant components of the multiorgan dysfunction syndrome that accompanies severe burns. Heparin-binding epidermal growth factor–like growth factor (HB-EGF) has been shown to reduce inflammation, preserve gut barrier function, and protect the lungs from acute injury in several models of intestinal injury; however, comparable effects of HB-EGF after burn injury have never been investigated. The present studies were based on the hypothesis that HB-EGF would reduce the severity of ALI and multiorgan dysfunction after scald burns in mice.
Materials and methods
Mice were randomized to sham, burn (25% of total body surface area with full thickness dorsal scald), and burn + HB-EGF groups. The HB-EGF group was pre-treated with two enteral doses of HB-EGF (1200 μg/kg/dose). Mice were resuscitated after injury and sacrificed at 8 h later. Their lungs were harvested for determination of pulmonary myeloperoxidase activity, wet:dry ratios, and terminal deoxynucleotidyl transferase dUTP nick end label and cleaved caspase 3 immunohistochemistry. Lung function was assessed using the SCIREQ Flexivent. Splenic apoptosis was quantified by Western blot for cleaved caspase 3, and intestinal permeability was measured using the everted gut sac method.
Results
Mice subjected to scald burn injury had increased lung myeloperoxidase levels, increased pulmonary and splenic apoptosis, elevated airway resistance and bronchial reactivity, and increased intestinal permeability compared with sham mice. These abnormalities were significantly attenuated in mice that were subjected to scald burn injury but treated with enteral HB-EGF.
Conclusions
These data suggest that HB-EGF protects mice from ALI after scald burn and attenuates the severity of postburn multiorgan dysfunction.
Keywords: Acute lung injury, Heparin-binding epidermal growth factor–like growth factor, Multiorgan dysfunction syndrome
1. Introduction
Severe burn injury represents a significant source of morbidity and mortality in the pediatric population, with scalds being the most predominant etiology of burn injury in children under the age of five according to the American Burn Association’s 2011 National Burn Repository. As with all burn injuries, scald burns are characterized by a massive hyper-metabolic response that is driven by an inflammatory cascade that occurs at both local and systemic levels [1]. A variety of pathways are involved in this process, collectively known as the systemic inflammatory response syndrome (SIRS), which if severe enough, ultimately contribute to remote organ injury. SIRS is often followed by the multiorgan dysfunction syndrome (MODS). Over time, given its unique sensitivity to systemic inflammation, the lung has become the sentinel organ for MODS investigation, and its study has helped to define the pathophysiological link between cutaneous thermal injury and remote organ injury.
Focusing on acute lung injury (ALI) after burns, early investigators noted that pulmonary neutrophil recruitment occurs early after burn injury [2]. Subsequent investigators described the link between burn injury and remote organ dysfunction, including ALI and acute respiratory distress syndrome, to rely primarily on the effects of neutrophil-mediated oxidative injury [3]. In this context, endotoxin-stimulated macrophage lineage cells within the local burn environment serve as the primary directors of remote neutrophil migration, oxidant production, and regulatory substance release. Recently, there has been a shift away from the isolated examination of the systemic effects of locally derived factors, such as monocyte-/macrophage-derived cytokines, after burn injury. Extrapolation of the gut’s response in a variety of injury models has allowed for the consideration of the gut as the primary motor of MODS. These investigations have demonstrated significant ALI using diverse models of intestinal ischemia, including intestinal ischemia/reperfusion (I/R) injury based on superior mesenteric artery occlusion, hemorrhagic shock and resuscitation (HS/R), and scald burn injury [4–7].
Recent investigations in our laboratory have demonstrated that enterally delivered heparin-binding epidermal growth factor–like growth factor (HB-EGF) reduces the severity of ALI after intestinal I/R [8]. This work suggested a potentially novel therapeutic role for HB-EGF in the prevention of SIRS after intestinal injury. HB-EGF is a member of the epidermal growth factor family that was initially identified in the conditioned medium of cultured human macrophages [9]. It is a 208 amino acid biologically active transmembrane protein that yields a 14–20 kDa soluble growth factor after extracellular proteolytic cleavage [10]. HB-EGF is produced by multiple cell types and acts as a potent mitogenic and chemoattractant protein for many cell types, including intestinal epithelial cells [10,11]. Using a model of intestinal I/R, we have demonstrated an increase in endogenous HB-EGF production in intestinal epithelial cells [12]. Furthermore, we have shown that exogenously administered HB-EGF protects intestines from injury in animal models of I/R injury, HS/R, and necrotizing enterocolitis [13–15].
Our prior studies suggest that HB-EGF works at the molecular level to decrease inflammation. We have shown that HB-EGF reduces nuclear factor kappa B activation [16], decreases the production of reactive oxygen species [17] and proinflammatory cytokines [18], and decreases the overproduction of inducible nitric oxide synthase and injurious nitric oxide [19] after intestinal injury. Based on these findings, as well as the similarity between thermal injury and direct intestinal I/R in inducing splanchnic vasoconstriction and ischemia [20], we hypothesized that enterally delivered HB-EGF would protect the lungs, and attenuate multiorgan dysfunction, after scald burn injury.
2. Materials and methods
2.1. Scald burn injury model
All animal procedures were approved by the Institutional Animal Care and Use Committee of the Research Institute at Nationwide Children’s Hospital (Protocol #AR10-00035). Eight- to 12-wk-old male C57BL/6 mice weighing 25–30 g were divided into three groups: (1) sham burn (sham), (2) scald burn (burn), and (3) scald burn with HB-EGF treatment (burn + HB-EGF). The sham and burn groups received no HB-EGF treatment. The burn + HB-EGF group received two gastric gavage doses of HB-EGF (1200 μg/kg/dose) diluted in 0.4 mL of phosphate buffered saline (PBS) at 12 h and 1 h before burn injury. Animals were anesthetized with 2.5% isoflurane, followed by intraperitoneal injection of ketamine (70 mg/kg) and xylazine (15 mg/kg). Their dorsal surfaces were shaved to ensure even contact with scalding water, and analgesia was achieved with intraperitoneal buprenorphine (0.5 mg/kg). Normal saline (1 mL) was then injected subcutaneously over the spinal column, to protect the spinal cord from scald injury. Mice were then placed supine into an insulated mold device, with an opening designed to expose ~25% of total body surface area (TBSA). Burn and burn + HB-EGF mice were submerged in 100°C water for 10 s. Sham mice were submerged in room temperature (RT) water for 10 s. After sham or burn injury, the dorsal skin was dried with a clean gauze. Each mouse then received intraperitoneal fluid resuscitation with Ringer’s lactate (1 mL). Mice were allowed to recover on a 37°C pad and were subsequently housed in a temperature-controlled environment for 8 h until the time of sacrifice.
2.2. Myeloperoxidase assay
Myeloperoxidase activity in the lung was measured as described by Netea et al. [21]. Lung tissue (75–150 mg) was homogenized for 30 s in potassium phosphate buffer (20 mM, pH 7.4, 4 mL). The homogenate was centrifuged at 20,800 g for 45 min at 4°C. The pellet was resuspended in potassium phosphate buffer (50 mM, pH 6, 4 mL) containing cetrimonium bromide (0.5 g/dL; Sigma-Aldrich, St. Louis, MO) and sonicated for 90 s at full power (Sonics Vibra Cell VCX 130; Sonics and Materials, Inc, Newtown, CT). The suspension was incubated in a 60°C water bath for 2 h and centrifuged for 10 min at 20,000 g. Supernatant of 25 μL was added to 725 μL of potassium phosphate buffer (50 mM, pH 6) containing o-Dianisidine (0.167 mg/mL; Sigma-Aldrich) and 0.0005% hydrogen peroxide (Fisher Scientific, Fair Lawn, NJ). Absorbance was measured at 460 nm at 1 and 3 min. Myeloperoxidase activity was calculated as follows: myeloperoxidase (MPO) activity (units per gram of tissue) = (ΔA460 × 13.5)/tissue weight (in grams), where ΔA460 equals the change in absorbance from 1 to 3 min. The coefficient 13.5 was empirically determined such that one unit of MPO activity was the amount of enzyme that reduced 1 μmol peroxide/min.
2.3. Terminal deoxynucleotidyl transferase dUTP nick end label staining
Apoptotic cells in the lung were identified by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) using an ApopTag Red in situ apoptosis detection kit (Chemicon International, Temecula, CA). Paraffin-embedded lung tissue sections were deparaffinized and pretreated with freshly diluted protein digesting enzyme for 15 min at RT. Equilibration buffer (75 μL/5 cm2) was applied directly to tissue sections and incubated for 10 s at RT. Excess liquid was gently tapped off the tissue sections, and a working strength of terminal deoxynucleotidyl transferase enzyme (55 μL/5 cm2) was applied to the sections and allowed to incubate in a humidified chamber at 37°C for 1 h. Slides were washed in three changes of PBS and incubated with anti-digoxigenin conjugate for 30 min at RT in the dark. The slides were then washed in four changes of 1× PBS for 2 min at RT and counterstained with a mounting media containing 4′,6-diamidino-2-phenylindole (Invitrogen, Eugene, OR). Positively stained apoptotic cells in the lung were counted randomly in ten 400× viewing fields.
2.4. Cleaved caspase 3 immunostaining
Apoptotic cells in the lung and spleen were identified by cleaved caspase 3 immunofluorescence staining. Deparaffinized sections were immersed in an autoclavable staining jar containing antigen retrieval solution (citric acid, pH 6, 0.01 M) and placed in a decloaking chamber for 30 min (15 psi at 120°C). The slides were allowed to cool to RT and then rinsed three times with distilled water. The blocking step was carried out by applying 10% bovine serum to each slide and incubating for 1 h at RT. After removing the blocking agent, rabbit anticleaved caspase 3 primary antibody (150 μL of a 1:400 dilution in 10% bovine serum/PBS + 0.1% Tween 20; Cell Signaling Technology, Danvers, MA) was applied to each slide and incubated at 4°C overnight. The primary antibody was removed by three 5-min rinses in PBS + 0.1% Tween 20. Anti-rabbit IgG-Cy3 secondary antibody (150 μL of a 1:500 dilution in 10% bovine serum; Jackson ImmunoResearch, West Grove, PA) was then applied to each slide and allowed to incubate for 1 h at RT. The secondary antibody was then removed with three 5-min rinses in PBS + 0.1% Tween 20. After removing excess liquid from the slides, they were mounted with ProLong Gold antifade reagent with 4′,6-diamidino-2-phenylindole mounting medium (Invitrogen, Grand Island, NY). Positively stained apoptotic cells were counted in 10 random 400× viewing fields.
2.5. Pulmonary function tests
Lung resistance and bronchial reactivity were measured using the SCIREQ Flexivent System (SCIREQ, Montreal, Canada) 8 h after burn injury. Mice were anesthetized with an intraperitoneal injection of ketamine (100 mg/mL) and xylazine (20 mg/mL). The trachea was then cannulated with a 20-gauge Luer-Tube adapter (#427564; Intramedic, Franklin Lakes, NJ) that was connected to a computer-controlled small animal ventilator (Flexivent; SCIREQ). Mice were ventilated with a tidal volume of 10 mL/kg at a frequency of 350 breaths/min and a positive end-expiratory pressure of 2 cm water. After stabilization on these settings, lung mechanics were measured twice using a forced oscillation technique. Pulmonary resistance was captured using a single compartment model for measuring respiratory mechanics. Forced oscillation perturbation “Quick Prime-3” gave measurements of airway resistance, total lung capacity, lung tissue resistance, and lung tissue elasticity simultaneously. Bronchial reactivity was assessed using escalating doses of aerosolized methacholine. A coefficient of determination of 0.95 was the lower limit for accepting a measurement.
2.6. Determination of pulmonary edema (wet:dry weight)
Left lungs were excised and weighed for determination of wet weight. Lungs were then placed in an incubator at 60°C, dried for 48 h, and reweighed to determine dry weight. The wet:dry ratio was then calculated for each lung, allowing for the comparison of mean ratios between groups.
2.7. Western blotting for cleaved caspase 3
Total cleaved caspase 3 levels in the spleen were determined by Western blotting. Splenic tissue samples were homogenized for 15 s at 20,000 rpm in a mixture of Phosphatase Inhibitor Cocktail 2 (1:100; Sigma-Aldrich, St Louis, MO), Halt Protease Inhibitor (1:100; Thermo Scientific, Rockford, IL), and radioimmunoprecipitation assay buffer with EDTA (Boston Bioproducts, Ashland, MA). The samples were centrifuged at 10,000 rpm for 20 min at 4 C, after which protein concentration was assayed using the BCA Protein Assay Reagent kit (Thermo Scientific, Rockford, IL). Protein lysis was achieved by boiling samples at 100°C for 5 min in sodium dodecyl sulfate buffer containing 5% β-mercaptoethanol. Samples (100 μg protein/sample) were separated by 12% sodium dodecyl sulfate/polyacrylamide gel electrophoresis before transfer to nitrocellulose membranes. The membranes were blocked with 5% skim milk at RT for 1 h and then incubated in primary cleaved caspase 3 antibodies (1:1000 dilution; Cell Signaling Technology, Danvers, MA) overnight at 4°C. The membranes were washed three times for 10 min each with 20 mmol/L Tris buffer, pH 7.4, containing 0.1% Tween 20 and then incubated in 2.5% skim milk containing a 1:10,000 dilution of secondary anti-rabbit IgG antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 h. The membranes were washed three times with 20 mmol/L Tris buffer, pH 7.4, containing 0.1% Tween 20. Protein bands were developed by enhanced chemifluorescence, scanned with Epson Perfection 4990 Photo (Long Beach, CA), and quantified using ImageJ software (U.S. National Institutes of Health, Bethesda, MD).
2.8. Intestinal permeability
Small intestinal mucosal barrier function was assessed using the ex vivo isolated everted sac method as we have previously described [22]. Briefly, 6-cm segments of terminal ileum were harvested, everted, and incubated in ice-cold Krebs-Henseleit bicarbonate buffer (KHBB buffer) at pH 7.4. Fluorescein-isothiocyanate dextran (FD4; molecular weight, 4000 Da) was used as a permeability probe. The everted gut sacs were gently distended by injecting 0.4 mL of KHBB and suspending the sacs in KHBB buffer with added FD4 (60 μg/mL) for 30 min. The incubation medium was maintained at 37°C and was continuously bubbled with a gas mixture containing 95% O2 and 5% CO2. The gut length (L) and diameter (D) were measured, and the intraluminal KHBB buffer (FD4ser) was collected and measured (intraluminal volume). Both FD4muc and FD4ser were measured with a fluorescence spectrophotometer (Spectra-Max Plus, Molecular Devices, CA). Gut permeability was expressed as the mucosal-to-serosal clearance of FD4 using the following formula: .
2.9. Statistical analyses
Sample sizes for multiple groups were determined by analysis of similar studies. Data are expressed as mean ± standard deviation. For all experiments except functional testing, between-group comparisons were performed using Student’s t-test followed by one-way analysis of variance (ANOVA). For lung resistance testing, groups were compared using one-way ANOVA with Bonferroni post hoc analysis. Methacholine challenge results were analyzed using two-way ANOVA with Bonferroni post hoc analysis, using the variables treatment and methacholine concentration. P values <0.05 were considered significant for all tests. Microsoft Excel 2011 software (Redmond, WA) or StatPlus Mac LE.2009 software (AnalystSoft Inc, Vancouver, BC) was used for all statistical evaluations.
3. Results
3.1. HB-EGF decreases lung MPO levels after burn injury
Lung MPO levels were determined as a measure of neutrophil sequestration. Scalded mice had significantly increased lung MPO activity compared with sham mice (7.6 ± 2.1 versus 3.4 ± 1.6 U/g; P = 0.006) (Fig. 1). Mice treated with HB-EGF had significantly decreased lung MPO activity compared with scalded mice that did not receive HB-EGF (3.2 ± 2.1 versus 7.6 ± 2.1 U/g; P = 0.003).
Fig. 1.

Pulmonary MPO levels. Lung MPO levels were determined 8 h after burn injury. *P = 0.006, **P = 0.003 using Student’s t-test, between-group analysis confirmed by one-way ANOVA with P = 0.002; n = 5 Sham, 6 Burn, and 7 Burn + HB-EGF.
3.2. HB-EGF decreases pulmonary apoptosis after burn injury
Apoptosis in the lungs was first evaluated using TUNEL staining. Relative to sham mice, those that underwent scald burn demonstrated an increase in apoptosis (1.14 ± 0.69 TUNEL-positive cells/high-power field [HPF] versus 0.4 ± 0.25 TUNEL-positive cells/HPF; P = 0.001) (Fig. 2). Treatment with HB-EGF led to decreased pulmonary apoptosis in scalded mice (0.61 ± 0.38 TUNEL-positive cells/HPF versus 1.14 ± 0.69 TUNEL-positive cells/HPF; P = 0.018). Secondary analysis using one-way ANOVA failed to confirm statistical significance in these findings (P = 0.06). We then performed immunostaining for cleaved caspase 3, which showed that scalded mice demonstrated significantly increased pulmonary apoptosis relative to sham (5.3 ± 0.5 versus 0.1 ± 0.1 cleaved caspase 3–positive cells/HPF; P = 0.0002), whereas scalded mice treated with HB-EGF had significantly decreased pulmonary apoptosis compared with scalded mice that did not receive HB-EGF (0.7 ± 0.5 versus 5.3 ± 1.9 cleaved caspase 3–positive cells/HPF; P = 0.00006) (Fig. 3). These findings were confirmed by one-way ANOVA.
Fig. 2.
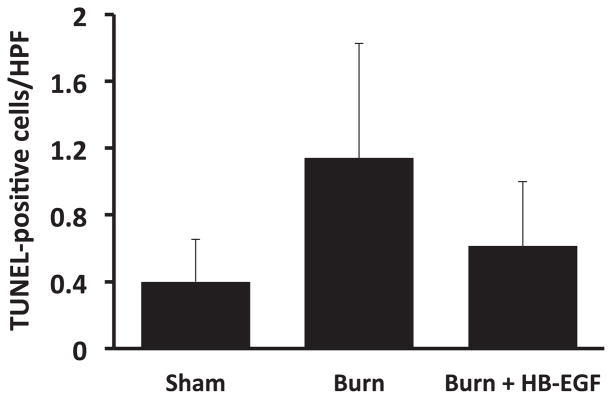
Quantification of pulmonary TUNEL staining. Each bar represents an average of 10 viewing fields/animal viewed at 400× magnification. Between-group comparisons were not significant using one-way ANOVA, P = 0.06; n = 5 Sham, 5 Burn, and 7 Burn + HB-EGF.
Fig. 3.

Pulmonary cleaved caspase 3 expression. (A) Caspase 3 immunohistochemistry, with positive cells stained in red. (B) Quantification of cleaved caspase 3 levels, with each bar representing an average of 10 viewing fields/animal viewed at 400× magnification. *P = 0.0002, **P = 0.00006 using Student’s t-test, between-group analysis confirmed by one-way ANOVA with P = 0.000002; n = 5 Sham, 6 Burn, and 7 Burn + HB-EGF.
3.3. HB-EGF prevents increased airway resistance and inducible bronchial reactivity after burn injury
Scalded mice demonstrated a significant increase in airway resistance relative to sham mice (Fig. 4A). Administration of HB-EGF before burn injury prevented this elevated airway resistance (P = 0.002). In a similar fashion, methacholine challenge revealed a significant increase in inducible bronchial reactivity in scalded mice relative to sham, which was significantly prevented by treatment with HB-EGF (P < 0.001) (Fig. 4B).
Fig. 4.
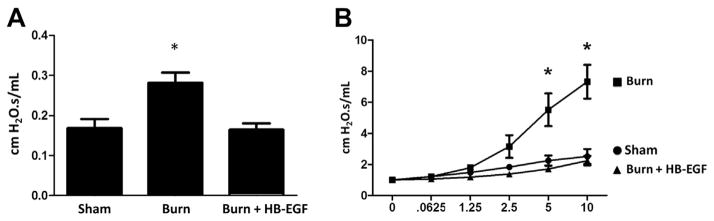
Pulmonary airway resistance and bronchial reactivity. (A) Proximal pulmonary airway resistance. *P = 0.002 using one-way ANOVA; n = 7 animals/group. (B) Inducible bronchial reactivity with methacholine challenge. *P < 0.001 using two-way ANOVA; n = 5 animals/group.
3.4. Burn injury does not lead to pulmonary edema at this time point
There were no differences in the degree of pulmonary edema between groups. Scalded mice did not demonstrate an increase in pulmonary edema relative to sham (wet:dry ratio, 4.43 ± 0.32 versus 4.49 ± 0.08), and HB-EGF pretreatment did not affect the degree of pulmonary edema in scalded mice (wet:dry ratio, 4.41 ± 0.13 versus 4.43 ± 0.32).
3.5. HB-EGF reduces splenic apoptosis after burn injury
Cleaved caspase 3 immunostaining revealed increased splenic apoptosis after burn injury, which was prevented by treatment with HB-EGF (Fig. 5A). Western blot analysis confirmed a significant increase in splenic cleaved caspase 3 levels in scalded mice relative to sham mice (percentage of sham activity, 4.1 ± 1.4 versus 1 ± 0.2; P = 0.0003) and a significant decrease in cleaved caspase 3 levels in scalded mice treated with HB-EGF compared with scalded mice that did not receive HB-EGF (percentage of sham activity, 2.1 ± 0.3 versus 4.1 ± 1.4; P = 0.006) (Figs. 5B and C).
Fig. 5.

Splenic cleaved caspase 3 expression. (A) Caspase 3 immunohistochemistry, with positive cells stained in red. (B) Western blot analysis for cleaved caspase 3, with β-actin levels as control. (C) Quantification of Western blot analysis for cleaved caspase 3. *P = 0.0003, **P = 0.006 using Student’s t-test, between-group analysis confirmed by one-way ANOVA with P = 0.00003; n = 6 animals/group.
3.6. HB-EGF prevents increased intestinal permeability after burn injury
There was a significant increase in intestinal permeability in scalded mice relative to sham mice (47.9 ± 26.9 versus 13.4 ± 7.7 mL/min/cm2; P = 0.006) (Fig. 6). Treatment of scalded mice with HB-EGF significantly prevented the increased intestinal permeability seen in scalded mice that did not receive HB-EGF (21.2 ± 13.5 versus 47.9 ± 26.9 mL/min/cm2; P = 0.013).
Fig. 6.

Intestinal permeability after burn injury. *P = 0.006, **P = 0.013 using Student’s t-test, between-group analysis confirmed by one-way ANOVA with P = 0.002; n = 7 Sham, 9 Burn, and 10 Burn + HB-EGF.
4. Discussion
ALI after severe burns continues to be a significant source of morbidity and mortality in the critically ill pediatric patient. Although the pathways by which cutaneous thermal injury results in remote organ dysfunction (MODS) continue to be more clearly elucidated, significant therapeutic targets have been difficult to identify. Therapies have been designed to target inflammation at the cutaneous and systemic level, with success largely limited to animal models. Although previous work from our laboratory demonstrated that topical application of HB-EGF to burn wounds led to acceleration of burn wound healing [23], the effects of HB-EGF on remote organs after scald burn injury have not been previously investigated.
Consistent with prior work defining the time course of pulmonary neutrophil sequestration [24,25], our model produced significant neutrophil sequestration 8 h after burn injury. Administration of HB-EGF led to significantly decreased pulmonary neutrophil sequestration as demonstrated by a significant decline in pulmonary MPO activity. Although neutrophil sequestration alone is not synonymous with pulmonary injury, the ability of the pulmonary circuit to house a massive number of neutrophils makes it uniquely susceptible to oxidant and enzymatic injury on neutrophil degranulation events or on “second hits” such as lipopolysaccharide exposure [26]. Thus, preventing pulmonary neutrophil sequestration should be beneficial.
Our analysis of pulmonary apoptosis after burn injury revealed a significant increase in pulmonary apoptosis after burn injury using cleaved caspase 3 immunostaining and a trend toward significance using TUNEL staining. With regard to pulmonary apoptosis as evaluated by TUNEL staining, our results are in accordance with those of Fukuzuka et al. [27] but contrary to those of Magnotti et al. [6]. These discrepant TUNEL findings are likely related to the size of burn, as Magnotti et al. used a 40% TBSA scald burn in rats to demonstrate a significant increase in alveolar apoptosis, whereas Fukuzuka et al. were unable to find a significant increase in pulmonary apoptosis using a 20% TBSA steam burn in mice. Although this would suggest that burn size is the primary factor influencing the progression of pulmonary alveolar apoptosis, we would argue that it is not only the size of burn that matters but also the temporal appropriateness of the assay used. We assert that the use of cleaved caspase 3 immunostaining and an 8-h postburn time point (as opposed to the 3-h time point used by the prior two authors) allowed for increased sensitivity of apoptosis, given the early role of caspase 3 relative to TUNEL in cellular senescence.
Our analysis of pulmonary function in scalded mice revealed a significant increase in proximal airway resistance that was effectively prevented with HB-EGF treatment. Furthermore, when subjected to higher doses of methacholine, a direct bronchoconstrictor challenge, scalded mice had a marked increase in airway reactivity compared with sham mice. Anatomically, these findings best represent flow at the bronchial level. Although increased proximal airway resistance could simply be because of airway edema, the results of our wet:dry ratios suggest that this is not the case. Rather, given the increase in inducible bronchial reactivity found with methacholine challenge, it is more likely that the increased airway resistance results from a state of increased bronchial smooth muscle tone secondary to the presence of arachidonic acid byproducts, as opposed to pulmonary edema. Although this physiology is certainly an established phenomenon in inhalational injuries, this has not been well described in isolated scald burns and raises intriguing questions.
The physiological link between cutaneous burn injury and remote lung injury likely relies on a complex interaction between various inflammatory cytokines and leukotrienes within the local pulmonary environment. For example, Finnerty et al. [28] described a significant elevation of interleukin-13 (IL-13) after burn injury in children. Prior murine models showed IL-13 to be a driving force of leukotriene-mediated bronchopulmonary hyper-reactivity and mucus accumulation [29]. Although the role of HB-EGF in this particular pathway remains uncertain, in vitro models of human bronchial epithelial cell repair have shown that IL-13 increases epidermal growth factor receptor phosphorylation and ultimately epithelial repair through the autocrine or paracrine release of HB-EGF [30]. Although we do not have direct evidence to support the action of enterally delivered HB-EGF at the bronchial epithelial level, future experiments will address this possibility.
To assess the effects of HB-EGF on MODS, we chose to examine splenic apoptosis, a commonly investigated parameter of multiorgan dysfunction in models of sepsis [31] and trauma [32]. We were able to show a significant increase in splenic apoptosis in burn-injured mice that was prevented by treatment with HB-EGF. These findings are in agreement with Fukuzuka et al. [27] who demonstrated increased splenic apoptosis after burn injury. Unlike these investigators, we were unable to demonstrate a significant increase in thymic apoptosis in our burn model (data not shown). Nonetheless, the ability of HB-EGF to prevent apoptosis in the spleen is significant. Further studies are needed to define the role of HB-EGF in the prevention of lymphocyte apoptosis in this model, to understand its potential impact on the modulation of innate and adaptive immunity after burn injury.
One of the most intriguing findings in our study is the ability of HB-EGF to significantly prevent the increased intestinal permeability seen after scald burn injury. Our finding of increased intestinal permeability after burn injury is in agreement with Herndon and Zeigler [20] who demonstrated a reduction in mesenteric blood flow with associated gut mucosal injury and bacterial translocation after thermal injury. Based on these findings, severe thermal injury likely leads to a state of hypovolemic shock resulting in significant splanchnic ischemia and serves as a mechanistic corollary to the intestinal ischemia induced by I/R and HS/R models.
To understand the potential therapeutic role of enterally administered HB-EGF in thermal injury, one must appreciate the well-established phenomenon of the reperfusion-injured gut serving as the motor of multiorgan dysfunction via release of proinflammatory mediators [33]. As described by Koike et al. [5] using a rodent model of intestinal I/R injury based on superior mesenteric artery occlusion, this phenomenon relies on the established sequence of splanchnic vaso-constriction and ischemia, with subsequent activation of intestinal phospholipase A2 and inflammatory mediator release. In accordance with our intestinal I/R injury findings [8], this group demonstrated an increase in circulating poly-morphonuclear priming and lung permeability, indicative of ALI [5]. They subsequently established the link between splanchnic hypoperfusion and distant organ injury to rely on the liberation of arachidonic acid from the gut, with the attendant release of leukotrienes, prostaglandins, thromboxane, and platelet activating factor into the mesenteric lymph [7]. This phenomenon was later confirmed in a rat scald burn model, in which significant increases in lung permeability, pulmonary neutrophil sequestration, and alveolar apoptosis were prevented with division of mesenteric lymphatics [6].
The unique ability of HB-EGF to protect the gut makes it an ideal agent for therapeutic investigation, and its use in a thermal injury model is based on the logical extrapolation of previous evidences accumulated in our laboratory. We have previously used animal models of I/R and HS/R to demonstrate the ability of HB-EGF to enhance intestinal restitution, preserve mesenteric microcirculatory blood flow, and protect the intestines from injury [13,14]. We have also demonstrated the ability of HB-EGF to protect the lungs after intestinal I/R [8]. Although we have not defined the specific link(s) between enteral administration of HB-EGF and the prevention of ALI after intestinal injury, it is certainly plausible that the link centers on the ability of HB-EGF to preserve intestinal integrity and downregulate the production of intestinal inflammatory byproducts in the face of mesenteric ischemia. Because we have previously demonstrated that radiolabeled 125I-HB-EGF administered via the intragastric route remains primarily within the gastrointestinal tract [34], we must assume that HB-EGF reduces remote organ dysfunction by its action within the gastrointestinal tract rather than by the generation of significant systemic levels of HB-EGF. The attenuation of multiorgan dysfunction demonstrated in our experiments would suggest that the intestine is the primary seat of pathology after thermal injury, as opposed to the thermal wound itself.
Our data reveal the ability of HB-EGF to protect remote organs from injury after scald burn, which is associated with its ability to preserve intestinal integrity in the setting of presumed hypovolemic shock and mesenteric ischemia. There are several limitations of our study that will need to be addressed to demonstrate translational utility in the future. Our investigations used a pretreatment design to demonstrate a proof of concept, as opposed to a therapeutic trial design. In addition, a relatively early 8-h time point was chosen based on prior scald burn studies to maximize our understanding of the effects of HB-EGF therapy on temporally known biological endpoints. However, changes in experimental endpoints at such an early stage may not portend clinical utility. The next phase of our investigations will examine administration of HB-EGF after thermal injury has already occurred and will investigate a broader array of postinjury time points.
Future studies will also focus on establishing the mechanism(s) by which preservation of gut integrity by HB-EGF attenuates remote organ injury. The implications of intestinal permeability will be assessed by mesenteric lymph node analysis for the presence of bacterial translocation. Inflammatory pathways within the intestine will be assessed by measurement of intestinal phospholipase A2, phosphorylated nuclear factor kappa B, and inducible nitric oxide synthase. Pulmonary leukotriene levels in bronchoalveolar lavage fluid will enhance our understanding of the effects of inflammatory byproducts within mesenteric lymph, as they pertain to the mechanisms underlying bronchial hyper-reactivity associated with thermal injury. Serum cytokine levels will supplement our understanding of inflammation at the systemic level. Through these future investigations, we hope to better understand the role of HB-EGF in the attenuation of ALI and MODS after thermal injury and ultimately its potential role as a novel therapeutic adjunct to the treatment of thermally injured patients.
Acknowledgments
The authors thank Laurie Goodchild, DVM, for her assistance in model development, Jixin Yang, MD, and Yanwei Su, MD, for their laboratory expertise, and Joshua Frazier, MD, for his thoughtful input throughout this project and assistance in the preparation of this manuscript.
References
- 1.Ward PA, Till GO. Pathophysiologic events related to thermal injury of skin. J Trauma. 1990;30:S75. doi: 10.1097/00005373-199012001-00018. [DOI] [PubMed] [Google Scholar]
- 2.Demling RH, LaLonde C, Liu YP, Zhu DG. The lung inflammatory response to thermal injury: relationship between physiologic and histologic changes. Surgery. 1989;106:52. [PubMed] [Google Scholar]
- 3.Solomkin JS. Neutrophil disorders in burn injury: complement, cytokines, and organ injury. J Trauma. 1990;30:S80. doi: 10.1097/00005373-199012001-00019. [DOI] [PubMed] [Google Scholar]
- 4.Anderson BO, Moore EE, Moore FA, et al. Hypovolemic shock promotes neutrophil sequestration in lungs by a xanthine oxidase-related mechanism. J Appl Physiol. 1991;71:1862. doi: 10.1152/jappl.1991.71.5.1862. [DOI] [PubMed] [Google Scholar]
- 5.Koike K, Moore EE, Moore FA, Kim FJ, Carl VS, Banerjee A. Gut phospholipase A2 mediates neutrophil priming and lung injury after mesenteric ischemia-reperfusion. Am J Physiol. 1995;268:G397. doi: 10.1152/ajpgi.1995.268.3.G397. [DOI] [PubMed] [Google Scholar]
- 6.Magnotti LJ, Xu DZ, Lu Q, Deitch EA. Gut-derived mesenteric lymph: a link between burn and lung injury. Arch Surg. 1999;134:1333. discussion 1340–1331. [PubMed] [Google Scholar]
- 7.Moore EE, Claude H, Organ Memorial lecture: splanchnic hypoperfusion provokes acute lung injury via a 5-lipoxygenase-dependent mechanism. Am J Surg. 2010;200:681. doi: 10.1016/j.amjsurg.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.James IA, Chen CL, Huang G, Zhang HY, Velten M, Besner GE. HB-EGF protects the lungs after intestinal ischemia/reperfusion injury. J Surg Res. 2010;163:86. doi: 10.1016/j.jss.2010.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Besner G, Higashiyama S, Klagsbrun M. Isolation and characterization of a macrophage-derived heparin-binding growth factor. Cell Regul. 1990;1:811. doi: 10.1091/mbc.1.11.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naglich JG, Metherall JE, Russell DW, Eidels L. Expression cloning of a diphtheria toxin receptor: identity with a heparin-binding EGF-like growth factor precursor. Cell. 1992;69:1051. doi: 10.1016/0092-8674(92)90623-k. [DOI] [PubMed] [Google Scholar]
- 11.Davis-Fleischer KM, Besner GE. Structure and function of heparin-binding EGF-like growth factor (HB-EGF) Front Biosci. 1998;3:d288. doi: 10.2741/a241. [DOI] [PubMed] [Google Scholar]
- 12.Xia G, Rachfal AW, Martin AE, Besner GE. Upregulation of endogenous heparin-binding EGF-like growth factor (HB-EGF) expression after intestinal ischemia/reperfusion injury. J Invest Surg. 2003;16:57. [PubMed] [Google Scholar]
- 13.El-Assal ON, Besner GE. HB-EGF enhances restitution after intestinal ischemia/reperfusion via PI3K/Akt and MEK/ERK1/2 activation. Gastroenterology. 2005;129:609. doi: 10.1016/j.gastro.2005.05.054. [DOI] [PubMed] [Google Scholar]
- 14.El-Assal ON, Radulescu A, Besner GE. Heparin-binding EGF-like growth factor preserves mesenteric microcirculatory blood flow and protects against intestinal injury in rats subjected to hemorrhagic shock and resuscitation. Surgery. 2007;142:234. doi: 10.1016/j.surg.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 15.Feng J, El-Assal ON, Besner GE. Heparin-binding epidermal growth factor-like growth factor decreases the incidence of necrotizing enterocolitis in neonatal rats. J Pediatr Surg. 2006;41:144. doi: 10.1016/j.jpedsurg.2005.10.018. discussion 144–149. [DOI] [PubMed] [Google Scholar]
- 16.Mehta VB, Besner GE. Inhibition of NF-kappa B activation and its target genes by heparin-binding epidermal growth factor-like growth factor. J Immunol. 2003;171:6014. doi: 10.4049/jimmunol.171.11.6014. [DOI] [PubMed] [Google Scholar]
- 17.Kuhn MA, Xia G, Mehta VB, Glenn S, Michalsky MP, Besner GE. Heparin-binding EGF-like growth factor (HB-EGF) decreases oxygen free radical production in vitro and in vivo. Antioxid Redox Signal. 2002;4:639. doi: 10.1089/15230860260220148. [DOI] [PubMed] [Google Scholar]
- 18.Rocourt DV, Mehta VB, Besner GE. Heparin-binding EGF-like growth factor decreases inflammatory cytokine expression after intestinal ischemia/reperfusion injury. J Surg Res. 2007;139:269. doi: 10.1016/j.jss.2006.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lara-Marquez ML, Mehta V, Michalsky MP, Fleming JB, Besner GE. Heparin-binding EGF-like growth factor down regulates proinflammatory cytokine-induced nitric oxide and inducible nitric oxide synthase production in intestinal epithelial cells. Nitric Oxide. 2002;6:142. doi: 10.1006/niox.2001.0393. [DOI] [PubMed] [Google Scholar]
- 20.Herndon DN, Zeigler ST. Bacterial translocation after thermal injury. Crit Care Med. 1993;21:S50. doi: 10.1097/00003246-199302001-00010. [DOI] [PubMed] [Google Scholar]
- 21.Netea MG, Fantuzzi G, Kullberg BJ, et al. Neutralization of IL-18 reduces neutrophil tissue accumulation and protects mice against lethal Escherichia coli and Salmonella typhimurium endotoxemia. J Immunol. 2000;164:2644. doi: 10.4049/jimmunol.164.5.2644. [DOI] [PubMed] [Google Scholar]
- 22.Zhang HY, Radulescu A, Besner GE. Heparin-binding epidermal growth factor-like growth factor is essential for preservation of gut barrier function after hemorrhagic shock and resuscitation in mice. Surgery. 2009;146:334. doi: 10.1016/j.surg.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cribbs RK, Luquette MH, Besner GE. Acceleration of partial-thickness burn wound healing with topical application of heparin-binding EGF-like growth factor (HB-EGF) J Burn Care Rehabil. 1998;19:95. doi: 10.1097/00004630-199803000-00002. [DOI] [PubMed] [Google Scholar]
- 24.Stengle J, Meyers R, Pyle J, Dries DJ. Neutrophil recruitment after remote scald injury. J Burn Care Rehabil. 1996;17:14. doi: 10.1097/00004630-199601000-00006. [DOI] [PubMed] [Google Scholar]
- 25.Hansbrough JF, Wikstrom T, Braide M, et al. Neutrophil activation and tissue neutrophil sequestration in a rat model of thermal injury. J Surg Res. 1996;61:17. doi: 10.1006/jsre.1996.0074. [DOI] [PubMed] [Google Scholar]
- 26.Moore EE, Moore FA, Harken AH, Johnson JL, Ciesla D, Banerjee A. The two-event construct of postinjury multiple organ failure. Shock. 2005;24(Suppl 1):71. doi: 10.1097/01.shk.0000191336.01036.fe. [DOI] [PubMed] [Google Scholar]
- 27.Fukuzuka K, Rosenberg JJ, Gaines GC, et al. Caspase-3-dependent organ apoptosis early after burn injury. Ann Surg. 1999;229:851. doi: 10.1097/00000658-199906000-00012. discussion 858–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finnerty CC, Herndon DN, Przkora R, et al. Cytokine expression profile over time in severely burned pediatric patients. Shock. 2006;26:13. doi: 10.1097/01.shk.0000223120.26394.7d. [DOI] [PubMed] [Google Scholar]
- 29.Vargaftig BB, Singer M. Leukotrienes mediate murine bronchopulmonary hyperreactivity, inflammation, and part of mucosal metaplasia and tissue injury induced by recombinant murine interleukin-13. Am J Respir Cell Mol Biol. 2003;28:410. doi: 10.1165/rcmb.2002-0032OC. [DOI] [PubMed] [Google Scholar]
- 30.Allahverdian S, Harada N, Singhera GK, Knight DA, Dorscheid DR. Secretion of IL-13 by airway epithelial cells enhances epithelial repair via HB-EGF. Am J Respir Cell Mol Biol. 2008;38:153. doi: 10.1165/rcmb.2007-0173OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hotchkiss RS, Swanson PE, Freeman BD, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27:1230. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 32.Hotchkiss RS, Schmieg RE, Jr, Swanson PE, et al. Rapid onset of intestinal epithelial and lymphocyte apoptotic cell death in patients with trauma and shock. Crit Care Med. 2000;28:3207. doi: 10.1097/00003246-200009000-00016. [DOI] [PubMed] [Google Scholar]
- 33.Hassoun HT, Kone BC, Mercer DW, Moody FG, Weisbrodt NW, Moore FA. Post-injury multiple organ failure: the role of the gut. Shock. 2001;15:1. doi: 10.1097/00024382-200115010-00001. [DOI] [PubMed] [Google Scholar]
- 34.Feng J, Mehta VB, El-Assal ON, Wu D, Besner GE. Tissue distribution and plasma clearance of heparin-binding EGF-like growth factor (HB-EGF) in adult and newborn rats. Peptides. 2006;27:1589. doi: 10.1016/j.peptides.2005.11.013. [DOI] [PubMed] [Google Scholar]