

Abstract
Chronic hepatitis C virus (HCV) infection is associated with B cell activation, although underlying mechanisms are unclear. To investigate B cell regulation during HCV infection, we measured bulk B cell CpG and Staphylococcus aureus Cowan-induced IgG Ab-secreting cell (ASC) frequency, HCV and tetanus-specific ASC frequency, BCR- and CD40L-dependent CD80/CD86 expression, and activation of memory CD4 cells. Immature transitional, naive, resting memory, mature activated, tissue-like memory, and plasma B cell subset frequencies, cell cycling, and intrinsic apoptosis were quantified. We observed intact or enhanced tetanus-specific and total IgG ASC frequency, serum IgG, BCR- and CD40L-dependent CD80/CD86 expression, and CD40L-dependent bulk B cell activation of memory CD4 cells in HCV infection. HCV-specific ASCs were observed in HCV-infected but not control subjects, although frequencies were lower compared with tetanus-specific cells. Immature transitional and mature activated B cell subset frequencies were increased in HCV-infected subjects, with immature transitional frequency associated with liver inflammation and serum B cell-activating factor. Mature activated B cells less commonly expressed Ki67, more commonly expressed Bcl2, and were more intrinsically resistant to apoptosis, whereas immature transitional B cells more commonly expressed Ki67, the latter associated with plasma HCV level. Taken together, these results indicate that in the setting of chronic HCV infection, a state of activation results in B cell subset skewing that is likely the result of alterations in homeostasis, cell cycling, and intrinsic resistance to apoptosis and that results in an overall intact or enhanced B cell response to BCR and CD40L.
Extrahepatic manifestations of chronic hepatitis C virus (HCV) infection occur in as many as 74% of HCV-infected individuals (1) and are thought to, in part, directly relate to B cell activation. Consequences of B cell activation include hypergammaglobulinemia, cryoglobulinemia, lymphoproliferative disorder, and autoantibodies (1–5). Hypergammaglobulinemia has been well described in the setting of both chronic HCV and HIV infection (6–10) and may be the consequence of many factors. The observation that HCV–HIV-coinfected individuals have greater levels of serum IgG than do HIV-monoinfected individuals and that these levels do not normalize with HIV therapy suggest the possibility of separate or additive mechanisms of B cell activation comparing HCV and HIV infection (7).
Analysis of peripheral B cell subset distribution in HIV infection has allowed for insight into the origin of B cell dysfunction and hypergammaglobulinemia. B cell subset alterations in the periphery of HIV-infected individuals include increased frequencies of activated and terminally differentiated B cells (11, 12), reduced frequencies of memory B cells (13), and increased frequencies of immature transitional B cells that are associated with CD4 T cell lymphopenia and serum IL-7 level (14, 15).
During chronic HCV infection, Ab-secreting cell (ASC) frequencies have been described as increased, consistent with polyclonal activation, whereas total B cell numbers are not substantially increased (6, 16). Increased serum Ig levels are thought to be composed of HCV-specific and other Ag-specific Abs (6). At the same time, frequencies of CD27+ B cells (considered to be memory B cells) are thought to be reduced in HCV infection (6). It has been proposed that in HCV infection, memory B cells may be more predisposed to undergo BCR-independent stimulation that results in Ab secretion as well as apoptosis. In this regard, HCV E2 has been shown to be capable of directly stimulating naive B cells via interaction with CD81 (17), although the significance of this in vivo is uncertain. Some direct evidence for B cell activation (increased expression of CD69 and CD86) during chronic HCV infection has been reported (17). However, other evidence suggests that there may not be an overall enhanced B cell activation state during chronic HCV infection and that specific factors may contribute to enhanced B cell frequencies and activation in only subsets of HCV-infected patients (16). Expanded cell subset analysis has suggested this may occur in the CD27− population, again suggesting activation through a mechanism independent of BCR/Ag.
Evaluation of B cell subset distribution in finer detail may help elucidate mechanisms of B cell activation in HCV infection. Functionally immature transitional B cells have been characterized by their short half lives, dominance during early phases of B cell reconstitution in the peripheral blood, and disposition to undergo apoptosis in response to BCR signaling (18). Immature transitional B cell numbers are associated with states of humoral immune deficiency, including peripheral immune reconstitution, common variable immune deficiency, X-linked lymphoproliferative disease, systemic lupus erythematosus, and HIV infection (15, 19, 20). The origin, functional role, and fate of these cells is not clear, but it has been suggested that mature B cells derive from one of two cellular pathways: one arising and maturing in the bone marrow and another in the periphery where immature transitional B cells transition into mature B follicular or marginal zone B cells (18). Mature activated and differentiated B cells in the setting of HIV infection have reduced proliferative capacity, plasmacytoid phenotypic features, and enhanced Ig-secreting capacity and are thought to contribute to the state of hypergammaglobulinemia in the setting of uncontrolled HIV infection (11).
To better understand B cell regulation during chronic HCV infection, we investigated B cell function and peripheral subset skewing. Results indicate intact or enhanced bulk B cell function and increased frequencies of immature transitional and mature activated B cell subsets. The latter subset frequencies are associated with altered cell cycling (immature transitional) and an intrinsic resistance to apoptosis (mature activated) that are likely driven by factors including liver inflammation, serum B cell-activating factor (BAFF) level, HCV itself, and Bcl2 expression.
Materials and Methods
Study subjects
Chronic HCV-infected subjects (n = 25 for bulk B cell functional and phenotypic assays, and an overlapping set of n = 25 for purified B cell subset, Ki67 expression, and Bcl2 expression assays) had detectable serum HCVAb for at least 6 mo, had detectable HCV RNA by PCR, and were not previously treated for HCV infection. Both HCV-infected and healthy control subjects (n = 25) provided written informed consent for venous blood sampling under approval of the institutional review boards for human studies at the Cleveland VA Medical Center and University Hospitals of Cleveland (Cleveland, OH). HCV-infected subjects were mainly infected with genotype 1 HCV (88.5%). HCV level ranged from 85,000–7,000,000 IU/ml. Alanine aminotransferase ranged from 16–302 U/l. Aspartate aminotransferase (AST)/platelet ratio index ranged from 0.07–1.31. One study subject had impaired renal function attributed to HCV-associated membranoproliferative glomerulonephtritis and immune complex deposition, whereas other subjects carried no clinical or laboratory diagnosis of exptrahepatic HCV manifestation (renal impairment, arthritis, monoclonal gammopathy). Rheumatoid factor (RF) had been previously tested on n = 23 HCV-infected subjects, and 13 of these were RF seropositive.
Cell isolation
PBMCs were prepared from fresh peripheral blood specimens using Isoprep (Fisher Scientific, Hudson, NH). Bulk B cells were prepared from PBMCs using a negative bead selection B cell Isolation Kit II (Miltenyi Biotec, Auburn, CA) depleting CD2, CD14, CD16, CD36, CD43, and CD235a (glycophorin A). Flow cytometric-based cell sorting of CD3-depleted PBMCs (RosetteSep Human CD3 cell depletion; StemCell Technologies, Vancouver, British Columbia, Canada) was performed using an FACSAria (BD Biosciences, San Jose, CA) housed inside a BSL2 Bioprotect II Biosafety Cabinet (Baker, Sanford, ME) within our Cleveland VA and Center for AIDS Research BSL2 core facility (Cleveland, OH).
Serum IgG, IgA, and IgM by ELISA, B cell total Ab ASC, and Ag ASC frequency by ELISPOT
Serum samples were collected using Vacutainer SST tubes (BD Biosciences) and frozen at −70°C. Serum samples were analyzed using the Total Human IgG, IgA, and IgM Elisa kits (Cygnus Technologies, Southport, NC).
Negatively selected B cells were stimulated in a 24-well plate for 4 d at 37°C with 2.5 μg/ml 2006 CpG (Operon, Huntsville, AL) and 1:10,000 Staphylococcus aureus Cowan (SAC) (EMD Chemicals, Gibbstown, NJ) in complete RPMI 1640 (penicillin/streptomycin and L-glut) with 10% FCS. ELI-SPOT plates (Whatman, Piscataway, NJ) were precoated with 10 μg/ml HCV core (aa 2–120), 10 μg/ml NS3 (aa1192-1457), and 10 μg/ml NS4 (aa 1569–1931) protein (Chiron, Emeryville, CA), 1:400 dilution of tetanus Ag (Wyeth, New York, NY), or 5 μg/ml goat anti-human κ and Λ Ab (Rockland, Gilbertsville, PA) in sterile PBS. Plates were blocked with 5% FCS in RPMI 1640 for2 h at room temperature, then precultured B cells were added. ASC frequency was measured after overnight incubation at 37°C, washing with PBS-0.025%Tween, and additional overnight incubation at 4°C with secondary Ab (100 μl 1:20,000 donkey anti-human IgG biotinylated Ab in 1% FCS in PBS-Tween; Jackson ImmunoResearch Laboratories, West Grove, PA). Plates were washed with PBS-0.025% Tween and incubated for 1 h at room temperature with 100 μl 1:2000 streptavidin-HRP (DakoCytomation, Carpinteria, CA) in PBS plus 1% BSA, washed with PBS, and developed with 200 μl 3-amino-9-ethylcarboazole. Spots were analyzed using an ELISPOT image analyzer (Cellular Technology Limited, Cleveland, OH).
BCR-dependent and CD40L-dependent B cell CD80/CD86 expression
A total of 300,000 PBMCs were incubated in a 96-well round-bottom plate for 3 d in the presence of media, 104 CD40L-expressing NIH 3T3 fibroblasts (supplied by Dr. Peter Heeger, Mount Sinai, New York, NY), 80 μg/ml goat anti-human IgG+IgA+IgM (IgGAM) (Jackson ImmunoResearch Laboratories), or CD40L-expressing 3T3 cells and anti-IgGAM. Cells were removed and stained with anti-CD19 allophycocyanin-Cy7, anti-CD27 APC, anti-CD80 PE-Cy5, and anti-CD86 PE (BD Biosciences) for flow cytometric analysis on an LSRII flow cytometer (BD Biosciences) with FACSDiva Software (BD Biosciences). CD40L dependence for activity was verified using CD40L-blocking Ab (Ancell Corporation, Bayport, MN) in comparison with isotype control.
B cell activation of allogeneic memory CD4 cells
Negatively selected B cells (1.5 × 105) were incubated on precoated IFN-γ ELISPOT plates for 3 d in the presence or absence of CD40L-expressing NIH 3T3 cells (104). Negatively selected memory CD4 T cells (Miltenyi Biotec) obtained from a single allogenic healthy control donor were added at 150,000 cells/well for the final 16 h of culture, and IFN-γ–secreting cell frequency was measured as previously described (21).
B and T lymphocyte frequency by flow cytometric analysis
Whole blood samples (200 μl) were stained with isotype control, anti-CD19 PE-Cy5 (or PerCP), anti-CD10 allophycocyanin, anti-CD20 allophycocyanin-Cy7, anti-CD27 PE (or PE-Cy7), anti-CD38 allophycocyanin (or PE-Cy7) (BD Biosciences), and anti-CD21 FITC (Beckman Coulter, Brea, CA) or PE (BD Biosciences) for 10 min at room temperature. Samples were incubated for 10 min with 2 ml 1:10 FACSLyse solution (BD Biosciences), then washed with 2 ml PBS with 0.01% BSA, then resuspended in 200 μl 1.0% paraformaldehyde solution (Electron Microscopy Science, Hatfield, PA) and 200 μl PBS with 0.01% BSA. Flow cytometric analysis was performed on an LSRII flow cytometer (BD Biosciences) with FACSDiva Software (BD Biosciences) for whole blood-based analysis of cell subset frequency. B lymphocytes were identified by forward and side scatter, and B cell subset frequencies were measured [proportion of CD19-gated cells that are CD10+ CD27− (immature transitional B cells), CD10− CD21+CD27− (naive B cells), CD10−CD21+CD27+ (resting memory B cells), CD20+CD21− CD27+ (mature activated B cells), CD10− CD21− CD27− (tissue like memory B cells), and CD20− CD38+ (plasma cells)].
For T lymphocyte phenotyping, cells were stained with anti-CD4 PerCP, anti-CD8 allophycocyanin, anti-CD45RA FITC, and anti-CCR7 PE-Cy7 (BD Biosciences). T lymphocytes were identified by forward and side scatter, and T cell subset frequencies were measured (proportion of CD4- or CD8-gated cells that are CD45RA−CCR7− [effector memory T cells], CD45RA−CCR7+ [central memory T cells], CD45RA+CCR7− [terminal effector T cells], and CD45RA+CCR7+ [naive T cells]).
Ki67 and Bcl2 intracellular flow cytometric analysis
Whole blood samples (200 μl) were stained with surface markers of B cell subsets as above for 10 min, then 2 ml 1:10 FACSLyse solution (BD Biosciences) was added for 10 min, followed by 2 ml 1:10 FACS Permeabilizing Solution 2 (BD Biosciences) for 10 min. Samples were washed in 2 ml PBS with 0.01% BSA and stained with isotype control, anti-Ki67 FITC, or anti-Bcl2 FITC (BD Biosciences) for 30 min. All of the above steps were completed at room temperature. B cell subset expression of Bcl2 and Ki67 was measured as proportion of cells staining positive, as well as by mean fluorescence intensity (MFI) of expression.
Serum IL-7 and BAFF by ELISA
Serum samples were stored at −70°C until analysis. IL-7 was measured using Quantikine HS Human IL-7 ELISA kit (R&D Systems, Minneapolis, MN). BAFF was measured using Human BAFF/BLyS/TNFSF13B Quantikine ELISA Kit (R&D Systems).
Flow-sorted B cell subset analysis for intrinsic cell death by Annexin V staining and flow cytometric analysis
Flow-sorted live B cell subsets were analyzed for cell death in direct ex vivo assays and in short-term culture (20 h at 37°C) by Annexin V staining. Purified B cell subsets were washed twice with 400 ml cold PBS, followed by addition of 100 μl cold 1× Annexin V binding buffer (BD Biosciences) and 1 μl Annexin V-FITC Ab (BD Pharmingen, San Diego, CA), then incubated 15 min at room temperature. Postincubation, 400 μl cold 1× Annexin V binding buffer was added, and samples were analyzed by flow cytometry within 1 h of staining.
Statistical analysis
We analyzed the data with SPSS for Windows, version 15.0 (SPSS, Chicago, IL) and Stata SE, version 9.2 (Stata, College Station, TX). We used conventional measures of central tendency and variability to describe the data. To compare continuous and categorical variables across groups, we used the Mann-Whitney U test or Kruskal-Wallis H test and Pearson’s χ2, respectively. We compared responses pre- and poststimulation with Wilcoxon’s signed-rank test and assessed the associations between continuous variables by Spearman’s rank correlation coefficient. All tests are two-sided, and a p value ≤0.05 was considered significant.
Results
HCV-specific IgG-secreting memory B cell frequency is increased in HCV infection, whereas total IgG-secreting memory cell frequency is comparable between HCV-infected and control subjects
As a first measure of bulk B cell function, we measured serum IgG level. Consistent with previously published data (6), serum IgG levels were increased in chronic HCV-infected compared with healthy control subject samples (Fig. 1A). No significant difference in serum IgA or IgM level was observed comparing HCV-infected and healthy control groups (not shown). To identify whether bulk peripheral B cells were altered in propensity to secrete Ab we evaluated B cell ASC capacity by culturing negatively selected B cells for 4 d with CpG 2006 and SAC. Cells were transferred to B cell ELISPOT plates for analysis of total IgG, tetanus protein Ag-specific, or HCV core, NS3, or NS4 recombinant protein-specific ASC frequency. We observed similar total IgG ASC frequencies comparing groups by this method (Fig. 1B). When evaluating tetanus Ag-specific IgG ASC frequency, we observed a trend toward an increase in HCV-infected subject samples (Fig. 1C), suggesting that a state of B cell activation may exist within the non-HCV Ag-specific B cell fraction or that HCV-infected subjects may be more likely to have recently received exposure to tetanus immunization. As expected, HCV protein Ag-specific ASC frequencies were observed in HCV-infected subjects and not healthy controls (Fig. 1D). However, the frequencies of these HCV-specific ASCs directed at each HCV Ag were lower than those directed at tetanus Ags. The phenomenon of low HCV-specific B cell frequency in comparison with that observed against other Ags is similar to data from our and other studies on HCV-specific T cells (22–26), perhaps suggesting inhibited, compartmentalized, or deleted HCV-specific ASCs.
FIGURE 1.

Increased serum IgG levels in chronic HCV infection. A, Serum samples (n = 10 healthy control subjects and n = 42 chronic HCV-infected subjects) were analyzed for total human IgG by ELISA. B, Negatively selected bulk B cells (5 × 104 or 1 × 105 as marked on x-axis) were analyzed for total IgG ASC frequency after 4 d culture with CpG 2006 (2.5 μg/ ml) and SAC (1:10,000) (n = 14 healthy controls and n = 14 HCV-infected subjects shown). C, Tetanus-specific IgG ASC (n = 13 healthy controls and n = 14 HCV-infected subjects shown). D, HCV-specific (HCV core, NS3, or NS4 recombinant protein) ASC (n = 11 healthy controls and n = 13 HCV-infected subjects shown).
CD40L- and BCR-induced bulk B cell activation is intact, and in some cases enhanced, in HCV infection
To directly explore ex vivo the functional response of B cells to BCR signaling and/or CD40L stimulus, we next evaluated PBMCs by flow cytometric analysis for CD19+CD27+-gated B cell CD80/ CD86 expression before and after 3 d of stimulation. As shown in Fig. 2 at baseline, CD27+ B cells expressed similar levels of CD80 and CD86 in the HCV-infected group when compared with healthy controls, although, notably, there were individuals in the HCV-infected group whose cells expressed higher levels of these markers of activation. Upon culture with CD40L presented on the surface of NIH 3T3 cells, HCV-infected and control B cells similarly upregulated CD80 and CD86 expression, and this was not further upregulated by the combined stimulus of CD40 and BCR (provided by anti-IgGAM). However, stimulus of the BCR alone yielded a significant increase in expression of CD80, and nearly significant difference for CD86, on B cells of HCV-infected compared with healthy control subjects (p = 0.03 and p = 0.07, respectively). Of note, BCR stimulation induced little upregulation of CD80 or CD86 in comparison with CD40L stimulation. Similar patterns of CD40L-induced CD80 and CD86 expression were observed on CD19+CD27−-gated cells, whereas no BCR-induced expression of these markers was observed on CD19+CD27− cells (data not shown). Taken together, these results indicate that in the setting of chronic HCV infection, bulk B cell activation is intact in response to CD40L signaling, whereas the memory CD27+ B cell response is modestly enhanced following BCR stimulation.
FIGURE 2.
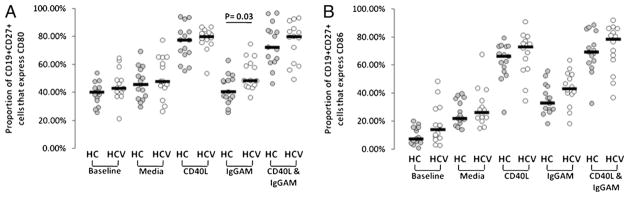
CD40L- and BCR-dependent memory B cell CD80 and CD86 expression. PBMCs (150,000/well) from healthy controls (n = 15) and HCV-infected subjects (n = 14) were cultured 3 d in the presence of media, CD40L, BCR stimulation in the form of anti-IgGAM, or the combination of stimuli. Cells were stained with CD19, CD27, CD80, and CD86, and CD19+CD27+ B cells were analyzed for expression of CD80 (A) or CD86 (B) by flow cytometric analysis.
CD40L-dependent B cell activation of memory CD4 cells is intact in chronic HCV infection
The ability of bulk B cells to activate memory T cells was next evaluated. CD40L enhanced the ability of bulk B cells to activate allogenic memory CD4 cells (Fig. 3A). Notably, this activation is not BCR dependent, as BCR stimulation did not provide B to T cell stimulatory activity (not shown). Allogenic memory CD4 cells from a single donor were activated by CD40L-stimulated B cells from HCV (n = 14) or healthy control (n = 15) subjects. As shown in Fig. 3B, CD40L-dependent B cell activation of allogenic memory CD4 cells was similar when comparing HCV-infected to healthy control subjects.
FIGURE 3.

CD40L-dependent B cell activation of memory CD4 cells is intact in HCV infection. A, B cells (150,000/well) from a healthy control donor were cultured in the presence or absence of CD40L-expressing NIH 3T3 cells for 72 h on IFN-γ ELISPOT plates, then allogenic memory CD4 T cells were added (150,000 cells/well) for 16 h, and wells were analyzed for IFN-γ secreting cell frequency. B, B cells (150,000 cells/well) from healthy control and HCV-infected subjects analyzed for CD40L-dependent allogenic memory CD4 cell activation as described in A. No signal was observed in the control well cultures in the absence of CD40L, B cells, or memory CD4 cells (not shown).
Enhanced immature transitional and mature activated B cell subset frequencies in chronic HCV infection
To evaluate whether peripheral B cell subset compartment alterations may explain hypergammagloblinemia and/or altered activation state, we analyzed peripheral blood samples from chronic HCV-infected and healthy control subjects for B cell frequencies and B cell subset frequencies by flow cytometry. Total percentages of peripheral B cells did not differ between healthy controls and HCV-infected subjects (Fig. 4A), in agreement with previous literature (6, 16), although a small subset of HCV-infected subjects had high frequencies of B cells. Subsets of human peripheral B cells can be defined by cell surface markers. Immature transitional B cells express CD19 and CD10 and lack of expression of CD27 (Fig. 4B). Increased frequencies of these cells have been described in the setting of advanced HIV-related disease, in the setting of immune reconstitution, and during autoimmune disease (15, 19, 20). Naive B cells lack expression of CD10 and CD27 and express CD19 and CD21. Resting memory B cells lack expression of CD10 and express CD19, CD21, and CD27. Mature activated memory B cells express CD19, CD20, and CD27 and lack expression of CD21. Tissue-like memory B cells lack expression of CD10, CD21, and CD27 and express CD19. Plasma cells express CD19 and CD38 and lack expression of CD20 and CD21. The proportion of B cells in the immature transitional stage was found to be increased in chronic HCV-infected subjects (p = 0.003; Fig. 4C). Additionally, increased proportions of B cells in the mature activated subset were observed in chronic HCV-infected subjects (p = 0.008; Fig. 4F). These altered frequencies appear driven by a subset of HCV-infected subjects with distinctly higher frequencies of these cells. Naive, tissue-like memory, plasma, and resting memory B cell frequencies did not significantly differ between HCV and healthy control subject groups, although subsets of HCV-infected subjects had substantially increased frequencies of tissue-like memory cells.
FIGURE 4.

Increased frequency of immature transitional and mature activated B cell subsets in peripheral blood of chronic HCV-infected subjects. Intravenous whole blood samples were stained with B cell phenotyping markers (CD10, CD19, CD20, CD21, CD27, and CD38) and analyzed for B cell subsets by flow cytometric analysis. Proportion of forward and side scattered lymphocytes that express CD19 are shown in A. B cell subset analysis was performed as shown in B. Proportions of CD19-gated cells that were CD10+CD27− (C, immature/transitional B cells), CD10−CD21+CD27− (D, naive B cells), CD10−CD21+CD27+ (E, resting memory B cells), CD10−CD21−CD27+ (F, mature/activated B cells), CD10−CD21−CD27− (G, tissue-like memory B cells), and CD20−CD38+ (H, plasma cells) were determined for HCV-infected (n = 24) and healthy control (n = 25) subjects.
Mature activated B cell Bcl2 expression is associated with decreased intrinsic apoptosis and decreased Ki67 expression, whereas immature transitional B cells more commonly express Ki67 in chronic HCV infection
Bcl2 expression has been observed to be increased in B cells of HCV-infected individuals (27, 28), and this is thought to provide resistance to apoptosis. To investigate whether enhanced immature transitional or mature activated B cell frequency is related to increased proportions of cells in cycle or reduced propensity to cell death, we next evaluated Ki67 and Bcl2 expression in B cell subsets. Mature activated and resting memory B cell subsets less commonly expressed Ki67 in HCV-infected subject samples as compared with controls (p = 0.03 and p = 0.05, respectively; Fig. 5), suggesting that these cells are less commonly in cycle in the context of HCV infection. Notably, immature transitional B cells quite commonly expressed Ki67 in a subset of HCV-infected subjects, contributing to the overall increase in Ki67 expression compared with corresponding B cells of healthy controls (p = 0.05).
FIGURE 5.

Ki67 expression tends to be decreased on mature activated and resting memory B cell subsets of HCV-infected subjects. Whole blood samples were stained with B cell subset markers and measured for intracellular KI-67 expression by flow cytometry. A, Immature transitional. B, Naive. C, Resting memory. D, Mature activated. E, Tissue-like memory. F, Plasma cells.
The antiapoptotic marker Bcl2 was found to be more commonly expressed in mature activated and tissue-like memory B cell subsets of HCV-infected compared with healthy control subjects (Fig. 6), although substantial heterogeneity was observed within the HCV-infected group. The MFI of Bcl2 expression was also increased in bulk CD19+ B cells in HCV-infected subject samples (median MFI 1263 versus 1371; p = 0.02), and MFI of Bcl2 expression was also increased within the naive B cell subset (median MFI 1152 versus 1305; p = 0.03), indicating propensity for increased Bcl2 expression on a number of B cell subsets in the setting of HCV infection.
FIGURE 6.

Increased BCL-2 expression on mature activated and tissue-like memory B cell subsets of HCV-infected subjects. Whole blood samples were stained with B cell subset markers and measured for BCL-2 expression by flow cytometry. A, Immature transitional. B, Naive. C, Resting memory. D, Mature activated. E, Tissue-like memory. F, Plasma cells.
To further evaluate intrinsic propensity to cell death, we isolated B cell subsets by flow cytometric-based cell sorting and evaluated baseline and unstimulated overnight cultured cells for Annexin staining. Although cell isolation by flow cytometric cell sorting itself may contribute to Annexin staining, we nonetheless found mature activated and resting memory B cell subsets to be more resistant to intrinsic pathway cell death in HCV-infected compared with healthy control subjects (p = 0.04 and p = 0.003, respectively; Fig. 7). In the case of mature activated cells, this is consistent with the observed increased proportion of cells expressing Bcl2 found in Fig. 6, and Bcl2 expression was negatively associated with day 1 B cell Annexin staining (r = −0.9, p = 0.04).
FIGURE 7.

Reduced intrinsic cell death propensity of mature activated and tissue-like memory B cell subsets from HCV-infected subjects. CD3-depleted PBMCs were prepared from peripheral blood samples and were stained with B cell subset markers (CD10, CD19, CD20, CD21, CD27, and CD38). B cell subsets were sorted by an FACSAria flow cytometer (BD Biosciences) and analyzed for baseline Annexin V staining as well as Annexin V staining after 20-h culture at 37°C. A, Immature transitional. B, Naive. C, Resting memory. D, Mature activated. E, Tissue-like memory. F, Plasma cells.
Immature transitional B cell frequency is positively correlated with serum AST and serum BAFF and negatively correlated with bulk B cell tetanus- and HCV-specific ASC frequency, whereas mature activated B cell frequency is positively correlated with CD40L+ IgGAM-induced bulk B cell CD80 expression
Next, we evaluated whether some of the above parameters were associated with each other in a subset of subjects on whom we had complete data sets. We also evaluated whether clinical parameters were associated with B cell numbers or function. Frequencies of B cells within each of the subsets were associated with each other in the following manner. Frequencies of naive B cells were negatively correlated with frequencies of resting memory and mature activated B cell frequencies in both healthy control (r = −0.93, p < 0.001; r = −0.52, p = 0.002, respectively) and HCV-infected (r = −0.56, p = 0.001; r = −0.73, p < 0.001, respectively) subjects, whereas naive B cell frequencies were negatively correlated with tissue-like memory B cell frequencies only in HCV-infected subjects (r = −0.54, p = 0.001), consistent with skewing of B cell subset frequencies shown in Fig. 4.
To consider clinical parameters that may contribute to B cell subset skewing, we evaluated markers of liver inflammation (serum AST and alanine aminotransferase), CD4 T cell (total CD4, naive, central memory, or effector memory) frequency, serum RF, or serum IL-7 level in relation to B cell subset frequency. Liver inflammation as measured by serum AST was positively correlated with immature transitional B cell subset frequency (r = +0.49, p = 0.009). Plasma HCV level positively correlated with proportions of immature transitional B cells that express Ki67 (r = +0.48, p = 0.02). RF testing was only available for nine subjects in whom B cell subset frequency testing was performed. Therefore, the significance of a correlation between RF and immature transitional B cell subset frequency (r = +0.79, p = 0.006) is currently unclear. CD4 numbers and serum IL-7 have been previously shown to associate with immature transitional B cell frequencies in the setting of HIV infection (14, 15). However, we found no association between these parameters and immature transitional B cell frequencies or absolute numbers in HCV-infected and healthy control subjects, although serum IL-7 levels were barely detectable in either group of subjects.
HCV infection is also associated with increased serum BAFF levels (29, 30), and BAFF is known to increase the survival of immature B cell subsets (31). We therefore evaluated serum BAFF and its relation to B cell subset frequencies. Consistent with prior observations, we observed increased serum BAFF levels in the HCV-infected group (Fig. 8; p = 0.04). We also observed a positive correlation between serum BAFF level and both immature transitional (r = +0.35, p = 0.05) and naive (r = +0.38, p = 0.03) B cell subset frequency in the setting of HCV infection, whereas we observed a negative correlation between BAFF level and resting memory B cell subset frequency (r = −0.48, p = 0.006).
FIGURE 8.

Serum BAFF level is positively correlated with immature transitional and naive B cell frequency and negatively correlated with resting memory B cell subset frequency. Serum BAFF level was measured by ELISA, and results comparing HCV-infected and healthy control subjects are shown in A, whereas results comparing BAFF level to peripheral B cell subset frequency were immature transitional (B), naive (C), resting memory (D), mature activated (E), tissue-like memory (F), and plasma cells (G).
The evaluation of relationships between B cell subset frequencies and bulk B cell function revealed negative correlations between immature transitional B cell and frequencies of tetanus-, NS3-, and NS4-specific ASC in HCV-infected subject samples (r = −0.64, −0.70, −0.66; p = 0.05, 0.04, 0.05, respectively). Notably, Ag-specific ASC frequency reflects bulk B cell function, as assays were performed with bulk B cells, so higher immature transitional frequency may just be reflective of a lower frequency of another subset. Finally, mature activated B cell subset frequency was positively correlated with bulk B cell CD80 expression in response to CD40L (r = +0.69, p = 0.03) and nearly IgGAM+CD40L (r = +0.81, p = 0.09).
Discussion
During chronic HCV infection, B cell function, as measured by the ability to secrete Ab (total ASC and tetanus-specific ASC after 4 d expansion), to upregulate CD80 and CD86 in response to BCR and CD40L stimulation, and to respond to CD40L and activate allogenic memory CD4 cells, appears intact. In fact, BCR-dependent CD80 expression and tetanus-specific ASC frequency appear enhanced. Although frequency HCV-specific ASC appears lower than expected in HCV-infected individuals, this observation may be reflective of virus-related rather than host-related effects. Taken together, these bulk B cell functional assays indicate intact and enhanced function in some cases. Whether this contributes to the state of hypergammaglobulinemia observed in this study, and published previously (6), is unclear.
We evaluated peripheral B cell subset frequencies in further detail to understand B cell subset skewing in the setting of chronic HCV infection. We found increased frequencies of immature transitional and mature activated B cell subsets, with considerable heterogeneity among the HCV-infected cohort. Peripheral subset skewing may be the result of compartmental redistribution, enhanced production due to specific or nonspecific activation stimulus, or alteration in cell death. Increased frequencies of immature transitional B cells have been found in the context of humoral immune deficiency, including peripheral immune reconstitution, common variable immune deficiency, X-linked lymphoproliferative disease, systemic lupus erythematosus, and HIV infection (15, 19, 20). The role of these cells in both health and disease is not clear, although immature transitional B cells can give rise to a subset of memory B cells that do not require germinal centers for production. These latter cells produce low-affinity natural Abs, including Abs against specific pathogens (32, 33) and thus are considered to be a first line of defense against certain infections. It has been recently shown that Ag-inexperienced cord blood human immature transitional B cells can, upon TLR9 ligand engagement, express AID and Blimp, genes necessary for hypersomatic mutation, class-switch recombination, and differentiation into plasma cells (34). Moreover, these stimulated cells can differentiate into plasma cells and produce IgG and IgM Abs, including Abs with antipneumococcal specificity (an Ag they have not seen). It has therefore been proposed that immature transitional B cells may serve as a precursor for plasma cells involved in innate Ab production. In the setting of HIV infection, their presence is associated with increased serum levels of IL-7 and CD4+ T cell lymphopenia, suggesting a homeostatic response may play a role in their overexpression in the context of advancing HIV disease. Because we find no association between serum IL-7 or CD4+ T cell frequency and immature transitional B cell frequency in this study, it is likely that other factors are involved in the presence of these cells in the setting of HCV infection. BAFF is one factor known to increase the survival of immature B cell subsets (31), and we in fact observed a positive correlation between serum BAFF level and immature transitional B cell subset frequency, suggesting this may be one factor involved in determining B cell subset skewing. Additionally, because we found a subset of HCV-infected subjects (and the group as a whole) with increased proportions of immature transitional B cells expressing Ki67, these cycling cells may arise as a result of HCV-induced turnover. Whether such increased turnover is a result of a homeostatic response to HCV-induced perturbations or release from bone marrow-inductive sites remains to be determined. As liver inflammation was observed in this study to be associated with immature transitional B cell subset frequency, host response to HCV may in part account for this observation. In fact, we also observed a positive correlation between proportions of immature transitional B cells expressing Ki67 and plasma HCV level.
In the setting of chronic HIV infection, differentiated B cells with reduced proliferative capacity, plasmacytoid phenotypic features, and enhanced Ig-secreting capacity are thought to contribute to the state of hypergammaglobulinemia (11). The mechanism underlying enhanced peripheral frequencies of these cells in HIV infection is thought to relate to a state of chronic immune activation and increased turnover that lead to an overrepresentation of this cell population that also has an increased tendency to undergo CD95-mediated apoptosis (12, 35). In contrast, in this paper, we find that in HCV infection, there is a reduced intrinsic tendency for cell death in resting memory and mature activated B cells, and this is associated with increased antiapoptotic factor (Bcl2) expression. Increased Bcl2 expression in B cells of HCV-infected individuals has been previously described (27, 28), and data in this study indicate expression is enhanced in mature activated, tissue-like memory, and, to some extent, even on naive B cell subsets. Whether enhanced frequencies of mature activated B cells have an increased propensity to undergo terminal differentiation and contribute to elevated serum IgG levels or other B cell associated abnormalities in the setting of chronic HCV infection is yet to be determined. Additionally, whether direct activation due to Ag drive or indirect activation is responsible for the origin of these cells is unknown. Certainly previous evidence suggests that HCV Ag may at least in part contribute to B cell activation in some HCV-infected individuals (36), although low frequencies of HCV-specific B cells detected in this study would suggest indirect activation is also a likely possibility.
Taken together, these data indicate that in chronic HCV infection, a state of immune activation is reflected by B cell subset skewing, that this skewing is likely in part mediated by alterations in Bcl2 expression, intrinsic apoptosis, serum BAFF level, and cell cycling, and that skewing and activation are associated with some enhancement in bulk B cell CD40L and BCR responsiveness. Whether any or all of these perturbations contribute to a state of hypergammaglobulinemia, lymphoproliferative disorder, or auto-immunity is presently unclear.
Acknowledgments
This work was supported by National Institutes of Health Grant AI 36219, National Institute of Allergy and Infectious Diseases Grant 1R21 AI066957, National Institute of Diabetes and Digestive and Kidney Diseases Grant DK068361, and the Case Western Reserve University Center for AIDS Research core facilities.
Abbreviations used in this paper
- ASC
Ab-secreting cell
- AST
aspartate aminotransferase
- BAFF
B cell-activating factor
- HCV
hepatitis C virus
- IgGAM
goat anti-human IgG+IgA+IgM
- MFI
mean fluorescence intensity
- RF
rheumatoid factor
- SAC
Staphylococcus aureus Cowan
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Cacoub P, Poynard T, Ghillani P, Charlotte F, Olivi M, Piette JC, Opolon P. Extrahepatic manifestations of chronic hepatitis C. MULTI-VIRC Group. Multidepartment Virus C. Arthritis Rheum. 1999;42:2204–2212. doi: 10.1002/1529-0131(199910)42:10<2204::AID-ANR24>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 2.Buskila D, Shnaider A, Neumann L, Lorber M, Zilberman D, Hilzenrat N, Kuperman OJ, Sikuler E. Musculoskeletal manifestations and autoantibody profile in 90 hepatitis C virus infected Israeli patients. Semin Arthritis Rheum. 1998;28:107–113. doi: 10.1016/s0049-0172(98)80043-7. [DOI] [PubMed] [Google Scholar]
- 3.Lamprecht P, Gause A, Gross WL. Cryoglobulinemic vasculitis. Arthritis Rheum. 1999;42:2507–2516. doi: 10.1002/1529-0131(199912)42:12<2507::AID-ANR2>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 4.Hansen KE, Arnason J, Bridges AJ. Autoantibodies and common viral illnesses. Semin Arthritis Rheum. 1998;27:263–271. doi: 10.1016/s0049-0172(98)80047-4. [DOI] [PubMed] [Google Scholar]
- 5.McMurray RW, Elbourne K. Hepatitis C virus infection and autoimmunity. Semin Arthritis Rheum. 1997;26:689–701. doi: 10.1016/s0049-0172(97)80005-4. [DOI] [PubMed] [Google Scholar]
- 6.Racanelli V, Frassanito MA, Leone P, Galiano M, De Re V, Silvestris F, Dammacco F. Antibody production and in vitro behavior of CD27-defined B-cell subsets: persistent hepatitis C virus infection changes the rules. J Virol. 2006;80:3923–3934. doi: 10.1128/JVI.80.8.3923-3934.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soriano-Sarabia N, Leal M, Delgado C, Molina-Pinelo S, De Felipe B, Ruiz-Mateos E, Sánchez-Quijano A, Lissen E, Vallejo A. Effect of hepatitis C virus coinfection on humoral immune alterations in naïve HIV-infected adults on HAART: a three year follow-up study. J Clin Immunol. 2005;25:296–302. doi: 10.1007/s10875-005-3864-1. [DOI] [PubMed] [Google Scholar]
- 8.Morris L, Binley JM, Clas BA, Bonhoeffer S, Astill TP, Kost R, Hurley A, Cao Y, Markowitz M, Ho DD, Moore JP. HIV-1 antigen-specific and -nonspecific B cell responses are sensitive to combination antiretroviral therapy. J Exp Med. 1998;188:233–245. doi: 10.1084/jem.188.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pahwa R, Good RA, Pahwa S. Prematurity, hypogammaglobulinemia, and neuropathology with human immunodeficiency virus (HIV) infection. Proc Natl Acad Sci USA. 1987;84:3826–3830. doi: 10.1073/pnas.84.11.3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shirai A, Cosentino M, Leitman-Klinman SF, Klinman DM. Human immunodeficiency virus infection induces both polyclonal and virus-specific B cell activation. J Clin Invest. 1992;89:561–566. doi: 10.1172/JCI115621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moir S, Malaspina A, Ogwaro KM, Donoghue ET, Hallahan CW, Ehler LA, Liu S, Adelsberger J, Lapointe R, Hwu P, et al. HIV-1 induces phenotypic and functional perturbations of B cells in chronically infected individuals. Proc Natl Acad Sci USA. 2001;98:10362–10367. doi: 10.1073/pnas.181347898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moir S, Malaspina A, Pickeral OK, Donoghue ET, Vasquez J, Miller NJ, Krishnan SR, Planta MA, Turney JF, Justement JS, et al. Decreased survival of B cells of HIV-viremic patients mediated by altered expression of receptors of the TNF superfamily. J Exp Med. 2004;200:587–599. [PubMed] [Google Scholar]
- 13.Titanji K, De Milito A, Cagigi A, Thorstensson R, Grützmeier S, Atlas A, Hejdeman B, Kroon FP, Lopalco L, Nilsson A, Chiodi F. Loss of memory B cells impairs maintenance of long-term serologic memory during HIV-1 infection. Blood. 2006;108:1580–1587. doi: 10.1182/blood-2005-11-013383. [DOI] [PubMed] [Google Scholar]
- 14.Malaspina A, Moir S, Chaitt DG, Rehm CA, Kottilil S, Falloon J, Fauci AS. IdiopathicCD4+ T lymphocytopenia isassociated withincreases in immature/transitional B cells and serum levels of IL-7. Blood. 2007;109:2086–2088. doi: 10.1182/blood-2006-06-031385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malaspina A, Moir S, Ho J, Wang W, Howell ML, O’Shea MA, Roby GA, Rehm CA, Mican JM, Chun TW, Fauci AS. Appearance of immature/transitional B cells in HIV-infected individuals with advanced disease: correlation with increased IL-7. Proc Natl Acad Sci USA. 2006;103:2262–2267. doi: 10.1073/pnas.0511094103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ni J, Hembrador E, Di Bisceglie AM, Jacobson IM, Talal AH, Butera D, Rice CM, Chambers TJ, Dustin LB. Accumulation of B lymphocytes with a naive, resting phenotype in a subset of hepatitis C patients. J Immunol. 2003;170:3429–3439. doi: 10.4049/jimmunol.170.6.3429. [DOI] [PubMed] [Google Scholar]
- 17.Rosa D, Saletti G, De Gregorio E, Zorat F, Comar C, D’Oro U, Nuti S, Houghton M, Barnaba V, Pozzato G, Abrignani S. Activation of naïve B lymphocytes via CD81, a pathogenetic mechanism for hepatitis C virus-associated B lymphocyte disorders. Proc Natl Acad Sci USA. 2005;102:18544–18549. doi: 10.1073/pnas.0509402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allman D, Pillai S. Peripheral B cell subsets. Curr Opin Immunol. 2008;20:149–157. doi: 10.1016/j.coi.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cuss AK, Avery DT, Cannons JL, Yu LJ, Nichols KE, Shaw PJ, Tangye SG. Expansion of functionally immature transitional B cells is associated with human-immunodeficient states characterized by impaired humoral immunity. J Immunol. 2006;176:1506–1516. doi: 10.4049/jimmunol.176.3.1506. [DOI] [PubMed] [Google Scholar]
- 20.Sims GP, Ettinger R, Shirota Y, Yarboro CH, Illei GG, Lipsky PE. Identification and characterization of circulating human transitional B cells. Blood. 2005;105:4390–4398. doi: 10.1182/blood-2004-11-4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anthony DD, Post AB, Valdez H, Peterson DL, Murphy M, Heeger PS. ELISPOT analysis of hepatitis C virus protein-specific IFN-gamma-producing peripheral blood lymphocytes in infected humans with and without cirrhosis. Clin Immunol. 2001;99:232–240. doi: 10.1006/clim.2001.5018. [DOI] [PubMed] [Google Scholar]
- 22.Valdez H, Anthony D, Farukhi F, Patki A, Salkowitz J, Heeger P, Peterson DL, Post AB, Asaad R, Lederman MM. Immune responses to hepatitis C and non-hepatitis C antigens in hepatitis C virus infected and HIV-1 coinfected patients. AIDS. 2000;14:2239–2246. doi: 10.1097/00002030-200010200-00004. [DOI] [PubMed] [Google Scholar]
- 23.Valdez H, Carlson NL, Post AB, Asaad R, Heeger PS, Lederman MM, Lehmann PV, Anthony DD. HIV long-term non-progressors maintain brisk CD8 T cell responses to other viral antigens. AIDS. 2002;16:1113–1118. doi: 10.1097/00002030-200205240-00004. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen TN, Lauer GM, Chung RT, Walker BD. Cellular immune response in HIV/HCV co-infection. Hepatology. 2000;32:270A. [Google Scholar]
- 25.Gruener NH, Lechner F, Jung MC, Diepolder H, Gerlach T, Lauer G, Walker B, Sullivan J, Phillips R, Pape GR, Klenerman P. Sustained dysfunction of antiviral CD8+ T lymphocytes after infection with hepatitis C virus. J Virol. 2001;75:5550–5558. doi: 10.1128/JVI.75.12.5550-5558.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wedemeyer H, He XS, Nascimbeni M, Davis AR, Greenberg HB, Hoofnagle JH, Liang TJ, Alter H, Rehermann B. Impaired effector function of hepatitis C virus-specific CD8+ T cells in chronic hepatitis C virus infection. J Immunol. 2002;169:3447–3458. doi: 10.4049/jimmunol.169.6.3447. [DOI] [PubMed] [Google Scholar]
- 27.Zignego AL, Ferri C, Giannelli F, Giannini C, Caini P, Monti M, Marrocchi ME, Di Pietro E, La Villa G, Laffi G, Gentilini P. Prevalence of bcl-2 rearrangement in patients with hepatitis C virus-related mixed cryoglobulinemia with or without B-cell lymphomas. Ann Intern Med. 2002;137:571–580. doi: 10.7326/0003-4819-137-7-200210010-00008. [DOI] [PubMed] [Google Scholar]
- 28.Kitay-Cohen Y, Amiel A, Hilzenrat N, Buskila D, Ashur Y, Fejgin M, Gaber E, Safadi R, Tur-Kaspa R, Lishner M. Bcl-2 rearrangement in patients with chronic hepatitis C associated with essential mixed cryoglobulinemia type II. Blood. 2000;96:2910–2912. [PubMed] [Google Scholar]
- 29.Fabris M, Quartuccio L, Sacco S, De Marchi G, Pozzato G, Mazzaro C, Ferraccioli G, Migone TS, De Vita S. B-Lymphocyte stimulator (BLyS) up-regulation in mixed cryoglobulinaemia syndrome and hepatitis-C virus infection. Rheumatology (Oxford) 2007;46:37–43. doi: 10.1093/rheumatology/kel174. [DOI] [PubMed] [Google Scholar]
- 30.Sène D, Limal N, Ghillani-Dalbin P, Saadoun D, Piette JC, Cacoub P. Hepatitis C virus-associated B-cell proliferation—the role of serum B lymphocyte stimulator (BLyS/BAFF) Rheumatology (Oxford) 2007;46:65–69. doi: 10.1093/rheumatology/kel177. [DOI] [PubMed] [Google Scholar]
- 31.Batten M, Groom J, Cachero TG, Qian F, Schneider P, Tschopp J, Browning JL, Mackay F. BAFF mediates survival of peripheral immature B lymphocytes. J Exp Med. 2000;192:1453–1466. doi: 10.1084/jem.192.10.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carsetti R, Rosado MM, Wardmann H. Peripheral development of B cells in mouse and man. Immunol Rev. 2004;197:179–191. doi: 10.1111/j.0105-2896.2004.0109.x. [DOI] [PubMed] [Google Scholar]
- 33.Weller S, Faili A, Garcia C, Braun MC, Le Deist F, de Saint FG, Basile G, Hermine O, Fischer A, Reynaud CA, Weill JC. CD40-CD40L independent Ig gene hypermutation suggests a second B cell diversification pathway in humans. Proc Natl Acad Sci USA. 2001;98:1166–1170. doi: 10.1073/pnas.98.3.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Capolunghi F, Cascioli S, Giorda E, Rosado MM, Plebani A, Auriti C, Seganti G, Zuntini R, Ferrari S, Cagliuso M, et al. CpG drives human transitional B cells to terminal differentiation and production of natural antibodies. J Immunol. 2008;180:800–808. doi: 10.4049/jimmunol.180.2.800. [DOI] [PubMed] [Google Scholar]
- 35.Ho J, Moir S, Malaspina A, Howell ML, Wang W, DiPoto AC, O’Shea MA, Roby GA, Kwan R, Mican JM, et al. Two over-represented B cell populations in HIV-infected individuals undergo apoptosis by different mechanisms. Proc Natl Acad Sci USA. 2006;103:19436–19441. doi: 10.1073/pnas.0609515103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Charles ED, Green RM, Marukian S, Talal AH, Lake-Bakaar GV, Jacobson IM, Rice CM, Dustin LB. Clonal expansion of immunoglobulin M+CD27+ B cells in HCV-associated mixed cryoglobulinemia. Blood. 2008;111:1344–1356. doi: 10.1182/blood-2007-07-101717. [DOI] [PMC free article] [PubMed] [Google Scholar]