

Abstract
The narrow species tropism of hepatitis C virus (HCV) limits animal studies. We found that pigtail macaque (Macaca nemestrina) hepatic cells derived from induced pluripotent stem cells support the entire HCV life cycle, although infection efficiency was limited by defects in the HCV cell entry process. This block was overcome by either increasing occludin expression, complementing the cells with human CD81, or infecting them with a strain of HCV with less-restricted requirements for CD81. Using this system, we can modify viral and host cell genetics to make pigtail macaques a suitable, clinically relevant model for the study of HCV infection.
Keywords: animal model, monkey, hepatitis C virus, replication
The hepatitis C virus (HCV) is responsible for over half of all liver cancers and the majority of liver transplants worldwide1. No HCV vaccine is available, and treatment is often ineffective and complicated by side effects and viral resistance2. HCV is only known to naturally infect humans and chimpanzees (reviewed in 3). Due to the moratorium on chimpanzee research, and associated ethical and financial issues, an alternative animal model is needed to study HCV in vivo pathogenesis and replication, and to develop antivirals and vaccines. Here, we sought to determine whether pigtail macaque (Macaca nemestrina, Mn) derived hepatocyte-like (MnHep) cells support HCV replication, with the ultimate goal of developing an immunocompetent nonhuman primate HCV animal model.
Several groups have recently shown that human induced pluripotent and embryonic stem cells can be differentiated into hepatic cells that support HCV infection4–7. We used a similar approach to differentiate MnHep cells from induced pluripotent stem cells (MniPSC)8 (Supplemental Figure 1). To test their ability to support HCV infection, MnHep and Huh-7.5 cells were challenged with infectious HCV derived in cell culture (HCVcc; HCV cell cultured systems reviewed in 9). MnHep cells supported HCVcc infection and replication, as gauged by increased intracellular HCV RNA over time and HCV E2 glycoprotein cell staining, both of which were inhibited by the HCV polymerase inhibitor 2′C-methyl-adenosine (2′CMA)10 (Figure 1A). However, this process was inefficient, as HCV RNA levels in MnHep cells were 13.1-fold lower than levels observed in positive control human Huh-7.5 cells. It should be noted that even approaching Huh-7.5 HCV infection levels is an achievement, as most primary cell culture systems allow infections that are dramatically less efficient. We believe that stem cell derived hepatic cells are simply better hosts for HCV replication than cultured primary cells. Indeed, we recently showed that human stem cell derived hepatic cells support HCV infection with similar efficiency as compared with Huh-7.5 cells11. HCV infected Huh-7.5 and MnHep cells secreted proportional amounts of HCV RNA (Figure 1B) that was shown to be associated with infectious virus on Huh-7.5 cells (Figure 1D), and the amount of HCV RNA secreted by Huh-7.5 and MnHep cells was proportional to the amount of infectious virus in the subsequent infection experiments. Thus, while MnHep cells support HCV infection, at least one step of the viral life cycle may be inefficient in these cells.
Figure 1. MnHep cells support HCV infection.
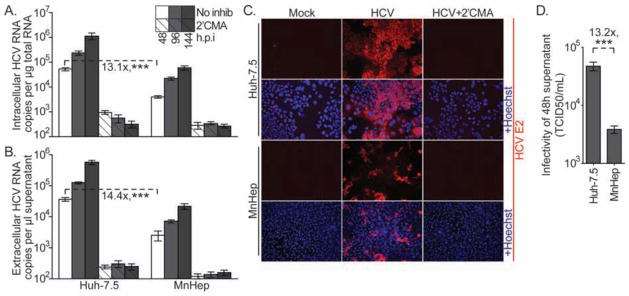
To gauge their capacity to support HCV infection, Huh7.5 and MnHep cells were challenged with wild type HCV that does not encode a reporter gene. At the indicated times post transfection (A) intracellular and (B) extracellular RNA was quantified by qRT-PCR. 2′CMA, an HCV polymerase inhibitor, was included in parallel infections to demonstrate replication independent RNA levels. Values represent the number of HCV RNA copies determined relative to a standard curve in relationship to the quantity of total RNA for intracellular samples, or volume of supernatant for extracellular samples. (C) At 96 hours post infection (h.p.i.), mock or HCV infected cells, with or without 2′CMA, were immunostained for the HCV E2 glycoprotein (red) and Hoechst counterstained (blue). (D) Infectious HCV in supernatants collected at 48 h.p.i. were quantified by limiting dilution assay on Huh-7.5 cells and expressed as 50% tissue culture infectious dose per ml (TCID50/ml). Means and standard deviations of three independent experiments, each performed in triplicate. *** P<0.001 (Mann-Whitney test)
HCV cell entry is one stage of the viral life cycle that influences both the tissue and species tropism of HCV infection12. To specifically test their capacity to support HCV cell entry, MnHep and Huh-7.5 cells were challenged with lentiviral particles bearing the HCV envelope glycoproteins (HCVpp) that encoded Gaussia luciferase (GLuc)(reviewed in 9). Following normalization to parallel VSVGpp infections to control for subtle differences in cell numbers, MnHep (expressing GFP alone as a negative control) were 7.9-fold less infectable than Huh-7.5 cells (Figure 2A). Importantly, the level of entry into MnHep cells was dependent on authentic HCV glycoprotein function, as infection was inhibited by an HCV E2 glycoprotein neutralizing antibody13.
Figure 2. Cellular and viral determinants of MnHep cell HCV entry capacity.

MnHep cells transduced to express GFP alone as a negative control or the indicated HCV cell entry factors were challenged with GLuc expressing (A) HCVpp or (B) HCVcc. Infections were performed in parallel with an HCV neutralizing antibody (Anti-E2). GLuc values were measured 48 h.p.i. for HCVpp or the indicated times for HCVcc, normalized to parallel VSVGpp infections, and set relative to infection of highly permissive Huh-7.5 cells. Three independent experiments, each performed with six replicates, and statistics are relative to GFP alone. Naïve Huh-7.5 and MnHep cells were challenged with mouse CD81 adapted HCVcc and (C) intracellular and (D) extracellular HCV RNA levels were assayed as described above. Three independent experiments, each performed in triplicate. *** P<0.001 (Mann-Whitney test)
Of the many cellular factors required for HCV cell entry, four influence HCV tropism (reviewed in 14). Cell type expression patterns of scavenger receptor class B type I (SR-BI), and claudin-1 (CLDN1) affect the tissue tropism of this process, while sequence differences in CD81 and occludin (OCLN) influence species tropism. All four of these factors were expressed in MnHep cells (Supplemental Figure 2A–C). Overexpression of the human versions of SR-BI and CLDN1 in these cells (monitored by immunoblot, Supplemental Figure 2E) did not enhance infection with HCVpp (Figure 2A) or HCVcc (Figure 2B). Thus, the Mn and human versions of these proteins were equally functional and present at saturating levels. Overexpression of either human or Mn OCLN enhanced infection with GLuc reporter expressing HCVpp and HCVcc (Figure 2A+B), which indicated that MnHep OCLN levels were limiting for HCV entry. It was not surprising that the OCLN proteins from both species functioned equally well, as sequences previously defined to be critical for this activity are similar (Supplemental Figure 4A) and both versions functioned equally in OCLN deficient human 786-O cells (Supplemental Figure 4B). Overexpression of human, but not Mn, CD81 enhanced HCVpp and HCVcc infection more than OCLN overexpression (Figure 2A and 2B). The Mn version of CD81 is identical to the African green monkey ortholog (Supplemental Figure 5A), which was previously shown to exhibit 25% of the function of the human protein15, and which we confirmed in human HepG2 cells (Supplemental Figure 5B). Co-transduction of human, but not Mn CD81, with either version of OCLN further enhanced HCVpp and HCVcc infection (Figure 2A+B). In summary, inefficient MnHep HCV cell entry caused by low OCLN expression levels and suboptimal activity of Mn CD81 limit could be overcome by genetic manipulation of these cells.
Bitzegeio et al. recently identified a mutant HCVcc that efficiently uses both human and mouse CD81 proteins16. This virus infected MnHep cells only 3.9-fold less efficiently than Huh-7.5 cells (Figure 2C) and these cells released proportional levels of HCV RNA (Figure 2D). Thus, based on our finding that wild type HCVcc infected MnHep cells 13.1-fold less efficiently than Huh-7.5 cells, the mouse CD81 adapted virus infects MnHep cells 3.4-fold better than wild type HCVcc (P< 0.001). These findings indicate that the low levels of HCV entry observed in MnHep cells can be overcome by genetic viral adaptation.
In summary, we show that hepatic cells derived from MniPSCs support HCV infection, which was further enhanced by modifications to both viral and cellular proteins. This significant finding challenges the historical assumption that only human and chimpanzee cells are susceptible to HCV. Indeed, a prior study failed to demonstrate HCV infection of rhesus macaques17. It is possible that the entry block described above was enough to prevent infection in this study. It is also feasible that the particularly strong replication kinetics of the JFH-1 strain used in our study were required to observably infect cells from macaques. Nevertheless, our study suggests that the pigtail macaque may indeed be a suitable model for studying HCV infection. Given the current restriction of chimpanzee research, the development of an alternative clinically relevant nonhuman model is critical for in vivo testing of novel HCV therapies. Our system provides a platform to study HCV-host interactions that influence the efficiency of HCV infection of these cells, which should lead to more tractable nonhuman animal models.
Supplementary Material
Acknowledgments
Grant Support:
M.S. was supported by the Robin Chemers Neustein Postdoctoral Fellowship. H.P.K. is a Markey Molecular Medicine investigator and the recipient of the Jose Carreras/E.D. Thomas Chair for Cancer Research, and NIH grants R01 HL098489 and P51 RR00016. V.G.E was supported by the Black Family Stem Cell Institute at the Icahn School of Medicine at Mount Sinai. M.J.E. was supported by NIH grants R01 DK095125, R00 AI077800, and R56 AI091792, an American Cancer Society Research Scholar Grant (RSG-12-176-01-MPC) and the Pew Charitable Funds (M.J.E.).
Footnotes
Disclosures: none
Writing assistance: none
Author contributions:
M.S., O.G., J.L.G., H.P.K, V.G.E., and M.E. designed the study and experiments. M.S., O.G., and W.E. acquired and analyzed data. V.G.E. and M.E. drafted the manuscript, which all authors critically reviewed.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brown RS., Jr Nature. 2005;436:973–78. doi: 10.1038/nature04083. [DOI] [PubMed] [Google Scholar]
- 2.Marks KM, et al. Antivir Ther. 2012;17:1119–31. doi: 10.3851/IMP2424. [DOI] [PubMed] [Google Scholar]
- 3.Bukh J. Gastroenterology. 2012;142:1279–1287. e3. doi: 10.1053/j.gastro.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 4.Roelandt P, et al. J Hepatol. 2012;57:246–51. doi: 10.1016/j.jhep.2012.03.030. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz RE, et al. Proc Natl Acad Sci U S A. 2012;109:2544–8. doi: 10.1073/pnas.1121400109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu X, et al. PLoS Pathog. 2012;8:e1002617. doi: 10.1371/journal.ppat.1002617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshida T, et al. Biochem Biophys Res Commun. 2011;416:119–24. doi: 10.1016/j.bbrc.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 8.Zhong B, et al. Stem Cells Dev. 2011;20:795–807. doi: 10.1089/scd.2010.0343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vieyres G, et al. Methods. 2013;59:233–248. doi: 10.1016/j.ymeth.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 10.Carroll SS, et al. J Biol Chem. 2003;278:11979–84. doi: 10.1074/jbc.M210914200. [DOI] [PubMed] [Google Scholar]
- 11.Goldman O, et al. Cell Stem Cell. 2013;12:748–60. doi: 10.1016/j.stem.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shulla A, et al. Curr Opin Virol. 2012;2:725–32. doi: 10.1016/j.coviro.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Law M, et al. Nat Med. 2008;14:25–7. doi: 10.1038/nm1698. [DOI] [PubMed] [Google Scholar]
- 14.Ploss A, et al. Nature. 2009;457:882–6. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flint M, et al. J Virol. 2006;80:11331–42. doi: 10.1128/JVI.00104-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bitzegeio J, et al. PLoS Pathog. 2010;6:e1000978. doi: 10.1371/journal.ppat.1000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bukh J, et al. J Viral Hepat. 2001;8:228–31. doi: 10.1046/j.1365-2893.2001.00284.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.