

Abstract
Lysine acetylation is an ancient, evolutionarily conserved, reversible post-translational modification. A multitude of diverse cellular functions are regulated by this dynamic modification, including energy and metabolism, protein folding, transcription, and translation. Gene expression can be manipulated through changes in histone acetylation status, and this process is controlled by the function of 2 opposing enzymes: histone acetyl transferases and histone deacetylases (HDACs). The zinc-dependent HDACs are a family of hydrolases that remove acetyl groups from lysines, and their function can be modulated by the action of small molecule ligands. Inhibition through competitive binding of the catalytic domain of these enzymes has been achieved by a diverse array of small molecule chemotypes. Structural biology has aided the development of potent, and in some cases highly isoform-selective, inhibitors that have demonstrated utility in a number of neurological disease models. Continued development and characterization of highly optimized small molecule inhibitors of HDAC enzymes will help refine our understanding of their function and, optimistically, lead to novel therapeutic treatment alternatives for a host of neurological disorders.
Electronic supplementary material
The online version of this article (doi:10.1007/s13311-013-0226-1) contains supplementary material, which is available to authorized users.
Keywords: Chromatin, Histone, HDACs, Acetylation, Zinc-dependent hydrolases, Isoform selective
Introduction
DNA is stored as chromatin and organized in 147 base pair segments around structural units called nucleosomes [1, 2]. The nucleosome is composed of an octamer of 4 core histones (H2A, H2B, H3, and H4), while a fifth histone, H1, serves as a linking motif. The N-terminal “tails” of the individual histones extend beyond the central core structural motif and can be covalently modified at specific loci by several distinct classes of enzymes that affect an array of post-translational modifications. These post-translational histone “tail” modifications include acetylation of lysines, methylation of lysines and arginines, phosphorylation of serines and threonines, ubiquitination and sumoylation of lysines, and adenosine diphosphate-ribosylation of glutamates. A subset of these histone modifications regulates the accessibility of DNA, which can affect gene expression. These “epigenetic” processes can regulate gene expression without modifying the primary DNA sequence. For instance, deacetylation of the ε-nitrogen of lysine residues in histone tails can induce a condensed (closed) and transcriptionally inactive chromatized DNA, whereas acetylation neutralizes positively-charged lysines, disrupting charge–charge interactions with the negatively-charged phosphates of DNA, leading to a relaxed (open) or transcriptionally active region [3]. The tightly balanced acetylation state of N-terminal histone tails is controlled by the function of 2 enzyme families, histone acetyl tranferases and histone deacetylases (HDACs), and is one of the most extensively studied epigenetic mechanisms (Fig. 1) [4].
Fig. 1.

The role of histone acetyl transferases (HATs) and histone deacetylases (HDACs) in modifying chromatin structure. Adapted with permission from Rikken Research publication
The catalytic activity of these enzymes can be modulated by small molecule activators and inhibitors resulting in equivalent effects on acetylation status, i.e., HDAC inhibitors (HDACi) and histone acetyl transferase activators increase histone acetylation. While primarily known for activity on histone substrates, it is increasingly recognized that HDACs catalyze the hydrolysis of acetylated lysine residues on nonhistone proteins, including transcription factor complex components. There are 2 types of HDACs defined by their hydrolytic mechanism of action: the metal (Zn2+)-dependent and nicotinamide adenine dinucleotide+-dependent HDACs (also known as Sirtuins).
The metal-dependent HDACs are divided into classes and subclasses based on their cellular localization, number of catalytic sites, domain organization, and homology to yeast deacetylase proteins (Fig. 2): class I (HDACs 1, 2, 3, and 8), class IIa (HDACs 4, 5, 7, and 9), class IIb (HDACs 6 and 10), and class IV (HDAC11) [5, 6]. As represented in Fig. 2, HDACs associate and regulate the acetylation of specific substrates and/or can often interact as components of multiprotein complexes [7, 8].
Fig. 2.

The zinc-dependent histone deacetylases (HDACs): classes, structural and functional domains, and cellular localization. Adapted with permission from [5], [9], and [10]. NRSE neuron-restrictive silencer element (also known as RE1) REST RE1-silencing transcription factor NRSF neuron-restrictive silencer factor SIN3A; CaMKII Ca2+/calmodulin-dependent protein kinases II CpG —C—phosphate—G— MECP2 methyl CpG binding protein 2 HSP90 heat shock protein 90; Ac acetyl K lysine P phospho
Class I HDACs are primarily localized in the nucleus; however, HDAC3 possesses a variable C-terminus with both nuclear import and export signals, which allows it to shuttle between the cytoplasm and nucleus. Class I HDACs are all expressed in the brain, with HDAC3 being the most prevalent, especially in cortex and hippocampus [11]. Class II HDACs are mainly localized in the cytoplasm, but they possess unique 14-3-3 binding sites at their N-termini, which control translocation in and out of the nucleus. While members of this class display little-to-no inherent catalytic activity as purified proteins, class IIa HDACs recruit higher-order protein complexes, often containing the HDAC3 and nuclear receptor co-repressor (NCoR)/ silencing mediator for retinoid or thyroid-hormone receptors (SMRT) domains to become catalytically competent [12, 13]. It has been hypothesized that class IIa HDACs serve as recruiters or “readers” to specific promoter regions, where HDAC3 would act as the deacetylase [13, 14]. Functionally, class IIb HDACs have been shown to modulate nonhistone substrates. For example, HDAC6 regulates α-tubulin and heat shock protein 90 acetylation (Fig. 2). The class IIa and IIb HDACs are tissue-specific, but are also expressed in the brain, with HDACs 4 and 5 being the most abundant, with minimal expression of HDACs 6, 7, 9, and 10 [11].
Evidence for aberrant epigenetic post-translational modifications is emerging as an important element in the pathogenesis of neurological disorders. While there is scant, direct, human genetic evidence implicating HDACs or their inhibition as a therapeutic approach in central nervous system (CNS) disorders [15, 16], several laboratories have demonstrated a key role for specific HDACs and the corresponding acetylation status in the brain. Specific HDAC isoform(s) have been shown to potentially play a role in schizophrenia [17, 18], Alzheimer’s disease (AD) [19], and Rubinstein–Taybi syndrome [20], and alterations in acetylation have been implicated in neurodegenerative disorders, including Huntington’s disease and Parkinson’s disease [21]. These data suggest that selective small molecule modulators of HDAC function could be beneficial in human neurological diseases. Indeed, preclinical evidence for the utility of HDACi to potentially treat a myriad of CNS disorders has accumulated rapidly over the last 5 years [10, 22]. For example, HDACi treatment has enhanced cognition in normal animals and reversed the cognitive deficits associated with aging and AD in several animal models [19]. As a potential therapeutic for psychiatric diseases, HDACi have ameliorated behavioral deficits associated with schizophrenia, autism, depression, bipolar disorder, and Rubinstein–Taybi syndrome in a number of animal models [20, 23–26]. Further, HDACi have shown utility in preclinical models of other neurological disorders, including Huntington’s disease, spinal muscular atrophy, Freidreich's ataxia, and amyotrophic lateral sclerosis [27, 28]. In addition to molecules targeting only HDAC activity, hybrid molecules incorporating dual agonistic and inhibitory activity for protein kinase C and HDACs, respectively, have been reported [29]. These molecules demonstrate dual pharmacological effects corresponding to their distinct binding activities, i.e., increasing amyloid precursor protein-α production, leading to amyloid-β40 clearance through protein kinase C activation and neuroprotection through HDAC inhibition, which could provide additive beneficial effects in AD. Thus, preclinical evidence suggests that HDACi, as a single agent or in combination therapies, could have a profound impact on an array of neurological disorders. However, to date, most inhibitors used in these studies are nonselective (inhibit ≥3 isoforms) and were developed for use in cancer. These small molecule inhibitors will have limited, if any, application in chronic CNS indications based on their clinical safety and toxicological profile. The chronic nature of many neurological disorders implies life-long use of HDACi and, consequently, the dose-dependent toxicity observed in the clinic must be mitigated to create a larger therapeutic window. The clinical dose-limiting toxicities of HDACi, such as thrombocytopenia, nausea, and fatigue [30–32], are attributed to targeting multiple HDACs (or a specific few isoforms) at the doses and schedules used [32, 33]. The development of highly potent and isoform selective inhibitors is critical to increase the therapeutic window by mitigating side effects caused by the inhibition of multiple HDACs. In addition, isoform-selective inhibitors will be key in refining our understanding regarding which HDAC isoform(s) are required for on-target efficacy. Herein, we review the efforts undertaken in academia and industry to design, synthesize, and characterize isoform selective inhibitors that may be applicable for broader use in non-oncological clinical settings, such as neurological disorders.
“First-generation” HDACi and Structure-based Design Considerations
Our understanding of HDAC inhibition by small molecules was initially informed by the first high-resolution crystal structure of a bacterial homolog of eukaryotic deacetylases, the histone deacetylase-like protein (hdlp) published in 1999 by Finnin et al. (Fig. 3a) [34]. The crystal structure obtained with the hydroxamic acid Trichostatin A (TSA; Fig. 3c) highlighted key elements of inhibitor–enzyme interactions, and displayed four prominent binding domains: 1) a surface binding domain, 2) an ~11-Å channel leading from the solvent exposed enzyme surface to 3) the catalytic zinc binding domain, and 4) an adjacent 14 Å internal cavity defined by several hydrophobic residues.
Fig. 3.
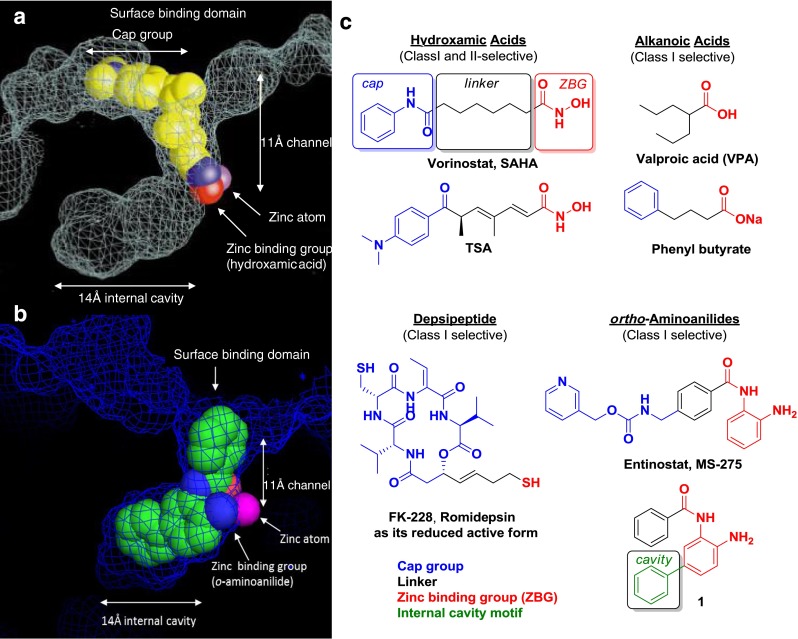
a X-ray crystal structure of bacterial histone deacetylase-like protein and Trichostatin A (TSA) (adapted with permission from [34]). b X-ray crystal structure of human histone deacetylase (HDAC) 2 and compound 1. c Prototypical HDAC inhibitors
As shown by these, and other, crystal structures (Fig. 3a, b), HDACi are often designed as structural mimics of the endogenous acetyl–lysine ligand, and, as a result, contain a surface binding or cap group, a hydrocarbon linking motif, and a zinc-binding group (ZBG). Newer HDACi, such as compound 1 (Fig. 3b, c), also optionally contain an internal cavity group. There are currently more than 20 HDACi in almost 400 active clinical trials. They can be divided in 4 main chemical classes, based on their zinc-binding element or capping group type: alkanoic acids, hydroxamic acids, depsipeptides (with thiol zinc-binding group), or macrocycles and ortho-aminoanilides (Fig. 3c). Examples of noncanonical ZBGs, such as trifluoromethyloxadiazoles or electrophilic ketones, have been reported in limited preclinical studies and will be discussed.
To date, 3 of the 4 HDACi chemotypes have completed human clinical trials and been successfully approved by the US Food and Drug Administration. Valproic acid (VPA; Fig. 3c), a weakly potent and class I-selective alkanoic acid, was first approved as an anti-epileptic drug in 1967 in France, and has since become one of the most widely prescribed anti-epileptic drugs worldwide. VPA has been sold and prescribed under a variety of brand names (e.g., Depakote, Depakene, Valparin) for the treatment of neurological disorders such as epilepsy, bipolar disorder, major depression, and schizophrenia. Valproate is unique as the only HDACi approved for neurological disorder indications, although its therapeutic effects most likely involve other modes of action beyond its’ marginal HDAC inhibitory activity [35]. Suberoylanilide hydroxamic acid (SAHA; vorinostat), was a first-in-class hydroxamic acid-based HDACi approved in 2006 under the tradename Zolinza for the treatment of cutaneous T-cell lymphoma. In 2009, romidepsin (Fig. 3c), a thiol-based depsipeptide, gained US Food and Drug Administration approval as Istodax, also for the treatment of cutaneous T-cell lymphoma.
Over the years, crystal structures of several ligand- and complex-bound human HDAC isoforms have been solved, refining our understanding of protein–protein (e.g., HDAC3/NCoR) and protein–ligand [e.g, HDAC2/N-(4-aminophenyl-3-yl)benzamide] interactions and enhancing structure-based drug design. Since the first high-resolution crystal structure of the bacterial hdlp reported by Finnin et al. [34], 13 independent structures comprising six human HDAC isozymes have been elucidated [36–45], including the class I and class II isoforms, with HDAC8 proving the most prolific, co-crystallized with a variety of ligand chemotypes (Table 1).
Table 1.
X-ray crystal structures of histone deacetylase (HDAC) isozymes and their corresponding ligands
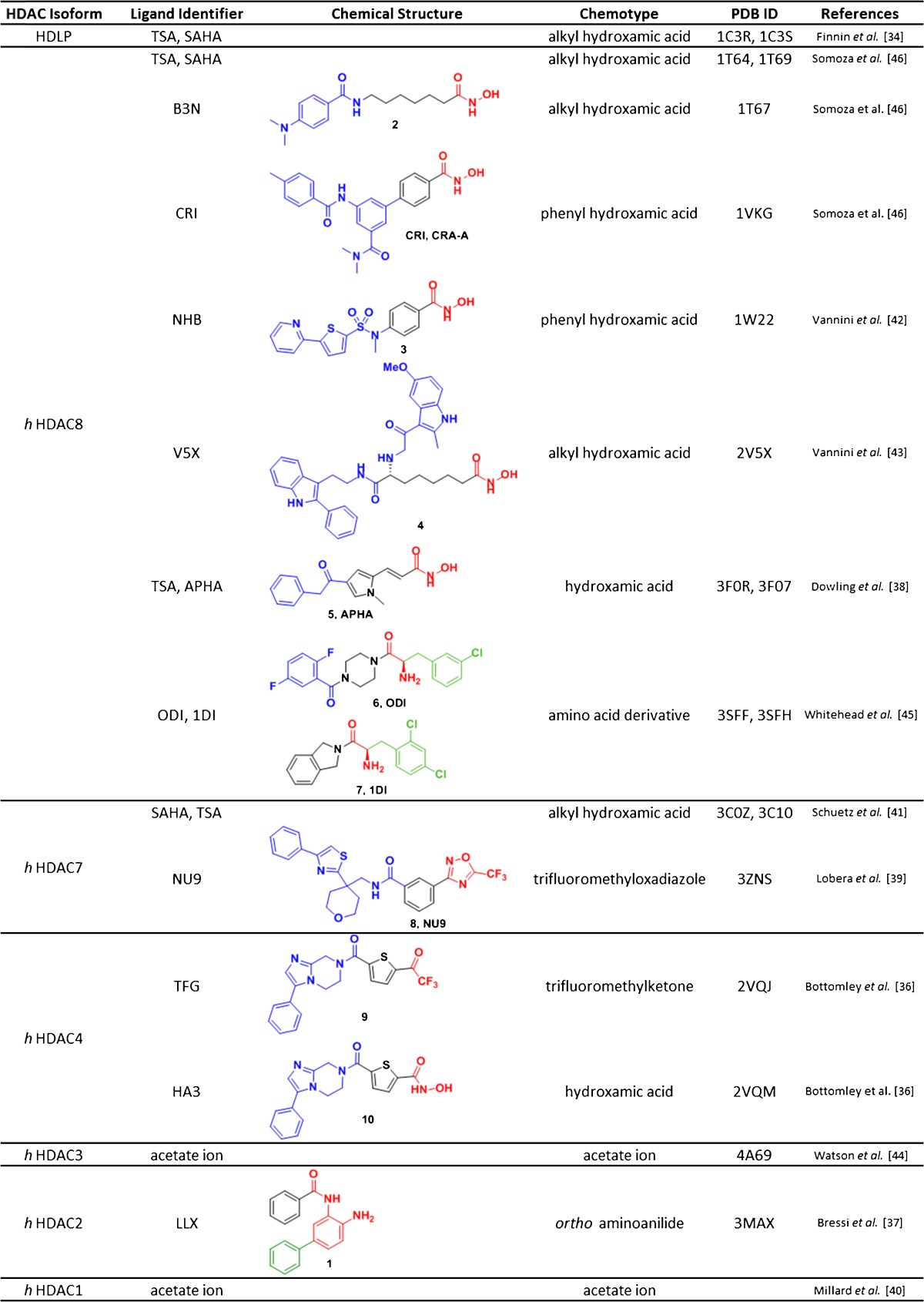
HDLP histone deacetylase-like protein TSA Trichostatin A; SAHA suberoylanilide hydroxamic acid; PDB protein data bank; B3N; CRI; NHB; V5X; APHA; ODI; 1DI; NU9; TFG; HA3; LLX
In 2004, Vannini et al. [42] and Somoza et al. [46] reported crystal structures of HDAC8 with hydroxamic acid inhibitors, such as SAHA, TSA, CRI (CRA-A), and NHB (compound 3). In 2008, Schuetz et al. [41] determined the structure of the HDAC7 catalytic domain with the hydroxamic acid inhibitors SAHA and TSA. Subsequently, the catalytic domain of HDAC4 bound to a trifluoromethylketone compound 9 or hydroxamic acid compound 10 inhibitor was reported by Bottomley et al. [36]. In 2010, Bressi et al. [37] determined the co-crystal structure of HDAC2 and compound 1 [37], an ortho-aminoanilide possessing a C-5 phenyl group that occupies the 14 Å internal cavity described in the original structure by Finnin et al. [34]. More recently, the work of Millard et al. [40] and Watson et al. [44] has elucidated the interactions of HDACs 1 and 3 with their corresponding co-repressor complexes in association with inositol tetraphosphate. These studies reveal the structural relation between the core enzymes and their higher-order complex partners, and the role of potential signaling molecules such as inositols on complex formation and enzymatic function.
High-resolution crystal structures provide several key insights, including amino acid differences in the catalytic binding domains, chelation interactions, and geometries across distinct zinc-binding motifs and structural interactions of the higher-order complexes partners. These observed differences, as well as predictive computational models, have informed, and will continue to inform, selective inhibitor design. This review will summarize the state-of-the-art in inhbitor design, key structural features, and efforts toward isoform-selective inhibitors within each of the HDACi chemical classes.
Short-chain Alkanoic Acids
Aliphatic acids and their conjugate bases were the first developed class of HDACi and also display the weakest HDAC inhibition activity (μM range; Fig. 4) [47]. The carboxylic acid is believed to serve as the zinc-binding moiety, while the hydrophobic alkyl chain, branched or linear, occupies the hydrophobic channel leading to the zinc atom. There is no direct X-ray crystallographic evidence to support this hypothesized binding mode for this class of inhibitors (see Table 1).
Fig. 4.
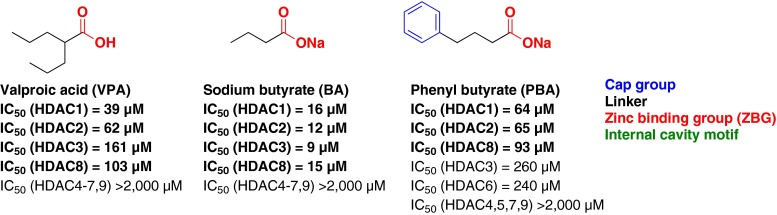
Short-chain alkanoic acid histone deacetylase inhibitors
VPA (Fig. 4) is the prototypical HDACi of this class [35]. VPA and its closely related analogs, such as sodium butyrate and phenyl butyrate (Fig. 4), are all weakly potent class I-selective HDACi (μM activity) [47]. The selectivity observed for this class of HDAC inhibitors is not well understood, as limited structure–activity relationship studies have been reported [48]. The carboxylic acid moiety is inherently a poor ZBG, but may have an influence on class I versus class II selectivity. The short alkyl chain linking motif proposed to occupy the 11-Å hydrophobic channel does not provide compensatory binding interactions to achieve improved potency. However, significant potency enhancements for this class have been achieved through the addition of surface binding motifs. Another important characteristic of the alkanoic acids are their physicochemical properties and the corresponding effects on blood–brain barrier (BBB) penetration. Pharmacokinetically, VPA is the most studied and has been shown to rapidly enter the CNS, even though it is more than 99 % ionized at physiological pH. The brain to plasma ratios of VPA range from 0.03 to 0.15 depending on the brain region [49], and is highly susceptible to both active uptake (medium-chain fatty acid-sensitive transporters) [50] and efflux (monocarboxylic acid-sensitive) transporters [51]. Owing to its modest brain penetration, weak HDAC inhibitory activity, and known neurological off-target effects, the alkanoic acid class of HDAC inhibitors present challenges and potential confounds when considering applications and the corresponding data interpretation as derived primarily from HDAC inhibition.
Hydroxamic Acids
Hydroxamic acids, also commonly called hydroxamates, are the most extensively investigated class of HDACi. The approved drug Zolinza (SAHA) belongs to this chemotype, and is recognized as one of the first and prototypical hydroxamic acid HDACi (Fig. 5). The hydroxamic acid moiety is a highly efficient metal chelator; therefore, most hydroxamic acids can be highly potent HDACi with IC50s in the low nanomolar range (Fig. 5). SAHA is an equipotent inhibitor of HDACs 1, 2, 3, and 6, with marginal activity on the other class I, II, and IV isoforms. Modification of the surface-binding (cap group) and linking motifs of hydroxamic acids led to the discovery of quisinostat [52] (IC50 <0.119 μM against HDACs 1–11) and pandacostat (Ki <1.4 μM against HDACs 1–9) [53] (Fig. 5), which display a broader HDAC inhibitory profile and represent the most potent pan-HDACis reported to date.
Fig. 5.

Hydroxamic acid histone deacetylase (HDAC) inhibitors. SAHA suberoylanilide hydroxamic acid; TSA Trichostatin A
Many hydroxamic acid-based HDACis are structurally related to SAHA, with variations in linking and capping motifs. The different linkers include substituted alkenyl (TSA), cinnamyl (belinostat, panobinostat), phenyl (givinostat), or heteroaryl (quisinostat) groups. The cap groups that extend beyond the surface of the enzyme vary in size and functionality, and have been exploited to impart isoform selectivity with the class II family.
HDAC6 selective Hydroxamic Acid Inhibitors
The first example of this approach, tubacin, was reported in 2003 by Haggarty et al. [54] as a highly potent and selective HDAC6 inhibitor (IC50 = 4 nM; Fig. 6). While structurally identical to SAHA in ZBG and linker motifs, the selectivity was attributed to specific interactions between the unique capping motif and the surface topology of HDAC6 [55].
Fig. 6.
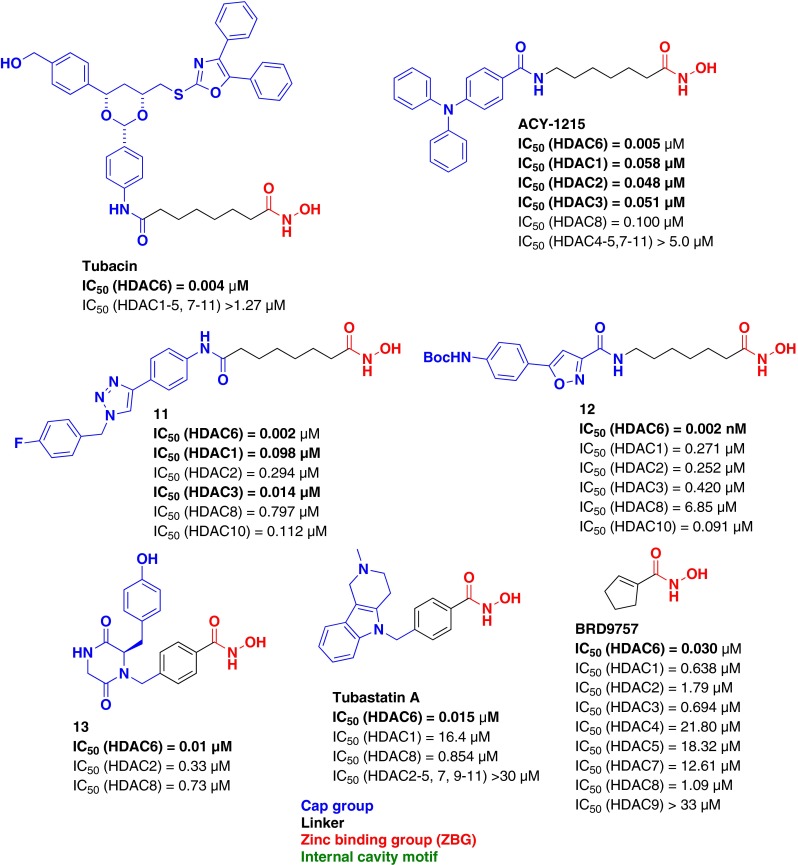
Histone deacetylase (HDAC) 6-selective hydroxamic acid inhibitors
Similarly, other groups have designed HDAC6-selective inhibitors relying on diverse capping moieties that are believed to confer selectivity (ACY-1215 [56], compound 11 [57], and compound 12 [58]). High potency (low nM or pM activity) and selectivity for HDAC6 vs other class I and IIa HDACs was obtained. Subsequently, a highly selective HDAC6 inhibitor, compound 13, possessing a phenyl linking motif and a chiral capping moiety was reported in 2009 by Smil et al. [59] The alternative phenyl linking motif takes advantage of the wider and shallower channel topology of HDAC6, while the cap group forms favorable surface interactions (IC50 = 0.010 μM) and confers HDAC6 selectivity (>40-fold vs all HDACs). An alternatively-capped, phenyl-linked HDAC6-selective inhibitor, tubastatin A (15 nM, >57-fold vs all HDACs), was described by Butler et al. in 2010 [60], and subsequent analogs with improved potency and selectivity were later reported [61]. In an effort to identify novel, selective, and highly efficient HDAC6 inhibitors, Wagner et al. [62] reported a linker-based strategy exploiting close contacts and structural differences between the various isoforms within the catalytic binding domain of HDAC6. BRD9757, cyclopentenyl hydroxamic acid, represents the most ligand-efficient HDAC6 selective inhibitor reported to date (ligE = 0.83) and demonstrates that linker elements alone can play a significant role in imparting isoform selectivity.
HDAC8 and HDAC6,8 Selective Hydroxamic Acid Inhibitors
Other “linkerless” or “capless” inhibitors with selectivity towards HDAC8 have been reported by KrennHrubec et al. (compound 14; Fig. 7) [63]. This compact (ligE = 0.38) HDAC8-selective inhibitor has ≥7-fold selectivity vs HDAC6, but displays weak inhibitory activity for HDAC8 (IC50 = 14.0 μM). In the same report, a more potent phenyl linked compound (15) was reported with limited counter-screening vs the other class I and class II isoforms. These HDACi were designed to take advantage of a large subpocket adjacent to the active site channel [64], which was observed in the crystal structure of HDAC8 bound with another phenyl linked hydroxamic acid, CRA-A (Table 1) [46].
Fig. 7.

Histone deacetylase 8 (HDAC8) and HDAC6, 8 selective hydroxamic acid inhibitors
Similarly, PCI-34051 (Fig. 7) was designed to maximize interactions with this secondary subpocket. PCI-034051, which possesses an indole-linking moiety, demonstrated >290-fold selectivity for HDAC8 over HDACs 1–3, 6, and 10 (HDAC8 IC50 = 10 nM) [65]. Several other HDAC8-selective inhibitors (compounds 16 and 17) have been reported, and possess a common, key structural feature: a meta-substituted phenyl linker [66, 67]. The meta-substitution pattern strategically positions the cap group to interact with the same subpocket. Following the same meta-substitution strategy, Olson et al. [68] recently published the first interclass-selective HDACi, BRD73954, that demonstrated selectivity for HDAC6 (member of class IIb) and HDAC8 (member of class I).
Hydroxamic acids have varied pharmacokinetic properties, but generally possess less optimal pharmacokinetic properties, including poor oral bioavailibility (<20 %) and high systemic clearance (T½ <1 h) [69]. In addition, SAHA has been reported to have poor brain penetration and suffers from P-glycoprotein efflux [70]. Co-administration of P-glycoprotein inhibitors with SAHA has been used to overcome the poor brain exposure of SAHA and increase brain concentrations [70]. Encouragingly, Envivo Pharmaceuticals reported the discovery of a brain penetrant hydroxamic acid (brain to plasma ratio of ~3.5 in rats), though no structure was described [71]. This report suggests that pharmacokinetic liabilities associated with hydroxamic acids can be overcome, and highly brain-penetrant compounds from this class of compounds are feasible. Further studies on a broader collection of compounds will be required to quantify and understand the entry, exposure, and residence time of hydroxamic acid HDACi in the brain.
Cyclic Peptides and Other Macrocycles
Members of this class of HDACi are characterized by large capping motifs derived from cyclic peptides, depsipeptides, or carbon-based macrocycles coupled with alkyl-linking motifs and a variety of ZBG. Most members of this class are either natural products or derivatives thereof [72]. The natural product romidepsin (FK-228; Fig. 8) [73] is a prototypical example of this family possessing a large depsipeptide-based cap group and an extended 4-carbon linker with a unique thiol zinc-chelating motif.
Fig. 8.

Cyclic peptides and macrocyclic histone deacetylase (HDAC) inhibitors
Despite the fact that it is one of the most potent HDACi known, with good selectivity for class I HDACs, very little is known about the structure–activity relationship governing its potency and selectivity in the capping region [74]. Because these large and diverse capping groups are believed to provide significant interactions with amino acid residues on the surface of the enzyme, members of the cyclic peptides or macrocyclic family of HDACi can possess a variety of ZBG (Fig. 8), including thiols (FK-228, largazole [75]), hydroxamic acids (compound 18 [76]), ketones (apicidin [77]), carboxylic acids (azumamide E [78]), and α-epoxy ketones (chlamydocin [79], trapoxin B [80]).
Most HDACi with macrocyclic capping moieties are highly potent, even when coupled with relatively poor metal chelating functionality (e.g., carboxylic acids, ketones) and display class I selectivity. For example, trapoxin B (an irreversible α-epoxy ketone) exhibits a 640-fold selectivity for HDAC1 vs HDAC6 with picomolar potency on HDAC1 (IC50 = 110 pM) [80]. In 2007, Maulucci et al. [78] reported a series of cyclic tetrapetides, the azumamides, of which the most selective analog, azumamide E, displayed 100-fold selectivity for HDACs 1, 2, and 3 (50–100 nM) vs all other HDAC isoforms. Similarily, apicidin, another cyclic tetrapeptide possessing a ketone-based ZBG, showed preferential selectivity for HDACs 1, 2, 3, and 8 over all other HDAC isoforms [53]. The class I selectivity displayed by this series of compounds is hypothesized to arise from the cap group similarity to a natural substrate of class I HDACs. Computational modeling suggests that 2 loops characteristic to class II HDACs located at the entrance to the catalytic binding domain clash with the sterically-demanding cyclic peptides [55]. Several groups have successfully designed hybrid inhibitors that combine the cyclic tetrapeptide scaffolds with more efficient ZBG, such as hydroxamic acids [76, 80]. An interesting example is a series of nonpeptidic chiral macrocycles, such as compound 18 (Fig. 8), designed based on the substructure of SAHA [81]. While most analogs showed potent activity against all 11 HDACs, a few diamide-based analogs, such as compound 18, show preferential inhibition of HDAC6 (4.4 nM, 55-fold selectivity) vs other HDAC isoforms.
Despite these encouraging results in obtaining isoform selectivity and potency, cyclic peptides are structurally more complex than other HDACi classes. This complexity presents synthetic challenges when attempting systematic exploration of cap group structure–activity relationships to improve not only their selectivity profiles, but also physicochemical properties (molecular weight, log P, etc.). When considering delivery into the CNS, the ability of these inhibitors to penetrate the BBB may be limited.
ortho-Aminoanilides
The ortho-aminoanilide family of HDACi is also broadly referred to as the benzamide family; however, the term benzamide describes only a subcategory of the larger ortho-aminoanilide family. Indeed, the ortho-aminoanilide chemotype is defined by the zinc-binding moiety highlighted in red (Fig. 9), while benzamide is defined by the linking phenyl (or benzene) ring and the adjacent amide motif, as illustrated in tacedinaline (CI-994) (Fig. 9). For instance, RGFP106 is classified as an ortho-aminoanilide, but not as a benzamide as the linking motif is an alkyl chain.
Fig. 9.

ortho-Aminoanilide histone deacetylase (HDAC) inhibitors
HDAC1, 2 and 3 Selective ortho-Aminoanilide Inhibitors
ortho-Aminoanilides, such as CI-994, are subclass I-selective inhibiting HDACs 1, 2, and 3, with no significant inhibition of HDAC8 or the class IIa and IIb HDAC isoforms. Despite the fact that HDAC 8 is a member of the class I HDACs, it is the least similar with only 30–34 % amino acid identity with HDACs 1, 2, and 3. Using a computational model, Vannini et al. [42] rationalized that a single amino acid difference—a leucine in HDACs 1, 2, and 3, and a tryptophan in HDAC8—located near the zinc-binding domain prevents the ortho-aminoanilide moiety from attaining an optimal chelation geometry to the zinc in HDAC8 [42]. This apparent steric clash imparts the subclass I selectivity for this family. MS-275 (entinostat, Syndax) and MGCD-0103 (mocetinostat, Methylgene) are other examples of highly potent HDAC 1, 2, and 3 inhibitors that display isosteric capping groups which take advantage of improved interactions on the enzyme surface of class I HDACs.
CI-994, which was investigated by Pfizer through phase II clinical studies as a single agent in oncology indications, is a good example of the potential of this series of compounds to cross the BBB. The brain pharmacokinetics of CI-994 were evaluated in Rhesus monkeys, and displayed a brain/plasma ratio of 0.15 and 0.45 based on Cmax and area under the curve, respectively [82]. In addition, CI-994 demonstrated a brain to cerebrospinal fluid ratio of 1.0 and exhibited prolonged residence times in brain and cerebrospinal fluid of up to 12.9 h. However, good brain exposure is not general to this series as an isotopalogue of MS-275 was studied using positron emission tomography in rodents and nonhuman primates, and was found to have low brain penetration (brain to plasma ratio of 0.12:0.15) [83].
HDAC1, 2 and HDAC2, 3 Selective ortho-Aminoanilide Inhibitors
Using crystallographic information from bacteria and human HDACs, Moradei et al. [84] exploited the 14 Å internal cavity adjacent to the catalytic zinc (Fig. 3a) to successfully design HDAC 1- and 2-selective inhibitors. Compound 19 (Fig. 9) is an analog of MS-275 with a 2-thienyl substituent para (C-5) to the chelating aniline, occupying the internal cavity and imparting increased potency (IC50 20 nM and 100 nM, respectively) and selectivity for HDACs 1 and 2 (200–1000-fold selective over all other HDAC isoforms). Compound 20 (aka Compound 60 or Merck 60), an analog of CI-994 with an identical modification, is equally as potent and selective (IC50 40 nM and 100 nM, 200–500-fold selective). This crucial and transformative discovery from Moradei et al. [84] has been widely used to impart selectivity within the class I HDACs utilizing an array of substituents projecting into the 14 Å internal cavity [85–87]. The binding of these types of ligands into this internal cavity was confirmed by Takeda’s published human HDAC2 crystal structure in complex with N-(4-aminobiphenyl-3-yl)benzamide (compound 1, Fig. 3b). The hydrophobic C-5 phenyl substituent fills the hydrophobic 14 Å internal cavity and achieves potent and selective inhibition of HDACs 1 and 2 over HDAC3 [37]. This substitution pattern has also been reported to produce HDAC1- vs HDAC2-selective compounds (>10-fold preference for HDAC1) [87]. However, the development of HDAC2-selective inhibitors using this approach or other strategies in this chemical series of compounds has proven highly elusive and challenging.
Interestingly, Boissinot et al. [88] recently reported the discovery of MI-192, a selective HDAC 2 and 3 inhibitor, obtained through modification of the surface-binding motif [88]. MI-192 displays high potency for HDACs 2 and 3 (30 nM and 16 nM, respectively), and excellent selectivity against all other isoforms (>4 μM on HDACs 1, 4, 6, 7, and 8), although it is difficult to ascertain the origin of the observed isoform selectivity for HDACs 2 and 3 vs HDAC1. This is the first report describing a small molecule capable of imparting differential binding in favor of HDAC2 vs HDAC1. Interestingly, both HDAC2 and HDAC3 have been reported to negatively regulate memory formation [89, 90]. In this context, a dual HDAC 2 and 3 inhibitor could provide the advantage of selectively targeting both relevant HDAC isoforms to promote cognitive enhancement with limited HDAC-related side effects [91]. However, no pharmacokinetic data were reported on this unique compound.
HDAC3 Selective ortho-Aminoanilide Inhibitors
Several groups have reported HDAC3-selective inhibitors within the ortho-aminoanilide family. Compound 21 [92], RGFP106, and RGFP136 [93] are ortho-aminoanilide HDAC3-selective inhibitors possessing a common saturated alkyl chain-linking motif (Fig. 9). These studies demonstrate that it is possible to achieve selectivity for HDAC3 through careful choice of linker and surface-binding motifs coupled with the ortho-aminoanilide zinc-binding motif. The latter 2 compounds, pimelic diphenylamide RGFP106 and RGFP136 (Repligen Corp), display interesting and distinct kinetic binding profiles for HDACs 1, 2, and 3. Indeed, owing to a slower off-rate for HDAC3, RGFP106 demonstrates 6-fold preferential binding for HDAC3 (Ki = 5 nM) vs HDAC1 (Ki = 32 nM). ortho-aminoanilides are known to display slow on/slow off binding kinetics, as described in an earlier report by Chou et al. [94], emphasizing the importance of careful analysis of kinetic parameters as significant shifts (up to 40-fold) in binding IC50s with 24-h incubation times can be observed. Remarkably, RGFP136 represents the first example of a selective HDACi used in vivo to demonstrate proof-of-concept in models of learning and memory enhancement and cognitive impairment (e.g., location-dependent and novel object recognition memory paradigms) [90]. However, reports examining the chemical stability and pharmacokinetic properties of this class of compounds have raised concerns about the applicability of these compounds in in vivo model systems [95]. More recently, Repligen Corp reported RGFP966 (Fig. 9) a heterocinnamyl-linked ortho-aminoanilide that displays potent (IC50 = 80 nM) and selective inhibition of HDAC3 [96]. RGFP966 possesses good brain penetration (brain/plasma = 0.45) and was used to enhance extinction of cocaine-seeking behavior in mice. The development of such selective and brain-penetrant HDACi is crucial to validate the safe application of isoform selective inhibitors in relevant disease models of neurological disorders and enhance our understanding on the role of the individual HDAC isoforms.
Other Zinc-binding Motifs
Over the years, novel zinc-binding motifs have emerged and could provide complementary selectivity profiles or open the field to potentially new brain-penetrant HDACi. In 2011, Whitehead et al. [45] reported the discovery of a series of class I-selective inhibitors, exemplified by compound 7 (Table 1; Fig. 10), which uses an amino acid derivative as a zinc-binding motif. This structurally unique small molecule inhibitor showed preferential inhibition of HDAC8 (IC50 = 90 nM) vs all other HDACs (>18-fold selectivity over HDACs 1, 2, and 6).
Fig. 10.

Histone deacetylase (HDAC) inhibitors with atypical zinc-binding motifs
In 2008, Jones et al. [97, 98] reported a series of 5-(trifluoroacetyl)thiophene-2-carboxamides as novel, potent, and selective class II HDACi exemplified by compound 9 (HDAC4 IC50 = 320 nM; HDAC6 IC50 = 310 nM). X-ray crystal structures of HDAC4 with compound 9 demonstrate these compounds are active site inhibitors and bind in their hydrated form. Wahhab et al. [99] reported the use of a sulfamide moiety as a zinc binder. The potency and selectivity of the reported inhibitors, such as compound 22, are believed to arise from the capping group linked to the ZBG by a long alkyl chain. In 2013, a class IIa-selective HDACi with a unique nonclassical chelating ZBG, a trifluoromethyloxadiazole, was reported (compound 8; Table 1; Fig. 10) [39]. This structurally unprecedented HDACi exhibited good potency for the class IIa HDACs 4, 5, 7, and 9 (19–126 nM) and approximately 20-fold selectivity vs the class I HDACs. Finally, in 2013, Ononye et al. [100] reported β-thujaplicin (a natural product from the tropolone family) derivatives with interesting HDAC isoform selectivity. While the tropolone functionality serves as the zinc-binding motif in compound 23, the β-substituent (a cyclopentyl) presumably gives rise to its exquisite HDAC2 selectivity and potency (0.04 nM, >3000-fold selectivity). This series of compounds represents the first report of highly potent and selective HDAC2 inhibitors. The β-substituted and O-methylated analog, compound 24, is a highly potent and selective HDAC8 inhibitor (8 nM, >1400-fold selectivity). It should be noted that no data were reported for HDACs 3, 7, 9, 10, or 11.
While the CNS penetration of these novel HDACi has not been investigated, they potentially offer new tools, with better selectivity and fewer off-target effects associated with the canonical ZBG, to evaluate the function of these isozymes in neurological disorders.
Perspectives
Increasing evidence implicates chromatin modifications, HDAC function, and/or acetylation status in neurological disorders [10]. There is a substantial body of preclinical evidence, neurobiological and pharmacological, to support the hypothesis that inhibition of the class I HDACs, particularly HDACs 2 and 3, could treat the cognitive deficits associated with a number of neurological disorders [19]. The treatment of most neurological disorders would imply the life-long, chronic use of a HDACi and, given the complexity of brain disorders, there are several critical considerations to be made regarding HDACi therapy. First, and foremost, the development of highly selective inhibitors for individual HDACs is key to understand which HDAC isoform(s) is (are) responsible for on-target efficacy and to mitigate potential mechanism-based dose-limiting toxicities. The demonstration of an acceptable safety profile is an ongoing hurdle and will be necessary for the consideration of HDACs or related epigenetic targets as a therapeutically viable approach in neurological disorders. Reports of selective HDACi have emerged in the last decade, including class-, subclass-, interclass-, and isoform-selective inhibitors. How these varied chemical classes of inhibitors and their corresponding selectivity profiles functionally translate in biological systems—for example histone acetylation, nonhistone acetylation, gene expression, and translation in specific cell types—presents an obvious field of continued research. Similarly, while structural biology has greatly enhanced our understanding of the origin of isoform selectivity by small molecule inhibitors and their interactions with the individual and isolated enzymes, insights into these interactions within higher-order complexes will be required. Indeed, recent reports on the protein–protein interactions involved and the signaling molecules associated within co-repressor complexes is shedding light on the structural nature and functional consequences of these higher-order systems. How the different chemical classes of inhibitors interact with and affect the structure and function of these higher-order complexes is an open question. These complexes may prove a more relevant system to interrogate small molecule binding events and may provide new opportunities for achieving isoform and/or “complex” selectivity. Finally, in order for a drug to reach its target in the brain, it must cross the BBB, and, unfortunately, only a few reports describe the brain permeability and pharmacokinetic properties of HDACi. However, these limited reports do suggest that many of the chemical series of inhibitors described in this review can be tailored to exhibit favorable brain pharmacokinetics. Exciting new chemical tools are still emerging and will catalyze new and ongoing biological and translational efforts to address the therapeutic potential of more selective HDACi in treating neurological disorders.
Electronic Supplementary Material
(PDF 1224 kb)
Acknowledgments
We thank Dr Taner Kaya (Stanley Center for Psychiatric Research) for providing computational assistance in the preparation of this manuscript.
The authors declare no conflict of interest. Full conflict of interest disclosures are available in the electronic supplementary material of this article.
Required Author Forms Disclosure forms provided by the authors are available with the online version of this article.
References
- 1.Khorasanizadeh S. The nucleosome: from genomic organization to genomic regulation. Cell. 2004;116:259–272. doi: 10.1016/S0092-8674(04)00044-3. [DOI] [PubMed] [Google Scholar]
- 2.Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–294. doi: 10.1016/S0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- 3.Sengupta N, Seto E. Regulation of histone deacetylase activities. J Cell Biochem. 2004;93:57–67. doi: 10.1002/jcb.20179. [DOI] [PubMed] [Google Scholar]
- 4.Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009;32:591–601. doi: 10.1016/j.tins.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodriquez M, Aquino M, Bruno I, et al. Chemistry and biology of chromatin remodeling agents: state of art and future perspectives of HDAC inhibitors. Curr Med Chem. 2006;13:1119–1139. doi: 10.2174/092986706776360905. [DOI] [PubMed] [Google Scholar]
- 7.Fass DM, Kemp MM, Schroeder FA, et al. Histone acetylation and deacetylation. Epigenet Regul Epigenomics. 2012;1:515–561. [Google Scholar]
- 8.Peart MJ, Smyth GK, van Laar RK, et al. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005;102:3697–3702. doi: 10.1073/pnas.0500369102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 10.Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- 11.Broide RS, Redwine JM, Aftahi N, et al. Distribution of histone deacetylases 1-11 in the rat brain. J Mol Neurosci. 2007;31:47–58. doi: 10.1007/BF02686117. [DOI] [PubMed] [Google Scholar]
- 12.Fischle W, Dequiedt F, Fillion M, et al. Human HDAC7 histone deacetylase activity is associated with HDAC3 in vivo. J Biol Chem. 2001;276:35826–35835. doi: 10.1074/jbc.M104935200. [DOI] [PubMed] [Google Scholar]
- 13.Fischle W, Dequiedt F, Hendzel MJ, et al. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell. 2002;9:45–57. doi: 10.1016/S1097-2765(01)00429-4. [DOI] [PubMed] [Google Scholar]
- 14.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11:384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 15.Kim T, Park JK, Kim HJ, Chung JH, Kim JW. Association of histone deacetylase genes with schizophrenia in Korean population. Psychiatry Res. 2010;178:266–269. doi: 10.1016/j.psychres.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 16.Tam GW, van de Lagemaat LN, Redon R, et al. Confirmed rare copy number variants implicate novel genes in schizophrenia. Biochem Soc Trans. 2010;38:445–451. doi: 10.1042/BST0380445. [DOI] [PubMed] [Google Scholar]
- 17.Guidotti A, Auta J, Davis JM, et al. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry. 2000;57:1061–1069. doi: 10.1001/archpsyc.57.11.1061. [DOI] [PubMed] [Google Scholar]
- 18.Simonini MV, Camargo LM, Dong E, et al. The benzamide MS-275 is a potent, long-lasting brain region-selective inhibitor of histone deacetylases. Proc Natl Acad Sci U S A. 2006;103:1587–1592. doi: 10.1073/pnas.0510341103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graff J, Tsai L-H. The potential of HDAC inhibitors as cognitive enhancers. Annu Rev Pharmacol Toxicol. 2013;53:311–330. doi: 10.1146/annurev-pharmtox-011112-140216. [DOI] [PubMed] [Google Scholar]
- 20.Abel T, Zukin RS. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr Opin Pharmacol. 2008;8:57–64. doi: 10.1016/j.coph.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saha RN, Pahan K. HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Diff. 2006;13:539–550. doi: 10.1038/sj.cdd.4401769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morris MJ, Monteggia LM. Unique functional roles for class I and class II histone deacetylases in central nervous system development and function. Int J Dev Neurosci. 2013;31:370–381. doi: 10.1016/j.ijdevneu.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foley AG, Gannon S, Rombach-Mullan N, et al. Class I histone deacetylase inhibition ameliorates social cognition and cell adhesion molecule plasticity deficits in a rodent model of autism spectrum disorder. Neuropharmacology. 2012;63:750–760. doi: 10.1016/j.neuropharm.2012.05.042. [DOI] [PubMed] [Google Scholar]
- 24.Machado-Vieira R, Ibrahim L, Zarate CA., Jr Histone deacetylases and mood disorders: epigenetic programming in gene-environment interactions. CNS Neurosci Ther. 2011;17:699–704. doi: 10.1111/j.1755-5949.2010.00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guidotti A, Auta J, Chen Y, et al. Epigenetic GABAergic targets in schizophrenia and bipolar disorder. Neuropharmacology. 2011;60:1007–1016. doi: 10.1016/j.neuropharm.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grayson DR, Kundakovic M, Sharma RP. Is there a future for histone deacetylase inhibitors in the pharmacotherapy of psychiatric disorders? Mol Pharmacol. 2010;77:126–135. doi: 10.1124/mol.109.061333. [DOI] [PubMed] [Google Scholar]
- 27.Schmalbach S, Petri S. Histone deacetylation and motor neuron degeneration. CNS Neurol Disord Drug Targets. 2010;9:279–284. doi: 10.2174/187152710791292684. [DOI] [PubMed] [Google Scholar]
- 28.Sleiman SF, Basso M, Mahishi L, et al. Putting the 'HAT' back on survival signalling: the promises and challenges of HDAC inhibition in the treatment of neurological conditions. Expert Opin Investig Drugs. 2009;18:573–584. doi: 10.1517/13543780902810345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kozikowski AP, Chen Y, Subhasish T, et al. Searching for disease modifiers-PKC activation and HDAC inhibition-a dual drug approach to Alzheimer's disease that decreases Aβ production while blocking oxidative stress. Chem Med Chem. 2009;4:1095–1105. doi: 10.1002/cmdc.200900045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fraczek J, Vanhaecke T, Rogiers V. Toxicological and metabolic considerations for histone deacetylase inhibitors. Expert Opin Drug Metab Toxicol. 2013;9:441–457. doi: 10.1517/17425255.2013.754011. [DOI] [PubMed] [Google Scholar]
- 31.Bishton MJ, Harrison SJ, Martin BP, et al. Deciphering the molecular and biologic processes that mediate histone deacetylase inhibitor-induced thrombocytopenia. Blood. 2011;117:3658–3668. doi: 10.1182/blood-2010-11-318055. [DOI] [PubMed] [Google Scholar]
- 32.Subramanian S, Bates SE, Wright JJ, Espinoza-Delgado I, Piekarz RL. Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals. 2010;3:2751–2767. doi: 10.3390/ph3092751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilting RH, Yanover E, Heideman MR, et al. Overlapping functions of HDAC1 and HDAC2 in cell cycle regulation and haematopoiesis. EMBO J. 2010;29:2586–2597. doi: 10.1038/emboj.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finnin MS, Donigian JR, Cohen A, et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 35.Rosenberg G. The mechanisms of action of valproate in neuropsychiatric disorders: can we see the forest for the trees? Cell Mol Life Sci. 2007;64:2090–2103. doi: 10.1007/s00018-007-7079-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bottomley MJ, Lo Surdo P, Di Giovine P, et al. Structural and functional analysis of the human HDAC4 catalytic domain reveals a regulatory structural zinc-binding domain. J Biol Chem. 2008;283:26694–26704. doi: 10.1074/jbc.M803514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bressi JC, Jennings AJ, Skene R, et al. Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg Med Chem Lett. 2010;20:3142–3145. doi: 10.1016/j.bmcl.2010.03.091. [DOI] [PubMed] [Google Scholar]
- 38.Dowling DP, Gantt SL, Gattis SG, Fierke CA, Christianson DW. Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry. 2008;47:13554–13563. doi: 10.1021/bi801610c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lobera M, Madauss KP, Pohlhaus DT, et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat Chem Biol. 2013;9:319–325. doi: 10.1038/nchembio.1223. [DOI] [PubMed] [Google Scholar]
- 40.Millard CJ, Watson PJ, Celardo I, et al. Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol Cell. 2013;51:57–67. doi: 10.1016/j.molcel.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schuetz A, Min J, Allali-Hassani A, et al. Human HDAC7 harbors a Class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity. J Biol Chem. 2008;283:11355–11363. doi: 10.1074/jbc.M707362200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vannini A, Volpari C, Filocamo G, et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci U S A. 2004;101:15064–15069. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vannini A, Volpari C, Gallinari P, et al. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep. 2007;8:879–884. doi: 10.1038/sj.embor.7401047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watson PJ, Fairall L, Santos GM, Schwabe JW. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature. 2012;481:335–340. doi: 10.1038/nature10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Whitehead L, Dobler MR, Radetich B, et al. Human HDAC isoform selectivity achieved via exploitation of the acetate release channel with structurally unique small molecule inhibitors. Bioorg Med Chem. 2011;19:4626–4634. doi: 10.1016/j.bmc.2011.06.030. [DOI] [PubMed] [Google Scholar]
- 46.Somoza JR, Skene RJ, Katz BA, et al. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure. 2004;12:1325–1334. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 47.Fass DM, Shah R, Ghosh B, et al. Short-chain HDAC inhibitors differentially affect vertebrate development and neuronal chromatin. ACS Med Chem Lett. 2011;2:39–42. doi: 10.1021/ml1001954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bora-Tatar G, Dayangac-Erden D, Demir AS, et al. Molecular modifications on carboxylic acid derivatives as potent histone deacetylase inhibitors: Activity and docking studies. Bioorg Med Chem. 2009;17:5219–5228. doi: 10.1016/j.bmc.2009.05.042. [DOI] [PubMed] [Google Scholar]
- 49.Loscher W, Nau H. Distribution of valproic acid and its metabolites in various brain areas of dogs and rats after acute and prolonged treatment. J Pharmacol Exp Ther. 1983;226:845–854. [PubMed] [Google Scholar]
- 50.Adkison KD, Shen DD. Uptake of valproic acid into rat brain is mediated by a medium-chain fatty acid transporter. J Pharmacol Exp Ther. 1996;276:1189–1200. [PubMed] [Google Scholar]
- 51.Cornford EM, Diep CP, Pardridge WM. Blood-brain barrier transport of valproic acid. J Neurochem. 1985;44:1541–1550. doi: 10.1111/j.1471-4159.1985.tb08793.x. [DOI] [PubMed] [Google Scholar]
- 52.Arts J, King P, Marien A, et al. JNJ-26481585, a novel "second-generation" oral histone deacetylase inhibitor, shows broad-spectrum preclinical antitumoral activity. Clin Cancer Res. 2009;15:6841–6851. doi: 10.1158/1078-0432.CCR-09-0547. [DOI] [PubMed] [Google Scholar]
- 53.Bradner JE, West N, Grachan ML, et al. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. 2010;6:238–243. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A. 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bieliauskas AV, Pflum MKH. Isoform-selective histone deacetylase inhibitors. Chem Soc Rev. 2008;37:1402–1413. doi: 10.1039/b703830p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Santo L, Hideshima T, Kung AL, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119:2579–2589. doi: 10.1182/blood-2011-10-387365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen Y, Lopez-Sanchez M, Savoy DN, et al. A series of potent and selective, triazolylphenyl-based histone deacetylases inhibitors with activity against pancreatic cancer cells and Plasmodium falciparum. J Med Chem. 2008;51:3437–3448. doi: 10.1021/jm701606b. [DOI] [PubMed] [Google Scholar]
- 58.Kozikowski AP, Tapadar S, Luchini DN, Kim KH, Billadeau DD. Use of the nitrile oxide cycloaddition (NOC) reaction for molecular probe generation: a new class of enzyme selective histone deacetylase inhibitors (HDACIs) showing picomolar activity at HDAC6. J Med Chem. 2008;51:4370–4373. doi: 10.1021/jm8002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smil DV, Manku S, Chantigny YA, et al. Novel HDAC6 isoform selective chiral small molecule histone deacetylase inhibitors. Bioorg Med Chem Lett. 2009;19:688–692. doi: 10.1016/j.bmcl.2008.12.045. [DOI] [PubMed] [Google Scholar]
- 60.Butler KV, Kalin J, Brochier C, et al. Rational Design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J Am Chem Soc. 2010;132:10842–10846. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kalin JH, Butler KV, Akimova T, Hancock WW, Kozikowski AP. Second-generation histone deacetylase 6 inhibitors enhance the immunosuppressive effects of Foxp3+ T-regulatory cells. J Med Chem. 2012;55:639–651. doi: 10.1021/jm200773h. [DOI] [PubMed] [Google Scholar]
- 62.Wagner FF, Olson DE, Gale JP, et al. Potent and selective inhibition of histone deacetylase 6 (HDAC6) does not require a surface-binding motif. J Med Chem. 2013;56:1772–1776. doi: 10.1021/jm301355j. [DOI] [PubMed] [Google Scholar]
- 63.Krennhrubec K, Marshall BL, Hedglin M, Verdin E, Ulrich SM. Design and evaluation of 'Linkerless' hydroxamic acids as selective HDAC8 inhibitors. Bioorg Med Chem Lett. 2007;17:2874–2878. doi: 10.1016/j.bmcl.2007.02.064. [DOI] [PubMed] [Google Scholar]
- 64.Estiu G, West N, Mazitschek R, et al. On the inhibition of histone deacetylase 8. Bioorg Med Chem. 2010;18:4103–4110. doi: 10.1016/j.bmc.2010.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Balasubramanian S, Ramos J, Luo W, et al. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia. 2008;22:1026–1034. doi: 10.1038/leu.2008.9. [DOI] [PubMed] [Google Scholar]
- 66.Suzuki T, Ota Y, Ri M, et al. Rapid discovery of highly potent and selective inhibitors of histone deacetylase 8 using click chemistry to generate candidate libraries. J Med Chem. 2012;55:9562–9575. doi: 10.1021/jm300837y. [DOI] [PubMed] [Google Scholar]
- 67.Tang W, Luo T, Greenberg EF, Bradner JE, Schreiber SL. Discovery of histone deacetylase 8 selective inhibitors. Bioorg Med Chem Lett. 2011;21:2601–2605. doi: 10.1016/j.bmcl.2011.01.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Olson DE, Wagner FF, Kaya T, et al. Discovery of the first histone deacetylase 6/8 dual inhibitors. J Med Chem. 2013;56:4816–4820. doi: 10.1021/jm400390r. [DOI] [PubMed] [Google Scholar]
- 69.Flipo M, Charton J, Hocine A, et al. Hydroxamates: relationships between structure and plasma stability. J Med Chem. 2009;52:6790–6802. doi: 10.1021/jm900648x. [DOI] [PubMed] [Google Scholar]
- 70.Palmieri D, Lockman PR, Thomas FC, et al. Vorinostat inhibits brain metastatic colonization in a model of triple-negative breast cancer and induces DNA double-strand breaks. Clin Cancer Res. 2009;15:6148–6157. doi: 10.1158/1078-0432.CCR-09-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Patzke H, Albayya FP, Bollen E, et al. The novel histone deacetylase inhibitor EVP-0334 is pro cognitive in rats. Behavioral Epigenetics 2010; University Massachusetts Boston Campus Center Boston, MA, USA [Abstract].
- 72.Seidel C, Schnekenburger M, Dicato M, Diederich M. Histone deacetylase modulators provided by Mother Nature. Genes Nutr. 2012;7:357–367. doi: 10.1007/s12263-012-0283-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Furumai R, Matsuyama A, Kobashi N, et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002;62:4916–4921. [PubMed] [Google Scholar]
- 74.Yurek-George A, Cecil AR, Mo AH, et al. The first biologically active synthetic analogues of FK228, the depsipeptide histone deacetylase inhibitor. J Med Chem. 2007;50:5720–5726. doi: 10.1021/jm0703800. [DOI] [PubMed] [Google Scholar]
- 75.Ying Y, Taori K, Kim H, Hong J, Luesch H. Total synthesis and molecular target of largazole, a histone deacetylase inhibitor. J Am Chem Soc. 2008;130:8455–8459. doi: 10.1021/ja8013727. [DOI] [PubMed] [Google Scholar]
- 76.Olsen CA, Ghadiri MR. Discovery of potent and selective histone deacetylase inhibitors via focused combinatorial libraries of cyclic alpha3beta-tetrapeptides. J Med Chem. 2009;52:7836–7846. doi: 10.1021/jm900850t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Darkin-Rattray SJ, Gurnett AM, Myers RW, et al. Apicidin: a novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc Natl Acad Sci U S A. 1996;93:13143–13147. doi: 10.1073/pnas.93.23.13143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maulucci N, Chini MG, Di Micco S, et al. Molecular insights into azumamide E histone deacetylases inhibitory activity. J Am Chem Soc. 2007;129:3007–3012. doi: 10.1021/ja0686256. [DOI] [PubMed] [Google Scholar]
- 79.De Schepper S, Bruwiere H, Verhulst T, et al. Inhibition of histone deacetylases by chlamydocin induces apoptosis and proteasome-mediated degradation of survivin. J Pharmacol Exp Ther. 2003;304:881–888. doi: 10.1124/jpet.102.042903. [DOI] [PubMed] [Google Scholar]
- 80.Furumai R, Komatsu Y, Nishino N, et al. Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. Proc Natl Acad Sci U S A. 2001;98:87–92. doi: 10.1073/pnas.98.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Auzzas L, Larsson A, Matera R, et al. Non-natural macrocyclic inhibitors of histone deacetylases: design, synthesis, and activity. J Med Chem. 2010;53:8387–8399. doi: 10.1021/jm101092u. [DOI] [PubMed] [Google Scholar]
- 82.Riva L, Blaney SM, Dauser R, et al. Pharmacokinetics and cerebrospinal fluid penetration of CI-994 (N-acetyldinaline) in the nonhuman primate. Clin Cancer Res. 2000;6:994–997. [PubMed] [Google Scholar]
- 83.Hooker JM, Kim SW, Alexoff D, et al. Histone deacetylase inhibitor MS-275 exhibits poor brain penetration: pharmacokinetic studies of [11C]MS-275 using positron emission tomography. ACS Chem Neurosci. 2010;1:65–73. doi: 10.1021/cn9000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Moradei OM, Mallais TC, Frechette S, et al. Novel aminophenyl benzamide-type histone deacetylase inhibitors with enhanced potency and selectivity. J Med Chem. 2007;50:5543–5546. doi: 10.1021/jm701079h. [DOI] [PubMed] [Google Scholar]
- 85.Heidebrecht RW, Jr, Chenard M, Close J, et al. Exploring the pharmacokinetic properties of phosphorus-containing selective HDAC 1 and 2 inhibitors (SHI-1:2) Bioorg Med Chem Lett. 2009;19:2053–2058. doi: 10.1016/j.bmcl.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 86.Witter DJ, Harrington P, Wilson KJ, et al. Optimization of biaryl Selective HDAC1&2 Inhibitors (SHI-1:2) Bioorg Med Chem Lett. 2008;18:726–731. doi: 10.1016/j.bmcl.2007.11.047. [DOI] [PubMed] [Google Scholar]
- 87.Methot JL, Chakravarty PK, Chenard M, et al. Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1:2) Bioorg Med Chem Lett. 2008;18:973–978. doi: 10.1016/j.bmcl.2007.12.031. [DOI] [PubMed] [Google Scholar]
- 88.Boissinot M, Inman M, Hempshall A, et al. Induction of differentiation and apoptosis in leukaemic cell lines by the novel benzamide family histone deacetylase 2 and 3 inhibitor MI-192. Leuk Res. 2012;36:1304–1310. doi: 10.1016/j.leukres.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 89.Guan J-S, Haggarty SJ, Giacometti E, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McQuown SC, Barrett RM, Matheos DP, et al. HDAC3 is a critical negative regulator of long-term memory formation. J Neurosci. 2011;31:764–774. doi: 10.1523/JNEUROSCI.5052-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Durham B. Novel histone deacetylase (HDAC) inhibitors with improved selectivity for HDAC2 and 3 protect against neural cell death. Biosci Horiz. 2012;5:hzs003. doi: 10.1093/biohorizons/hzs003. [DOI] [Google Scholar]
- 92.Chen Y, He R, Chen Y, et al. Studies of benzamide- and thiol-based histone deacetylase inhibitors in models of oxidative-stress-induced neuronal death: identification of some HDAC3-Selective Inhibitors. Chem Med Chem. 2009;4:842–852. doi: 10.1002/cmdc.200800461. [DOI] [PubMed] [Google Scholar]
- 93.Rai M, Soragni E, Chou CJ, et al. Two new pimelic diphenylamide HDAC inhibitors induce sustained frataxin upregulation in cells from Friedreich's ataxia patients and in a mouse model. PLoS One. 2010;5:e8825. doi: 10.1371/journal.pone.0008825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chou CJ, Herman D, Gottesfeld JM. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J Biol Chem. 2008;283:35402–35409. doi: 10.1074/jbc.M807045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Beconi M, Aziz O, Matthews K, et al. Oral administration of the pimelic diphenylamide HDAC inhibitor HDACi 4b is unsuitable for chronic inhibition of HDAC activity in the CNS in vivo. PLoS One. 2012;7:e44498. doi: 10.1371/journal.pone.0044498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Malvaez M, McQuown Susan C, Rogge George A, et al. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc Natl Acad Sci U S A. 2013;110:2647–2652. doi: 10.1073/pnas.1213364110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ontoria JM, Altamura S, Di Marco A, et al. Identification of novel, selective, and stable inhibitors of class II histone deacetylases. Validation studies of the inhibition of the enzymatic activity of HDAC4 by small molecules as a novel approach for cancer therapy. J Med Chem. 2009;52:6782–6789. doi: 10.1021/jm900555u. [DOI] [PubMed] [Google Scholar]
- 98.Jones P, Bottomley MJ, Carfi A, et al. 2-Trifluoroacetylthiophenes, a novel series of potent and selective class II histone deacetylase inhibitors. Bioorg Med Chem Lett. 2008;18:3456–3461. doi: 10.1016/j.bmcl.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 99.Wahhab A, Smil D, Ajamian A, et al. Sulfamides as novel histone deacetylase inhibitors. Bioorg Med Chem Lett. 2009;19:336–340. doi: 10.1016/j.bmcl.2008.11.081. [DOI] [PubMed] [Google Scholar]
- 100.Ononye SN, VanHeyst MD, Oblak EZ, et al. Tropolones as lead-like natural products: the development of potent and selective histone deacetylase inhibitors. ACS Med Chem Lett. 2013;4:757–761. doi: 10.1021/ml400158k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 1224 kb)