

Summary
Tissue-resident memory T (Trm) cells represent a population of memory CD8+ T cells that can act as first responders to local infection. The mechanisms regulating the formation and maintenance of intestinal Trm cells remain elusive. Here we showed that transforming growth factor-β (TGF-β) controlled both stages of gut Trm cell differentiation through different mechanisms. During the formation phase of Trm cells, TGF-β signaling inhibited the migration of effector CD8+ T cells from the spleen to the gut by dampening the expression of integrin α4β7. During the maintenance phase, TGF-β was required for the retention of intestinal Trm cells at least in part through the induction of integrins αEβ7 and α1, as well as CD69. Thus, the cytokine acts to control cytotoxic T cell differentiation in lymphoid and peripheral organs.
Introduction
CD8+ memory T cells have long been divided into two circulating populations termed central memory (Tcm) and effector memory (Tem). Tcm cells migrate between the secondary lymphoid organs and blood, and represent a long-lived population of memory cells with a superior proliferative capacity upon pathogen re-encounter. Tem cells traffic through spleen, blood and peripheral tissues, and maintain immediate effector potential (Masopust et al., 2001; Sallusto et al., 1999). There is accumulating evidence to support the existence of a third population of CD8+ memory T cells that never returns to the circulation (Mueller et al., 2012; Sheridan and Lefrancois, 2011). These tissue-resident memory (Trm) cells are a self-sustaining population within non-lymphoid tissues and are phenotypically distinct from Tem cells. Trm cells have been identified in a variety of tissues including the skin, dorsal root ganglia, brain, salivary glands, vagina, stomach, kidney, pancreas, heart and gut (Casey et al., 2012; Gebhardt et al., 2009; Gebhardt et al., 2011; Hawke et al., 1998; Hofmann and Pircher, 2011; Jiang et al., 2012; Masopust et al., 2010; Tang and Rosenthal, 2010; Wakim et al., 2010). In many tissues, Trm cells are required for optimal protection against subsequent local infection (Gebhardt et al., 2009; Jiang et al., 2012; Mackay et al., 2012; Schenkel et al., 2013; Wakim et al., 2008).
Intestinal Trm cells are relatively well characterized. During both local and systemic infections, CD8+ effector T cells develop gut-homing capacity through the upregulation of the integrin α4β7 and the chemokine receptor CCR9 (Johansson-Lindbom et al., 2003; Kim et al., 1997; Masopust et al., 2010; Masopust et al., 2001; Mora et al., 2003; Stagg et al., 2002). Integrin α4β7 binds to the mucosal addressin cell adhesion molecule-1 (MAdCAM-1), which is expressed by a subset of vascular endothelial cells in the intestinal lamina propria (Berlin et al., 1995; Berlin et al., 1993). Antibody blocking of this interaction prevents the migration of effector T cells to the gut (Bargatze et al., 1995; Hamann et al., 1994). However, the signals that control the expression of integrin α4β7 on effector T cells remain largely unidentified. Interestingly, during acute viral infection, there is a narrow window (day 4.5 to 7 post infection) for splenic effector CD8+ T cells to express a sufficient amount of integrin α4β7 to mediate migration to the intestine (Masopust et al., 2010). Whether the intestinal trafficking capacity of CD8+ T cells is under similar control during chronic infection remains largely unexplored.
Upon arrival in the intestinal tissues including both the lamina propria (LP) and intraepithelial lymphocyte (IEL) compartments, CD8+ T cells quickly acquire novel surface phenotypes such as the downregulation of integrin α4β7 and upregulation of CD103 (integrin αE) and the lectin CD69 (Casey et al., 2012; Masopust et al., 2010). Integrin αEβ7 binds to E-cadherin, which is constitutively expressed by intestinal epithelial cells. This interaction may mediate the tethering of IELs to the intestinal epithelium (Cepek et al., 1994). Trm cells that lack CD103 expression are defective in the long-term maintenance in the intestinal IEL compartment (Casey et al., 2012; Schon et al., 1999). CD69 inhibits the expression of S1P1 (Shiow et al., 2006), which mediates the egress of T cells from lymphoid organs (Cyster and Schwab, 2012). In addition, integrin α1 (the α subunit of very late antigen-1, VLA-1) is highly expressed on gut IEL cells (Choy et al., 1990) and may mediate the retention of CD8+ T cells in the mucosal tissues (Meharra et al., 2000; Roberts et al., 1999; Sandoval et al., 2013). Thus the expression of CD103, CD69 and integrin α1 together may contribute to the retention of Trm cells within the gut.
It has been known for more than two decades that transforming growth factor-β (TGF-β) signaling greatly enhances the expression of CD103 on activated T cells in vitro (Kilshaw and Murant, 1990). In vivo, TGF-β promotes CD103 expression on CD8+ effector T cells in the gut in a graft versus host disease model (El-Asady et al., 2005). Recently, it was shown that TGF-β is required for the differentiation of CD103+CD69+ CD8+ T cell within the IEL compartment at the effector phase of an immune response to acute lymphocytic choriomeningitis virus (LCMV) infection (Casey et al., 2012). Therefore, it is generally believed that TGF-β signaling is essential for the retention of intestinal Trm cells through the induction of CD103 expression. However, because T cell-specific deletion of TGF-β receptor early in development (Tgfbr2f/f CD4-cre) leads to an early onset lethal autoimmune disease (Li et al., 2006; Marie et al., 2006), all previous work employed a transgenic mouse model carrying a dominant negative form of TGF-β receptor II (dnTGF-βRII) in T cells (Gorelik and Flavell, 2000). Presumably due to the limitations of dnTGF-βRII mice (Ishigame et al., 2013; Sanjabi and Flavell, 2010), there is no direct evidence to support the role of TGF-β signaling in the long-term maintenance of the intestinal Trm compartment during the memory phase of an immune response. Beside the induction of CD103 expression, there is no evidence to support other functions that TGF-β signaling may have in the Trm cells.
Recently, we introduced a different model of T cell-specific conditional deletion of TGF-βRII employing Cre driven by the distal promoter of the gene encoding the kinase Lck (dLck-cre) in which deletion begins after T cell positive selection and continues slowly in the periphery (Zhang and Bevan, 2012). In adult Tgfbr2f/f dLck-cre mice, although the vast majority of T cells lack TGF-βRII expression, no signs of autoimmunity are observed. To study the function of TGF-β signaling to T cells in the formation and maintenance of intestinal Trm compartment during viral infection, we generated Tgfbr2f/f dLck-cre (hereafter referred to as Tgfbr2−/−) P14 TCR transgenic mice, that carry CD8+ T cells specific for the Db-GP33–41 LCMV epitope. Here, we report that virus specific CD8+ T cells express a markedly increased amount of TGF-βRII during chronic compared to acute LCMV infection. During acute LCMV Armstrong infection, TGF-β signaling is essential for the maintenance of intestinal Trm compartment at least in part through the induction of CD103, CD69, and integrins β7 and α1. To our surprise, during chronic LCMV Clone 13 infection, the Tgfbr2−/− Trm population is apparently normal in spite of the similar defects in the expression of CD103, CD69, and integrins β7 and α1 in the absence of TGF-β signaling. To solve this obvious discrepancy, we demonstrate that during chronic LCMV infection, the expression of integrin α4β7 and the α4β7-dependent trafficking of effector CD8+ T cells from the secondary lymphoid organs to the gut are substantially enhanced in the absence of TGF-β signaling. Thus, during the prolonged infection with LCMV Clone 13, effectors able to traffic to the gut are continually generated. Therefore, TGF-β plays location and stage-specific roles in the formation and maintenance of the intestinal Trm compartment. In the secondary lymphoid organs during the formation phase, TGF-β inhibits effector CD8+ T cells from migrating to the gut while in the gut during the maintenance phase, TGF-β signaling promotes CD8+ T cell residency.
Results
Enhanced TGF-βRII expression on CD8+ T cells during chronic LCMV infection
To investigate the role of TGF-β signaling to CD8+ T cells during acute and chronic viral infection, we adoptively transferred equal numbers of congenically marked control and Tgfbr2−/− naïve P14 T cells into wild type (WT) mice followed by LCMV Armstrong or Clone 13 infection. Interestingly, the surface expression of TGF-βRII on control P14 cells was substantially increased in chronically infected mice compared to that of acutely infected mice (Figure 1A, 1B and S1A). The enhanced expression of TGF-βRII in Clone 13 infected mice was a long-lasting effect (Figure 1B). The lack of staining of TGF-βRII on Tgfbr2−/− P14 T cells confirmed the efficient deletion mediated by dLck-cre. Consistent with a previous report (Tinoco et al., 2009), the phosphorylation of Smad 2 and Smad 3 downstream of TGF-β signaling was greatly increased in the P14 T cells isolated from LCMV Clone 13 infected mice compared to that from LCMV Armstrong infected mice (Figure S1A). Together, these results indicated that antigen specific CD8+ T cells received a stronger TGF-β signal during chronic LCMV infection at least in part due to enhanced surface expression of TGF-βRII.
Figure 1. Response in the spleen of TGF-βRII-deficient CD8+ T cells following acute and chronic LCMV infection.

Naïve P14 T cells were purified from control (CD45.1+) and TGF-βRII-deficient (CD45.1+CD90.1+) mice, mixed at a 1:1 ratio, and 104 cells transferred into sex-matched B6 recipients followed by LCMV Armstrong or Clone 13 infection. The expression of TGF-βRII on P14 T cells is shown in A and B. Representative FACS plots of P14 T cells are shown in C. The ratio of control vs TGF-βRII-deficient P14 cells is shown in D (combined data from 4 independent experiments). Each symbol in B and D represents an individual recipient mouse. *, p<0.05 (Student t-test). Data in B and D are represented as mean +/− SEM. See also Figure S1.
Slightly reduced expansion of Tgfbr2−/− P14 T cells
Previous reports using dnTGF-βRII transgenic mice, suggested that with decreased TGF-β signaling, the expansion of antigen-specific CD8+ T cells was significantly increased in response to both acute and chronic infections due to reduced apoptosis of effector T cells (Sanjabi et al., 2009; Tinoco et al., 2009). During chronic LCMV infection, dnTGF-βRII CD8+ T cells appeared less functionally exhausted, exhibiting increased cytokine production and cytotoxicity and decreased expression of PD-1 (Tinoco et al., 2009). In sharp contrast, in our system based on conditional deletion of the TGF-β receptor, CD8+ T cell expansion in the spleen was slightly but consistently decreased in the absence of TGF-β signaling (Figure 1C, 1D and Figure S1B). P14 expansion was robust for both control and Tgfbr2−/− populations in these experiments: 38.2±3.1% (Armstrong, n=13) and 27.8±2.7% (Clone 13, n=11) of total CD8+ T cells were donor P14 T cells at day 8 post infection. IFN-γ and IL-2 production and the expression of PD-1 were comparable between control and Tgfbr2−/− P14 T cells following acute and chronic viral infection (data not shown and Figure S1C). In one (Tinoco et al., 2009) but not other previous reports (Boettler et al., 2012; Garidou et al., 2012), blocking TGF-β signaling resulted in accelerated clearance of LCMV clone 13 infection. In our system, viral antigens were present in tissues for a prolonged period following LCMV Clone 13 infection (Figure S1D). Thus, TGF-β signaling slightly promoted the expansion of CD8+ T cells during LCMV infection while it was not critically involved in the functional exhaustion of CD8+ T cells during persistent viral infection.
Defective maintenance of Tgfbr2−/− P14 cells in the gut following acute, but not chronic infection
To investigate the function of TGF-β signaling in the formation and maintenance of intestinal Trm cells, an equal number of naïve P14 T cells from congenically marked control and Tgfbr2−/− mice were co-transferred into WT mice followed by LCMV Armstrong or Clone 13 infection. At different time points following the infection, donor P14 T cells in the lamina propria and IEL compartment of the small intestine were examined. At the peak of the response following acute LCMV Armstrong infection, comparable numbers of control and Tgfbr2−/− P14 T cells accumulated in the lamina propria and IEL compartment (Figure 2A, 2B and 2C). However, shortly after LCMV Armstrong clearance (day 14 post infection), the number of Tgfbr2−/− P14 T cells declined in the intestinal tissues. At day 35 to 36 post infection, the number of Tgfbr2−/− cells were reduced more than 45 fold compared to the co-transferred control P14 cells in the intestinal IEL compartment. A similar, but less dramatic decrease of the Tgfbr2−/− cell population in the lamina propria was also observed (Figure 2A and 2B). Notably, at the same time and in the same mice, there were no severe defects in the number of Tgfbr2−/− cells in the spleen (Figure 1C, 1D and S1B).
Figure 2. Defective maintenance of TGF-βRII-deficient CD8+ T cells in the gut following acute, but not chronic LCMV infection.

Similar experimental setup as in Figure 1. Data from LCMV Armstrong infected mice are shown in A, B and C, and from LCMV Clone 13 infected mice in D, E and F. At the indicated time post infection, lymphocytes were purified from the lamina propria and IEL compartment of the recipient mice. Representative FACS plots are shown in A and D. The ratio of control vs TGF-βRII-deficient P14 cells is shown in B and E (combined data from 4 independent experiments). The percentage of P14 T cells in CD8β+ IEL cells is shown in C and F (combined data from 4 independent experiments). Each symbol in B and E represents an individual recipient mouse. Data in B, C, E and F are represented as mean +/− SEM. See also Figure S2.
In sharp contrast to the picture with acute LCMV infection, no significant (p>0.2) decrease was observed for the Tgfbr2−/− P14 T cell population in the intestinal lamina propria and IEL compartment following LCMV Clone 13 infection (Figure 2D, 2E and 2F). Importantly, the percentage of control and Tgfbr2−/− P14 T cells in the CD8+ IEL compartment during chronic infection was comparable to that of control P14 cells during acute infection (Figure 2C and 2F). In addition, the percentage of total CD8+ T cells in the IEL compartment remained elevated for a prolonged period following chronic LCMV infection (Figure S2). Therefore, TGF-β unresponsive CD8+ T cells exhibited severe defects in the maintenance of intestinal Trm cells following acute, but not chronic LCMV infection.
Defective retention of TGF-βRII-deficient T cells in the gut
To elucidate the underlying mechanisms leading to the dramatically different outcomes of the gut Trm cell population following acute versus chronic LCMV infection, the phenotype of P14 T cells isolated from the small intestine was characterized. Compared to dnTGF-βRII T cells from the gut, that express reduced, but readily detectable amounts of CD103 (Casey et al., 2012; El-Asady et al., 2005), CD103 expression on Tgfbr2−/− P14 T cells was absent. The expression of CD69 was slightly, but consistently reduced on Tgfbr2−/− P14 T cells (Figure 3). Furthermore, the expression of integrin β7 was substantially decreased on Tgfbr2−/− cells from the IEL compartment at day 14 and 24 post-infection (Figure 3B and 3C). Consistent with a previous report (Casey et al., 2012), the expression of CD103 on control P14 T cells was reduced from Clone 13 compared with Armstrong infected mice. Moreover, defective expression of integrin α1 was observed on Tgfbr2−/− P14 T cells in the IEL compartment (Figure S3). Thus, the defective expression of integrins αEβ7, α1 and CD69 likely result in the severely impaired retention of Tgfbr2−/− CD8+ T cells in the gut following acute LCMV infection. However, similar defects were exhibited by the intestinal Tgfbr2−/− P14 cells from chronically infected mice (Figure 3 and S3). Therefore, the surface molecules associated with the retention of Trm cells were similarly defective on Tgfbr2−/− P14 cells from both acutely and chronically infected animals.
Figure 3. TGF-βRII-deficient cells in the gut exhibit reduced expression of markers associated with the retention.

Similar experimental setup as in Figure 1. On day 8 (A), 14 (B) and 24 (C) post LCMV infection, lymphocytes were purified from the lamina propria and IEL compartment of the recipient mice. The expression of integrin β7, CD103 and CD69 on the P14 T cells were determined by flow cytometry. Data are representative of 4 independent experiments. See also Figure S3.
To directly test the retention of Tgfbr2−/− T cells in the gut, we modified the protocol used to purify IEL (see Methods). In short, a more gentle isolation step was added before the standard vigorous step. Thus, the IEL were divided into a loosely attached (from the gentle step) and a tightly attached (from the vigorous step) population. Compared to control P14 T cells, Tgfbr2−/− P14 cells were enriched in the loose IEL population following both acute and chronic LCMV infection (Figure 4). Considering the facts that control and Tgfbr2−/− T cells exhibited comparable apoptosis (Figure S4A) and proliferation (Figure S4B), we concluded that defective retention of Tgfbr2−/− T cells was observed following both acute and chronic LCMV infection. These results left open the question: Why was the Tgfbr2−/− CD8+ Trm cell population in the gut maintained normally during chronic LCMV infection?
Figure 4. Defective retention of TGF-βRII-deficient T cells in the gut IEL.

Similar experimental setup as in Figure 1. At day 8 and 12 post LCMV Arm infection and day 8 and 15 post LCMV Cl13 infection, IEL cells were purified following a two-step protocol. The ratio of control vs TGF-βRII-deficient P14 T cells in the tight and loose IEL populations is shown. Each pair of symbols represents an individual recipient mouse. Combined data from 3 independent experiments are shown. See also Figure S4.
Increased expression of integrin α4β7 on Tgfbr2 −/− P14 cells in the spleen and peripheral blood
To answer the above question, we examined the P14 T cells from the secondary lymphoid organs, that were the major source of intestinal Trm cells. Interestingly, we found that the expression of integrin β7 and α4β7 was increased on Tgfbr2−/− T cells from the spleen (Figure 5A) and peripheral blood (Figure 5B). The effect was more dramatic during LCMV Clone 13 infection compared to Armstrong infection (Figure 5A and 5B). To address the possibility that naïve TGF-βRII-deficient P14 T cells were already programmed to express higher level of α4β7 than control P14 cells upon activation, the effect of TGF-β signaling on WT P14 T cells was directly tested in vitro. As shown in Figure S5, TGF-β dramatically inhibited the expression of α4β7 on activated WT P14 T cells in vitro. We concluded that TGF-β signaling downregulated the expression of integrin α4β7 on effector CD8+ T cells.
Figure 5. Increased expression of integrin α4β7 on TGF-βRII-deficient CD8+ T cells in the spleen and peripheral blood.
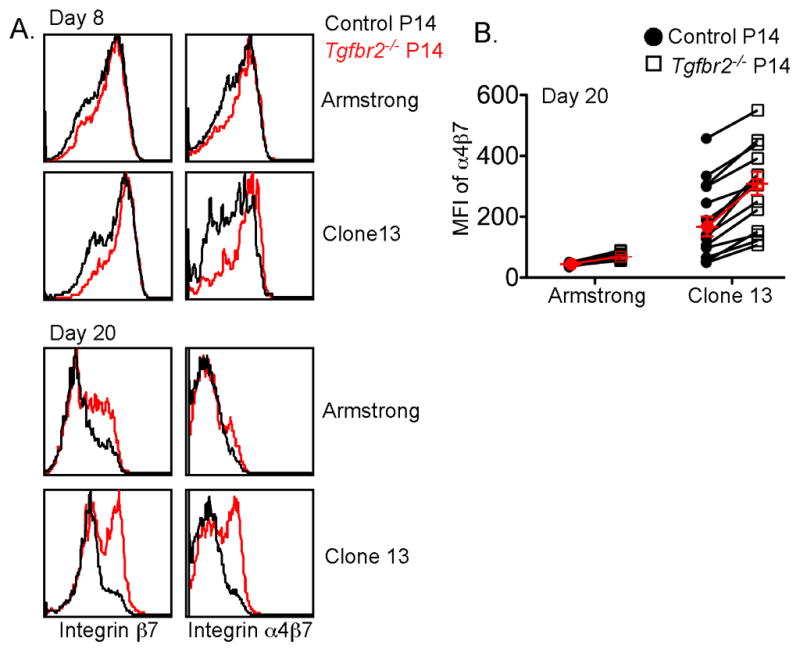
Similar experimental setup as in Figure 1. On day 8 and 20 post infection, the expression of integrin α4β7 and β7 by P14 T cells in the spleen (A) and peripheral blood (B) was examined. Each pair of symbols in (B) represents an individual recipient mouse. Red symbols represent the mean +/− SEM of 14–15 mice. Data are representative of 5 independent experiments. See also Figure S5.
Integrin α4β7 is required for the trafficking of activated T cells to the gut (Bargatze et al., 1995; Hamann et al., 1994). Thus, to explain the different outcomes in the maintenance of the gut Trm cells following acute versus chronic LCMV infection we reasoned as follows: During acute LCMV infection, virus was cleared around day 8 following infection. Under these conditions both control and Tgfbr2−/− CD8+ T cells quickly lost the expression of integrin α4β7 and the capacity to migrate to the gut. However, during chronic LCMV infection, virus persisted for a longer time. TGF-β signaling significantly inhibited the expression of integrin α4β7 giving Tgfbr2−/− CD8+ T cells a greatly enhanced capacity to migrate to the gut. In the intestinal tissues, TGF-β was required for the expression of integrin αEβ7 and the optimal upregulation of CD69 and integrin α1 so that TGF-β unresponsive T cells were poorly retained within the intestine. Therefore, during acute infection, without continuous recruits from secondary lymphoid organs, Tgfbr2−/− T cells quickly disappeared from the intestinal compartment. On the other hand, during chronic infection, enhanced continued migration of CD8+ T cells to the gut counterbalanced the defective maintenance of gut Trm cells in the absence of TGF-β signaling.
Increased migration of Tgfbr2 −/− CD8+ T cells to the gut following LCMV Clone 13 infection
To directly test the above hypothesis, we examined the gut-homing capacity of control and Tgfbr2−/− P14 T cells following acute and chronic infection. At day 9 following infection, pooled lymphocytes from the spleen and lymph nodes, containing a mix of control and Tgfbr2−/− P14 effector T cells, were transferred into naïve mice. At the time of transfer, dramatically enhanced expression of integrin α4β7 was observed only for the Tgfbr2−/− T cells from LCMV Clone 13 infected mice (Figure 6A). Nineteen hours after the cell transfer, donor control and Tgfbr2−/− P14 T cells in different tissues were quantified. Control and Tgfbr2−/− T cells from both LCMV Armstrong and Clone 13 infected mice showed comparable homing capacity to the spleen (Figure 6B). Control and Tgfbr2−/− T cells from acute LCMV infected mice exhibited a minimal, but comparable homing capacity to the intestinal IEL compartment. In sharp contrast, Tgfbr2−/− CD8+ T cells from chronic LCMV Clone 13 infected mice exhibited greatly enhanced homing capacity to the intestinal IEL compartment (Figure 6B). Similar results were observed when P14 T cells were transferred into infection-matched recipient mice (Figure S6).
Figure 6. Dramatically increased migration of TGF-βRII-deficient CD8+ T cells to the gut following LCMV Clone 13 infection.

Similar experimental setup as in Figure 1. On day 9 post infection, 2x107 pooled lymphocytes from spleen and lymph nodes were transferred into each naïve B6 mouse. 19 hours later, recipient mice were sacrificed and the number of transferred P14 T cells from different tissues was determined by flow cytometry and cell counting. (A) The expression of integrin α4β7 and β7 on the donor P14 T cells before secondary transfer. (B) The number of recovered P14 cells from the spleen and IEL compartment. Each pair of symbols represents an individual recipient mouse. Data are representative of two independent experiments. See also Figure S6.
Next, we tested whether the increased expression of integrin α4β7 resulted in the enhanced gut-homing capacity and maintenance of normal numbers of Tgfbr2−/− T cells from chronic LCMV infected mice. To this end, naïve P14 T cells from congenically marked control and Tgfbr2−/− mice were mixed and transferred into WT recipients followed by LCMV Clone 13 infection (Figure S7A). Starting at day 10 post-infection, integrin α4 blocking antibody or control antibody was administrated every other day. As shown in Figure 7A, anti-integrin α4 antibody efficiently blocked the surface staining of integrin α4β7. Anti-integrin α4 antibody inhibits the migration of lymphocytes to the mucosal tissues (Bargatze et al., 1995; Hamann et al., 1994). If our hypothesis were correct, blocking integrin α4 would lead to the defective maintenance of Tgfbr2−/− T cells in the intestine of LCMV Clone 13 infected mice. Indeed, we observed that the Tgfbr2−/− T cell population from the IEL compartment was significantly decreased in anti-integrin α4 antibody, but not control Ig treated animals (Figure 7B and 7C). Notably, anti-integrin α4 antibody treatment had minimal effects on splenic T cells (Figure 7C). To rule out the involvement of other integrins containing the α4 subunit, such as α4β1, the effect of anti-α4β7 blocking antibody was directly tested. Indeed, preincubation with α4β7 blocking antibody, but not control Ig, completely abolished the migration capacity of P14 T cells to the gut following LCMV clone 13 infection (Figure S7B and S7C). Furthermore, in contrast to the expression pattern of β7, the expression of integrin β1 was comparable between control and Tgfbr2−/− T cells following both acute and chronic LCMV infection (data not shown). Taken together, we concluded that the increased expression of integrin α4β7 led to the enhanced migration of Tgfbr2−/− T cells to the gut. Therefore, during chronic LCMV infection, Tgfbr2−/− P14 T cells exhibited enhanced and prolonged gut-homing capacity that compensated for the defective maintenance of the gut Trm cell population in the absence of TGF-β signaling.
Figure 7. Blocking integrin α4 results in defective maintenance of TGF-βRII-deficient CD8+ T cells in the gut following chronic LCMV infection.
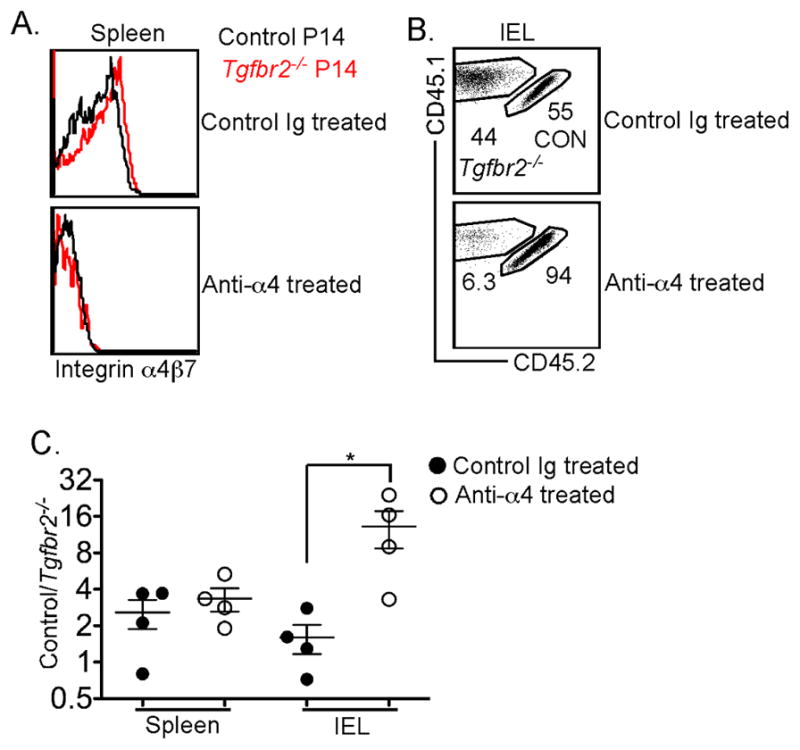
Naïve P14 T cells were purified from control (CD45.1+CD45.2+) and TGF-βRII-deficient (CD45.1+) mice, mixed at a 1:1 ratio, and 104 cells transferred into sex-matched B6 recipients followed by LCMV Clone 13 infection. Starting at day 10 post infection, 400μg anti-integrin α4 antibody (clone PS/2) or control Ig were injected intraperitoneally every other day (Figure S7A). The expression of integrin α4β7 on splenic P14 T cells is shown in A. Representative FACS profiles of the P14 T cells in the IEL compartment are shown in B. (C) The ratio of control vs Tgfbr2−/− P14 T cells in spleen and IEL is shown. Each symbol in C represents an individual mouse. *, p<0.05 (Student t-test). Data in C are represented as mean +/− SEM. See also Figure S7 and S8.
Decreased Tgfbr2−/− Trm population in the IEL compartment after clearance of chronic LCMV infection
Following LCMV clone 13 infection, WT mice control and eliminate virus in most tissues around two months after infection (Lauterbach et al., 2007; Sevilla et al., 2000; Wherry et al., 2003). To further validate our hypothesis, we examined the IEL compartment after the clearance of LCMV clone 13 infection. Elimination of the virus reduced the amounts of viral antigen and the population of activated CD8+ T cells that was the major CD8+ T cell population with efficient gut-homing capacity. If our hypothesis were correct, after the clearance of chronic viral infection, Tgfbr2−/− P14 T cells would decline in the intestinal IEL compartment similar to the situation after acute viral infection. Indeed, we found that Tgfbr2−/− P14 T cells were significantly reduced compared to control cells in the IEL compartment at 71 days and further reduced at day 105 post infection with LCMV Clone 13 (Figure S8).
Discussion
Our results have revealed two distinct functions of TGF-β signaling during the formation and maintenance of the intestinal Trm cell population during systemic viral infection. In secondary lymphoid organs, TGF-β signaling to T cells inhibits the upregulation of integrin α4β7 and thus the intestinal homing capacity of effector CD8+ T cells. In the gut, TGF-β signaling is critical for the maintenance of Trm cells through the induction of integrins αEβ7, α1 and CD69.
The TGF-β mediated suppression of α4β7 expression and gut homing was most apparent following chronic infection with LCMV Clone 13. During acute LCMV Armstrong infection, the enhanced migration of Tgfbr2−/− P14 cells to the gut was not substantial, but still present. Tgfbr2−/− P14 cells expressed slightly but consistently higher level of integrin α4β7 during LCMV Armstrong infection. At an early time point (day 8) following LCMV Armstrong infection, the ratio of control/Tgfbr2−/− P14 T cells was significantly lower for the IEL compartment compared to that of the spleen (control/Tgfbr2−/− =2.42±0.27 for spleen; 1.21±0.10 for IEL). Therefore, TGF-β unresponsive P14 T cells had an enhanced ability to traffic to the gut IEL compartment during both acute and chronic LCMV infection. However, following acute LCMV Armstrong infection, responding T cells lost the gut-homing capacity shortly after clearance of the virus, which limited the role of TGF-β signaling to a narrow window of active infection. Only following chronic infection TGF-β played a major and extended role during the formation of intestinal Trm cells.
TGF-β signaling was also essential for the maintenance of intestinal Trm cells. Although TGF-β is generally accepted as a key cytokine to induce the Trm cell phenotype and promote Trm cell retention, previous studies focused on the effector, but not memory phase of an immune response (Casey et al., 2012; El-Asady et al., 2005). There is no evidence in the literature to show that lacking or diminishing TGF-β responsiveness leads to the loss of Trm cells in the memory phase of an immune response. Here we showed that the molecules involved in Trm cell retention, such as integrins αEβ7, α1 and CD69, were dramatically reduced and the long-term maintenance of Trm cells was severely defective in the absence of TGF-β signaling. As a caveat, it should be noted that the effects of TGF-β signaling on the expression of integrin β7 might not be direct because in CD103 deficient mice, the expression of integrin β7 on the gut IEL lymphocytes was also significantly reduced (Schon et al., 1999). CD103 deficient P14 T cells exhibit similar, but less dramatic defects to those exhibited by TGF-β unresponsive P14 T cells (Casey et al., 2012; Schon et al., 1999). Thus, the defective induction of CD103 alone cannot fully explain the defects in TGF-β unresponsive resident T cells. The expression of integrin α1 may contribute to the retention of effector T cells in the mucosal tissues (Meharra et al., 2000; Roberts et al., 1999; Sandoval et al., 2013). In addition, CD69 inhibits the function of S1P1 (Cyster and Schwab, 2012), which mediate T cell egress from the lymphoid organs. Therefore, diminished expression of integrins αEβ7, α1 and CD69 together may explain the defective maintenance of TGF-β unresponsive T cells in the gut. Although more comprehensive analysis is needed to evaluate the homeostasis of TGF-βRII-deficient Trm cells, no apparent defects in proliferation and apoptosis were observed. Interestingly, TGF-βRII-deficient Trm cells were consistently enriched in the loosely attached IEL population following both acute and chronic LCMV infection. Together, these data suggest that promoting retention is the main function of TGF-β signaling in gut Trm cells. The short life span (3–5 days) of epithelial cells along the small intestine villi is accompanied by the constant division of progenitor cells in the crypts, moving of newly generated epithelial cells towards the tips of villi and shedding of the epithelial cells into the lumen from the tips (Clevers and Batlle, 2013). The retention of Trm cells in such a dynamic microenvironment is critical to maintain the cell population. Our data suggest that the downstream targets of TGF-β signaling cooperatively mediate the retention of gut Trm cells.
It has been reported previously that CD8+ T cells carrying a dominant-negative TGF-βRII transgene exhibit enhanced expansion during both acute and chronic infections thought to be due to increased survival (Sanjabi et al., 2009; Tinoco et al., 2009). In contrast, we consistently observed a slightly reduced accumulation of TGF-βRII-deficient CD8+ T cells in response to a variety of pathogens. There are several possibilities that may account for the opposite outcomes of two different models for suppression of TGF-β signaling. First, the dnTGF-βRII transgene is driven by the CD4 promoter (Gorelik and Flavell, 2000) that is activated at the double positive (DP) thymocyte stage of T cell development. Distal Lck-cre is activated after thymocyte positive selection (Hale et al., 2010; Zhang et al., 2005). Whether interfering with TGF-β signaling during DP thymocyte development has a long lasting effect on mature naïve T cell expansion remains unknown. Second, the dnTGF-βRII transgene is of human origin. Direct transfer of CD8+ T cells carrying the transgene into WT recipient mice results in the rapid rejection of the donor cells (Sanjabi and Flavell, 2010). This may be the main reason why in previous reports utilizing dnTGF-βRII mice, only the effector, but not the memory phase of an immune response was studied. Transferring dnTGF-βRII T cells into CD11c-dnTGF-βRII transgenic mice overcomes the problem of rapid rejection of the donor cells (Sanjabi and Flavell, 2010) but such recipient mice contain altered populations of dendritic cells and NK cells (Laouar et al., 2005; Laouar et al., 2008), which may affect the CD8+ T cell response. Third, the results from direct infection of dnTGF-βRII T cell transgenic mice have a potential caveat. Although dnTGF-βRII T cell transgenic mice remain healthy for 3–4 months (Gorelik and Flavell, 2000), the asymptomatic young adult mice may harbor a pro-inflammatory environment with an unexpected impact on the CD8+ T cell response. Indeed, recent studies have questioned some conclusions based on the data obtained from dnTGF-βRII mice following chronic LCMV infection (Boettler et al., 2012; Garidou et al., 2012). Furthermore, a recent publication has documented functions of the dnTGF-βRII transgene that are independent of endogenous TGF-βRII signaling, which are responsible for the enhanced expansion of effector CD8+ T cells and the development of transformed memory T cells (Ishigame et al., 2013). In contrast, in our system, a small number of control and TGF-βRII deficient naïve P14 T cells were co-transferred into WT recipient mice followed by LCMV infection so that the function of TGF-β signaling to CD8+ T cells could be examined under more physiological conditions. It is also to be noted that the inhibition of TGF-β signaling by the dnTGF-βRII transgene is not complete (Gorelik and Flavell, 2000). Thus it is that a significant number of dnTGF-βRII expressing CD8+ T cells differentiate into CD103+ cells within the intestinal IEL compartment (Casey et al., 2012; El-Asady et al., 2005). On the contrary, in our experiments TGF-βRII-deficient CD8+ T cells completely lost the ability to upregulate CD103 expression within the intestinal epithelium.
It has been reported that during chronic infection, intestinal Trm CD8+ T cells exhibit a decreased ability to upregulate Trm signature molecules including CD103 and CD69 (Casey et al., 2012). It has been proposed that persistent antigen stimulation inhibits the acquisition of Trm signatures in the peripheral tissues except for the brain (Casey et al., 2012), where antigen stimulation is required for CD103 induction (Wakim et al., 2010). Our results suggest another possible explanation. Compared to the narrow window for responding CD8+ T cells to migrate to the gut during an acute infection, there is a prolonged period of migration to the gut during chronic infection. Therefore, the CD103− cells observed in the intestinal IEL compartment during LCMV Clone 13 infection may represent recent arrivals from the secondary lymphoid organs. The prolonged period of migration to the gut may also explain the interesting phenomenon that total CD8+ T cells persist at a higher level in the IEL compartment from chronically infected mice compared to that from acutely infected mice.
Together, our data demonstrate that TGF-β signaling inhibits the migration of effector CD8+ T cells from the secondary lymphoid organs to the intestine by controlling the expression of integrin α4β7. In the intestinal tissues, TGF-β signaling is required for the long-term maintenance of Trm cells at least in part through the induction of integrins αEβ7, α1 and CD69. The mechanisms underlying the tissue and stage-specific function of TGF-β signaling warrant future investigation, which may represent another example of context-dependent outcomes of TGF-β signaling (Massague, 2012).
Methods
Mice and Viruses
Tgfbr2f/f dLck-cre mice were as described (Zhang and Bevan, 2012). C57BL/6 (stock no. 000664) mice were obtained from The Jackson Laboratory and a colony of DbGP33–41 TCR transgenic (P14) mice was maintained at our specific pathogen-free animal facilities at the University of Washington (Seattle, WA). All recipient mice were used at 6 to 12 wk of age. All experiments were done in accordance with the University of Washington Institutional Animal Care and Use Committee guidelines. Mice were infected intraperitoneally with 2x105 pfu LCMV Armstrong or intravenously with 2x106 pfu LCMV Clone 13. Viruses were grown and quantified as described (Ahmed et al., 1984).
Adoptive Transfer
Naive P14 T cells were isolated from spleen and lymph nodes using a CD8 isolation kit (Miltenyi) following the manufacturer’s instruction but with the addition of biotin-αCD44 antibody during the biotin antibody cocktail incubation step. The indicated number of naïve P14 T cells was transferred intravenously into appropriate recipient mice. For the in vivo migration assay, at indicated time post infection, 2–3x107 total lymphocytes from pooled spleen and lymph nodes were transferred intravenously into sex-matched recipient mice. In some experiments, cells were incubated with 500μg blocking or control antibody in 300μl PBS on ice for 1 hour and then transferred into recipients without washing. Nineteen hours later, donor P14 T cells were quantified from the spleen and gut IEL compartment of the recipient mice.
Purification of the IEL
For most experiments, the standard protocol was followed. Briefly, small pieces of the small intestine were stirred at 800 rpm for 20 minutes in HBSS buffer containing 1mM dithiothreitol and 10% FBS followed by a 15 second high speed vortex. The released cells were purified by gradient centrifugation using 44% and 67% Percoll. For the experiments shown in Figure 4, a more gentle isolation step was performed before the standard vigorous step. Small pieces of the small intestine were stirred at 400 rpm for 10 minutes in HBSS buffer in the absence of dithiothreitol. The vortex step was omitted for the gentle isolation. The released cells from the gentle (loosely attached IEL) and the standard (tightly attached IEL) steps were subjected to gradient centrifugation and FACS analysis.
Antibodies and Flow Cytometry
Single-cell suspensions were prepared from the spleen, lymph nodes and small intestine after perfusion of the animal at the indicated time-points post infection. Cells were typically stained with antibodies specific for CD8, CD103, CD90.1, CD69, CD49a (integrin α1), integrin β7, integrin α4β7, TCRβ, CD45.1, CD45.2, and PD-1 (eBioscience, Biolegend and BD) and TGF-βRII (R&D Systems). For in vivo antibody blocking, anti-integrin α4 antibody (CD49d, clone PS/2) was obtained from the UCSF monoclonal antibody core. Anti-integrin α4β7 (clone DATK-32) was a generous gift from Dr. Eugene Butcher. Cells were analyzed using a FACSCanto (BD) and analyzed using FLOWJO (TreeStar) software.
Supplementary Material
Acknowledgments
We thank Dr. Eugene C. Butcher for providing the α4β7 antibody; Drs. Erin Williams and Jason Netland for the production and titration of LCMV; and Xiao-Cun Pan and Mary Chase for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MB. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J Exp Med. 1984;160:521–540. doi: 10.1084/jem.160.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargatze RF, Jutila MA, Butcher EC. Distinct roles of L-selectin and integrins alpha 4 beta 7 and LFA-1 in lymphocyte homing to Peyer’s patch-HEV in situ: the multistep model confirmed and refined. Immunity. 1995;3:99–108. doi: 10.1016/1074-7613(95)90162-0. [DOI] [PubMed] [Google Scholar]
- Berlin C, Bargatze RF, Campbell JJ, von Andrian UH, Szabo MC, Hasslen SR, Nelson RD, Berg EL, Erlandsen SL, Butcher EC. alpha 4 integrins mediate lymphocyte attachment and rolling under physiologic flow. Cell. 1995;80:413–422. doi: 10.1016/0092-8674(95)90491-3. [DOI] [PubMed] [Google Scholar]
- Berlin C, Berg EL, Briskin MJ, Andrew DP, Kilshaw PJ, Holzmann B, Weissman IL, Hamann A, Butcher EC. Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell. 1993;74:185–195. doi: 10.1016/0092-8674(93)90305-a. [DOI] [PubMed] [Google Scholar]
- Boettler T, Cheng Y, Ehrhardt K, von Herrath M. TGF-beta blockade does not improve control of an established persistent viral infection. Viral Immunol. 2012;25:232–238. doi: 10.1089/vim.2011.0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, Lucas PJ, Artis D, Wherry EJ, Hogquist K, et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol. 2012;188:4866–4875. doi: 10.4049/jimmunol.1200402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepek KL, Shaw SK, Parker CM, Russell GJ, Morrow JS, Rimm DL, Brenner MB. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the alpha E beta 7 integrin. Nature. 1994;372:190–193. doi: 10.1038/372190a0. [DOI] [PubMed] [Google Scholar]
- Choy MY, Richman PI, Horton MA, MacDonald TT. Expression of the VLA family of integrins in human intestine. J Pathol. 1990;160:35–40. doi: 10.1002/path.1711600109. [DOI] [PubMed] [Google Scholar]
- Clevers H, Batlle E. SnapShot: the intestinal crypt. Cell. 2013;152:1198–1198. e1192. doi: 10.1016/j.cell.2013.02.030. [DOI] [PubMed] [Google Scholar]
- Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol. 2012;30:69–94. doi: 10.1146/annurev-immunol-020711-075011. [DOI] [PubMed] [Google Scholar]
- El-Asady R, Yuan R, Liu K, Wang D, Gress RE, Lucas PJ, Drachenberg CB, Hadley GA. TGF-{beta}-dependent CD103 expression by CD8(+) T cells promotes selective destruction of the host intestinal epithelium during graft-versus-host disease. J Exp Med. 2005;201:1647–1657. doi: 10.1084/jem.20041044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garidou L, Heydari S, Gossa S, McGavern DB. Therapeutic blockade of transforming growth factor beta fails to promote clearance of a persistent viral infection. J Virol. 2012;86:7060–7071. doi: 10.1128/JVI.00164-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol. 2009;10:524–530. doi: 10.1038/ni.1718. [DOI] [PubMed] [Google Scholar]
- Gebhardt T, Whitney PG, Zaid A, Mackay LK, Brooks AG, Heath WR, Carbone FR, Mueller SN. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature. 2011;477:216–219. doi: 10.1038/nature10339. [DOI] [PubMed] [Google Scholar]
- Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–181. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- Hale JS, Ames KT, Boursalian TE, Fink PJ. Cutting Edge: Rag deletion in peripheral T cells blocks TCR revision. J Immunol. 2010;184:5964–5968. doi: 10.4049/jimmunol.1000876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann A, Andrew DP, Jablonski-Westrich D, Holzmann B, Butcher EC. Role of alpha 4-integrins in lymphocyte homing to mucosal tissues in vivo. J Immunol. 1994;152:3282–3293. [PubMed] [Google Scholar]
- Hawke S, Stevenson PG, Freeman S, Bangham CR. Long-term persistence of activated cytotoxic T lymphocytes after viral infection of the central nervous system. J Exp Med. 1998;187:1575–1582. doi: 10.1084/jem.187.10.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann M, Pircher H. E-cadherin promotes accumulation of a unique memory CD8 T-cell population in murine salivary glands. Proc Natl Acad Sci U S A. 2011;108:16741–16746. doi: 10.1073/pnas.1107200108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishigame H, Mosaheb MM, Sanjabi S, Flavell RA. Truncated Form of TGF-betaRII, But Not Its Absence, Induces Memory CD8+ T Cell Expansion and Lymphoproliferative Disorder in Mice. J Immunol. 2013 doi: 10.4049/jimmunol.1300397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature. 2012;483:227–231. doi: 10.1038/nature10851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson-Lindbom B, Svensson M, Wurbel MA, Malissen B, Marquez G, Agace W. Selective generation of gut tropic T cells in gut-associated lymphoid tissue (GALT): requirement for GALT dendritic cells and adjuvant. J Exp Med. 2003;198:963–969. doi: 10.1084/jem.20031244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilshaw PJ, Murant SJ. A new surface antigen on intraepithelial lymphocytes in the intestine. Eur J Immunol. 1990;20:2201–2207. doi: 10.1002/eji.1830201008. [DOI] [PubMed] [Google Scholar]
- Kim SK, Reed DS, Heath WR, Carbone F, Lefrancois L. Activation and migration of CD8 T cells in the intestinal mucosa. J Immunol. 1997;159:4295–4306. [PubMed] [Google Scholar]
- Laouar Y, Sutterwala FS, Gorelik L, Flavell RA. Transforming growth factor-beta controls T helper type 1 cell development through regulation of natural killer cell interferon-gamma. Nat Immunol. 2005;6:600–607. doi: 10.1038/ni1197. [DOI] [PubMed] [Google Scholar]
- Laouar Y, Town T, Jeng D, Tran E, Wan Y, Kuchroo VK, Flavell RA. TGF-beta signaling in dendritic cells is a prerequisite for the control of autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2008;105:10865–10870. doi: 10.1073/pnas.0805058105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterbach H, Truong P, McGavern DB. Clearance of an immunosuppressive virus from the CNS coincides with immune reanimation and diversification. Virol J. 2007;4:53. doi: 10.1186/1743-422X-4-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455–471. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, Heath WR, Carbone FR, Gebhardt T. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci U S A. 2012;109:7037–7042. doi: 10.1073/pnas.1202288109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity. 2006;25:441–454. doi: 10.1016/j.immuni.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, Wang J, Casey KA, Barber DL, Kawamura KS, et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med. 2010;207:553–564. doi: 10.1084/jem.20090858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;291:2413–2417. doi: 10.1126/science.1058867. [DOI] [PubMed] [Google Scholar]
- Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meharra EJ, Schon M, Hassett D, Parker C, Havran W, Gardner H. Reduced gut intraepithelial lymphocytes in VLA1 null mice. Cell Immunol. 2000;201:1–5. doi: 10.1006/cimm.2000.1630. [DOI] [PubMed] [Google Scholar]
- Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, Von Andrian UH. Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature. 2003;424:88–93. doi: 10.1038/nature01726. [DOI] [PubMed] [Google Scholar]
- Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T Cell Subsets, Migration Patterns, and Tissue Residence. Annu Rev Immunol. 2012 doi: 10.1146/annurev-immunol-032712-095954. [DOI] [PubMed] [Google Scholar]
- Roberts AI, Brolin RE, Ebert EC. Integrin alpha1beta1 (VLA-1) mediates adhesion of activated intraepithelial lymphocytes to collagen. Immunology. 1999;97:679–685. doi: 10.1046/j.1365-2567.1999.00812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- Sandoval F, Terme M, Nizard M, Badoual C, Bureau MF, Freyburger L, Clement O, Marcheteau E, Gey A, Fraisse G, et al. Mucosal imprinting of vaccine-induced CD8(+) T cells is crucial to inhibit the growth of mucosal tumors. Sci Transl Med. 2013;5:172ra120. doi: 10.1126/scitranslmed.3004888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjabi S, Flavell RA. Overcoming the hurdles in using mouse genetic models that block TGF-beta signaling. J Immunol Methods. 2010;353:111–114. doi: 10.1016/j.jim.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjabi S, Mosaheb MM, Flavell RA. Opposing effects of TGF-beta and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity. 2009;31:131–144. doi: 10.1016/j.immuni.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkel JM, Fraser KA, Vezys V, Masopust D. Sensing and alarm function of resident memory CD8(+) T cells. Nat Immunol. 2013;14:509–513. doi: 10.1038/ni.2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon MP, Arya A, Murphy EA, Adams CM, Strauch UG, Agace WW, Marsal J, Donohue JP, Her H, Beier DR, et al. Mucosal T lymphocyte numbers are selectively reduced in integrin alpha E (CD103)-deficient mice. J Immunol. 1999;162:6641–6649. [PubMed] [Google Scholar]
- Sevilla N, Kunz S, Holz A, Lewicki H, Homann D, Yamada H, Campbell KP, de La Torre JC, Oldstone MB. Immunosuppression and resultant viral persistence by specific viral targeting of dendritic cells. J Exp Med. 2000;192:1249–1260. doi: 10.1084/jem.192.9.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan BS, Lefrancois L. Regional and mucosal memory T cells. Nat Immunol. 2011;12:485–491. doi: 10.1038/ni.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiow LR, Rosen DB, Brdickova N, Xu Y, An J, Lanier LL, Cyster JG, Matloubian M. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature. 2006;440:540–544. doi: 10.1038/nature04606. [DOI] [PubMed] [Google Scholar]
- Stagg AJ, Kamm MA, Knight SC. Intestinal dendritic cells increase T cell expression of alpha4beta7 integrin. Eur J Immunol. 2002;32:1445–1454. doi: 10.1002/1521-4141(200205)32:5<1445::AID-IMMU1445>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Tang VA, Rosenthal KL. Intravaginal infection with herpes simplex virus type-2 (HSV-2) generates a functional effector memory T cell population that persists in the murine genital tract. J Reprod Immunol. 2010;87:39–44. doi: 10.1016/j.jri.2010.06.155. [DOI] [PubMed] [Google Scholar]
- Tinoco R, Alcalde V, Yang Y, Sauer K, Zuniga EI. Cell-intrinsic transforming growth factor-beta signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity. 2009;31:145–157. doi: 10.1016/j.immuni.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakim LM, Waithman J, van Rooijen N, Heath WR, Carbone FR. Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science. 2008;319:198–202. doi: 10.1126/science.1151869. [DOI] [PubMed] [Google Scholar]
- Wakim LM, Woodward-Davis A, Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci U S A. 2010;107:17872–17879. doi: 10.1073/pnas.1010201107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DJ, Wang Q, Wei J, Baimukanova G, Buchholz F, Stewart AF, Mao X, Killeen N. Selective expression of the Cre recombinase in late-stage thymocytes using the distal promoter of the Lck gene. J Immunol. 2005;174:6725–6731. doi: 10.4049/jimmunol.174.11.6725. [DOI] [PubMed] [Google Scholar]
- Zhang N, Bevan MJ. TGF-beta signaling to T cells inhibits autoimmunity during lymphopenia-driven proliferation. Nat Immunol. 2012;13:667–673. doi: 10.1038/ni.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.