

Abstract
Epigenetic marks are modifications of DNA and histones. They are considered to be permanent within a single cell during development, and are heritable across cell division. Programming of neurons through epigenetic mechanisms is believed to be critical in neural development. Disruption or alteration in this process causes an array of neurodevelopmental disorders, including autism spectrum disorders (ASDs). Recent studies have provided evidence for an altered epigenetic landscape in ASDs and demonstrated the central role of epigenetic mechanisms in their pathogenesis. Many of the genes linked to the ASDs encode proteins that are involved in transcriptional regulation and chromatin remodeling. In this review we highlight selected neurodevelopmental disorders in which epigenetic dysregulation plays an important role. These include Rett syndrome, fragile X syndrome, Prader–Willi syndrome, Angelman syndrome, and Kabuki syndrome. For each of these disorders, we discuss how advances in our understanding of epigenetic mechanisms may lead to novel therapeutic approaches.
Electronic supplementary material
The online version of this article (doi:10.1007/s13311-013-0227-0) contains supplementary material, which is available to authorized users.
Keywords: Autism, Neurodevelopmental disorders, Rett, Prader-Willi, Angelman, Fragile X, Methylation, Acetylation, Histones
Introduction
Autism, first described by Kanner in 1943 [1], is one of the most frequently diagnosed neurodevelopmental disorders. Autism spectrum disorders (ASDs), encompassing classical autism, Asperger syndrome, childhood disintegrative disorder, Rett syndrome (RTT), and pervasive developmental disorder not otherwise specified, are an important group of neurodevelopmental disorders. ASDs are characterized by impaired language development, abnormal social interactions, and stereotyped and repetitive behaviors, and can be diagnosed reliably in children around 2–3 years of age. Epidemiological studies suggest that there is an increase in occurrence of ASD, with an estimated prevalence of about 1 in 88 children [2] and about 1 % in the general population [3]. The prevalence is higher in male children (about 1 in 54) than in female children (about 1 in 250). Environmental factors, prenatal maternal inflammatory disease, and genetics have all been implicated as causative factors in ASDs [4, 5].
Genetic studies in the last decade, primarily using high-density array comparative genomic hybridization, have identified several genetic loci where copy number abnormalities have been associated with ASDs [6]. This accounts for about 20 % of cases. Twin and family studies confirm the importance of genetics in ASDs. The risk of recurrence of ASD in a family with a single affected child is between 2 and 18 % [7–11]. A recent study by Hallmayer et al. [12] provided rigorous quantitative estimates of genetic heritability in a large cohort of twin pairs with autism. In male twins, the estimated concordance was 77 % for monozygotic pairs and 31 % for dizygotic pairs. In female twins, the concordance was 50 % for monozygotic pairs and 36 % for dizygotic pairs. This suggests that susceptibility to ASD has both a genetic component and a significant environmental component [12].
Epigenetics and ASDs
“Epi-” is derived from the Greek for “over” or “above”, and the term “epigenetics”, coined by Waddington, refers to mechanisms “above” the DNA sequences that regulate gene expression [13]. The sequence of a person’s genome alone may be inadequate when we try to understand the combined effect of genetic make up and environment on the phenotype of children with developmental disorders. Regulation of neuronal structure and function through epigenetic mechanisms is believed to be critical in the development of the nervous system. Disruption or alteration of this process may cause an array of neurodevelopmental disorders, including ASDs [14]. The role of epigenetics in autism has only emerged in the last few years, and represents a growing area of research [15].
DNA in the genome is organized as chromatin in tightly packed 3-dimensional structures, composed of nucleosomes in different states of compaction. The nucleosome consists of a 146 base-pair (bp) DNA segment wrapped around an octamer of core histone proteins made up of a pair of H3–H4 dimers and a pair of H2A–H2B dimers, connected by linker DNA. Epigenetic modifications include biochemical modifications of DNA (methylation of cytosine residues at CpG islands) and post-translational modification of histone proteins. These epigenetic marks are considered to be permanent within a cell all through development, and are accurately replicated in daughter cells through mitosis. Epigenetic modifications affect DNA–protein interactions resulting in large-scale changes in chromatin structure or modulation of gene transcription.
Genomic Imprinting in ASD
One specific form of epigenetic regulation of gene expression is genomic imprinting. The term genomic imprinting was first coined by Helen Crouse in 1960 [16] and applies to epigenetic regulation of gene expression in a parent-of-origin-specific manner. In imprinted regions, either the paternal or the maternal allele of selected genes is silenced. Normal development requires bi-parental inheritance and expression of genes from both chromosomes. A gene is “maternally imprinted” if the maternal allele is silenced, and “paternally imprinted” if the paternal allele is silenced. The imprinting pattern is established in germ cells. Many genes are ubiquitously imprinted in the body; however, there are genes that have tissue-specific or activity-specific imprinting patterns [17]. An extreme example of genomic imprinting is X-inactivation in females, the process by which 1 of the 2 X-chromosomes is completely silenced and forms the Barr body [18].
Genomic imprinting patterns are determined by the specificity of DNA methylation and post-translational modification of histones. One characteristic of imprinted genes is the presence of differentially methylated regions (DMRs), where the 2 parental chromosomes are differentially marked by DNA methylation [19]. DMRs act as imprinting control regions (ICRs), loci that are required for imprinting of all genes within a cluster [20]. De novo DNA cytosine-5-methyltransferase-3-alpha catalyzes methylation of cytosine residues within CpG dinucleotides in both germ cells and in pre-implantation embryos. Imprinted genes are thought to play an important role in embryonic development [21]. Expression of imprinted genes is high in both brain and placenta. There are approximately 70 known imprinted genes in humans. More maternally methylated DMRs and ICRs have been identified than paternally methylated DMRs and ICRs [22]. In mice, about 1300 imprinted genes have been identified, and another 350 genes are transcribed with a parent-of-origin allelic bias in a sex-specific manner [23]. Many of these imprinted loci have been shown to influence neuronal differentiation, behavior, or susceptibility to neurological disease [24–26]. Evidence linking genomic imprinting to neurodevelopmental disorders including ASD comes from the study of Prader–Willi syndrome (PWS) and Angelman syndrome (AS). Although clinically distinct, both disorders are caused by mutations within chromosome 15q11-13 (PWS/AS region). The phenotype is dependent on which parental chromosome carries the genetic modification. Analysis of chromosome 15q11-13 and its orthologous chromosome 7 in mice has provided insight into the molecular mechanisms of imprinting in the PWS/AS region [27, 28].
Angelman Syndrome
AS was described by Harry Angelman in 1965. AS is characterized by delayed development, intellectual disability, speech impairment, epilepsy, puppet-like ataxic movement, prognathism, tongue protrusion, paroxysms of laughter, and abnormal sleep patterns. Mutation or deletion of the maternal gene ubiquitin protein ligase E3A (UBE3A) causes AS. UBE3A encodes a homologous to E6-associated protein C terminus domain E3 ubiquitin ligase. Deletions (~6 Mb in size, including UBE3A) in the maternal chromosome 15q11-13 account for about 75 % of AS cases, and point mutations in maternal UBE3A account for 10–15 % of cases [29–31]. The remaining cases are caused by uniparental disomy or mutations within the imprinting control regions. In humans and mice, UBE3A is bi-allelically expressed in most tissues. In neurons, owing to tissue-specific paternal imprinting, the maternal allele alone is expressed, while the paternal allele is epigenetically silenced [32]. Analysis of “knock-in” mice in which a UBE3AYFP (yellow fluorescent protein) fusion gene was inserted into the UBE3A locus revealed that UBE3A was enriched in nuclei and dendrites of neurons. Study of YFP expression in the brain when the fusion gene was maternally or paternally inherited clearly demonstrated paternal imprinting of UBE3A in neurons of the hippocampus, cortex, thalamus, olfactory bulb, and cerebellum [33]. Biallelic expression was seen in glial cells.
The molecular mechanism by which the paternal UBE3A gene is imprinted is complex (Fig. 1). The UBE3A promoter region is unmethylated in both the maternal and paternal chromosomes. This implies that differential methylation of the UBE3A promoter is not the mechanism for paternal imprinting in the brain. Antisense RNA-mediated epigenetic silencing of many genes has been reported [34]. The UBE3A antisense RNA transcript (UBE3A-ATS) is thought to mediate the silencing of the paternal UBE3A allele [35]. UBE3A-ATS is a large transcript (500– ∼ 1000 kb) that extends through the SNRPN–SNURF–UBE3A gene cluster in chromosome 15q11-13. The UBE3A-ATS is expressed exclusively from the paternal allele in neurons; in these same neurons, the sense UBE3A messenger RNA (mRNA) is expressed from the maternal allele. UBE3A-ATS is an atypical RNA polymerase II transcript, and functions in cis to suppress paternal UBE3A expression [36]. The paternal antisense transcript is positively regulated by the PWS imprinting center (PWS-IC) and deletion of this PWS-IC causes increased expression of the paternal sense UBE3A mRNA [37]. Thus, the paternal PWS-IC promotes expression of genes from the paternal SNRPN–SNURF–UBE3A cluster, while at the same time repressing paternal UBE3A gene expression via an antisense RNA mechanism [38]. Inhibition of UBE3A-ATS expression is a potential therapeutic target for patients with AS carrying a mutant maternal allele [38, 39]. In an alternate model, it has been proposed that expression of UBE3A-ATS induces histone modifications within the paternal UBE3A locus that aborts transcriptional elongation of sense UBE3A, resulting in truncated, inactive UBE3A mRNAs [31].
Fig. 1.
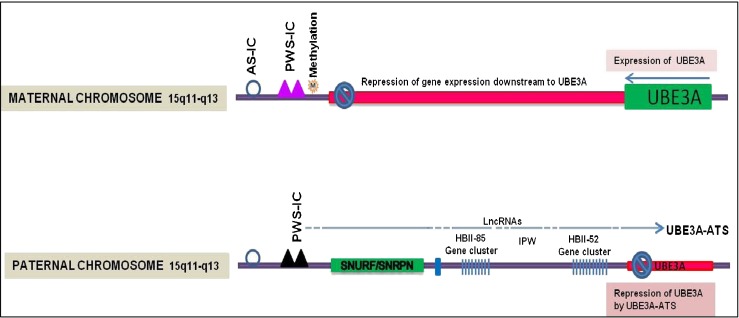
Diagram of the imprinted maternal and paternal chromosome 15q11-q13 regions containing Prader–Willi/Angelman Syndrome (PWS–AS) locus. This locus controls maternal and paternal specific expression of (A) protein-coding genes ubiquitin protein ligase E3A (UBE3A) and SNURF/SNRPN = SNRPN upstream reading frame / small nuclear ribonucleoprotein polypeptide N (green); (B) long noncoding RNA (lncRNA) HBII-85, imprinted in Prader-Willi (IPW), HBII-52 (blue); and (C) UBE3A-antisense RNA transcript (ATS) (0.5–1.0 Mb in length) that overlaps the UBE3A gene. The PWS/AS imprinting centers (AS-IC and PWS-IC) have parent specific epigenetic marks that regulate gene expression. During development, the AS-IC (open circle) establishes and maintains the PWS-IC on the maternal chromosome. Methylation at PWS-IC (pink triangles) on the maternal chromosome suppresses expression of genes downstream of the PWS-IC center (red bar). On the paternal chromosome, differential methylation of PWS-IC results in paternal expression of lncRNAs, including the UBE3A-ATS, which represses UBE3A gene expression (red bar) [27–29]
In a recent study, Huang et al. [40] used a novel strategy to discover drugs that unsilence an imprinted paternal UBE3A allele. They used the UBE3A–YFP knock-in mouse model as a reporter of UBE3A imprinting [33]. In cultured embryonic cortical neurons prepared from these mice, UBE3A–YFP expression was suppressed when UBE3A–YFP was paternally inherited (UBE3Am+/pYFP), but was expressed when UBE3A–YFP was maternally inherited (UBE3AmYFP/p+). They used neurons from UBE3Am+/pYFP animals to screen for drugs that upregulated YFP fluorescence. Unexpectedly, topoisomerase inhibitors (including toptecan and irinotecan) were found to unsilence the paternal UBE3A allele. They then tested topotecan in an AS mouse model (maternally-derived UBE3A-null allele) and found that topotecan up-regulated expression of active UBE3A in cultured neurons from AS mice. When given intrathecally (intraventricular injections or injection into the spinal subarachnoid space) into AS mice, topotecan, unsilenced and upregulated expression of the paternal UBE3A allele in several regions of the nervous system. This effect was long lasting, suggesting that these were potential therapeutic agents for treatment of AS.
Prader-Willi Syndrome
PWS is characterized by hypotonia and failure to thrive in the immediate postnatal period. Appearing later are the more typical features of hyperphagia resulting in obesity, hypogonadism, mild-to-moderate mental retardation, behavior problems, and autistic features. The prevalence of PWS is 1 in 10,000 to 1 in 22,000 [41]. Microdeletion (5–6-Mb region) in the paternal chromosome 15q11.2-13 accounts for 75 % of patients with PWS. Approximately 20 % of PWS patients have maternal uniparental disomy, where both copies of chromosome 15 derive from the mother. A mutation within the PWS-IC is seen in almost 5 % of patients. Less than 1 % of patients have a chromosomal rearrangement resulting in a breakpoint within or deletion of the paternal 15q11.2-q13 region [42, 43]. Each of these genetic lesions leads to the loss of differential methylation at PWS-IC, resulting in the loss of expression of paternally-derived alleles. Deficiency in these paternal transcripts is implicated in PWS, but the function of most of the encoded proteins is not completely understood. Lack of the paternally expressed SNORD116 small nucleolar RNA has been shown to result in a PWS or PWS-like phenotype [44].
Duplication of Chromosome 15q11-13
Inherited maternal duplication (dup 15) or triplication (isodicentric extranumerary chromosome, idic15) of chromosomal region 15q11-q13 are important genetic causes of ASD. Imbalance of 15q11-13 dosage accounts for about 1–2 % of ASD cases. This can disrupt normal parental homologue pairing, DNA methylation patterns, and gene expression patterns within 15q11-13. Features of autism, along with cognitive impairment, anxiety, and hyperactivity are seen in 15q duplication syndrome [45–50]. Penetrance of the autism phenotype is complete in patients with idic15. Paternally-inherited duplication of this region very rarely predisposes an individual to ASD [51–53]. Studies in postmortem brain samples have shown that, despite increased maternal dosage, the expression of imprinted genes within this region (SNRPN, NDN, small nucleolar RNAs) is reduced. The 15q11-13 region also contains non-imprinted gamma-aminobutyric acid (GABA) receptor genes (GABRB3, GABRA5, and GABRG3). The expression of these genes is also reduced. In contrast, studies in transformed lymphocytes and postmortem brain tissue from idic15 cases indicate that UBE3A expression (mRNA and protein) is increased, proportional to the increase in gene dosage [54]. It is suggested that the epigenetic alteration due to long-range chromatin organization might contribute to these gene expression abnormalities [55].
Fragile X Syndrome
Fragile X syndrome (FXS), caused by mutation of the X-linked FMR1 gene, is one of the most studied monogenic autism spectrum disorders. FXS was the first genetic disorder defined as being caused by a new type of mutation—expansion of an unstable trinucleotide repeat sequence [56]. The prevalence of FXS is about 1:4000 among men and 1:8000 among women [57, 58]. Behavioral signs of FXS include autistic features, moderate-to-severe intellectual disability, seizures, hypersensitivity to sensory stimuli, anxiety, attention deficit, hyperactivity, and motor incoordination. Somatic features include macro-orchidism, microcephaly, elongated face, and large, everted ears [59]. Mutation of FMR1 results in silencing of the FMR1 gene [60]. The FMR1 gene encodes fragile X mental retardation protein (FMRP), an mRNA-binding protein that is highly expressed in neurons. FMRP functions to negatively regulate synaptic protein synthesis in the brain. Current models propose that loss of FMRP leads to a dysregulated increase in synaptic protein synthesis, causing defects in synaptic plasticity and function, resulting in dendritic pathology [61].
The FMR1 gene contains 17 exons spanning 38 kb of chromosome Xq27.3. The full-length mRNA is about 4.4 Kb, although multiple transcripts are generated by alternative splicing of exons. FRMP is a 632-amino acid protein with a molecular mass of 80 kDa. The translation initiation site of FMR1 is located 69 bp downstream of a CGG repeat-containing region in the 5′ untranslated region (UTR) of exon 1. A CpG island is located about 250 bp upstream from this CGG repeat [62].
The most common mutation causing FXS is an expansion of the CGG repeat within the 5’-UTR to more than 200 CGG repeats. In normal individuals the CGG repeat length is not constant, but usually ranges from 6 to 54 [61]. This CGG repeat has the inherent property of meiotic instability because of its secondary structure. The expansion is thought to happen in the germline meiotic divisions or postmeiotically during early divisions of the zygote [63]. In men with FXS with >200 CGG repeats, epigenetic changes drive the suppression of FMR1 gene transcription through a unique mechanism involving hypermethylation of the CGG repeats within the 5’-UTR and the CpG island within the FMR1 promoter region. This results in transcriptional shut-down of the FMR1 gene and deficiency of the FMRP protein. Chemical modifications of histones—histone H3 tail deacetylation, histone H3K9 methylation, and histone H3K4 demethylation—have been reported (Fig. 2) [64]. Treatment of FXS cells with 5′-azadeoxycytidine, a DNA methyltransferase inhibitor, results in DNA demethylation and acetylation of histones H3 and H4, and FMR1 transcriptional reactivation [65, 66].
Fig. 2.

The FMR1 gene contains a promoter region (violet box) flanked by an upstream CpG island and a <45-repeat CGG tract (blue oval), which is followed by the transcription start site. Recognized domains include a nuclear localization signal (NLS) , K-homology domains (KH1 and KH2) , an arginine–glycine–glycine box (RGG), and a nuclear export signal (NES). In normal patients, in the FMR1 promoter region, there are high levels of histone acetylation and H3K4 methylation resulting in a euchromatic state for FMR1 gene expression. In fragile X syndrome (FXS), the presence of >200 CGG repeats induces methylation of histones (H3K9) in the promoter region. This results in DNA methylation of promoter and the upstream CpG island resulting in a heterochromatized state and FMR1 inactivation
A CGG repeat length in the 55–200 range is called a premutation because of the likelihood of expansion into a full mutation. Premutation alleles are relatively frequent in the general population, with an estimated prevalence of 1 in 259 women and 1 in 813 men [67]. Unlike the full mutation, the premutation results in increased FMR1 mRNA concentration and a toxic effect on cells via an RNA gain-of-function mechanism. Repeat lengths in the premutation range are not associated with hypermethylation of the upstream CpG Island. Females with premutation alleles are at greater risk of developing premature ovarian failure, chronic muscle pain, and thyroid disease compared with the general population. Individuals over the age of 50 years with premutation alleles are at risk of developing a neurodegenerative disorder called fragile X-associated tremor/ataxia syndrome (FXTAS). FXTAS is characterized by a progressive action tremor, gait ataxia, and cognitive decline [68].
FMR1 gene expression appears to be linked to both the cellular differentiation state and epigenetic status [66]. In a study of chorionic villus samples from male fetuses carrying the full FMR1 mutation at different gestational ages, it was found that the FXS mutation does not affect FMR1 gene expression in early embryogenesis (gastrulation). This suggests that epigenetic silencing of the gene happens at later developmental stages [69]. Human embryonic stem cells derived from FXS patient embryos express FMR1 in undifferentiated cells despite the presence of a full mutation, indicating the absence of epigenetic marks. This is further supported by studies in induced pluripotent stem (iPS) cell lines from fibroblasts of individuals carrying the fragile X mutation [70, 71]. In somatic cells (FXS fibroblasts) that were reprogrammed to pluripotency, DNA hypermethylation and histone modifications were preserved, leading to FMR1 gene repression. This indicated that the epigenetic marks were permanent, once established in somatic cells, and maintained in iPS cells.
Various mechanisms have been proposed for the epigenetic silencing of the FMR1 gene in FXS. It has been shown that the CGG repeats in FMR1 RNA readily form hairpins under physiological conditions. The RNA hairpin involves the 97 uninterrupted CGG repeats within the expansion. This double-stranded CGG–RNA hairpin does not activate interferon–inducible protein kinase, which is activated by a wide range of double–stranded RNAs. It is cleaved by the human Dicer enzyme, generating small interfering RNAs (siRNAs) [72, 73]. siRNAs generated from the FMR1 RNA hairpin after Dicer processing are thought to associate with the “RNA-induced initiator of transcriptional gene silencing” complex containing Ago1 (homologue of the fission yeast protein, Argonaute), Chp1 (a heterochromatin-associated chromo domain protein), and Tas3 (a novel protein). siRNAs contained within this complex bind to their homologous regions of CGG repeats in DNA, and recruit de novo DNA methyltransferase(s) and histone methyltransferases. This leads to DNA methylation and histone modification, resulting in heterochromatin formation and transcription suppression [72, 73]. A recent study examined DNA methylation levels at nearly 500,000 sites throughout the genome using peripheral blood and fibroblast-derived iPS cells from FXS patients [70]. No other differentially methylated loci were found. One would expect that there are other CGG tracts in the genome that activate RNA-induced transcriptional silencing resulting in methylation of specific target genes, but these are not observed [70].
Methyl-binding Proteins and RTT
DNA methylation, the primary epigenetic modification of eukaryotic genomes, involves addition of a methyl group at the 5-position of cytosine in CpG dinucleotides. DNA methylation plays an important role during mammalian development. DNA methylation influences cell fate through various mechanisms including control of specific gene expression, genomic imprinting, and X-chromosome inactivation [74, 75]. Methylation of promoter regions prevents the binding of transcription factors to DNA, resulting in altered gene expression [76, 77]. DNA methylation can also alter chromatin structure by recruitment of specialized proteins that bind to the methyl-CpG (mCpG) and induce gene silencing. Nuclear proteins that recognize mCpG in DNA are known as mCpG binding domain (MBD) proteins. These proteins repress transcription through the recruitment of co-repressor complexes that alter chromatin structure [78–80]. The MBD family includes MeCP2, MBD1, MBD2, and MBD4, which share the capacity for binding symmetrically to methylated CpGs via the MBD. Three other MBD family proteins, MBD3, MBD5 and MBD6, do not bind methylated DNA [81–83].
MeCP2 mutation causes distinct disorders namely: 1) RTT, resulting from MeCP2 loss of function; and 2) MeCP2 duplication syndrome, caused by increased MeCP2 expression. These disorders are described below.
Rett Syndrome
Loss-of-function mutations of MeCP2 account for over 90 % of cases of RTT. RTT is an X-linked dominant neurological disorder affecting predominantly females and has an estimated prevalence of 1 in 10,000–15,000 females [84]. MeCP2 mutation in males usually causes a severe neonatal encephalopathy with early lethality, but specific mutations are associated with an X-linked mental retardation phenotype. Severity of the presenting phenotype even in classic RTT females is variable, even among individuals with the same mutation. This phenotypic variability may be mediated by skewed X inactivation [85–87]. Symptoms of RTT are generally recognized around 6–18 months of age following an apparently normal period of early development. Regression of previously acquired skills and deceleration of head growth are key features of the disease in conjunction with stereotypic hand movements [88–90]. Cognitive, social, and language impairment, a variety of motor and breathing abnormalities, seizures, microcephaly, gastrointestinal dysfunction, scoliosis, kyphosis, and spasticity are observed in patients with RTT. Pathological studies of human RTT cases have shown a reduction in brain size, small neuronal soma size, and an increase in cell packing density [91–94]. There is a significant reduction in the dendritic branching of neurons in specific layers of the frontal, motor, temporal, and limbic cortex [95].
MeCP2 is a 53-kD nuclear protein that contains 1) an MBD that binds to methylated CpG dinucleotides; 2) transcriptional repression domain that binds to chromatin remodeling factors; 3) AT-hook domain that binds AT-rich DNA; and 4) a C-terminal domain alpha and beta, which can bind to naked DNA and to RNA splicing factors (Fig. 3a) [96–99]. Most MeCP2 mutations in RTT occur de novo, while familial cases are very rare. The mutations are dispersed throughout the MeCP2 domains and the majority of them are of missense or nonsense mutation in the coding sequences, which alter MeCP2 protein function.
Fig. 3.

a Schematic diagram of MeCP2 domain structure and protein interactions. mCpG binding domain (MBD) [amino acids (aa) 78–162]; transcriptional repression domain (TRD) (aa 207–310) the transcription repressor domain. There are 2 nuclear localization signals (NLS), 1 between the MBD and TRD, and the other within the TRD. Three AT-hook domains have been identified: AT-hook 1 (aa 185–194), AT-hook 2 (aa 265–272) in TRD domain, and AT-hook 3 in the C-terminus. Many factors interact with these MeCP2 domains mediating specific MeCP2 functions like chromatin structure modulation, gene activation, and gene repression. b Model of the dual role of MeCp2 as a transcriptional repressor and activator. MeCp2 competes with histone H1 for sites within linker DNA resulting in a heterochromatin structure and transcriptional repression. In this process, MeCP2 interacts with 5mC = 5-methylcytosine and ATRX = alpha thalassemia / mental retardation X-linked through its MDB domain, while the AT-hook domain in the TRD interacts with AT-rich DNA. MeCP2 bound to 5mC recruits transcription repressors and co-repressors [mSin3, histone deacetylase (HDAC)] resulting in transcriptional down-regulation. When MeCP2 binds hydroxymethyl cytosine (5hmC), it recruits transcription co-activators such as cycline adenosine monophosphate response element-binding protein (CREB), to activate target gene transcription. 5mCpG = 5-methyl cytosine phospho guanosine; coREST = corepressor of REST (RE1 silencing transcription factor); CREB = cAMP response element-binding protein; RNA Pol II = RNA polymerase II
MeCP2 Duplication Syndrome
While loss of MeCP2 function causes RTT, increased MeCP2 activity resulting from gene duplication (or triplication) causes MeCP2 duplication syndrome [100–102]. Males with MeCP2 duplication disorder display severe mental retardation, hypotonia, epilepsy, and progressive spasticity. Macrocephaly is observed in more severe cases of MeCP2 duplication syndrome and with MeCP2 triplication. Because of preferential activation of the normal X chromosome, females with MeCP2 gene duplications are often phenotypically normal. As observed in RTT, the severity of symptoms among male patients with MeCP2 duplication varies considerably. Even between brothers with identical MeCP2 gene duplication, the levels of clinical severity can differ significantly [101]. There is limited neuroradiological data available from patients with MeCP2 duplication syndrome; hence, little information on brain abnormalities associated with this neurological disorder. In the small number of cases where magnetic resonance imaging scans have been performed, generalized cortical atrophy and mildly enlarged lateral ventricles have been reported, suggesting neuronal loss [100, 103, 104]. In a single patient where results of postmortem neuropathological examination have been reported, diffuse neuronal loss in the cortex and minor disturbance of cortical lamination was observed [105]. Two lines of MeCP2 transgenic mice have been generated and serve as models of MeCP2 duplication syndrome. In one of these transgenic lines the human MeCP2 cDNA was targeted to the Tau locus [106], which is active only in mature neurons. While mice heterozygous for the MeCP2 transgene have no abnormalities, homozygous transgenic mice display progressive neurological dysfunction, but have a normal lifespan. Weaknesses of this line as a model of MeCP2 duplication syndrome include the disruption of the Tau gene (and therefore Tau function), as well as expression of MeCp2 under the control of the Tau promoter, which, unlike the endogenous MeCP2 promoter, is not active in glia. The second set of transgenic lines was generated by Huda Zhogbi’s laboratory using a large genomic PAC clone containing the MeCP2 locus [107]. Depending on the line, MeCP2 is expressed at 1–7 times the normal levels in the brain. These mice are normal at birth, but then develop progressive neurological deficits, including seizures and hypoactivity, the severity of which correlate with the level of transgene expression [107]. It deserves mention that diagnosis of MeCP2 duplication as a common cause of progressive mental retardation in human patients is recent and was prompted by the discovery of severe neurological deficits in MeCP2-overexpressing mice.
MeCP2 protein is abundantly expressed in neurons and may function as a transcriptional modulator via different mechanisms: inhibition of transcription factor binding, formation of chromatin loops, and influencing alternative RNA splicing [108]. Initial functional studies on MeCP2 identified its ability to bind selectively to DNA that contains a single mCpG pair and inhibit transcription from both methylated and nonmethylated DNA templates [109, 110]. This process was shown to be mediated through the interaction of the transcriptional repression domain with a co-repressor complex containing the transcriptional repressor mSin3A and histone deacetylases [111, 112]. The role of MeCP2 as an important regulator of neuronal gene expression is expanding, and it has been shown that MeCP2 also acts as an activator of gene expression in the hypothalamus of mice [113]. The MeCP2 gene is alternatively spliced to generate two different proteins with distinct translation initiation sites and N-termini [114]. Except for a 21-amino acid region unique to the e1 isoform and a 9-amino acid region unique to e2, both isoforms are identical. The Mecp2–e2 isoform is not expressed in nonmammalian vertebrates and is 10 times less abundant than the e1 isoform in the postnatal brain [115]. Although Mecp2–e1 and Mecp2–e2 display distinct expression patterns within different brain areas and developmental stages [115], the functional difference between the two isoforms has not been adequately investigated and the general assumption is that there is none. Contrary to common belief, a recent study found that the expression of both MeCP2 isoforms is differentially regulated during neuronal death, with e2 levels increasing and e1 decreasing in neurons primed to die [116]. Moreover, it was shown that elevated expression of the MeCP2–e2 isoform, but not the MeCP2–e1 isoform, promotes neuronal death when overexpressed and that knockdown of the e2 form has a neuroprotective effect [116].
MeCP2 represses expression of the brain-derived neurotrophic factor (BDNF) gene by binding selectively to BDNF promoter III [117, 118]. Phosphorylation of MeCP2, a direct result of neuronal depolarization or activity, modulates binding of MeCP2 to the BDNF promoter and BDNF gene transcription. This suggested a potential mechanism (regulation of gene expression by neuronal activity) by which MeCP2 mutation could lead to the developmental abnormalities observed in neurons [117, 118]. Recently, it was shown that MeCP2 is acetylated by p300 and that SIRT1 mediates its deacetylation [119]. This suggests a functional interplay between 2 critical epigenetic regulators, MeCP2 and SIRT1, modulating MeCP2 binding to the BDNF promoter in specific brain regions. MeCP2 is thought to suppress transcription when it binds to methylated sites in gene promoters. However, gene expression studies have not identified many candidate genes with highly methylated promoters available for MeCP2 regulation. MeCP2 may alter gene expression through interaction with methylated sites outside of gene promoter regions, collapsing and altering chromatin structure [108, 120]. MeCP2, bound to introns of specific target genes, has been shown to promote gene expression [121]. Recently, MeCP2 has shown to contain three AT-hook-like domains over a stretch of 250 amino acids, similar to high mobility group A DNA-binding proteins. In a truncating mutation, MeCP2–R270X, one of these conserved AT-hooks, is disrupted leading to altered chromatin structure [98]. In this way, differences in MeCP2-binding domains, the sites where they bind, and the protein complexes that are recruited, can differentially regulate downstream gene expression (Fig. 3b).
The expression of MeCP2 itself may be modulated by the nucleosomal binding protein, high mobility group N1 protein (HMGN1). HMGN1 affects chromatin structure and has been shown to down-regulate MECP2 [122]. In this study, alterations in HMGN1 levels have shown to alter chromatin structure and histone modifications in the MeCP2 promoter region. Further, HMGN1-/- mice display hypoactivity and abnormal social behavior, similar to other mouse models of ASDs. It is possible that the neurological phenotype of HMGN1 knock-out mice may be due to MeCP2 down-regulation. Data from the study of MeCP2 mutant mice suggest that over-expression and repression of MeCP2 affects approximately 85 % of the 2582 genes studied [108]. Genes whose expression is altered by MeCP2 mutation include GABRB3, UBE3A, BDNF, and DLX5, all important in neural developmental [55, 85].
In a recent study, Mellén et al. [123] showed that 5-hydroxymethyl cytosine (5hmC) epigenetic marks are enriched in active genes and their contribution to gene activation depends critically on cell type [123]. The authors identified MeCP2 as the major 5hmC-binding protein in the brain. MeCP2 binds mCpG-containing and 5hmC-containing DNA with similar high affinities. The R133C MeCP2 mutation (causing RTT) preferentially inhibits 5hmC binding. These findings support a model in which 5hmC and MeCP2 constitute a cell-specific epigenetic mechanism for gene expression (Fig. 3b).
Epigenetic alteration of MeCP2 gene function was also indicated in the development of autism and reduced levels of MeCP2 were found to correlate with increased MECP2 promoter methylation levels in frontal cortex samples from males with autism [124–126]. This suggests that epigenetic alterations at the MECP2 promoter could contribute to the development of ASD.
Very little information is available on the biological effect of mutation in other MBDs. Mbd1 mutant (Mbd1-/-) mice exhibit several core deficits frequently associated with autism, including defects in learning, abnormal social interaction, anxiety, and abnormal brain serotonin activity [127]. Htr2c is a serotonin receptor, the expression of which is directly regulated by Mbd1. Mbd1 binds to the Htr2c gene promoter, and the loss of Mbd1 leads to elevated expression of Htr2c [127]. In one study, missense and deletion mutations in MBD genes, including MBD 1, MBD 2, MBD 3, and MBD4, were seen in children with autism [128]. Another recent study suggests that the MBD gene family may be important in rare and private genetic causes of autism, and identified patients with potentially detrimental alterations in MBD6 and SETDB1 genes [129].
Kabuki Syndrome
Kabuki syndrome (KS) (OMIM#147920) was first described by 2 groups from Japan [130, 131], with an estimated prevalence of 1 in 32,000. KS is characterized by distinctive facial features with long palpebral fissures and arched eyebrows, microcephaly, and mild-to-moderate mental retardation. Other somatic features include postnatal dwarfism, a broad and depressed nasal tip, large prominent ear lobes, a cleft or high-arched palate, scoliosis, short fifth finger, persistence of finger pads, radiographic abnormalities of the vertebrae, hands, and hip joints, and recurrent otitis media in infancy. Recently, exome sequencing identified mutations in the myeloid/lymphoid or mixed-lineage leukemia 2 (MLL2) gene (also known as KMT2D gene) as the cause of KS [132, 133]. The majority of cases are sporadic, but some inherited cases suggest that KS is an autosomal dominant disorder. More than 30 different mutations in MLL2 have been defined, most being nonsense or frameshift mutations, which are predicted to result in haploinsufficiency [134]. Mutation of MLL2 only accounts for about 70 % of cases of KS suggesting genetic heterogeneity [135, 136]. Recently, deletions or point mutations in the lysine (K)-specific demethylase 6A (KDM6A) gene, which encodes a histone demethylase that interacts with MLL2, were found in patients with KS [137]. In these patients, a de novo microdeletion or complete deletion of KDM6A gene, located on the X chromosome, was identified. KDM6A is one of few X-linked genes that escapes X inactivation [138].
The MLL2 gene, located at chromosome 12q13, is primarily a nuclear protein and is expressed in most adult tissues, with the exception of the liver [139]. MLL2 encodes a large (5537-amino acid) nuclear protein containing multiple domains, including an AT-hook DNA-binding domain, a DHHC-type zinc finger domain, 5 plant homeodomain (PHD) zinc fingers, a catalytic histone methyltransferase SET domain, a post-SET domain, and a RING-type zinc finger [139]. MLL2 has histone methyltransferase activity and methylates the Lys-4 position of histone H3. The complexity of histone methylation is such that it can either activate or repress genes based on the position and status of methylation [140, 141]. Methylated histone H3 Lys 4 (H3K4me) is associated with gene activation and the trimethylated H3K4 (H3K4me3) is a genome-wide mark that surrounds the transcription start sites of up to 75 % of all human genes in several different cells lines and embryonic stem cells [134]. MLL2 directly methylates histone H3 through its SET domain [142, 143]. MLL2 is part of the ASC-2/NCOA6 complex (ASCOM), which possesses histone methylation activity and is involved in transcriptional co-activation. ASCOM has been shown to be a transcriptional regulator of the beta-globin and estrogen receptor genes. MLL2 participates in retinoic acid receptor signaling by promoting retinoic acid-responsive gene transcription [144]. Activation of the retinoic acid receptor leads to ligand-dependent recruitment of ASCOM and subsequent transient H3-lysine 4 methylation of the retinoid-responsive promoter region in vivo. MLL2 also facilitates neuronal differentiation of NT2/D1 cells by activating differentiation-specific genes, such as HOXA1–3 and NESTIN [145]. Mice lacking the MLL2 gene in adult forebrain excitatory neurons display impaired hippocampus-dependent memory. Gene expression analysis in MLL2 knock-out animals revealed that 152 genes were down-regulated in the hippocampal dentate gyrus region, a result of the deficit in histone 3 lysine 4 di- and trimethylation [145]. Mouse knockdown studies have demonstrated that MLL2 is involved in the regulation of adhesion-related cytoskeletal events, which might affect cell growth and survival [142]. The MLL2 gene mutations associated with KS results in the disruption of histone methylation pattern associated with gene expression, leading to abnormalities in growth and development. KS is thus another example of a genetic syndrome caused by mutation of an epigenetic gene [146].
Potential Epigenetic Therapies
In this article, we have described some of the complex interactions between epigenetic factors and genetic alterations in neurodevelopmental disorders. As a result of progress in this field, treatment based on epigenetics has been done in model organisms (Table 1). In an AS mouse model, inhibition of UBE3A–ATS, a long noncoding RNA, is, potentially, an important therapeutic target [38]. Screening for compounds using a transgenic mouse model led to the identification of 12 topoisomerase I inhibitors and 4 topoisomerase II inhibitors that unsilenced the paternal UBE3A allele. Among those compounds, Topotecan had the highest potential in up-regulating UBE3A gene expression in spinal cord neurons for 12 weeks after the termination of treatment. It is important to note that the all the compounds tested are approved for human use in cancer treatment, suggesting a potential for the immediate human use in the treatment of AS [37, 40]. In PWS, the CpG island of the PWS-IC is methylated and hypoacetylated resulting in the repression of genes on the maternal allele. Demethylation of PWS-IC with DNA methyltransferase inhibitor 5-azadeoxycytidine in PWS cells results in the expression of imprinted genes from the maternal allele. The restoration of maternal gene activity was shown to be associated with the increased H4 acetylation at PWS-IC [147]. This study provided the basis for the epigenetic activation of maternally-silenced genes as a potential treatment of PWS.
Table 1.
Summary of molecular defects and potential epigenetic therapeutic targets in selected neurodevelopmental (NDD) and autism spectrum disorders (ASD)
| Disorders | Molecular defect | Potential epigenetic targets | Ref. |
|---|---|---|---|
| NDD | |||
| Down syndrome | Trisomy for chromosome 21—results in overexpression of genes leading to abnormal brain development | Goal—Suppress expression from one chromosome 21 | [157] |
| • Whole chromosome silencing in trisomic neurons | |||
| • Insertion of an inducible X-inactive specific transcript (XIST) into DYRK1A locus in induced pluripotent stem cells from a Down syndrome patient | |||
| • XIST-mediated silencing reversed the deficits in neuronal proliferation and neural rosette formation | |||
| • Potential novel approach to therapy in Down syndrome | |||
| Kabuki syndrome | Mutations in the myeloid/lymphoid or mixed-lineage leukemia 2 (MLL2) gene, a methyltransferase that methylates histone H3 at Lys-4 | Goal—Promote methylation of H3K4 | [144–146] |
| • Histone methylating agents or MLL2 mimics | |||
| • Histone deacetylase (HDAC) inhibitors have been shown to up-regulate H3K4 methylation | |||
| Fragile X syndrome | Expansion of a CGG trinucleotide repeat (>200 triplet) results in hypermethylation of the 5’-untranslated region and the upstream CpG island within the promoter of FMR1 gene resulting in transcriptional repression of the FMR1 gene | Goal—Transcriptional reactivation of FMR1 | [65, 66, 158] |
| • DNA demethylation agent 5-azadeoxycytidine | |||
| • Histone hyperacetylation agents such as 4-phenylbutyrate, sodium butyrate, and trichostatin A | |||
| • Non-epigenetic approaches based on synaptic biology | |||
| ASD | |||
| Rett syndrome | MeCP2 mutation resulting in loss of MeCP2 function | Goal—Normalize MeCP2 levels | [148, 150] |
| • Viral delivery of MeCP2 complementary DNA under native promoter to restore physiological levels of MeCP2 protein | |||
| • Insufficient evidence that epigenetic modifiers (HDAC inhibitors or histone acetyltransferase inhibitors) augment MeCP2-mediated function | |||
| Angelman syndrome | Mutation or deletion of the maternal UBE3A gene | Goal—Restore UBE3A expression in neurons | [38, 40] |
| • Unsilence the imprinted paternal UBE3A allele—topotecan | |||
| • Inhibition of paternal UBE3A antisense RNA transcript expression | |||
| Prader–Willi syndrome | Mutation in the paternal chromosome 15q11.2–13; loss of expression of paternally-derived genes | • Identifying the epigenetic architecture in this region will help in identifying novel therapeutic targets | [42] |
UBE3A = ubiquitin protein ligase E3A
Similarly, MeCP2 is an important epigenetic regulator in neuronal cells. One possible therapeutic approach for RTT may be selective activation of the silenced wild-type X-chromosome. Such an unsilencing approach shows promise in the case of AS. Amelioration of symptoms in a mouse model of RTT was recently described using intracranially-delivered adeno-associated virus to drive expression of MeCP2 from its own promoter, supporting the concept of gene therapy for RTT [148]. Given that elevated MeCP2 expression causes neurological issues of its own, however, this approach is not without challenges. Another approach that has been tested with success in mice is the transplantation of bone marrow from healthy animals into mutant MeCP2 mice that were irradiated [149]. Rescue was found to be due to engraftment of wild-type microglia in the mutant mouse brain. An alternative to targeting MeCP2 is to target molecules downstream of MeCP2 function. One such target is BDNF [118, 150]. Indeed, increasing BDNF expression and signaling has been reported to have beneficial effects in RTT mouse models [151, 152]. Another growth factor that could be targeted is insulin-like growth factor-1 (IGF-1), which is known to play important roles in the development and function of the nervous system. An increased level of IGF-binding protein-3 has been described in MeCp2 knockout mice and RTT patients, which could lead to reduced IGF-1 signaling [153]. Administration of IGF-1 has been tested in MeCP2 knockout mouse model and shown to reverse RTT disease symptoms [154]. Much work on the potential deleterious effects of such treatment approaches is required before these are translated into humans.
In the case of FXTAS, where premutation CGG expansion within the 5’-UTR of the FMR1 gene results in increased FMR1 mRNA expression, a recent study performed in a Drosophila model of the disorder reported that over-expression of specific histone deacetylases (HDACs) suppresses neurodegeneration [155]. In this study, administration of histone acetyltransferase inhibitors, which would be expected to increase HDAC activity, reduced FMR1 mRNA expression in premutation carrier cell lines and extended lifespan in permutation CGG repeat-expressing Drosophila. It is tempting to speculate that such inhibitors will have utility in treating human patients with FXTAS. The effect of HDAC inhibitors has already been tested in human patients with FXS, where the CGG expansion results in silencing of FMR1 gene transcription. Behavioral symptoms associated with FXS were alleviated with valproic acid, a HDAC inhibitor commonly used to treat epilepsy [156]. Treatment of lymphoblastoid cells from FXS patients was found to reactivate FXS gene transcription.
Conclusions
In this review, we have outlined the importance of epigenetic modifications (DNA methylation, histone acetylation and methylation, chromatin remodeling) in neurodevelopmental disorders, including ASDS. Examples of such disorders are AS and PWS, FXS, RTT, and KS. Mutation of genes encoding proteins that are involved in DNA methylation and proteins that interpret epigenetic markings (MBDs) may lead to ASDs. Copy number variations, which are identified in a significant percentage or patients with developmental disorders, may also lead to alterations in the fine structure of the brain through epigenetic effects (as illustrated by maternal 15q duplication syndrome). Epigenetic effects may also be important in the regulation of specific candidate genes that are critical for brain development. Better understanding of epigenetic mechanisms in ASD and neurodevelopmental disorders will ultimately help in the development of novel treatment options. A number of potential therapeutic approaches are also described here. Strategies include pharmacological means of unsilencing imprinted genes, and strategies targeting specific epigenetic mechanisms (HDAC and HAT inhibitors). In these strategies, one major issue that has to be addressed is the effect of such treatments on expression of other genes (off-target effects) and potential long-term side effects of such therapies.
Electronic Supplementary Material
(PDF 1724 kb)
Acknowledgments
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
References
- 1.Kanner L. Autistic disturbances of affective contact. Nervous Child. 1943;2:217–250. [PubMed] [Google Scholar]
- 2.Autism and Developmental Disabilities Monitoring Network Surveillance Year 2008 Principal Investigators; Centers for Disease Control and Prevention. Prevalence of autism spectrum disorders—Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008. MMWR Surveill Summ 2012;30:61:1–19. [PubMed]
- 3.Brugha TS, McManus S, Bankart J, et al. Epidemiology of autism spectrum disorders in adults in the community in England. Arch Gen Psychiatry. 2011;68:459–465. doi: 10.1001/archgenpsychiatry.2011.38. [DOI] [PubMed] [Google Scholar]
- 4.Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 5.Dietert RR, Dietert JM, Dewitt JC. Environmental risk factors for autism. Emerg Health Threats J. 2011;4:10.3402. doi: 10.3402/ehtj.v4i0.7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sebat J, Lakshmi B, Malhotra D, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Folstein S, Rutter M. Genetic influences and infantile autism. Nature. 1977;265:726–728. doi: 10.1038/265726a0. [DOI] [PubMed] [Google Scholar]
- 8.Folstein S, Rutter M. Infantile autism: a genetic study of 21 twin pairs. J Child Psychol Psychiatry. 1977;18:297–321. doi: 10.1111/j.1469-7610.1977.tb00443.x. [DOI] [PubMed] [Google Scholar]
- 9.Steffenburg S, Gillberg C, Hellgren L, Andersson L, Gillberg IC, Jakobsson G, Bohman M. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J Child Psychol Psychiatry. 1989;30:405–416. doi: 10.1111/j.1469-7610.1989.tb00254.x. [DOI] [PubMed] [Google Scholar]
- 10.Wahlström J, Steffenburg S, Hellgren L, Gillberg C. Chromosome findings in twins with early-onset autistic disorder. Am J Med Genet. 1989;32:19–21. doi: 10.1002/ajmg.1320320105. [DOI] [PubMed] [Google Scholar]
- 11.Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- 12.Hallmayer J, Cleveland S, Torres A, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011;68:1095–1102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waddington CH. The epigenotype. Endeavour. 1942;1:18–20. [Google Scholar]
- 14.Fagiolini M, Jensen CL, Champagne FA. Epigenetic influences on brain development and plasticity. Curr Opin Neurobiol. 2009;19:207–212. doi: 10.1016/j.conb.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.IACC/OARC Autism Spectrum Disorder Research Publications Analysis Report. The Global Landscape of Autism Research. July 2012 Office of Autism Research Coordination (OARC), National Institute of Mental Health and Thomson Reuters, Inc. on behalf of the Interagency Autism Coordinating Committee (IACC).
- 16.Crouse HV. The controlling element in sex chromosome behavior in sciara. Genetics. 1960;45:1429–1443. doi: 10.1093/genetics/45.10.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–93. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 18.Sha K. A mechanistic view of genomic imprinting. Annu Rev Genomics Hum Genet. 2008;9:197–216. doi: 10.1146/annurev.genom.122007.110031. [DOI] [PubMed] [Google Scholar]
- 19.Morey C, Avner P. Genetics and epigenetics of the X chromosome. Ann N Y Acad Sci. 2010;1214:E18–E33. doi: 10.1111/j.1749-6632.2010.05943.x. [DOI] [PubMed] [Google Scholar]
- 20.Ferguson-Smith AC. Genomic imprinting: the emergence of an epigenetic paradigm. Nat Rev Genet 2011;12:565-575. [DOI] [PubMed]
- 21.Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001;2:21–32. doi: 10.1038/35047554. [DOI] [PubMed] [Google Scholar]
- 22.Abramowitz LK, Bartolomei MS. Genomic imprinting: recognition and marking of imprinted loci. Curr Opin Genet Dev. 2012;22:72–78. doi: 10.1016/j.gde.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gregg C, Zhang J, Weissbourd B, Luo S, Schroth GP, Haig D, Dulac C. High resolution analysis of parent-of-origin allelic expression in the mouse brain. Science. 2010;329:643–648. doi: 10.1126/science.1190830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Butler MG. Genomic imprinting disorders in humans: a mini-review. J Assist Reprod Genet. 2009;26:477–486. doi: 10.1007/s10815-009-9353-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keverne B. Monoallelic gene expression and mammalian evolution. Bioessays. 2009;31:1318–1326. doi: 10.1002/bies.200900074. [DOI] [PubMed] [Google Scholar]
- 26.Tollkuhn J, Xu X, Shah NM. A custody battle for the mind: evidence for extensive imprinting in the brain. Neuron. 2010;67:359–362. doi: 10.1016/j.neuron.2010.07.026. [DOI] [PubMed] [Google Scholar]
- 27.Buiting K. Prader–Willi syndrome and Angelman syndrome. Am J Med Genet Part C Semin Med Genet. 2010;153C:365–376. doi: 10.1002/ajmg.c.30273. [DOI] [PubMed] [Google Scholar]
- 28.Nicholls RD, Knepper JL. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu Rev Genomics Hum Genet. 2001;2:153–175. doi: 10.1146/annurev.genom.2.1.153. [DOI] [PubMed] [Google Scholar]
- 29.Van Buggenhout G, Fryns JP. Angelman syndrome (AS, MIM 105830) Eur J Hum Genet. 2009;17:1367–1373. doi: 10.1038/ejhg.2009.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lalande M, Calciano MA. Molecular epigenetics of Angelman syndrome. Cell Mol Life Sci. 2007;64:947–960. doi: 10.1007/s00018-007-6460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mabb AM, Judson MC, Zylka MJ, Philpot BD. Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 2011;34:293–303. doi: 10.1016/j.tins.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JT, Bartolomei MS. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell. 2013;152:1308–1323. doi: 10.1016/j.cell.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 33.Dindot SV, Antalffy BA, Bhattacharjee MB, Beaudet AL. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum Mol Genet. 2008;17:111–118. doi: 10.1093/hmg/ddm288. [DOI] [PubMed] [Google Scholar]
- 34.Verona RI, Mann MR, Bartolomei MS. Genomic imprinting: intricacies of epigenetic regulation in clusters. Annu Rev Cell Dev Biol. 2003;19:237–259. doi: 10.1146/annurev.cellbio.19.111401.092717. [DOI] [PubMed] [Google Scholar]
- 35.Rougeulle C, Cardoso C, Fontés M, Colleaux L, Lalande M. An imprinted antisense RNA overlaps UBE3A and a second maternally expressed transcript. Nat Genet. 1998;19:15–16. doi: 10.1038/ng0598-15. [DOI] [PubMed] [Google Scholar]
- 36.Yamasaki K, Joh K, Ohta T, et al. Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum Mol Genet. 2003;12:837–847. doi: 10.1093/hmg/ddg106. [DOI] [PubMed] [Google Scholar]
- 37.Chamberlain SJ, Brannan CI. The Prader-Willi syndrome imprinting center activates the paternally expressed murine Ube3a antisense transcript but represses paternal Ube3a. Genomics. 2001;73:316–322. doi: 10.1006/geno.2001.6543. [DOI] [PubMed] [Google Scholar]
- 38.Meng L, Person RE, Beaudet AL. Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum Mol Genet. 2012;21:3001–3012. doi: 10.1093/hmg/dds130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chamberlain SJ, Chen PF, Ng KY, et al. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc Natl Acad Sci U S A. 2010;107:17668–17673. doi: 10.1073/pnas.1004487107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang HS, Allen JA, Mabb AM, et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature. 2011;481:185–189. doi: 10.1038/nature10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee PD. Disease management of Prader-Willi syndrome. Expert Opin Pharmacother. 2002;3:1451–1459. doi: 10.1517/14656566.3.10.1451. [DOI] [PubMed] [Google Scholar]
- 42.Horsthemke B, Wagstaff J. Mechanisms of imprinting of the Prader-Willi/Angelman region. Am J Med Genet A. 2008;146A:2041–2052. doi: 10.1002/ajmg.a.32364. [DOI] [PubMed] [Google Scholar]
- 43.Bittel DC, Butler MG. Prader-Willi syndrome: clinical genetics, cytogenetics and molecular biology. Expert Rev Mol Med. 2005;7:1–20. doi: 10.1017/S1462399405009531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duker AL, Ballif BC, Bawle EV, et al. Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. Eur J Hum Genet. 2010;18:1196–1201. doi: 10.1038/ejhg.2010.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berdasco M, Esteller M. Genetic syndromes caused by mutations in epigenetic genes. Hum Genet. 2013;132:359–383. doi: 10.1007/s00439-013-1271-x. [DOI] [PubMed] [Google Scholar]
- 46.Schroer RJ, Phelan MC, Michaelis RC, et al. Autism and maternally derived aberrations of chromosome 15q. Am J Med Genet. 1998;76:327. doi: 10.1002/(sici)1096-8628(19980401)76:4<327::aid-ajmg8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 47.Wang NJ, Liu D, Parokonny AS, Schanen NC. High-resolution molecular characterization of 15q11-q13 rearrangements by array comparative genomic hybridization (array CGH) with detection of gene dosage. Am J Hum Genet. 2004;75:267–281. doi: 10.1086/422854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schanen NC. Epigenetics of autism spectrum disorders. Hum Mol Genet. 2006;15:R138–R150. doi: 10.1093/hmg/ddl213. [DOI] [PubMed] [Google Scholar]
- 50.Hogart A, Leung KN, Wang NJ, et al. Chromosome 15q11-13 duplication syndrome brain reveals epigenetic alterations in gene expression not predicted from copy number. J Med Genet. 2009;46:86–93. doi: 10.1136/jmg.2008.061580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith SE, Zhou YD, Zhang G, Jin Z, Stoppel DC, Anderson MP. Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci Transl Med. 2011;3:103ra97. doi: 10.1126/scitranslmed.3002627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cook EH, Jr, Lindgren V, Leventhal BL, et al. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am J Hum Genet. 1997;60:928–934. [PMC free article] [PubMed] [Google Scholar]
- 53.Bolton PF, Dennis NR, Browne CE, et al. The phenotypic manifestations of interstitial duplications of proximal 15q with special reference to the autistic spectrum disorders. Am J Med Genet. 2001;105:675–685. doi: 10.1002/ajmg.1551. [DOI] [PubMed] [Google Scholar]
- 54.Di Rocco A, Loggini A, Di Rocco M, et al. Paradoxical worsening of seizure activity with pregabalin in an adult with isodicentric 15 (IDIC-15) syndrome involving duplications of the GABRB3, GABRA5 and GABRG3 genes. BMC Neurol. 2013;13:43. doi: 10.1186/1471-2377-13-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hogart A, Nagarajan RP, Patzel KA, Yasui DH, Lasalle JM. 15q11-13 GABAA receptor genes are normally biallelically expressed in brain yet are subject to epigenetic dysregulation in autism-spectrum disorders. Hum Mol Genet. 2007;16:691–703. doi: 10.1093/hmg/ddm014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cummings CJ, Zoghbi HY. Trinucleotide repeats: mechanisms and pathophysiology. Annu Rev Genomics Hum Genet. 2000;1:281–328. doi: 10.1146/annurev.genom.1.1.281. [DOI] [PubMed] [Google Scholar]
- 57.Coffee B, Keith K, Albizua I, et al. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet. 2009;85:503–514. doi: 10.1016/j.ajhg.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Santoro MR, Bray SM, Warren ST. Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu Rev Pathol. 2012;7:219–245. doi: 10.1146/annurev-pathol-011811-132457. [DOI] [PubMed] [Google Scholar]
- 59.Baker JK, Seltzer MM, Greenberg JS. Behaviour problems, maternal internalising symptoms and family relations in families of adolescents and adults with fragile X syndrome. J Intellect Disabil Res. 2012;56:984–995. doi: 10.1111/j.1365-2788.2012.01580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Verkerk AJ, Pieretti M, Sutcliffe JS, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 61.Bhakar AL, Dölen G, Bear MF. The pathophysiology of fragile X (and what it teaches us about synapses) Annu Rev Neurosci. 2012;35:417–443. doi: 10.1146/annurev-neuro-060909-153138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heitz D, Rousseau F, Devys D, et al. Isolation of sequences that span the fragile-X and identification of a fragile X-related CpG island. Science. 1991;251:1236–1239. doi: 10.1126/science.2006411. [DOI] [PubMed] [Google Scholar]
- 63.Hirst MC, White PJ. Cloned human FMR1 trinucleotide repeats exhibit a length-and orientation-dependent instability suggestive of in vivo lagging strand secondary structure. Nucleic Acids Res. 1998;26:2353–2358. doi: 10.1093/nar/26.10.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pietrobono R, Tabolacci E, Zalfa F, et al. Molecular dissection of the events leading to inactivation of the FMR1 gene. Hum Mol Genet. 2005;14:267–277. doi: 10.1093/hmg/ddi024. [DOI] [PubMed] [Google Scholar]
- 65.Tabolacci E, Pietrobono R, Moscato U, Oostra BA, Chiurazzi P, Neri G. Differential epigenetic modifications in the FMR1 gene of the fragile X syndrome after reactivating pharmacological treatments. Eur J Hum Genet. 2005;13:641–648. doi: 10.1038/sj.ejhg.5201393. [DOI] [PubMed] [Google Scholar]
- 66.Sheridan SD, Theriault KM, Reis SA, et al. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS One. 2011;6:e26203. doi: 10.1371/journal.pone.0026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jacquemont S, Hagerman RJ, Leehey MA, et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA. 2004;291:460–469. doi: 10.1001/jama.291.4.460. [DOI] [PubMed] [Google Scholar]
- 68.Hagerman RJ, Leavitt BR, Farzin F, et al. Fragile-X-associated tremor/ataxia syndrome (FXTAS) in females with the FMR1 premutation. Am J Hum Genet. 2004;74:1051–1056. doi: 10.1086/420700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Penagarikano O, Mulle JG, Warren ST. The pathophysiology of fragile x syndrome. Annu Rev Genomics Hum Genet. 2007;8:109–129. doi: 10.1146/annurev.genom.8.080706.092249. [DOI] [PubMed] [Google Scholar]
- 70.Alisch RS, Wang T, Chopra P, Visootsak J, Conneely KN, Warren ST. Genome-wide analysis validates aberrant methylation in fragile X syndrome is specific to the FMR1 locus. BMC Med Genet. 2013;14:18. doi: 10.1186/1471-2350-14-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Urbach A, Bar-Nur O, Daley GQ, Benvenisty N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell. 2010;6:407–411. doi: 10.1016/j.stem.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Handa V, Saha T, Usdin K. The fragile X syndrome repeats form RNA hairpins that do not activate the interferon-inducible protein kinase, PKR, but are cut by Dicer. Nucleic Acids Res. 2003;31:6243–6248. doi: 10.1093/nar/gkg818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Verdel A, Jia S, Gerber S, et al. RNAi-mediated targeting of heterochromatin by the RITS complex. Science. 2004;303:672–676. doi: 10.1126/science.1093686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science. 2001;293:1068–1070. doi: 10.1126/science.1063852. [DOI] [PubMed] [Google Scholar]
- 75.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 76.Hashimshony T, Zhang J, Keshet I, Bustin M, Cedar H. The role of DNA methylation in setting up chromatin structure during development. Nat Genet. 2003;34:187–192. doi: 10.1038/ng1158. [DOI] [PubMed] [Google Scholar]
- 77.Lorincz MC, Dickerson DR, Schmitt M, Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat Struct Mol Biol. 2004;11:1068–1075. doi: 10.1038/nsmb840. [DOI] [PubMed] [Google Scholar]
- 78.Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol. 1998;18:6538–6547. doi: 10.1128/mcb.18.11.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fraga MF, Ballestar E, Montoya G, Taysavang P, Wade PA, Esteller M. The affinity of different MBD proteins for a specific methylated locus depends on their intrinsic binding properties. Nucleic Acids Res. 2003;31:1765–1774. doi: 10.1093/nar/gkg249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rauch C, Trieb M, Wibowo FR, Wellenzohn B, Mayer E, Liedl KR. Towards an understanding of DNA recognition by the methyl-CpG binding domain 1. J Biomol Struct Dyn. 2005;22:695–706. doi: 10.1080/07391102.2005.10507036. [DOI] [PubMed] [Google Scholar]
- 81.Hendrich B, Tweedie S. The methyl-CpG binding domain and the evolving role of DNA methylation in animals. Trends Genet. 2003;19:269–277. doi: 10.1016/S0168-9525(03)00080-5. [DOI] [PubMed] [Google Scholar]
- 82.Roloff TC, Ropers HH, Nuber UA. Comparative study of methyl-CpG-binding domain proteins. BMC Genomics. 2003;4:1. doi: 10.1186/1471-2164-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fournier A, Sasai N, Nakao M, Defossez PA. The role of methyl-binding proteins in chromatin organization and epigenome maintenance. Brief Funct Genomics. 2012;11:251–264. doi: 10.1093/bfgp/elr040. [DOI] [PubMed] [Google Scholar]
- 84.Wan M, Lee SS, Zhang X, et al. Rett syndrome and beyond:recurrent spontaneous and familial MECP2 mutations at CpG hotspots. Am J Hum Genet. 1999;65:1520–1529. doi: 10.1086/302690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moretti P, Zoghbi HY. MeCP2 dysfunction in Rett syndrome and related disorders. Curr Opin Genet Dev. 2006;16:276–281. doi: 10.1016/j.gde.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 86.Gonzales ML, LaSalle JM. The role of MeCP2 in brain development and neurodevelopmental disorders. Curr Psychiatry Rep. 2010;12:127–134. doi: 10.1007/s11920-010-0097-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 88.Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett's syndrome: report of 35 cases. Ann Neurol. 1983;14:471–479. doi: 10.1002/ana.410140412. [DOI] [PubMed] [Google Scholar]
- 89.Dunn HG. Neurons and neuronal systems involved in the pathophysiologies of Rett syndrome. Brain Dev. 2001;23(Suppl. 1):S99–S100. doi: 10.1016/s0387-7604(01)00354-0. [DOI] [PubMed] [Google Scholar]
- 90.Dunn HG, MacLeod PM. Rett syndrome: review of biological abnormalities. Can J Neurol Sci. 2001;28:16–29. doi: 10.1017/s0317167100052513. [DOI] [PubMed] [Google Scholar]
- 91.Bauman ML, Kemper TL, Arin DM. Pervasive neuroanatomic abnormalities of the brain in three cases of Rett's syndrome. Neurology. 1995;45:1581–1586. doi: 10.1212/wnl.45.8.1581. [DOI] [PubMed] [Google Scholar]
- 92.Bauman ML, Kemper TL, Arin DM. Microscopic observations of the brain in Rett syndrome. Neuropediatrics. 1995;26:105–108. doi: 10.1055/s-2007-979737. [DOI] [PubMed] [Google Scholar]
- 93.Belichenko PV, Oldfors A, Hagberg B, Dahlström A. Rett syndrome: 3-D confocal microscopy of cortical pyramidal dendrites and afferents. Neuroreport. 1994;5:1509–1513. [PubMed] [Google Scholar]
- 94.Jentarra GM, Olfers SL, Rice SG, et al. Abnormalities of cell packing density and dendritic complexity in the MeCP2 A140V mouse model of Rett syndrome/X-linked mental retardation. BMC Neurosci. 2010;11:19. doi: 10.1186/1471-2202-11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Armstrong DD. Neuropathology of Rett syndrome. J Child Neurol. 2005;20:747–753. doi: 10.1177/08830738050200090901. [DOI] [PubMed] [Google Scholar]
- 96.Guy J, Cheval H, Selfridge J, Bird A. The role of MeCP2 in the brain. Annu Rev Cell Dev Biol. 2011;27:631–652. doi: 10.1146/annurev-cellbio-092910-154121. [DOI] [PubMed] [Google Scholar]
- 97.Nan X, Ng HH, Johnson CA, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 98.Baker SA, Chen L, Wilkins AD, Yu P, Lichtarge O, Zoghbi HY. An AT-hook domain in MeCP2 determines the clinical course of Rett syndrome and related disorders. Cell. 2013;152:984–996. doi: 10.1016/j.cell.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hite KC, Adams VH, Hansen JC. Recent advances in MeCP2 structure and function. Biochem Cell Biol. 2009;87:219–227. doi: 10.1139/o08-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Van Esch H. MECP2 duplication syndrome. Mol Syndromol. 2012;2:128–136. doi: 10.1159/000329580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Peters SU, Hundley RJ, Wilson AK, Carvalho CM, Lupski JR, Ramocki MB. Brief report: regression timing and associated features in MECP2 duplication syndrome. J Autism Dev Disord. 2013;43:2484–2490. doi: 10.1007/s10803-013-1796-9. [DOI] [PubMed] [Google Scholar]
- 102.Ramocki MB, Tavyev YJ, Peters SU. The MECP2 duplication syndrome. Am J Med Genet A. 2010;152A:1079–1088. doi: 10.1002/ajmg.a.33184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Himada S, Okamoto N. Ito et al. MECP2 duplication syndrome in both genders. Brain Dev. 2013;35:411–419. doi: 10.1016/j.braindev.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 104.Lugtenberg D, Kleefstra T, Oudakker AR, et al. Structural variation in Xq28: MECP2 duplications in 1% of patients with unexplained XLMR and in 2% of male patients with severe encephalopathy. Eur J Hum. Genet. 2009;17:444–453. doi: 10.1038/ejhg.2008.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Van Esch H, Bauters M, Ignatius J, et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet. 2005;77:442–453. doi: 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Luikenhuis S, Giacometti E, Beard CF, Jaenisch R. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc Natl Acad Sci U S A. 2004;101:6033–6038. doi: 10.1073/pnas.0401626101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Collins AL, Levenson JM, Vilaythong AP, et al. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet. 2004;13:2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- 108.Yasui DH, Peddada S, Bieda MC, et al. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc Natl Acad Sci U S A. 2007;104:19416–19421. doi: 10.1073/pnas.0707442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Singh J, Saxena A, Christodoulou J, Ravine D. MECP2 genomic structure and function: insights from ENCODE. Nucleic Acids Res. 2008;36:6035–6047. doi: 10.1093/nar/gkn591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Meehan RR, Lewis JD, Bird AP. Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res. 1992;20:5085–5092. doi: 10.1093/nar/20.19.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lewis JD, Meehan RR, Henzel WJ, et al. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69:905–914. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- 112.Jones PL, Veenstra GJ, Wade PA, et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 113.Chahrour M, Jung SY, Shaw C, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mnatzakanian GN, Lohi H, Munteanu I, et al. A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat Genet. 2004;36:339–341. doi: 10.1038/ng1327. [DOI] [PubMed] [Google Scholar]
- 115.Dragich JM, Kim YH, Arnold AP, Schanen NC. Differential distribution of the MeCP2 splice variants in the postnatal mouse brain. J Comp Neurol. 2007;501:526–542. doi: 10.1002/cne.21264. [DOI] [PubMed] [Google Scholar]
- 116.Dastidar SG, Bardai FH, Ma C, et al. Isoform-specific toxicity of Mecp2 in postmitotic neurons: suppression of neurotoxicity by FoxG1. J Neurosci. 2012;32:2846–2855. doi: 10.1523/JNEUROSCI.5841-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhou Z, Hong EJ, Cohen S, et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent BDNF transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chen WG, Chang Q, Lin Y, et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- 119.Zocchi L, Sassone-Corsi P. SIRT1-mediated deacetylation of MeCP2 contributes to BDNF expression. Epigenetics. 2012;7:695–700. doi: 10.4161/epi.20733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Horike S, Cai S, Miyano M, Cheng JF, Kohwi-Shigematsu T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat Genet. 2005;37:31–40. doi: 10.1038/ng1491. [DOI] [PubMed] [Google Scholar]
- 121.Yasui DH, Xu H, Dunaway KW, Lasalle JM, Jin LW, Maezawa I. MeCP2 modulates gene expression pathways in astrocytes. Mol Autism. 2013;4:3. doi: 10.1186/2040-2392-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Abuhatzira L, Shamir A, Schones DE, Schäffer AA, Bustin M. The chromatin-binding protein HMGN1 regulates the expression of methyl CpG-binding protein 2 (MECP2) and affects the behavior of mice. J Biol Chem. 2011;D286:42051–42062. doi: 10.1074/jbc.M111.300541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mellén M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–1430. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nagarajan RP, Patzel KA, Martin M, et al. MECP2 promoter methylation and X chromosome inactivation in autism. Autism Res. 2008;1:169–178. doi: 10.1002/aur.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nagarajan RP, Hogart AR, Gwye Y, Martin MR, LaSalle JM. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics. 2006;1:e1–11. doi: 10.4161/epi.1.4.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Grafodatskaya D, Chung B, Szatmari P, Weksberg R. Autism spectrum disorders and epigenetics. J Am Acad Child Adolesc Psychiatry. 2010;49:794–809. doi: 10.1016/j.jaac.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 127.Allan AM, Liang X, Luo Y, et al. The loss of methyl-CpG binding protein 1 leads to autism-like behavioral deficits. Hum Mol Genet. 2008;17:2047–2057. doi: 10.1093/hmg/ddn102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cukier HN, Rabionet R, Konidari I, et al. Novel variants identified in methyl-CpG-binding domain genes in autistic individuals. Neurogenetics. 2010;11:291–303. doi: 10.1007/s10048-009-0228-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cukier HN, Lee JM, Ma D, et al. The expanding role of MBD genes in autism: identification of a MECP2 duplication and novel alterations in MBD5, MBD6, and SETDB1. Autism Res. 2012;5:385–397. doi: 10.1002/aur.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kuroki Y, Suzuki Y, Chyo H, Hata A, Matsui I. A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J Pediatr. 1981;99:570–573. doi: 10.1016/s0022-3476(81)80256-9. [DOI] [PubMed] [Google Scholar]
- 131.Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, Kajii T. Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J Pediatr. 1981;99:565–569. doi: 10.1016/s0022-3476(81)80255-7. [DOI] [PubMed] [Google Scholar]
- 132.Ng SB, Bigham AW, Buckingham KJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bokinni Y. Kabuki syndrome revisited. J Hum Genet. 2012;57:223–227. doi: 10.1038/jhg.2012.28. [DOI] [PubMed] [Google Scholar]
- 134.Micale L, Augello B, Fusco C, et al. Mutation spectrum of MLL2 in a cohort of Kabuki syndrome patients. Orphanet J Rare Dis. 2011;6:38. doi: 10.1186/1750-1172-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Priolo M, Micale L, Augello B, et al. Absence of deletion and duplication of MLL2 and KDM6A genes in a large cohort of patients with Kabuki syndrome. Mol Genet Metab. 2012;107:627–629. doi: 10.1016/j.ymgme.2012.06.019. [DOI] [PubMed] [Google Scholar]
- 136.Banka S, Veeramachaneni R, Reardon W, et al. How genetically heterogeneous is Kabuki syndrome: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur J Hum Genet. 2012;20:381–388. doi: 10.1038/ejhg.2011.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lederer D, Grisart B, Digilio MC, et al. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am J Hum Genet. 2012;90:119–124. doi: 10.1016/j.ajhg.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Greenfield A, Carrel L, Pennisi D, et al. The UTX gene escapes X inactivation in mice and humans. Hum Mol Genet. 1998;7:737–742. doi: 10.1093/hmg/7.4.737. [DOI] [PubMed] [Google Scholar]
- 139.Prasad R, Zhadanov AB, Sedkov Y, et al. Structure and expression pattern of human ALR, a novel gene with strong homology to ALL-1 involved in acute leukemia and to Drosophila trithorax. Oncogene. 1997;15:549–560. doi: 10.1038/sj.onc.1201211. [DOI] [PubMed] [Google Scholar]
- 140.Shilatifard A. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr Opin Cell Biol. 2008;20:341–348. doi: 10.1016/j.ceb.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Shi Y, Whetstine JR. Dynamic regulation of histone lysine methylation by demethylases. Mol Cell. 2007;25:1–14. doi: 10.1016/j.molcel.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 142.Issaeva I, Zonis Y, Rozovskaia T, et al. Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol Cell Biol. 2007;27:1889–1903. doi: 10.1128/MCB.01506-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Vicent GP, Nacht AS, Font-Mateu J, et al. Four enzymes cooperate to displace histone H1 during the first minute of hormonal gene activation. Genes Dev. 2011;25:845–862. doi: 10.1101/gad.621811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Guo C, Chang CC, Wortham M, et al. Global identification of MLL2-targeted loci reveals MLL2's role in diverse signaling pathways. Proc Natl Acad Sci U S A. 2012;109:17603–17608. doi: 10.1073/pnas.1208807109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dhar SS, Lee SH, Kan PY, et al. Trans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes Dev. 2012;26:2749–2762. doi: 10.1101/gad.203356.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kerimoglu C, Agis-Balboa RC, Kranz A, et al. Histone-methyltransferase MLL2 (KMT2B) is required for memory formation in mice. J Neurosci. 2013;33:3452–3464. doi: 10.1523/JNEUROSCI.3356-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Saitoh S, Wada T. Parent-of-origin specific histone acetylation and reactivation of a key imprinted gene locus in Prader-Willi syndrome. Am J Hum Genet. 2000;66:1958–1962. doi: 10.1086/302917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Garg SK, Lioy DT, Cheval H, et al. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J Neurosci. 2013;33:13612–13620. doi: 10.1523/JNEUROSCI.1854-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Derecki NC, Cronk JC, Lu Z, et al. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484:105–109. doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Martinowich K, Hattori D, Wu H, et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 151.Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 152.Ogier M, Wang H, Hong E, Wang Q, Greenberg ME, Katz DM. Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. J Neurosci. 2007;27:10912–10917. doi: 10.1523/JNEUROSCI.1869-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Itoh M, Ide S, Takashima S, et al. Methyl CpG-binding protein 2 (a mutation of which causes Rett syndrome) directly regulates insulin-like growth factor binding protein 3 in mouse and human brains. J Neuropathol Exp Neurol. 2007;66:117–123. doi: 10.1097/nen.0b013e3180302078. [DOI] [PubMed] [Google Scholar]
- 154.Tropea D, Giacometti E, Wilson NR, et al. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci U S A. 2009;106:2029–2034. doi: 10.1073/pnas.0812394106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Todd PK, Oh SY, Krans A, et al. Histone deacetylases suppress CGG repeat-induced neurodegeneration via transcriptional silencing in models of fragile X tremor ataxia syndrome. PLoS Genet. 2010;6:e1001240. doi: 10.1371/journal.pgen.1001240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Torrioli M, Vernacotola S, Setini C, et al. Treatment with valproic acid ameliorates ADHD symptoms in fragile X syndrome boys. Am J Med Genet A. 2010;152A:1420–1427. doi: 10.1002/ajmg.a.33484. [DOI] [PubMed] [Google Scholar]
- 157.Jiang J, Jing Y, Cost GJ, et al. Translating dosage compensation to trisomy 21. Nature. 2013;500:296–300. doi: 10.1038/nature12394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gross C, Berry-Kravis EM, Bassell GJ. Therapeutic strategies in fragile X syndrome: dysregulated mGluR signaling and beyond. Neuropsychopharmacology. 2012;37:178–195. doi: 10.1038/npp.2011.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 1724 kb)