

Abstract
cAMP is a universal second messenger. Its signalling is mediated by protein kinase A, Epac and certain types of ion channels in mammalians. cAMP signalling is involved in many physiological processes ranging from vision to the control of insulin secretion, pacemaker activity and gene transcription and therefore selective pharmacological interference is of medical interest. Whereas selective inhibitors of PKA and selective activators of Epac are well established, no inhibitors of Epac were available until recently. Here the action of four of the novel Epac inhibitors was analysed by biophysical means. ESI-05 is confirmed as a selective inhibitor of Epac2. No direct action of Brefeldin A on Epac could be demonstrated. ESI-09 and HJC0197 were found to act as chemicals with general protein denaturing properties and do not act on Epac selectively.
Three classes of proteins sense cAMP concentrations by cyclic nucleotide binding (CNB) domains in mammalians1,2. These are protein kinase A (PKA), ion channels with CNB domains and Epac proteins. The expression of ion channels with CNB domains is mainly restricted to the heart and the visual and olfactory system. They function in synchronising the pacemaker activity and in generating the electrophysiological response to light and odour. PKA and Epac are expressed in a wide range of tissues and function in many processes of signal transduction, including regulation of gene transcription and stimulation of insulin secretion. In these tissues the physiological response to elevated cAMP levels is often mediated by a concerted action of PKA and Epac.
Binding of cAMP to the regulatory subunits of PKA results in the release of the catalytic kinase subunits, which then phosphorylate downstream targets1. These targets include various cellular proteins, among which transcriptions factors of the CREB family, which mediate effects of PKA on gene regulation. Epac proteins are Guanine Nucleotide Exchange Factors (GEFs) for the small G-proteins Rap1 and Rap2 (ref. 3,4). G-proteins cycle between an inactive GDP-bound state and an active GTP-bound state. GEFs catalyse the exchange of GDP for GTP. In the GTP-bound conformation G-proteins interact with effector proteins that transmit the signal5. In mammalians two Epac genes, Epac1 and Epac2, exist. Both proteins have a similar domain organisation, but Epac2 contains an additional N-terminal CNB domain. In the inactive conformation the N-terminal CNB domain is positioned face-to-face to the second CNB domain6,7. However, the N-terminal CNB domain is neither required to maintain the inactive conformation nor is cAMP binding to the N-terminal CNB domain required to induce activation of Epac2 (ref. 7). Epac1-mediated signalling is involved in the control of cell adhesion processes and Epac2 is known to stimulate insulin secretion by pancreatic islets and to contribute to the control of blood pressure8.
The ability to interfere selectively with PKA- and Epac-mediated signalling is of both biological and pharmacological interest. Biologically this ability facilitates unravelling the involved signalling routes. Pharmacologically, this ability allows a more direct and restricted interference with physiological effects. Selective inhibition of PKA can be achieved by kinase inhibitors such as H-89 (ref. 9,10) and selective activation of Epac by the cyclic nucleotide analogue 8-pCPT-2′-O-Me-cAMP (ref. 11). Recent research has identified several inhibitors of Epac. Brefeldin A, a known inhibitor of the large ArfGEFs12,13, was shown to antagonise Epac2 mediated signalling in vivo14. Furthermore, Tsalkova et al. have identified several Epac inhibitors by screening a library of 14,400 drug-like molecules for the ability to compete with cAMP for binding to Epac2 (ref. 15). Finally Courilleau et al. have indentified CE3F4 (5,7-bis(bromanyl)-6-fluoranyl-2-methyl-1,2,3,4-tetrahydroquinoline-1-carbaldehyde) in a screen of 640 small molecules for their ability to inhibit Epac1 catalysed exchange activity towards Rap1 (ref. 16). Here, I have revaluated the commercially available Epac inhibitors ESI-05 (ref. 15), ESI-09 (ref. 15), HJC0197 (ref. 17) and Brefeldin A (ref. 14) by a detailed biophysical study of their effects on Epac1 and Epac2.
Results
ESI-05 is an Epac2 selective inhibitor
Exchange activity of Epac towards Rap can be determined in vitro by the use of a fluorescence-based assay18. Rap1 is pre-loaded with the fluorescent GDP analogue mGDP. The fluorescence intensity of Rap bound mGDP is approximately twice as intense as of mGDP in solution. Therefore, in the presence of an excess unlabelled GDP the nucleotide exchange reaction causes a decay in fluorescence19. The speed of the decay reflects exchange activity. To analyse the selectivity of ESI-05 the effect of ESI-05 on Epac1, Epac2 and RapGEF6 catalysed nucleotide exchange was monitored (Fig. 1). Although RapGEF6 is a GEF for Rap related to Epac, it contains pseudo CNB (ψCNB) domains instead of CNB domains. These domains are not able to bind cAMP (ref. 20,21) and were removed from the RapGEF6 construct used here (Fig. 1a). Addition of ESI-05 to Epac2fl but not to Epac2Δ280 reduces the exchange activity in the presence of 100 μM cAMP (Fig. 1b,c). Epac2Δ280 is lacking the N-terminal CNB domain and the DEP domain (Fig. 1a). No inhibitory effect was observed when ESI-05 was added to Epac1 activated with 100 μM cAMP or to RapGEF6 (Fig. 1d,e). This confirms the classification of ESI-05 as an Epac2-selective inhibitor by Tsalkova et al. (ref. 15).
Figure 1.

(a) Domain organisation of Epac1, Epac2 and RapGEF6. The borders of the used construct as well as the name by with the are referred to in the text are indicated. DEP, Dishevelled, Egl-10, Pleckstrin domain; CNB, cyclic nucleotide binding domain; REM, Ras Exchange Motif; RA, Ras Association domain; CDC25-HD, CDC25-homology domain; ψCNB, pseudo cyclic nucleotide binding domain; PDZ, PSD-95, Dlg, ZO domain. (b), (c), (d), (e) Nucleotide exchange activity of Epac2fl, Epac2Δ280, Epac1 and RapGEF6 in the presence of various concentrations of ESI-05 as indicated. The activities of Epac2fl Epac2Δ280 and Epac1 were recorded in the presence of 100 μM cAMP except for the traces labelled “no cAMP”. The data are fitted as single exponential decay with off-set (red lines) to obtain the rate constants kobs. Each panel shows one experiments out of three replicates. (f) kobs of Epac2fl catalysed exchange activity in the presence of 100 μM cAMP (open circles) or 10 μM cAMP (filled circles) were plotted against the concentration of ESI-05. The IC50 points (0.2 μM and 3 μM) are indicated. The red dotted line corresponds to the exchange activity in the absence of cAMP. The titration was repeated once.
The exchange activity can be quantified by fitting the time dependency of the fluorescence signal as single exponential decay with offset18. The obtained rate constant kobs is then a measure for GEF activity. kobs values for Epac2fl induced activity were recorded for different concentrations of ESI-05 at 10 μM and 100 μM cAMP (Fig. 1f). Epac2fl is activated by cAMP with an AC50 of 20 μM (ref. 7). Epac2fl is thus not fully activated at a concentration of 10 μM cAMP (Fig. 1f). ESI-05 was used at a maximal concentration of 10 μM due to its limited solubility in aqueous solutions. Dilution of ESI-05 into reaction buffer to a final concentration of 100 μM resulted in a milky suspension. At 10 μM concentration ESI-05 is not able to reach full inhibition of Epac2fl (Fig. 1f). Full inhibition would correspond to the activity of Epac2fl in the absence of cAMP (red dotted line in Fig. 1f). However, IC50 values of ESI-05 in the presence of 10 μM and 100 μM cAMP can be estimated to be 0.2 μM and 3 μM, respectively. The IC50 value was estimated as the inflexion point of the curve by curve fitting and found to be in agreement with graphical estimation. The IC50 values differ roughly by a factor 10, which is expected for an inhibitor that competes with cAMP. As the titrations were performed in the presence of 10 μM and 100 μM cAMP and as cAMP activates Epac2 fl with an AC50 of 20 μM (ref. 7), ESI-05 can be estimated to bind Epac2fl with an apparent affinity of approximately 1 μM.
ESI-09 and HJC0197 have protein denaturing properties
In accordance with its orange colour, ESI-09 shows an absorption maximum at about 400 nm. HJC0197 is colourless with an absorption maximum at about 300 nm (Supplementary Fig. 1a). The spectroscopic properties of ESI-09 are incompatible with the use of mGDP. mGDP requires excitation at about 370 nm and recording of fluorescence at about 450 nm19. ESI-09 is expected to act as an inner filter that absorbs the excitation light beam and the mGDP-emitted fluorescence. Indeed, the emission intensity of mGDP is reduced to less than 10% in the presence of 100 μM ESI-09 (Supplementary Fig. 1b). This problem can be overcome by the use of GDP-bodipy, whose fluorescence is recorded at 512 nm after excitation at 485 nm.
ESI-09 and HJC0197 interfere with Epac2fl, Epac1 and RapGEF6 induced exchange activity (Fig. 2). The effects are concentration-dependent but exhibit abnormalities in particular in the case of Epac1 and RapGEF6. Exchange activity of Epac1 induced by 100 μM cAMP is only little affected by ESI-09 if applied at 50 μM but abolished if applied at 100 μM (Fig. 2b). A similar behaviour is observed with HJC0197 (Fig. 2e). In the presence of ESI-09 or HJC0197 the fluorescence decay caused by RapGEF6 mediated nucleotide exchange does not fit a single exponential decay. Instead, an initially fast decay turns into slow decay (Fig. 2c,f and Supplementary Fig. 2). Such behaviour indicates the loss of exchange activity over time. Furthermore, the effects on Epac do not depend on the cAMP concentration as shown for Epac2fl and ESI-09 (Supplementary Fig. 3). ESI-09 even inhibits the intrinsic, cAMP independent exchange activity of Epac2fl (Supplementary Fig. 3). ESI-09 and HJC0197 thus do not act as a competitive inhibitor.
Figure 2. Exchange activity of Epac2fl (a, d) Epac1 (b, e) and RapGEF6 (c,f) upon addition of the indicated concentrations of ESI-09 (a, b, c) and HJC0197 (d, e, f).

The activities of Epac2fl and Epac1 were recorded in the presence of 100 μM cAMP except for the traces labelled “no cAMP”. Each panel shows one experiment out of three replicates.
The effect of ESI-09 and HJC0197 on protein stability was analysed in Thermal Shift Assays (TSA). The assay is based on the property of the fluorophore Sypro Orange to bind hydrophobic moieties of proteins22. The emission intensity of Sypro Orange is enhanced in the hydrophobic environment of the protein, which allows monitoring of protein denaturation. Melting curves of Epac1, Epac2fl, RapGEF6, Rlf, GST and lysozyme were recorded at different concentrations of ESI-09, HJC0197 or ESI-05 (Fig. 3). Rlf is a GEF that belongs, together with Epac and RapGEF6, to the CDC25-homology domain containing family of GEFs, but acts on the small G-protein Ral and not Rap. GST and lysozyme were chosen as completely unrelated proteins. Two main effects are observed. At concentrations of 25 μM and higher, ESI-09 and HJC0197 often induce high fluorescence intensities at low temperatures, which indicate the presence of unfolded protein (Fig. 3). Second, the melting curves are shifted to lower temperatures with increasing concentrations of ESI-09 or HJC0197, indicative for reduced protein stability (Fig. 3). Both effects are often overlaid (Fig. 3). ESI-09 and HJC0197 thus show protein destabilising and denaturing properties, which likely explain their apparent inhibitory effects.
Figure 3. Melting curves of Epac1, Epac2fl, RapGEF6, Rlf, GST and lysozyme in the presence of ESI-09 (a) HJC0197 (b) or ESI-05 (c).

Melting curves of Epac were determined in the presence of 100 μM cAMP. For ESI-09 and HJC0197 melting curves were determined at 1 μM, 2.5 μM, 10 μM, 25 μM, 50 μM and 100 μM and for ESI-05 at 0.1 μM, 0.15 μM, 1 μM, 2.5 μM, 10 μM and 25 μM. If two or more curves were highly overlapping the curves corresponding to the lower concentrations were omitted for clarity of presentation. The concentrations are indicated next to the curves in μM. Each melting curve was determined at least in duplicate.
Brefeldin A does not act on Epac directly
In vivo Brefeldin A was used at concentrations up to 100 μM to inhibit Epac mediated effects14,23,24. Brefeldin A affects Golgi apparatus associated processes at concentrations of about 5 μM (ref. 25,26), for example 4 μM are sufficient to inhibit protein secretion in hepatocytes and 40 μM inhibit protein synthesis by 30% (ref. 25). This effects are attributed to the inhibition of ArfGEFs by Brefeldin A12,13. At concentration up to 100 μM Brefeldin A does not inhibit exchange activity of Epac1 or Epac2fl in vitro (Fig. 4), suggesting that Brefeldin A does not act on Epac directly.
Figure 4. Exchange activity of Epac2fl (a) and Epac1 (b) upon addition of the indicated concentrations of Brefeldin A in the presence of 100 μM cAMP except for the traces labelled “no cAMP”.
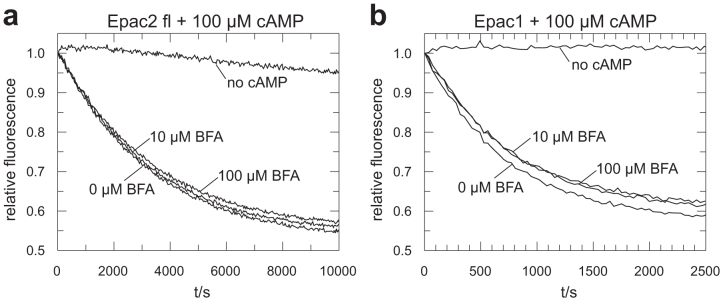
Each panel shows one experiment out of two replicates.
Discussion
This study confirms the inhibitory properties of ESI-05. Tsalkova et al. have suggested that ESI-05 binds in the interface between the two CNB domains of Epac2 and thus stabilised the inactive conformation by “gluing” both CNB domains together15. This model is supported by the inability of ESI-05 to inhibit cAMP induced exchange activity of Epac2Δ280, which is lacking the N-terminal CNB domain and the competitive nature of the inhibition (Fig. 1a,c). Furthermore, this mechanism of action explains the inability of ESI-05 to inhibit Epac1 that contains only one CNB domain. However, the competition does not have to be direct. Binding of ESI-05 to region outside the cAMP binding site could stabilise the inactive conformation that is incompatible with cAMP binding. In this way ESI-05 and cAMP would compete by non overlapping binding sites “indirectly” for Epac2 binding.
ESI-05 binds Epac2fl with an approximated apparent affinity of 1 μM, which is 20 times higher than the affinity of cAMP. This classifies ESI-05 as a rather potent inhibitor. The use of ESI-05 in biological systems may be limited by its rather low solubility in aqueous solution. This study suggests that ESI-05 can be applied at concentrations of up to 10 μM. Care should be taken if ESI-05 is diluted from stock solution in DMSO into aqueous solution as ESI-05 readily sticks to plastic surfaces if (local) concentrations reach the maximal solubility. On the other hand, the hydrophobic properties of ESI-05 should facilitate uptake across cell membranes and thereby improve bioavailability.
This study questions the ability of ESI-09 and HJC0197 to selectively inhibit Epac. Both compounds do not only interfere with Epac1 and Epac2 but also with RapGEF6-mediated exchange activity (Fig. 2). Tsalkova et al. have identified ESI-09 and a compound related to HJC0197 in a screen for compounds which compete with cAMP for binding to Epac2 (ref. 15). Such competition is not possible in the case of RapGEF6 since the used construct does not contain CNB domains and since the activity was measured in the absence of cAMP. It may therefore be that ESI-09 and HJC0197 impinge on the catalytic CDC25-homology domain directly. However, this study suggests that the “inhibitory” properties of ESI-09 and HJC0197 rather originate from a general protein destabilising effect. The data presented in Figure 2 for Epac1 and RapGEF6 do not support an inhibitory mechanism that is based on a reversible interaction between ESI-09 or HJC0197 and the protein. This is reflected in the concentration dependency and the impossibility to describe the exchange kinetics as single exponential. These abnormalities are less pronounced for Epac2fl.
In line with the previous, the TSA analysis demonstrates clear protein destabilising properties of ESI-09 and HJC0197. The tested proteins differ in their sensitivity towards the presence of these compounds. Epac1, Epac2, RapGEF6 and lysozyme seem to be in particular sensitive, as the addition of ESI-09 or HJC0197 at concentrations of 25 μM or higher causes high fluorescence intensities at low temperature. This suggests immediate denaturation upon addition of the compounds. Rlf and GST seem to be more robust. The effect of the compounds at low temperature is small. This allows recording melting curves with a clear transition temperature. Increasing concentrations of the compound shift the melting point to lower temperature.
This study strongly suggests that ESI-09 and HJC0197 do not function as selective inhibitors of Epac. Instead, both compounds seem to have unspecific protein denaturing effect. It may be that a (weak) effect of specific inhibition is overlaid by the general protein denaturing effects, but both effects would occur in the same concentration range. In any case ESI-09 and HJC0197 affect Epac independent of the cAMP concentration and even inhibit the cAMP independent intrinsic activity (Supplementary Figure 3). The protein denaturing effect can explain why ESI-09 and a related compound of HJC0197 were picked up in the original screen performed by Tsalkova et al (ref. 15). Protein denaturing compounds would have been picked up as false positives, as they would have seemed to have competed cAMP out of the binding pocket. To validate initial hits Tsalkova et al. have used a similar activity assay as described here15. As shown in Fig. 2 the recordings of such an assay can give the impression of inhibition. This impression might have been enhanced by data processing. Tsalkova et al. report fluorescence traces normalised to time point zero15. As Tsalkova et al. have used ESI-09 in combination with mGDP such a normalisation would likely have resulted in apparently almost constant fluorescence trace due to the inner filter effect of ESI-09.
The denaturing properties of ESI-09 and HJC0197 were demonstrated in the TSA with a set of six proteins. TSA can therefore be used as a simple and low-cost way to eliminate false positive hits obtained from high throughput screens. By screening a whole library e.g. with lysozyme at only one temperature the library could even be cleared from compounds with obvious protein destabilising effects prior to the general use of the library in a multitude of drug screens.
In conclusion, four recently indentified and commercially available inhibitors of Epac were characterised biophysically. A direct action of Brefeldin A on Epac could not be demonstrated. The reported effects of Brefeldin A on Epac signalling in vivo are thus likely indirect. It remains to be determined whether these effects are originated from a link between ArfGEF mediated signalling and Epac or by a yet unknown target of Brefeldin A. ESI-09 and HJC0197 have general protein denaturing properties, which likely explain the apparent inhibitory effects on Epac. The use of ESI-09 and HJC0197 as Epac inhibitors is therefore not recommended. ESI-05 was confirmed as a direct and selective inhibitor of Epac2. ESI-05 binds Epac2 with about 20-fold higher affinity than cAMP. A recent study by Chen et al. describes several Epac2 inhibitors derived from ESI-05 (ref. 27). Improvements of ESI-05, as for example an improved solubility, are therefore likely possible.
Methods
Reagents
ESI-05 (4-Methylphenyl-2,4,6-trimethylphenylsulfone), ESI-09 (3-[5-(tert.-Butyl)isoxazol-3-yl]-2-[2-(3-chlorophenyl)hydrazono]-3-oxopropanenitrile) and HJC0197 (4-Cyclopentyl-2-(2, 5-dimethylbenzylsulfanyl)-6-oxo-1,6-dihydropyrimidine-5-carbonitrile) were from Biolog Life Science Institute (Bremen, Germany), GDP-bodipy (G-22360) from Invitrogen, life technologies and Sypro Orange Brefeldin A, cAMP, GDP and human lysozyme (L1667) from Sigma-Aldrich (St. Louis, USA). ESI-05, ESI-09 and HJC0197 stocks solutions of 50 mM were prepared in DMSO, diluted in reaction buffer to 2 × final concentration in glass vials and mixes 1:1 with the reaction mixture. It was ensured that a final concentration of 0.2% DMSO was not exceeded.
Constructs and protein expression
The following constructs were used for the expression and purification of recombinant proteins: Rap1B (aa 1–167, Homo sapiens) in ptac (ref. 28); Epac1 (aa 149–881, Homo sapiens), Epac2fl (aa 1–993, Mus musculus), Epac2Δ280 (aa 280–993, Mus musculus) and GST in pGEX4T and RapGEF6 (aa 410–1137, Homo sapiens) in a modified version of pET20b. Proteins from the plasmids ptac and pGEX4T were expressed in the bacterial strain CK600K and from the plasmid pET20b in BL21DE3,Rosetta. Bacteria were grown at 25°C and protein production was induced with 100 μM IPTG at OD600 of 0.8. After culturing over night bacteria were harvested by centrifugation, washed once with 0.9% NaCl and stored at −20°C if desired or re-suspended in the respective buffer and lysed by sonification for 3 minutes. Lysates were cleared by centrifugation at 50.000 × g.
For the purification of Rap bacteria were lysed in 32 mM TrisHCl pH 7.6, 5 mM MgCl2 and 5 mM β-mercapthoethanol (buffer 1) and cleared lysates were loaded to an 800 ml Q-Sepharose column equilibrated in buffer1. The column was washed with buffer1 and eluted in a 4 litre gradient form 0 mM to 300 mM NaCl in buffer 1. Fractions were subjected to SDS-PAGE and Rap containing fractions were pooled. Protein was precipitated from these fractions with ammonium sulphate, re-suspended in buffer 1 containing 50 mM NaCl and loaded to a Superdex 75 26/60 gelfiltration column. Rap containing fractions were identified by SDS-PAGE analysis and concentrated.
For the purification of Epac-proteins bacteria were lysed in 50 mM TrisHCl pH 7.5, 50 mM NaCl, 5 mM EDTA, 5% glycerol and 5 mM β-mercapthoethanol (buffer A) and cleared lysates were loaded to a 30 ml glutathione column. The column was washed with 6 column volumes of buffer A, 10 volumes of 50 mM TrisHCl pH 7.5, 400 mM NaCl, 5% glycerol and 5 mM β-mercapthoethanol (buffer B), 12 volumes of 50 mM TrisHCl pH 7.5, 100 mM KCl, 10 mM MgCl2, 5% glycerol, 5 mM β-mercapthoethanol and 250 μM ATP (buffer C) at low flow rates and with 2 volumes of 50 mM TrisHCl pH 7.5, 50 mM NaCl, 10 mM CaCl2, 5% glycerol and 5 mM β-mercapthoethanol (buffer D), loaded with 25 ml of buffer D containing 80 units thrombin (Serva), incubated over night and eluted with buffer D. Protein containing fractions were pooled concentrated and loaded to a Superdex 200 16/60 gelfiltration column equilibrated with 50 mM TrisHCl pH 7.5, 50 mM NaCl, 2% glycerol and 5 mM β-mercapthoethanol (buffer E). Fractions were analysed by SDS-PAGE and Epac containing fractions were concentrated.
For the purification of GST bacteria were lysed in buffer A and cleared lysates were loaded to a 50 ml glutathione column. The column was washed with 6 volumes of buffer A, 10 volumes of buffer B, 2 volumes of buffer E and eluted in buffer E containing 20 mM glutathione. Protein containing fractions were concentrated and loaded to a Superdex 75 26/60 column equilibrated with buffer E.
For the purification of RapGEF6 bacteria were lysed in 50 mM HEPES pH 8.0, 500 mM NaCl, 5 mM Imidazole and 1 mM β-mercapthoethanol (buffer α). Cleared lysates were loaded to a 30 mM Ni-NTA column equilibrated in buffer α. The column was washed with 6 volumes buffer α and eluded in a gradient from 5 mM to 252,5 mM Imidazole in buffer α. RapGEF6 containing fractions were pooled, dialysed against 50 mM TrisHCl pH 7.5, 500 mM NaCl and 5 mM β-mercapthoethanol (buffer β), concentrated and loaded to a Superdex 200 16/60 column equilibrated with buffer β.
Purity of the proteins was commonly 95% or higher as judged from SDS-PAGE analysis (supplementary figure 4).
Determination of GEF activity
Rap1B was loaded with the fluorescent GDP analogue 2'-/3'-O-(N-methylanthraniloyl)-guanosine diphosphate (mGDP) or GDP-bodipy as described previously for mGDP18. The exchange reactions were performed in 50 mM TrisHCl pH 7.5, 50 mM NaCl, 5 mM MgCl2, 5 mM DTT, 5% glycerol at 20°C, with 200 nM G-protein and 150 nM Epac1, 200 nM Epac2 or 150 nM RapGEF6 and started by the addition of 20 μM GDP. The final reaction volume was 700 μl, and the reaction was performed in Quartz Cuvettes (Hellma, Müllheim, Germany). Four reactions were monitored in a Carry Eclipse fluorescence spectrometer in parallel. The time-dependent fluorescence signal obtained was fitted as a single exponential decay with off-set in the program GraFit3.0 to obtain the rate constant (kobs) (ref. 18).
To analyse the inhibitory propertied of ESI-05 towards Epac2, kobs were plotted against the concentration of ESI-05. The inflexion point of the curve (IC50) was estimated by fitting the concentration dependency of kobs to (α-β)*(([Epac2] + [ESI-05] + IC50)/2 − (([Epac2] + [ESI-05] + IC50)2/4 − [Epac2]*[ESI-05])0.5) + β*[Epac2], where [ESI-05] is the concentration of ESI-05, [Epac2] the concentration of Epac2, which was 0.2 μM and α and β are constants of proportionality. α was set to 6.6*10−5 s−1M−1 assuming that the exchange activity of maximal inhibition is identical to the exchange activity of Epac2 in the absence of cAMP.
Thermal shift assay
TSA was performed in 50 mM TrisHCl, pH 7.5, 50 mM NaCl, 2.5% glycerol and 5 mM DTT with a final protein concentration of 0.2 g/l and a total reaction volume of 25 μl in 96 well real time PCR plates. 100 μM cAMP was added to Epac1 and Epac2fl. Sypro Orange was used at 10 × concentration assuming the stock solution supplied by the manufacturer being 5,000 ×. ESI-09 or HJC0197 were added at 1 μM, 2.5 μM, 10 μM, 25 μM, 50 μM and 100 μM and ESI-05 at 0.1 μM, 0.25 μM, 1 μM, 2.5 μM, 10 μM and 25 μM ensuring that the final concentration of DMSO was not exceeding 0.2%. In the absence of ESI-09 and HJC0197 no difference was observed in the absence or presence of 0.2% DMSO. Fluorescence traces were recorded in BioRad CFX96 Real-Time PCR Detection system with λex = 560 nm − 590 nm and λem = 610 − 650 nm over a temperature range from 11°C to 80°C ramped in increments of 0.5°C with plateau times of 30 s.
Author Contributions
H.R. had written the manuscript and performed the experiments described therein.
Supplementary Material
supplementary information
Acknowledgments
I thank Marije Rensen-de Leeuw for excellent technical assistance and M. Popovic, B. Ponsioen and J.L. Bos for critical reading the manuscript. This work was supported by the TI Pharma Project T3-106 to J.L. Bos.
References
- Beavo J. A. & Brunton L. L. Cyclic nucleotide research -- still expanding after half a century. Nat. Rev. Mol. Cell Biol. 3, 710–718 (2002). [DOI] [PubMed] [Google Scholar]
- Rehmann H., Wittinghofer A. & Bos J. L. Capturing cyclic nucleotides in action: snapshots from crystallographic studies. Nat. Rev. Mol. Cell Biol. 8, 63–73 (2007). [DOI] [PubMed] [Google Scholar]
- de Rooij J. et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396, 474–477 (1998). [DOI] [PubMed] [Google Scholar]
- Kawasaki H. et al. A family of cAMP-binding proteins that directly activate Rap1. Science 282, 2275–2279 (1998). [DOI] [PubMed] [Google Scholar]
- Vetter I. R. & Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science 294, 1299–1304 (2001). [DOI] [PubMed] [Google Scholar]
- Rehmann H. et al. Structure and regulation of the cAMP-binding domains of Epac2. Nat. Struct. Biol. 10, 26–32 (2003). [DOI] [PubMed] [Google Scholar]
- Rehmann H., Das J., Knipscheer P., Wittinghofer A. & Bos J. L. Structure of the cyclic-AMP-responsive exchange factor Epac2 in its auto-inhibited state. Nature 439, 625–628 (2006). [DOI] [PubMed] [Google Scholar]
- Gloerich M. & Bos J. L. Epac: defining a new mechanism for cAMP action. Annu. Rev. Pharmacol. Toxicol. 50, 355–375 (2010). [DOI] [PubMed] [Google Scholar]
- Chijiwa T. et al. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5- isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem 265, 5267–5272 (1990). [PubMed] [Google Scholar]
- Hidaka H., Watanabe M. & Kobayashi R. Properties and use of H-series compounds as protein kinase inhibitors. Methods Enzymol 201, 328–339 (1991). [DOI] [PubMed] [Google Scholar]
- Enserink J. M. et al. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol 4, 901–906 (2002). [DOI] [PubMed] [Google Scholar]
- Helms J. B. & Rothman J. E. Inhibition by brefeldin A of a Golgi membrane enzyme that catalyses exchange of guanine nucleotide bound to ARF. Nature 360, 352–354 (1992). [DOI] [PubMed] [Google Scholar]
- Donaldson J. G., Finazzi D. & Klausner R. D. Brefeldin A inhibits Golgi membrane-catalysed exchange of guanine nucleotide onto ARF protein. Nature 360, 350–352 (1992). [DOI] [PubMed] [Google Scholar]
- Zhong N. & Zucker R. S. cAMP acts on exchange protein activated by cAMP/cAMP-regulated guanine nucleotide exchange protein to regulate transmitter release at the crayfish neuromuscular junction. J. Neurosci. 25, 208–214 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsalkova T. et al. Isoform-specific antagonists of exchange proteins directly activated by cAMP. Proc. Natl. Acad. Sci. U. S. A 109, 18613–18618 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courilleau D. et al. Identification of a tetrahydroquinoline analog as a pharmacological inhibitor of the cAMP-binding protein Epac. J. Biol. Chem. 287, 44192–44202 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. et al. 5-Cyano-6-oxo-1,6-dihydro-pyrimidines as potent antagonists targeting exchange proteins directly activated by cAMP. Bioorg. Med. Chem. Lett. 22, 4038–4043 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehmann H. Characterization of the activation of the Rap-specific exchange factor Epac by cyclic nucleotides. Methods Enzymol 407, 159–173 (2006). [DOI] [PubMed] [Google Scholar]
- Lenzen C., Cool R. H. & Wittinghofer A. Analysis of intrinsic and CDC25-stimulated guanine nucleotide exchange of p21ras-nucleotide complexes by fluorescence measurements. Methods Enzymol 255, 95–109 (1995). [DOI] [PubMed] [Google Scholar]
- Liao Y. et al. RA-GEF, a Novel Rap1A Guanine Nucleotide Exchange Factor Containing a Ras/Rap1A-associating Domain, Is Conserved between Nematode and Humans. J. Biol. Chem. 274, 37815–37820 (1999). [DOI] [PubMed] [Google Scholar]
- Kuiperij H. B. et al. Characterisation of PDZ-GEFs, a family of guanine nucleotide exchange factors specific for Rap1 and Rap2. Biochim. Biophys. Acta 1593, 141–149 (2003). [DOI] [PubMed] [Google Scholar]
- Pantoliano M. W. et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screen. 6, 429–440 (2001). [DOI] [PubMed] [Google Scholar]
- Park J. H. et al. Glucagon-like peptide-1 enhances glucokinase activity in pancreatic beta-cells through the association of Epac2 with Rim2 and Rab3A. Endocrinology 153, 574–582 (2012). [DOI] [PubMed] [Google Scholar]
- Mironov S. L., Skorova E. Y. & Kugler S. Epac-mediated cAMP-signalling in the mouse model of Rett Syndrome. Neuropharmacology 60, 869–877 (2011). [DOI] [PubMed] [Google Scholar]
- Misumi Y. et al. Novel blockade by brefeldin A of intracellular transport of secretory proteins in cultured rat hepatocytes. J. Biol. Chem. 261, 11398–11403 (1986). [PubMed] [Google Scholar]
- Fujiwara T., Oda K., Yokota S., Takatsuki A. & Ikehara Y. Brefeldin A causes disassembly of the Golgi complex and accumulation of secretory proteins in the endoplasmic reticulum. J. Biol. Chem. 263, 18545–18552 (1988). [PubMed] [Google Scholar]
- Chen H. et al. Identification and characterization of small molecules as potent and specific EPAC2 antagonists. J. Med. Chem. 56, 952–962 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann C., Horn G., Spaargaren M. & Wittinghofer A. Differential interaction of the ras family GTP-binding proteins H-Ras, Rap1A, and R-Ras with the putative effector molecules Raf kinase and Ral-guanine nucleotide exchange factor. J. Biol. Chem. 271, 6794–6800 (1996). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary information