

Abstract
Myostatin, a member of the TGF superfamily, is sufficient to induce skeletal muscle atrophy. Myostatin-induced atrophy is associated with increases in E3-ligase atrogin-1 expression and protein degradation and decreases in Akt/mechanistic target of rapamycin (mTOR) signaling and protein synthesis. Myostatin signaling activates the transcription factor Smad3 (Small Mothers Against Decapentaplegic), which has been shown to be necessary for myostatin-induced atrogin-1 expression and atrophy; however, it is not known whether Smad3 is sufficient to induce these events or whether Smad3 simply plays a permissive role. Thus, the aim of this study was to address these questions with an in vivo model. To accomplish this goal, in vivo transfection of plasmid DNA was used to create transient transgenic mouse skeletal muscles, and our results show for the first time that Smad3 expression is sufficient to stimulate atrogin-1 promoter activity, inhibit Akt/mTOR signaling and protein synthesis, and induce muscle fiber atrophy. Moreover, we propose that Akt/mTOR signaling is inhibited by a Smad3-induced decrease in microRNA-29 (miR-29) expression and a subsequent increase in the translation of phosphatase and tensin homolog (PTEN) mRNA. Smad3 is also sufficient to inhibit peroxisome proliferator-activated receptor-γ coactivator-1α (PGC1α) promoter activity and to increase FoxO (Forkhead Box Protein, Subclass O)-mediated signaling and the promoter activity of plasminogen activator inhibitor 1 (PAI-1). Combined, this study provides the first evidence that Smad3 is sufficient to regulate many of the events associated with myostatin-induced atrophy and therefore suggests that Smad3 signaling may be a viable target for therapies aimed at preventing myostatin-induced muscle atrophy.
Skeletal muscle mass is ultimately determined by the balance between protein synthesis and protein degradation (1). Thus, a net decrease in protein synthesis and/or a net increase in protein degradation can lead to a reduction in muscle mass, otherwise known as muscle atrophy. Muscle atrophy occurs in many different conditions, such as cancer cachexia, aging, heart failure, obesity, mechanical unloading, chronic obstructive pulmonary disease, renal disease, and HIV, and can markedly impact mobility, whole-body metabolism, disease resistance, and quality of life (2–4). Interestingly, these atrophic conditions are also associated with an elevation in the expression of myostatin, a member of the TGF superfamily and a negative regulator of muscle growth (5–12). This suggests that myostatin may play a role in the etiology of the muscle atrophy associated with these conditions. Indeed, elevated myostatin has been shown to induce atrophy and fibrosis and impair regeneration in skeletal muscle (13–16). As such, the inhibition of myostatin signaling has been proposed as a promising strategy for attenuating atrophy, increasing muscle mass, reducing fibrosis, and improving muscle regeneration in various pathological conditions (17).
Myostatin signaling is triggered by the binding of the mature myostatin peptide to plasma membrane-associated activin type IIA and IIB receptors (ActRIIA/IIB) (18). Activation of the ActRIIA/IIB, in turn, recruits and activates serine/threonine type 1 receptor kinases called activin receptor-like kinase-4 (ALK4) and ALK5, which subsequently phosphorylate the transcription factors Smad2 (Small Mothers Against Decapentaplegic) and Smad3 (18). Smad2 and Smad3 are also phosphorylated in response to TGF-β binding to TGF-β type II receptors (TGF-βRII) and to activins binding to ActRIIA/IIB, leading to the activation of ALK5 and ALK4/7, respectively (19, 20). Interestingly, both TGF-β and activin A are also implicated in skeletal muscle atrophy and fibrosis (21, 22). Once phosphorylated, Smad2 and Smad3 form a heterotrimeric complex by binding to another Smad2 or Smad3 and Smad4. This complex then translocates to the nucleus to positively or negatively regulate gene transcription by associating with various transcription factors, coactivators, and corepressors (23). Despite significant progress being made in identifying the various disease models in which myostatin levels are elevated, much less is known about the role of Smad signaling and the downstream transcriptional targets that mediate myostatin's atrophic effect on skeletal muscle in vivo (24).
Sartori et al (25) recently demonstrated that overexpression of a constitutively active (c.a.)-ALK5 mutant in vivo increased the promoter activity of the muscle-specific ubiquitin E3 ligase atrogin-1 gene and induced muscle fiber atrophy via a Smad3-dependent mechanism. Furthermore, Smad3 has been found to be necessary for myostatin-induced increases in atrogin-1 and atrophy in cultured myotubes (26, 27). These data suggest that myostatin-induced muscle fiber atrophy is, in part, due to a Smad3-mediated increase in atrogin-1 expression and increased ubiquitin proteasome-mediated protein degradation (27–29). Although Smad3 signaling may be necessary for these events, it remains to be determined whether Smad3 is sufficient to activate atrogin-1 expression and induce muscle fiber atrophy in vivo or whether Smad3 merely plays a permissive role in these events. Moreover, further investigation is required to identify other potential Smad signaling targets that may also play a role in myostatin-induced muscle atrophy.
Myostatin-induced muscle atrophy may be related not only to an increase in protein degradation but also to a decrease in protein synthesis. Indeed, cell culture studies have shown that increased myostatin is sufficient to induce a decrease in protein synthesis, whereas inhibition of myostatin signaling increases rates of protein synthesis in cell culture and in vivo (27, 30–35). Myostatin may inhibit protein synthesis by promoting the atrogin-1–mediated degradation of ribosomal proteins and translation initiation factors and/or by the inhibition of signaling through the Akt/mechanistic target of rapamycin (mTOR) pathway (26, 27, 34, 36, 37). Importantly, Smad3 signaling has been shown to be necessary for the myostatin-induced inhibition of the Akt/mTOR pathway (24, 26); however, the mechanism responsible for Smad3's effect on skeletal muscle Akt/mTOR signaling remains to be determined.
Results from recent studies may provide a potential link between Smad3 and Akt/mTOR signaling that involves the microRNA-29 (miR-29) family and the tumor suppressor, phosphatase and tensin homolog deleted on chromosome 10 (PTEN). Specifically, Smad3 has been shown to be necessary and sufficient to inhibit miR-29 expression in cultured myoblasts by directly binding to the miR-29 promoter (38). Importantly, miR-29 is known to regulate the translation of PTEN mRNA with the inhibition of miR-29 leading to increased PTEN protein expression (39–41). These data raise the possibility that myostatin-induced increases in Smad3 signaling could inhibit miR-29 expression, resulting in increased PTEN mRNA translation and a subsequent inhibition of Akt/mTOR signaling. To date, however, no studies have examined whether Smad3 is sufficient to inhibit the miR-29 expression, decrease PTEN mRNA translation, or inhibit Akt/mTOR signaling and protein synthesis in skeletal muscle in vivo.
Therefore, the overall aim of this study was to fill many of the aforementioned gaps in our knowledge regarding Smad3 signaling in skeletal muscle in vivo. Specifically, we aimed to determine whether Smad3 was sufficient to 1) regulate the promoters of genes involved in protein degradation and fibrosis, 2) inhibit the translation of PTEN mRNA, 3) inhibit Akt/mTOR signaling, 4) inhibit protein synthesis, and 5) induce muscle fiber atrophy in skeletal muscle.
Materials and Methods
Animals
Female FVB/N mice, 8 to 10 weeks old, were used for all conditions. Mice were housed under a 12-hour light, 12-hour dark cycle with ad libitum access to food and water unless otherwise stated. Before all surgical procedures, mice were anesthetized with an ip injection of ketamine (100 mg/kg) and xylazine (10 mg/kg). After tissue extraction, the mice were euthanized by cervical dislocation. All methods were approved by the Institutional Animal Care and Use Committee of the University of Wisconsin-Madison.
Plasmid constructs and purification
For all experiments, Smad3 overexpression was achieved by the in vivo transfection, or cotransfection, of a pCMV-3X hemagglutinin (HA)-tagged Smad3 plasmid construct (30 μg). LacZ (encoded by the pCMVβ plasmid) or green fluorescent protein (GFP) (encoded by the pEGFP-C3 plasmid) were cotransfected as control conditions. All other plasmid DNA constructs used for cotransfection in this study are listed in Supplemental Table 1 (published on The Endocrine Society's Journals Online website at http://mend.endojournals.org). Plasmid DNA was amplified in DH5α Escherichia coli, purified with an EndoFree plasmid kit (QIAGEN), and resuspended in sterile PBS.
In vivo transfection via electroporation
Sterile plasmid DNA was transfected into mouse tibialis anterior (TA) muscles by electroporation as described previously (42, 43). In brief, mice were anesthetized, and a small incision was made through the skin covering the TA muscle. A 27-gauge needle was used to inject plasmid DNA solution into the proximal (6 μL) and distal (6 μL) ends of the muscle belly. Details regarding the amount of DNA injected for each specific plasmid are provided in Supplemental Table 1. After the injections, electric pulses were applied through 2 stainless steel pin electrodes (1-cm gap; Harvard Apparatus) laid on top of the proximal and distal myotendinous junctions. Eight 20-millisecond square-wave electric pulses at a frequency of 1 Hz were delivered with an ECM 830 electroporation unit (BTX-Harvard Apparatus) with a field strength of 160 V/cm. After the electroporation procedure, the incision was closed with Vetbond surgical glue. At 3 or 7 days after transfection, muscles were collected and subjected to the procedures described below.
Luciferase reporter assays
For all measurements of reporter luciferase activities, cotransfected muscles were collected at 3 days after transfection. Muscles were homogenized with a Polytron homogenizer in passive lysis buffer (Promega), and firefly and Renilla luciferase activities were measured with a FLUOstar Optima luminometer (BMG Labtech) by using the dual-luciferase reporter assay kit (Promega) as described in the manufacturer's instructions. All firefly luciferase activities were normalized to the Renilla luciferase activity in the same sample.
Western blot analysis
Frozen tissues were homogenized with a Polytron homogenizer (Kinematica AG) for 20 seconds in ice-cold Western blot buffer (40mM Tris [pH 7.5], 1mM EDTA, 5mM EGTA, 0.5% Triton X-100, 25mM β-glycerophosphate, 25mM NaF, 1mM Na3VO4, 10 μg/ml leupeptin, and 1mM phenylmethylsulfonyl fluoride), and the whole homogenate was used for further analysis. Sample protein concentrations were determined with a detergent compatible protein assay kit (Bio-Rad Laboratories), and equivalent amounts of protein from each sample were dissolved in Laemmli buffer and subjected to electrophoretic separation on SDS-PAGE acrylamide gels as described previously (44). Electrophoresis was terminated when the dye front ran off the bottom of the gel. After electrophoretic separation, proteins were transferred to a polyvinylidene difluoride membrane, blocked with 5% powdered milk in Tris-buffered saline containing 0.1% Tween 20 (TBST) for 1 hour followed by an overnight incubation at 4°C with primary antibody dissolved in TBST containing 1% BSA. The rabbit anti-total p70 (1:1000), rabbit anti-total Smad3 (1:1000), and rabbit anti-GFP (1:1000) primary antibodies were obtained from Cell Signaling Technology, whereas the rabbit anti-phospho-p70(T389) (1:3000) and rat anti-HA (1:2000) primary antibodies were obtained from Santa Cruz Biotechnology and Roche, respectively. After the overnight incubation, the membranes were washed for 30 minutes in TBST and then probed with a peroxidase-conjugated secondary antibody for 1 hour at room temperature. Peroxidase-conjugated antirabbit and peroxidase-conjugated antirat antibodies were purchased from Vector Laboratories. After 30 minutes of washing in TBST, the blots were developed on film using regular enhanced chemiluminescence reagent (Pierce) or enhanced chemiluminescence prime reagent (Amersham). Once the appropriate image was captured, the membranes were stained with Coomassie Blue to verify equal loading in all lanes. Densitometric measurements were carried out using National Institutes of Health ImageJ (http://rsb.info.nih.gov/nih-image/).
Immunohistochemical analysis of rates of protein synthesis and muscle fiber cross-sectional area
For in vivo measurements of protein synthesis using the SUnSET (surface sensing of translation) technique, mice were anesthetized at 7 days after transfection and then given an ip injection of 0.04 μmol/g puromycin dissolved in 100 μL PBS, as previously described (43, 45). At exactly 30 minutes after the injection, TA muscles were excised and immediately submerged in optimal cutting temperature compound (Tissue-Tek; Sakura) at resting length and frozen in liquid nitrogen-chilled isopentane. Midbelly cross-sections (10 μm thick) were taken perpendicular to the long axis of the muscle with a cryostat and immediately fixed in −20°C acetone for 10 minutes. Sections were warmed to room temperature for 5 minutes and then incubated in PBS for 15 minutes, followed by a 1-hour incubation in solution A (PBS with 0.5% BSA and 0.5% Triton X-100) containing antimouse IgG Fab (1:10; Jackson ImmunoResearch). After three 5-minutes washes with PBS, samples were incubated for 1 hour with solution A containing primary antibodies (mouse IgG2a Fc 2A monoclonal antipuromycin [clone 12D10, 1:1000] (46), rabbit IgG polyclonal antilaminin [1:500; Sigma-Aldrich], and either chicken IgY polyclonal anti-LacZ [1:300; Abcam] or rat IgG1 monoclonal anti-HA [clone 3F10, 1:200; Roche]). Sections were washed with PBS and then incubated for 1 hour with solution A containing secondary antibodies (DyLight 594-conjugated antimouse IgG Fc 2a [1:500], Alexa 350-conjugated goat antirabbit IgG [1:1000; Invitrogen], and either fluorescein isothiocyanate [FITC]-conjugated bovine antichicken IgY [1:100; Santa Cruz Biotechnology] or FITC-conjugated goat antirat IgG [1:100; Santa Cruz, Biotechnology]). Finally, the sections were washed with PBS, and fluorescence images were captured with a Nikon DS-QiMc camera on a Nikon 80i epifluorescence microscope with tetramethylrhodamine isothiocyanate (puromycin), FITC (LacZ or HA), and 4′,6-diamidino-2-phenylindole (laminin) cubes. The monochrome images were merged with Nikon NIS Elements D image analysis software. Measurements of the average cross-sectional area (CSA) and the average puromycin signal intensity were obtained from randomly selected LacZ-transfected or HA-tagged Smad3-transfected fibers, and an equal number of nontransfected fibers, by tracing the peripheral laminin stain. To calculate changes in fiber CSA and rates of protein synthesis, the CSA and intensity of the puromycin signal in LacZ- and HA-Smad3-positive fibers was expressed relative to the mean CSA and intensity obtained in nontransfected fibers from the same muscle section, respectively. All analyses were performed by investigators that were blinded to the sample identification.
Statistical analysis
All values are expressed as means (+SEM in graphs). Student's 2-tailed unpaired t tests were used for all 2-group comparisons. Three-group comparisons were performed using ANOVA, followed by Student-Newman-Keuls post hoc analysis. Differences between groups were considered significant if P ≤ .05. All data analysis was performed using GraphPad Prism version 5.0 (GraphPad Software Inc).
Results
Before examining the effect of Smad3 on the activity of specific target gene promoters, we first wanted to confirm that the Smad3 encoded by our plasmid construct was transcriptionally active when transfected into skeletal muscle in vivo. To do this, we cotransfected Smad3 with a Smad binding element (SBE) luciferase reporter construct containing 12 Smad CAGA binding motifs. As shown in Figure 1, Smad3 induced a large increase in the activity of the SBE reporter compared with the control condition, confirming that the Smad3 construct was transcriptionally active. As mentioned earlier, when activated by myostatin or TGF-β, phosphorylated Smad3 forms a heterotrimer with another Smad3 or Smad2 and a Smad4 before entering the nucleus. To confirm that the Smad3-induced increase in the SBE reporter activity was dependent on the transfected Smad3 interacting with endogenous Smads, we also cotransfected the SBE reporter with a Smad3 construct containing a D408H mutation in the C-terminal MH2 domain, which lacks the ability to interact with Smads 2, 3, and 4 (47). As expected, the transfection of the D408H mutant did not significantly alter the SBE reporter activity (Figure 1). Together, these results confirm that our Smad3 construct was transcriptionally active in skeletal muscle in vivo and that this transcriptional activity required interaction with other endogenous Smads.
Figure 1.

The effect of Smad3 on Smad-mediated transcriptional activity in skeletal muscle. Mouse TA muscles were cotransfected with LacZ as a control, Smad3 or Smad3 D408H, an SBE firefly (FF) luciferase reporter (SBE FF), and pRL-SV40 Renilla (Ren) luciferase reporter. At 72 hours after transfection, the muscles were collected and FF and Ren luciferase activities were measured by a dual-luciferase assay. Measurements of the relative light units produced by FF luciferase were normalized to that produced by Ren luciferase, and this ratio was expressed as a percentage of the values obtained from the LacZ-transfected muscles. Values are the mean + SEM; n = 3–6 per group. *, Significantly different from the values obtained in LacZ-transfected and Smad3 D408H-transfected muscles.
Effect of Smad3 on atrophy-related gene promoter activity
Atrogin-1 and muscle ring finger 1 (MuRF1) are E3 ligases involved in the ubiquitination and degradation of proteins by the ubiquitin proteasome system and have been shown to play an important role in mediating skeletal muscle atrophy (48–50). Moreover, atrogin-1 and to a lesser extent MuRF1 are upregulated by myostatin (27, 51). Recent studies have shown that although Smad3 is not required for the myostatin-induced increase in MuRF1 expression, Smad3 is necessary for an increase in atrogin-1 expression (25, 27). It remains to be determined, however, whether Smad3 is sufficient to activate atrogin-1 expression in skeletal muscle in vivo. We therefore cotransfected Smad3 with an atrogin-1 promoter reporter construct and found that Smad3 was sufficient to induce a 1.8-fold increase in atrogin-1 promoter activity (Figure 2A). Next, we examined the MuRF1 promoter and found that although it was markedly activated by a constitutively active mutant of FoxO3 (Forkhead Box Protein, Subclass O, c.a.-FoxO3), Smad3 was not sufficient to induce an increase in MuRF1 promoter activity (Figure 2B). Recently, it was suggested that Smad2 might play a role in the myostatin-induced expression of MuRF1 (27). We therefore cotransfected Smad2 with the MuRF1 promoter reporter but found no Smad2-induced activation of MuRF1 promoter activity (Supplemental Figure 1). Together, these data show that although Smad3 is sufficient to activate atrogin-1, other signaling mechanisms are required for MuRF1 promoter activation.
Figure 2.

The effect of Smad3 on the activity of the atrogin-1, MuRF1, and PGC1α promoters in skeletal muscle. A, Mouse TA muscles were cotransfected with LacZ as a control or Smad3, an atrogin-1 promoter firefly (FF) luciferase reporter (Atrogin-1 FF), and pRL-SV40 Renilla luciferase reporter (Ren). B, Mouse TA muscles were cotransfected with LacZ, Smad3 or c.a.-FoxO3, a MuRF1 promoter firefly luciferase reporter (MuRF1 FF), and Ren. C, Mouse TA muscles were cotransfected with LacZ or Smad3, a PGC1α promoter FF luciferase reporter (PGC1α FF), and Ren. At 72 hours after transfection, the muscles were collected and FF and Ren luciferase activities were measured by a dual-luciferase assay. Measurements of the relative light units produced by FF luciferase were normalized to that produced by Ren luciferase, and this ratio was expressed as a percentage of the values obtained from the LacZ-transfected muscles. Values are the mean + SEM; n = 5–10 per group. *, Significantly different from the values obtained in LacZ-transfected muscles; #, significantly different from the values obtained in LacZ- and Smad3-transfected muscles.
Peroxisome proliferator-activated receptor-γ coactivator-1α (PGC1α) is a master regulator of genes encoding proteins involved in mitochondrial biogenesis, oxidative metabolism, and protection from oxidative stress (52). Recently, it has also been suggested that PGC1α could play a role in muscle atrophy by preventing the induction of certain atrophy-related genes (53, 54). Interestingly, overexpression of myostatin in skeletal muscle decreases PGC1α expression, whereas skeletal muscles from myostatin-knockout mice and mice treated with antimyostatin antibodies have elevated levels of PGC1α expression (13, 55, 56). Given that the PGC1α gene promoter region contains multiple putative Smad binding sites (57), we next examined whether Smad3 was sufficient to inhibit PGC1α promoter activity in skeletal muscle. As shown in Figure 2C, Smad3 was sufficient to decrease PGC1α promoter activity by 50%, indicating that myostatin-induced Smad3 signaling may play a direct role in reducing PGC1α in skeletal muscle.
Effect of Smad3 on fibrosis-related gene promoter activity
Muscle fibrosis is another common feature of conditions that involve muscle atrophy/wasting, and myostatin and TGF-β have been shown to regulate the expression of fibrosis-related genes (14, 58, 59). Plasminogen activator inhibitor 1 (PAI-1), a TGF-β-responsive gene that contains multiple Smad binding sites in its proximal promoter region, is involved in the inhibition of plasminogen activation and, thus, plasmin-mediated fibrinolytic activity (60, 61). Moreover, increased PAI-1 expression is associated with muscle atrophy, fibrosis, and impaired muscle regeneration (62, 63). To determine whether Smad3 was sufficient to activate the PAI-1 promoter in skeletal muscle, we cotransfected muscles with Smad3 and a PAI-1 promoter reporter construct. As shown in Figure 3A, Smad3 transfection induced a 3.5-fold increase in PAI-1 promoter activity, demonstrating that Smad3 is indeed sufficient to activate PAI-1 promoter in vivo.
Figure 3.

Smad3 activates the PAI-1 promoter, inhibits the miR-29 promoter, and activates the PTEN 3′-UTR and FoxO response element reporters in skeletal muscle. Mouse TA muscles were cotransfected with LacZ as a control or Smad3, pRL-SV40 Renilla (Ren) luciferase reporter, and the PAI-1 promoter (A), miR-29b/c promoter firefly (B), PTEN 3′-UTR (C), or FoxO response element (FoxO RE FF) firefly (FF) (D) luciferase reporters. At 72 hours after transfection, the muscles were collected and FF and Ren luciferase activities were measured by a dual-luciferase assay. Measurements of the relative light units produced by FF luciferase were normalized to that produced by Ren luciferase, and this ratio was expressed as a percentage of the values obtained from the LacZ-transfected muscles. Values are the mean + SEM; n = 3–10 per group. *, Significantly different from the values obtained in LacZ-transfected muscles.
Members of the miR-29 family (miR-29a-c) also play a significant role in the regulation of tissue fibrosis by inhibiting the translation of mRNAs that encode proteins such as collagens, fibrillins, and elastin (64, 65). miR-29 expression is down-regulated in skeletal muscle of mdx mice and human Duchenne muscular dystrophy patients, both of which have elevated levels of TGF-β (66, 67). Importantly, the miR-29 gene proximal promoter contains several Smad binding sites, and TGF-β signaling has been shown to inhibit miR-29 expression (38). Moreover, it has recently been demonstrated that the TGF-β–induced inhibition of miR-29 expression occurs in a Smad3-dependent manner in cultured muscle cells (38). Thus, we sought to determine whether Smad3 was sufficient to inhibit miR-29 gene promoter activity in skeletal muscle in vivo. To this end, we cotransfected Smad3 with a miR-29b/c promoter luciferase reporter construct and found that Smad3 inhibited the miR-29b/c promoter activity by 85% compared with the control condition (Figure 3B). Taken together, these results demonstrate that Smad3 is sufficient to regulate the promoter activity of genes associated with fibrosis in skeletal muscle.
Effect of Smad3 on PTEN translation and Akt/mTOR signaling
In addition to the role of miR-29 in regulating the translation of mRNAs involved in fibrosis, recent studies have also shown that miR-29 can inhibit the translation of PTEN, an antagonist of the Akt/mTOR signaling pathway (39–41). These findings were of particular interest given recent studies showing that increased myostatin expression is sufficient to inhibit Akt/mTOR signaling, whereas reduced myostatin results in an increase in Akt/mTOR signaling (25, 26, 32, 34, 37, 68). Thus, our finding that Smad3 was sufficient to markedly inhibit miR-29 promoter activity (Figure 3B) led us to hypothesize that the Smad3-induced inhibition of the miR-29 promoter would be associated with increased PTEN mRNA translation and an inhibition of Akt/mTOR signaling. To test this hypothesis, we first cotransfected Smad3 with a reporter construct containing a basic promoter and the gene encoding firefly luciferase followed immediately by a sequence encoding the PTEN mRNA 3′-untranslated region (UTR), which contains the predicted binding motif for miR-29a-c (UGGUGCUA) (39). Therefore, a Smad3-induced decrease in miR-29 expression should result in reduced binding of miR-29 to the PTEN 3′-UTR and increased PTEN mRNA translation as evidenced by an increase in luciferase activity. As shown in Figure 3C, Smad3 transfection resulted in an ∼2.2-fold increase in luciferase activity compared with the control condition, suggesting that Smad3 is sufficient to induce an increase in PTEN mRNA translation.
Next, we wanted to determine whether the Smad3-induced increase in PTEN translation was associated with reduced Akt signaling. FoxO transcription factors are direct targets of Akt with a reduction in Akt signaling leading to reduced FoxO phosphorylation, increased FoxO translocation into the nucleus, and increased FoxO-mediated transcriptional activity (69–71). Therefore, as an indirect measure of Smad3-induced changes in Akt activity, we next examined whether the in vivo transfection of Smad3 would induce an increase in FoxO transcriptional activity. To accomplish this, we cotransfected Smad3 with a FoxO response element luciferase reporter construct, which contains multiple FoxO binding motifs (TTGTTTAC). As shown in Figure 3D, Smad3 induced a 50% increase in FoxO transcriptional activity consistent with a Smad3-induced decrease in Akt activity.
Because Akt is an upstream regulator of mTOR signaling, we next examined whether Smad3 was sufficient to decrease mTOR signaling in vivo. To do this, we cotransfected Smad3 with glutathione S-transferase (GST)-tagged p70 (GST p70) (42). Muscles were then analyzed by Western blot for changes in the phosphorylation of GST p70 on the threonine 389 residue (GST P-p70[T389]) as a marker of mTOR signaling. As shown in Figure 4, Smad3 reduced GST P-p70(T389) phosphorylation by ∼50% compared with control muscles, indicating that Smad3 was sufficient to inhibit mTOR signaling. Taken together, these data are consistent with the hypothesis that Smad3 inhibits miR-29 expression, which in turn leads to an increase in PTEN translation and a corresponding inhibition of the Akt and mTOR signaling.
Figure 4.

Smad3 is sufficient to inhibit mTOR signaling in skeletal muscle. Mouse TA muscles were cotransfected with GFP as a control or Smad3 and GST-tagged p70 (GST p70). At 72 hours after transfection, the muscles were collected and subjected to Western blot analysis with the indicated antibodies. The phosphorylated to total protein ratio for GST P-p70(T389) was calculated and expressed as a percentage of the values obtained in the GFP control samples. Values are the mean + SEM; n = 4–6 per group. *, Significantly different from the values obtained in GFP-transfected muscles.
Effect of Smad3 on protein synthesis and muscle fiber CSA
Finally, given our data showing that Smad3 was sufficient to increase atrogin-1 promoter activity and to decrease Akt/mTOR signaling, we next determined whether Smad3 expression would be sufficient to induce muscle atrophy and whether this would be associated with a decrease in muscle fiber protein synthesis. Thus, we transfected Smad3 into mouse TA muscles and collected them at 7 days after transfection for measurements of muscle fiber CSA and protein synthesis rates. Analysis of changes in muscle fiber protein synthesis rates was performed with our recently developed and validated immunohistochemical SUnSET technique, which measures the rate of incorporation of the antibiotic puromycin into newly synthesized peptides (72). Using this technique, we found that the rate of protein synthesis in Smad3-transfected fibers was 16.9% lower than that in LacZ-transfected fibers (Figure 5, D–F, and Supplemental Figure 2, C and D), indicating that Smad3 is sufficient to inhibit muscle fiber protein synthesis. Furthermore, the CSA of Smad3-transfected fibers was 13.7% smaller than LacZ-transfected fibers (Figure 5, A–C, and Supplemental Figure 2, A and B). Taken together, these data show that Smad3 is sufficient to induce skeletal muscle fiber atrophy in vivo and that this may, in part, be due to a Smad3-induced decrease in muscle fiber protein synthesis.
Figure 5.
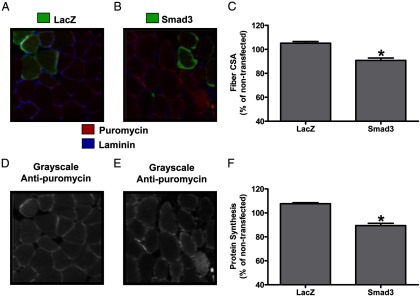
Smad3 is sufficient to inhibit protein synthesis and induce atrophy in skeletal muscle. Mouse TA muscles were transfected with LacZ as a control or HA-tagged Smad3. At 7 days after transfection, mice were injected with puromycin 30 minutes before the collection of the muscles. Muscles were then subjected to IHC for rates of protein synthesis (puromycin, red), laminin (blue), and LacZ or the HA tag (green). A and B, Representative merged images of anti-LacZ (A) and anti-HA (B) and antilaminin and antipuromycin signals. D and E, Grayscale images of the signal for puromycin shown in A and B, respectively. C and F, Fiber CSA (C) and puromycin staining intensity (F) in LacZ-transfected and HA-tagged Smad3-transfected fibers, expressed relative to the mean value obtained in nontransfected (control) fibers from the same section. Values are the mean + SEM; n = 153–460 fibers per group from 6 independent samples per group. *, Significantly different from the values obtained in LacZ-transfected fibers.
Discussion
This is the first study to demonstrate that Smad3 is sufficient to regulate the promoters of critical atrophy- and fibrosis-related genes and to induce a decrease in protein synthesis and CSA in skeletal muscle fibers in vivo. Furthermore, this study presents the first evidence that Smad3 is sufficient to inhibit mTOR signaling in vivo and proposes a possible mechanism to explain the previously published findings that myostatin induces a decrease in mTOR signaling, ie, a Smad3-induced decrease in miR-29 expression and a subsequent increase in PTEN mRNA translation and inhibition of Akt/mTOR pathway signaling. Overall, these results suggest that Smad3 may be a viable target of therapies to ameliorate many of the detrimental effects of increased myostatin and/or TGF-β expression on skeletal muscle mass and fibrosis.
In this study, we found that Smad3 was sufficient to induce a 1.8-fold increase in the activity of an atrogin-1 promoter reporter. This finding is very similar to the study of Sartori et al (25), which found that the expression of a constitutively active mutant of ALK5 (c.a.-AKL5) induced an ∼2.2-fold increase in the activity of the same atrogin-1 promoter construct used in our study. Our results are also consistent with previous cell culture studies showing that Smad3 is necessary for myostatin's effect on atrogin-1 expression (27). It is unclear, however, whether this Smad3-induced activation of the atrogin-1 gene is due to direct Smad3 binding to the atrogin-1 promoter and/or to Smad3-mediated activation of FoxO signaling (69, 70). Examination of the mouse, rat, and human atrogin-1 proximal promoter sequences published by Zhao et al (73) and GenBank (accession no. AY929855.1), shows the presence of 2 previously reported FoxO binding elements (ATAAACA and ATAAATA) (69) and also multiple putative SBEs (CAGA and GTCT). Thus, activation of the atrogin-1 promoter could potentially be induced by direct binding of Smad3, a Smad3-induced increase in FoxO DNA binding, and/or the formation of a Smad3/FoxO transcriptional complex (74). Interestingly, although the MuRF1 promoter was markedly activated by c.a.-FoxO3 (Figure 2B), the 50% increase in FoxO transcriptional activity induced by Smad3 expression was not sufficient to activate the MuRF1 promoter. This suggests that the MuRF1 promoter may require a relatively large increase in FoxO signaling and/or that it needs some other myostatin-induced binding partner for activation. Nevertheless, this finding is also consistent with the study of Sartori et al (25), which showed that expression of c.a.-ALK5 did not activate the MuRF1 promoter. Recently, it was proposed that perhaps MuRF1 is activated by Smad2 (27); however, we found no activation of MuRF1 promoter activity when coexpressed with Smad2 in vivo (Supplemental Figure 1), again suggesting that other factors play a role in the activation of MuRF1. Combined, these aforementioned points indicate that Smad3 is necessary and sufficient for the activation of atrogin-1, but not MuRF1, promoter activity in skeletal muscle in vivo.
PGC1α is a master regulator of genes regulating mitochondrial biogenesis and fusion, oxidative metabolism, and antioxidant enzymes. Recently, PGC1α has also been implicated in the repression of atrophy-related genes, such as atrogin-1, by inhibiting the transcriptional activities of FoxOs and nuclear factor kappa-light-chain-enhancer of activated β-cells (53, 54, 75, 76). Therefore, a decrease in PGC1α expression could contribute to muscle atrophy via a reduced repression of atrophy genes and/or indirectly by impairing mitochondrial function and fusion and increasing energy and oxidative stress (77, 78). Importantly, increased myostatin expression has been shown to decrease PGC1α expression, whereas myostatin knockout and antibody-induced inhibition of myostatin signaling increases PGC1α expression (13, 55, 56). Myostatin has also recently been shown to impair normal mitochondrial morphology and function in cultured cells (79). Because the PGC1α gene promoter region contains multiple Smad binding sites (57), this suggests that Smad3 signaling may play a role in the myostatin-induced decrease in PGC1α expression in skeletal muscle. Our data show for the first time that Smad3 is sufficient to markedly inhibit PGC1α promoter activity in skeletal muscle in vivo, suggesting that a Smad3-induced decrease in PGC1α expression could contribute, in part, to myostatin-induced muscle atrophy and mitochondrial dysfunction.
Myostatin signaling has recently been linked to the inhibition of the Akt/mTOR signaling pathway, with increased myostatin reducing Akt phosphorylation and markers of mTOR signaling, and reduced myostatin increasing Akt phosphorylation and mTOR signaling (26, 32–34, 37). To date, however, the mechanism behind myostatin's inhibition of Akt/mTOR signaling has remained elusive. PTEN is an antagonist of the Akt/mTOR pathway by competing for phosphatidylinositol-3,4,5 trisphosphate (PIP3) and reducing the recruitment of Akt to the membrane for phosphorylation and activation (80). Recent studies have shown that members of the miR-29 family are able to bind to the 3′-UTR of PTEN mRNA and inhibit its translation in cultured nonmuscle cells, whereas miR-29 inhibition leads to increased PTEN protein (39–41). Importantly, Smad3 has been shown to inhibit the expression of miR-29 in cultured myoblasts (38). Combined, these data led us to hypothesize that the previously reported myostatin-induced inhibition of Akt/mTOR signaling might be mediated via a Smad3-induced inhibition of miR-29 expression and a subsequent increase in the translation of PTEN mRNA. Our findings, summarized in Figure 6, show that Smad3 was sufficient to 1) inhibit miR-29 promoter activity, 2) increase the activity of the PTEN–3′-UTR reporter (indicating increased PTEN mRNA translation), 3) increase FoxO-mediated transcriptional activity (a surrogate measure of decreased Akt activity), and 4) decrease mTOR signaling. Thus, the results of this study are entirely consistent with this hypothesis and may, for the first time, explain how myostatin mediates a reduction in Akt/mTOR pathway signaling and increase FoxO signaling in skeletal muscle in vivo. Further mechanistic work is now required to confirm this proposed myostatin/Smad3/miR-29/PTEN/Akt/mTOR signaling pathway at the RNA and protein levels using different model systems.
Figure 6.

A summary of the proposed mechanism by which Smad3 inhibits Akt/mTOR signaling and induces decreased protein synthesis and increased protein degradation in skeletal muscle in vivo. Based on this study, and other previous studies, we propose that Smad3 inhibits miR-29 expression, which leads to increase PTEN mRNA translation and then inhibition of Akt and mTOR signaling. Our results also show that Smad3 is sufficient to activate the atrogin-1 promoter and increase FoxO transcriptional activity. In addition, Smad3 activates the PAI-1 promoter and decreases the activity of the PGC1α promoter. Combined, these Smad3-induced events may result in increased protein degradation, reduced protein synthesis, increased fibrosis, impaired mitochondrial function, and increased energy and oxidative stress.
Myostatin has also been shown to induce a decrease in protein synthesis in cultured myotubes (27, 30, 34). Using our recently developed IHC SUnSET technique for determining rates of protein synthesis at the level of the single muscle fiber, we were able to demonstrate that Smad3 is sufficient to induce a decrease in muscle fiber protein synthesis in vivo. This Smad3-induced effect on protein synthesis is most likely due to increased atrogin-1–mediated degradation of ribosomal proteins and translation initiation factors and a decrease in Akt/mTOR signaling (26, 27, 30, 37). We also investigated whether Smad3 would induce muscle fiber atrophy in vivo, and our experiments revealed that Smad3 was indeed sufficient to induce skeletal muscle fiber atrophy. Our finding of an ∼14% reduction in fiber CSA compared with controls is similar to the range of atrophy previously reported for in vivo transient overexpression of myostatin (15%) and c.a.-ALK4 and -5 (∼20%) over a similar time period (13, 25). Moreover, our result is consistent with previous data showing that Smad3 is necessary for mediating myostatin-induced atrophy in myotubes and in muscle fibers (25, 27). Combined, these results suggest that Smad3 does not merely play a permissive role in myostatin-induced atrophy but that it is both necessary and sufficient to induce muscle fiber atrophy in vivo.
Although our results clearly show that the in vivo overexpression of Smad3 is sufficient to regulate several critical atrophy-related gene promoters, inhibit muscle fiber protein synthesis, and induce muscle fiber atrophy, there are some potential limitations to this approach. First, although the atrogin-1, PGC1α, PAI-1, and miR-29 promoters are known to contain putative SBEs, we have not directly determined that the transfected Smad3 did indeed bind to these sites, thus leaving open the possibility that Smad3 induced some other transcription factors that ultimately activated or inhibited these promoters. Second, the overexpression of Smad3 in individual muscle fibers is likely to induce changes in gene promoter activity, in part, due to a mass action effect and thus may not fully represent changes that occur under more physiological conditions. Thus, futures studies are therefore needed to confirm the direct binding of Smad3 to the relevant SBEs contained in the endogenous genes and further determine the role of changes in Smad3 abundance and/or Smad3 phosphorylation to myostatin-induced muscle atrophy under more physiological conditions.
Finally, it was recently reported by Ge et al (81) that Smad3-null mice displayed an atrophic phenotype with increased protein ubiquitination and presumably increased protein degradation. These findings would seem to contradict the results of the current study by suggesting that Smad3 in fact plays a role in the inhibition of protein degradation and muscle atrophy; however, several factors make a comparison between these two studies problematic. First, although the absolute muscle mass of Smad3-null mice is indeed smaller, when normalized to the significantly smaller body mass, there is no difference from wild-type control mice (81). This suggests that there is impairment in the overall development of the Smad3-null mice and not necessarily the presence of muscle atrophy per se. Furthermore, Smad3-null mice also have impaired satellite cell function, decreased IGF-1 expression, and increased myostatin expression (81). Thus, any increase in protein degradation in these mice could be due to a number of factors including increased myostatin signaling and the subsequent activation of Smad2 transcriptional activity. Therefore, given these issues, it is difficult to draw firm conclusions about the role of Smad3 in differentiated skeletal muscle from these Smad3-null mice. Based on this, we do not believe that the results of Ge et al (81) contradict the results of our study.
Conclusion
In summary, this study demonstrates that Smad3 is sufficient to induce many of the key events that occur in skeletal muscle in response to increased levels of myostatin and TGF-β. Moreover, these results suggest that Smad3 does not simply play a permissive role, but rather, it is both necessary and sufficient to mediate many of the effects of myostatin and TGF-β on skeletal muscle in vivo. Based on this, and other previous studies, we propose that myostatin inhibits Akt/mTOR signaling by a Smad3-induced inhibition of miR-29 expression and increased PTEN translation. Overall, our results suggest the Smad3 signaling may be a viable target for therapies aimed at preventing myostatin-induced muscle atrophy. Given that Smad3 is likely to have other roles in skeletal muscle cells besides just mediating the negative effects of myostatin and TGF-β, further investigation is now required to determine the specific transcription factors, coactivators, and corepressors that interact with Smad3 in response to myostatin stimulation. Identifying these specific molecular interactions may allow for the development of small-molecule inhibitors or genetic therapies to disrupt these interactions and thus attenuate or prevent the adverse effects of increased myostatin/TGF-β signaling while maintaining other Smad3 interactions required for normal cell function. Such interventions could have significant benefits for atrophic conditions such as cancer cachexia, aging, heart failure, obesity, mechanical unloading, chronic obstructive pulmonary disease, renal disease, and HIV.
Acknowledgments
This work was supported by National Institutes of Health Grants AR057347 (to T.A.H.), and AR063256 (to C.A.G. and T.A.H.) and Muscular Dystrophy Association Grant 69637 (to F.M.H.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ActRIIA/IIB
- activin type IIA and IIB receptors
- ALK4
- activin receptor-like kinase-4
- c.a.
- constitutively active
- CSA
- cross-sectional area
- FITC
- fluorescein isothiocyanate
- FoxO
- Forkhead Box Protein, Subclass O
- GFP
- green fluorescent protein
- GST
- glutathione S-transferase
- HA
- hemagglutinin
- miR-29
- microRNA-29
- mTOR
- mechanistic target of rapamycin
- MuRF1
- muscle ring finger 1
- PAI-1
- plasminogen activator inhibitor 1
- PGC1α
- peroxisome proliferator-activated receptor-γ coactivator-1α
- PTEN
- tumor suppressor, phosphatase and tensin homolog deleted on chromosome 10
- SBE
- Smad binding element
- Smad
- Small Mothers Against Decapentaplegic
- SUnSET
- surface sensing of translation
- TA
- tibialis anterior
- TBST
- Tris-buffered saline containing 0.1% Tween 20
- TGF-βRII
- TGF-β type II receptor
- UTR
- untranslated region.
References
- 1. Goodman CA, Mayhew DL, Hornberger TA. Recent progress toward understanding the molecular mechanisms that regulate skeletal muscle mass. Cell Signal. 2011;23:1896–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech. 2013;6:25–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Srikanthan P, Karlamangla AS. Relative muscle mass is inversely associated with insulin resistance and prediabetes. Findings from the Third National Health and Nutrition Examination Survey. J Clin Endocrinol Metab. 2011;96:2898–2903 [DOI] [PubMed] [Google Scholar]
- 4. Lynch GS. Tackling Australia's future health problems: developing strategies to combat sarcopenia: age-related muscle wasting and weakness. Intern Med J. 2004;34:294–296 [DOI] [PubMed] [Google Scholar]
- 5. Aversa Z, Bonetto A, Penna F, et al. Changes in myostatin signaling in non-weight-losing cancer patients. Ann Surg Oncol. 2012;19:1350–1356 [DOI] [PubMed] [Google Scholar]
- 6. Léger B, Derave W, De Bock K, Hespel P, Russell AP. Human sarcopenia reveals an increase in SOCS-3 and myostatin and a reduced efficiency of Akt phosphorylation. Rejuvenation Res. 2008;11:163B–175B [DOI] [PubMed] [Google Scholar]
- 7. Bish LT, George I, Maybaum S, Yang J, Chen JM, Sweeney HL. Myostatin is elevated in congenital heart disease and after mechanical unloading. PLoS ONE. 2011;6:e23818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hittel DS, Berggren JR, Shearer J, Boyle K, Houmard JA. Increased secretion and expression of myostatin in skeletal muscle from extremely obese women. Diabetes. 2009;58:30–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gustafsson T, Osterlund T, Flanagan JN, et al. Effects of 3 days unloading on molecular regulators of muscle size in humans. J Appl Physiol. 2010;109:721–727 [DOI] [PubMed] [Google Scholar]
- 10. Ju CR, Chen RC. Serum myostatin levels and skeletal muscle wasting in chronic obstructive pulmonary disease. Respir Med. 2012;106:102–108 [DOI] [PubMed] [Google Scholar]
- 11. Verzola D, Procopio V, Sofia A, et al. Apoptosis and myostatin mRNA are upregulated in the skeletal muscle of patients with chronic kidney disease. Kidney Int. 2011;79:773–782 [DOI] [PubMed] [Google Scholar]
- 12. Gonzalez-Cadavid NF, Taylor WE, Yarasheski K, et al. Organization of the human myostatin gene and expression in healthy men and HIV-infected men with muscle wasting. Proc Natl Acad Sci U S A. 1998;95:14938–14943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Durieux AC, Amirouche A, Banzet S, et al. Ectopic expression of myostatin induces atrophy of adult skeletal muscle by decreasing muscle gene expression. Endocrinology. 2007;148:3140–3147 [DOI] [PubMed] [Google Scholar]
- 14. Li ZB, Kollias HD, Wagner KR. Myostatin Directly Regulates Skeletal Muscle Fibrosis. J Biol Chem. 2008;283:19371–19378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu J, Li Y, Shen W, et al. Relationships between transforming growth factor-β1, myostatin, and decorin: implications for skeletal muscle fibrosis. J Biol Chem. 2007;282:25852–25863 [DOI] [PubMed] [Google Scholar]
- 16. Elkasrawy M, Immel D, Wen X, Liu X, Liang LF, Hamrick MW. Immunolocalization of myostatin (GDF-8) following musculoskeletal injury and the effects of exogenous myostatin on muscle and bone healing. J Histochem Cytochem. 2012;60:22–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Han HQ, Zhou X, Mitch WE, Goldberg AL. Myostatin/activin pathway antagonism: molecular basis and therapeutic potential. Int J Biochem Cell Biol. 2013;pii:S1357–S2725 [DOI] [PubMed] [Google Scholar]
- 18. Elkina Y, von Haehling S, Anker SD, Springer J. The role of myostatin in muscle wasting: an overview. J Cachexia Sarcopenia Muscle. 2011;2:143–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Walton KL, Makanji Y, Harrison CA. New insights into the mechanisms of activin action and inhibition. Mol Cell Endocrinol. 2012;359:2–12 [DOI] [PubMed] [Google Scholar]
- 20. Tsuchida K, Nakatani M, Uezumi A, Murakami T, Cui X. Signal transduction pathway through activin receptors as a therapeutic target of musculoskeletal diseases and cancer. Endocr J. 2008;55:11–21 [DOI] [PubMed] [Google Scholar]
- 21. Mendias CL, Gumucio JP, Davis ME, Bromley CW, Davis CS, Brooks SV. Transforming growth factor-β induces skeletal muscle atrophy and fibrosis through the induction of atrogin-1 and scleraxis. Muscle Nerve. 2012;45:55–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee SJ, Lee YS, Zimmers TA, et al. Regulation of muscle mass by follistatin and activins. Mol Endocrinol. 2010;24:1998–2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ross S, Hill CS. How the Smads regulate transcription. Int J Biochem Cell Biol. 2008;40:383–408 [DOI] [PubMed] [Google Scholar]
- 24. Schiaffino S, Dyar KA, Ciciliot S, Blaauw B, Sandri M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013;280:4294–4314 [DOI] [PubMed] [Google Scholar]
- 25. Sartori R, Milan G, Patron M, et al. Smad2 and 3 transcription factors control muscle mass in adulthood. Am J Physiol Cell Physiol. 2009;296:C1248–C1257 [DOI] [PubMed] [Google Scholar]
- 26. Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol. 2009;296:C1258–C1270 [DOI] [PubMed] [Google Scholar]
- 27. Lokireddy S, McFarlane C, Ge X, et al. Myostatin induces degradation of sarcomeric proteins through a Smad3 signaling mechanism during skeletal muscle wasting. Mol Endocrinol. 2011;25:1936–1949 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28. Lokireddy S, Mouly V, Butler-Browne G, et al. Myostatin promotes the wasting of human myoblast cultures through promoting ubiquitin-proteasome pathway-mediated loss of sarcomeric proteins. Am J Physiol Cell Physiol. 2011;301:C1316–C1324 [DOI] [PubMed] [Google Scholar]
- 29. Lokireddy S, Wijesoma IW, Sze SK, McFarlane C, Kambadur R, Sharma M. Identification of atrogin-1-targeted proteins during the myostatin-induced skeletal muscle wasting. Am J Physiol Cell Physiol. 2012;303:C512–C529 [DOI] [PubMed] [Google Scholar]
- 30. Taylor WE, Bhasin S, Artaza J, et al. Myostatin inhibits cell proliferation and protein synthesis in C2C12 muscle cells. Am J Physiol Endocrinol Metab. 2001;280:E221–E228 [DOI] [PubMed] [Google Scholar]
- 31. Welle S, Bhatt K, Pinkert CA. Myofibrillar protein synthesis in myostatin-deficient mice. Am J Physiol Endocrinol Metab. 2006;290:E409–E415 [DOI] [PubMed] [Google Scholar]
- 32. Welle S, Burgess K, Mehta S. Stimulation of skeletal muscle myofibrillar protein synthesis, p70 S6 kinase phosphorylation, and ribosomal protein S6 phosphorylation by inhibition of myostatin in mature mice. Am J Physiol Endocrinol Metab. 2009;296:E567–E572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Welle S, Mehta S, Burgess K. Effect of postdevelopmental myostatin depletion on myofibrillar protein metabolism. Am J Physiol Endocrinol Metab. 2011;300:E993–E1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rodriguez J, Vernus B, Toubiana M, et al. Myostatin inactivation increases myotube size through regulation of translational initiation machinery. J Cell Biochem. 2011;112:3531–3542 [DOI] [PubMed] [Google Scholar]
- 35. Hulmi JJ, Oliveira BM, Silvennoinen M, et al. Muscle protein synthesis, mTORC1/MAPK/Hippo signaling, and capillary density are altered by blocking of myostatin and activins. Am J Physiol Endocrinol Metab. 2013;304:E41–E50 [DOI] [PubMed] [Google Scholar]
- 36. Lagirand-Cantaloube J, Offner N, Csibi A, et al. The initiation factor eIF3-f is a major target for Atrogin1/MAFbx function in skeletal muscle atrophy. EMBO J. 2008;27:1266–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Amirouche A, Durieux AC, Banzet S, et al. Down-regulation of Akt/mammalian target of rapamycin signaling pathway in response to myostatin overexpression in skeletal muscle. Endocrinology. 2009;150:286–294 [DOI] [PubMed] [Google Scholar]
- 38. Zhou L, Wang L, Lu L, Jiang P, Sun H, Wang H. Inhibition of miR-29 by TGF-beta-Smad3 signaling through dual mechanisms promotes transdifferentiation of mouse myoblasts into myofibroblasts. PLoS ONE. 2012;7:e33766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tumaneng K, Schlegelmilch K, Russell RC, et al. YAP mediates crosstalk between the Hippo and PI(3)K–TOR pathways by suppressing PTEN via miR-29. Nat Cell Biol. 2012;14:1322–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kong G, Zhang J, Zhang S, Shan C, Ye L, Zhang X. Upregulated microRNA-29a by hepatitis B virus X protein enhances hepatoma cell migration by targeting PTEN in cell culture model. PLoS ONE. 2011;6:e19518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang C, Bian Z, Wei D, Zhang JG. miR-29b regulates migration of human breast cancer cells. Mol Cell Biochem. 2011;352:197–207 [DOI] [PubMed] [Google Scholar]
- 42. Goodman CA, Miu MH, Frey JW, et al. A phosphatidylinositol 3-kinase/protein kinase B-independent activation of mammalian target of rapamycin signaling is sufficient to induce skeletal muscle hypertrophy. Mol Biol Cell. 2010;21:3258–3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goodman CA, Mabrey DM, Frey JW, et al. Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB J. 2011;25:1028–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Goodman CA, Frey JW, Mabrey DM, et al. The role of skeletal muscle mTOR in the regulation of mechanical load-induced growth. J Physiol. 2011;589:5485–5501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Goodman CA, Kotecki JA, Jacobs BL, Hornberger TA. Muscle fiber type-dependent differences in the regulation of protein synthesis. PLoS ONE. 2012;7:e37890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6:275–277 [DOI] [PubMed] [Google Scholar]
- 47. Schiro MM, Stauber SE, Peterson TL, et al. Mutations in protein-binding hot-spots on the hub protein Smad3 differentially affect its protein interactions and Smad3-regulated gene expression. PLoS ONE. 2011;6:e25021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bodine SC, Latres E, Baumhueter S, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708 [DOI] [PubMed] [Google Scholar]
- 49. Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci U S A. 2001;98:14440–14445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Foletta VC, White LJ, Larsen AE, Léger B, Russell AP. The role and regulation of MAFbx/atrogin-1 and MuRF1 in skeletal muscle atrophy. Pflügers Arch. 2011;461:325–335 [DOI] [PubMed] [Google Scholar]
- 51. McFarlane C, Plummer E, Thomas M, et al. Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-κB-independent, FoxO1-dependent mechanism. J Cell Physiol. 2006;209:501–514 [DOI] [PubMed] [Google Scholar]
- 52. Handschin C. Regulation of skeletal muscle cell plasticity by the peroxisome proliferator-activated receptor γ coactivator 1α. J Recept Signal Transduct Res. 2010;30:376–384 [DOI] [PubMed] [Google Scholar]
- 53. Sandri M, Lin J, Handschin C, et al. PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci U S A. 2006;103:16260–16265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Brault JJ, Jespersen JG, Goldberg AL. Peroxisome Proliferator-activated receptor γ coactivator 1α or 1β overexpression inhibits muscle protein degradation, induction of ubiquitin ligases, and disuse atrophy. J Biol Chem. 2010;285:19460–19471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shan T, Liang X, Bi P, Kuang S. Myostatin knockout drives browning of white adipose tissue through activating the AMPK-PGC1α-Fndc5 pathway in muscle. FASEB J. 2013;27:1981–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Murphy KT, Koopman R, Naim T, et al. Antibody-directed myostatin inhibition in 21-mo-old mice reveals novel roles for myostatin signaling in skeletal muscle structure and function. FASEB J. 2010;24:4433–4442 [DOI] [PubMed] [Google Scholar]
- 57. Yadav H, Quijano C, Kamaraju AK, et al. Protection from obesity and diabetes by blockade of TGF-[beta]/Smad3 signaling. Cell Metab. 2011;14:67–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Samarakoon R, Overstreet JM, Higgins PJ. TGF-β signaling in tissue fibrosis: redox controls, target genes and therapeutic opportunities. Cell Signal. 2013;25:264–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhou L, Lu H. Targeting fibrosis in Duchenne muscular dystrophy. J Neuropathol Exp Neurol. 2010;69:771–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Suelves M, Vidal B, Ruiz V, et al. The plasminogen activation system in skeletal muscle regeneration: antagonistic roles of urokinase-type plasminogen activator (uPA) and its inhibitor (PAI-1). Front Biosci. 2005;10:2978–2985 [DOI] [PubMed] [Google Scholar]
- 61. Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF[beta]-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17:3091–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Naderi J, Bernreuther C, Grabinski N, et al. Plasminogen activator inhibitor type 1 up-regulation is associated with skeletal muscle atrophy and associated fibrosis. Am J Pathol. 2009;175:763–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Koh TJ, Bryer SC, Pucci AM, Sisson TH. Mice deficient in plasminogen activator inhibitor-1 have improved skeletal muscle regeneration. Am J Physiol Cell Physiol. 2005;289:C217–C223 [DOI] [PubMed] [Google Scholar]
- 64. van Rooij E, Sutherland LB, Thatcher JE, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A. 2008;105:13027–13032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. He Y, Huang C, Lin X, Li J. MicroRNA-29 family, a crucial therapeutic target for fibrosis diseases. Biochimie. 2013;95:1355–1359 [DOI] [PubMed] [Google Scholar]
- 66. Greco S, De Simone M, Colussi C, et al. Common micro-RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemia. FASEB J. 2009;23:3335–3346 [DOI] [PubMed] [Google Scholar]
- 67. Goldstein JA, McNally EM. Mechanisms of muscle weakness in muscular dystrophy. J Gen Physiol. 2010;136:29–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lipina C, Kendall H, McPherron AC, Taylor PM, Hundal HS. Mechanisms involved in the enhancement of mammalian target of rapamycin signalling and hypertrophy in skeletal muscle of myostatin-deficient mice. FEBS Lett. 2010;584:2403–2408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sandri M, Sandri C, Gilbert A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Stitt TN, Drujan D, Clarke BA, et al. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell. 2004;14:395–403 [DOI] [PubMed] [Google Scholar]
- 71. Rena G, Guo S, Cichy SC, Unterman TG, Cohen P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J Biol Chem. 1999;274:17179–17183 [DOI] [PubMed] [Google Scholar]
- 72. Goodman CA, Hornberger TA. Measuring protein synthesis with SUnSET: a valid alternative to traditional techniques? Exerc Sport Sci Rev. 2013;41:107–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhao W, Wu Y, Zhao J, Guo S, Bauman WA, Cardozo CP. Structure and function of the upstream promotor of the human Mafbx gene: The proximal upstream promotor modulates tissue-specificity. J Cell Biochem. 2005;96:209–219 [DOI] [PubMed] [Google Scholar]
- 74. Gomis RR, Alarcón C, He W, Wang Q, Seoane J, Lash A, Massagué J. A FoxO-Smad synexpression group in human keratinocytes. Proc Natl Acad Sci U S A. 2006;103:12747–12752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang H, Liu D, Cao P, Lecker S, Hu Z. Atrogin-1 affects muscle protein synthesis and degradation when energy metabolism is impaired by the anti-diabetic drug, berberine. Diabetes. 2010;59:1879–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Eisele PS, Salatino S, Sobek J, Hottiger MO, Handschin C. The peroxisome proliferator-activated receptor γ coactivator 1α/β (PGC-1) coactivators repress the transcriptional activity of NF-κB in skeletal muscle cells. J Biol Chem. 2013;288:2246–2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Romanello V, Sandri M. Mitochondrial biogenesis and fragmentation as regulators of muscle protein degradation. Curr Hypertens Rep. 2010;12:433–439 [DOI] [PubMed] [Google Scholar]
- 78. Powers SK, Smuder AJ, Criswell DS. Mechanistic links between oxidative stress and disuse muscle atrophy. Antioxid Redox Signal. 2011;15:2519–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Liu Y, Cheng H, Zhou Y, et al. Myostatin induces mitochondrial metabolic alteration and typical apoptosis in cancer cells. Cell Death Dis. 2013;4:e494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chalhoub N, Baker SJ. 2009 PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ge X, McFarlane C, Vajjala A, et al. Smad3 signaling is required for satellite cell function and myogenic differentiation of myoblasts. Cell Res. 2011;21:1591–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]