

Abstract
Several studies on 5S ribosomal DNA (5S rDNA) have been focused on a subset of the following features in mostly one organism: number of copies, pseudogenes, secondary structure, promoter and terminator characteristics, genomic arrangements, types of non-transcribed spacers and evolution. In this work, we systematically analyzed 5S rDNA sequence diversity in available metazoan genomes, and showed organism-specific and evolutionary-conserved features. Putatively functional sequences (12 766) from 97 organisms allowed us to identify general features of this multigene family in animals. Interestingly, we show that each mammal species has a highly conserved (housekeeping) 5S rRNA type and many variable ones. The genomic organization of 5S rDNA is still under debate. Here, we report the occurrence of several paralog 5S rRNA sequences in 58 of the examined species, and a flexible genome organization of 5S rDNA in animals. We found heterogeneous 5S rDNA clusters in several species, supporting the hypothesis of an exchange of 5S rDNA from one locus to another. A rather high degree of variation of upstream, internal and downstream putative regulatory regions appears to characterize metazoan 5S rDNA. We systematically studied the internal promoters and described three different types of termination signals, as well as variable distances between the coding region and the typical termination signal. Finally, we present a statistical method for detection of linkage among noncoding RNA (ncRNA) gene families. This method showed no evolutionary-conserved linkage among 5S rDNAs and any other ncRNA genes within Metazoa, even though we found 5S rDNA to be linked to various ncRNAs in several clades.
Keywords: 5S rRNA, homologous genes, noncoding RNA, secondary structure, paralogs, birth-and-death evolution
Introduction
The evolution of 5S ribosomal DNA (5S rDNA) has been studied in some groups of organisms, mainly within genera or within families (for example, Martins and Wasko (2004); Rooney and Ward (2005); Vierna et al. (2009; Freire et al. (2010); Perina et al. (2011); Vizoso et al. (2011)). Nevertheless, several intriguing features, such as high conservation along evolution in contrast to high intragenomic divergence, a plastic genomic organization and linkage to other genes, make this multigene family an interesting issue in evolutionary genetics that deserves a large-scale analysis.
5S rDNA (as well as other ribosomal genes) is expected to display low intragenomic divergence levels owing to the occurrence of homogenizing mechanisms (unequal crossing-overs and gene conversions) that are favored by the tandem arrangement of these genes and lead to so-called concerted evolution (reviewed in Eickbush and Eickbush (2007)). However, many reports have been recently published in which the concerted evolution model did not explain the intragenomic divergence found in some organisms, mainly (but not exclusively) within the non-transcribed spacer (NTS) region (Rooney and Ward, 2005; Fujiwara et al., 2009; Vierna et al., 2009; Freire et al., 2010; Úbeda-Manzanaro et al., 2010; Perina et al., 2011; Vizoso et al., 2011). Other evolutionary models (birth-and-death evolution; mixed process of concerted and birth-and-death evolution (Nei and Rooney, 2005)) have been proposed to drive the evolution of 5S rDNA (Rooney and Ward, 2005; Fujiwara et al., 2009; Vierna et al., 2009; Freire et al., 2010; Úbeda-Manzanaro et al., 2010; Perina et al., 2011; Vizoso et al., 2011).
5S rDNA is present in a variable number of repeats (usually, hundreds of copies) in each genome. These repeats can occur in tandem forming long arrays in some species, whereas in other cases they are dispersed throughout the genome. In some organisms, 5S rDNA repeats have been found linked to other noncoding RNA (ncRNA) gene families, such as small nuclear RNAs (snRNAs) (Vahidi et al., 1988; Nilsen et al., 1989; Zeng et al., 1990; Keller et al., 1992; Pelliccia et al., 2001; Cross and Rebordinos, 2005; Manchado et al., 2006; Marz et al., 2008; Freire et al., 2010; Vierna et al., 2011; Vizoso et al., 2011) or to protein-coding genes such as histones (Eirin-Lopez et al., 2004).
Although linkages of 5S rDNA to other ncRNAs have been shown also in bacteria (Gongadze, 2011), protists (Drouin and Tsang, 2012) and plants (Wicke et al., 2011; Layat et al., 2012) for longer time scales, the animal linkages of ncRNAs seem not to be stable over long evolutionary time scales. They appear to be the result of stochastic processes within genomes with no effect on fitness, even though this has not been demonstrated (see Drouin and Moniz de Sá (1995) for a review). Interestingly, 5S rDNA repeats can show different organization modes in the same species (Little and Braaten, 1989), and their transposition could be frequent within genomes, as reported by Drouin and Moniz de Sá (1995); Kalendar et al. (2008); Cohen et al. (2010).
Reports on the evolution of 5S rDNA in various animal and fungi groups have been published during the last few years, and all (Martins and Wasko, 2004; Vierna et al., 2009; Freire et al., 2010; Úbeda-Manzanaro et al., 2010; Perina et al., 2011; Vizoso et al., 2011) except one (Rooney and Ward, 2005) have relied on data obtained from PCR-cloning-sequencing techniques. Even though these procedures are appropriate when working with non-model organisms, they may not give a complete picture of the features and diversity of this multigene family. Fortunately, this can be solved by using genome project data, when available. Here, we have obtained a huge set of animal 5S rDNA candidate sequences, which were carefully filtered according to stringent criteria. Additionally, we gathered a set of U1 small nuclear DNA sequences from the same metazoan genomes, that were used in the linkage analysis between 5S rRNA and other ncRNAs.
Materials and methods
Sequence data
Previously known 5S rRNA and U1 snRNA sequences were taken from Rfam (Gardner et al., 2011) and selected previous studies (Marz et al., 2008; Vierna et al., 2009). These sequences (available from the electronic supplement http://www.rna.uni-jena.de/supplements/5SRNA/index.html) were used as an initial query in the development of a candidate pool (see below). The source, composition, download dates, assembly status, coverage, real number of nucleotides and expected number of nucleotides (from the animal genome size database (Gregory, 2012)) of all genomes analyzed are listed in the electronic supplement as well.
Homology search for 5S rRNAs and U1 snRNAs
Development of a candidate pool
First, we used blast (Altschul et al., 1990) with a low E-value <10−4 to get as many 5S rRNA and U1 snRNA candidates as possible. Overlapping hits were merged and extended 50 nt in both directions, manually viewed using emacs ralee mode (RNA ALignment Editor in Emacs) (Griffiths-Jones, 2005) and cut into their expected length. Consensus sequences of each alignment block and species were added to the query data set. We repeated this blast search with the same parameters and the collection step for all organisms until no new reliable candidates were found.
Sequence conservation
After having studied in detail previously reported 5S rRNA and U1 snRNA sequences, we selected four conserved motifs in animals for each ncRNA, Figure 1. Subsequently, we wrote rnabob descriptors, which characterized the conserved motifs (boxes and Sm-binding site) and their allowed distances (Figure 1, right). We decided against a covariance model, as we did not want a high variability and speed up the analysis. To detect divergent 5S rRNAs, we allowed point mutations to occur in one of the boxes, and variable distances between motifs. Additionally, box Z for the 5S rRNA and Sm-binding site for U1 snRNA were used in six species because of its huge initial candidate set: Homo sapiens, Pongo pygmaeus, Macaca mulatta, Bos taurus, Pteropus vampyrus and Saccoglossus kowalevskii. Candidates that did not fulfill these criteria were not discarded but marked with a demerit for further analysis.
Figure 1.

Known conserved motifs of 5S rRNA (top) and U1 snRNA (bottom) according to previous reports, and motifs used in the filtering step. For each motif, a specific number of mutations was accepted. Allowed distances between motifs are displayed on the left. Red motifs were used to filter sequence candidates in all species. Green motifs were used as additional filters in some species (H. sapiens, P. pygmaeus, M. mulatta, B. taurus, P. vampyrus and S. kowalevskii) because the number of candidates was too large. Whereas function of U1 boxes are diverse (Box A recognizes 5′-splice site in mRNA precursors (Zhuang and Weiner, 1986, and references therein); Box B is part of the U1-70K protein-binding site (Query et al., 1989); Box C is part of the U1-A protein-binding region (Scherly et al., 1989) and the Sm protein-binding region, named ‘domain A' (Branlant et al., 1982)), those 5S rRNA boxes are essential for transcription of 5S rRNA itself (Pieler et al., 1987; Bogenhagen, 1993; Hall, 2005). A full color version of this figure is available at the Heredity journal online.
Structure conservation
In a second step, we examined the secondary structure of the candidates. If RNAfold (Hofacker, 2003) did not fold the sequences instantly into the expected structure as depicted in Figure 1 (observed for all candidates manually), we used constraint folding RNAfold -C. The constraints used are all individually displayed at the Supplemental Page. Alternatively, we created alignments of the previously reported sequences, given in Figure 1 using clustalw (Larkin et al., 2007) and RNAalifold (Hofacker, 2007). Other candidates were marked with additional penalty for further analysis.
Manual inspection
For each organism, alignments were manually examined for irregularities, such as insertions/deletions, indicating a non-functionality.
Final genes and grouping
Finally, each candidate received a classification: satisfying all our filtering criteria indicated that functionality of 5S rRNA sequences was highly likely (Table 1, A-type). If sequence or structure contained slight variations (single point mutations that affected the secondary structure only slightly), then the candidate was declared as B-type.
Table 1. Number of identified 5S rRNAs.
| Spec. | No. of Cop. | No. of A | No. of B | No. of Q | No. of Diff. | No. of Blocks | No. of Clus. | |
|---|---|---|---|---|---|---|---|---|
| hsa | 1.1f | 18 | 18 | 0 | 0 | 3 | 1 | 1 |
| ppy | 1.1f | 6 | 5 | 0 | 1 | 4 | 1 | 0 |
| mac | 1.1f | 12 | 11 | 0 | 1 | 4 | 1 | 0 |
| cjc | 1.2c | 10 | 6 | 0 | 4 | 7 | 3 | 2 |
| tsy | 1.6s | 15 | 5 | 7 | 3 | 15 | 10 | 0 |
| oga | 1.0s | 20 | 8 | 8 | 4 | 17 | 4 | 1 |
| mmu | 1.2f | 42 | 41 | 0 | 1 | 6 | 1 | 0 |
| rno | 1.2f | 13 | 6 | 7 | 0 | 13 | 5 | 1 |
| dor | 2.0s | 27 | 27 | 0 | 0 | 6 | 2 | 0 |
| str | 1.1s | 5 | 5 | 0 | 0 | 2 | 1 | 0 |
| cpo | 1.4s | 67 | 57 | 8 | 2 | 53 | 3 | 0 |
| opr | 0.6s | 7 | 6 | 1 | 0 | 3 | 2 | 2 |
| ocu | 0.9s | 26 | 25 | 1 | 0 | 9 | 1 | 3 |
| tbe | 0.9s | 8 | 4 | 4 | 0 | 6 | 5 | 1 |
| fca | 0.7s | 36 | 30 | 6 | 0 | 18 | 2 | 3 |
| cfa | 1.2f | 10 | 8 | 0 | 2 | 9 | 1 | 1 |
| vpa | 1.1s | 8 | 7 | 1 | 0 | 5 | 3 | 0 |
| ttr | 1.2s | 52 | 52 | 0 | 0 | 15 | 1 | 6 |
| bta | 1.2f | 10 | 8 | 0 | 2 | 8 | 1 | 0 |
| ssc | 1.4f | 5 | 3 | 2 | 0 | 5 | 5 | 0 |
| eca | 1.3f | 5 | 3 | 2 | 0 | 5 | 5 | 0 |
| mlu | 0.8s | 30 | 24 | 2 | 4 | 18 | 3 | 3 |
| pva | 1.2s | 28 | 17 | 0 | 11 | 20 | 1 | 8 |
| eeu | 1.1s | 16 | 16 | 0 | 0 | 5 | 1 | 1 |
| sar | 1.0s | 17 | 7 | 9 | 1 | 16 | 10 | 0 |
| laf | 1.0s | 45 | 24 | 17 | 4 | 36 | 2 | 1 |
| ete | 1.3s | 12 | 10 | 1 | 1 | 12 | 5 | 0 |
| pca | 1.3s | 10 | 9 | 1 | 0 | 4 | 2 | 1 |
| dno | 1.1s | 7 | 7 | 0 | 0 | 3 | 2 | 0 |
| cho | 1.7s | 22 | 5 | 9 | 8 | 17 | 4 | 1 |
| mdo | 1.3f | 18 | 17 | 0 | 1 | 9 | 2 | 0 |
| meu | 1.4s | 19 | 2 | 16 | 1 | 19 | 5 | 1 |
| oan | 1.4f | 23 | 20 | 3 | 0 | 8 | 3 | 0 |
| tgu | 1.0f | 17 | 16 | 0 | 1 | 16 | 2 | 1 |
| gga | 1.1f | 6 | 5 | 1 | 0 | 4 | 3 | 1 |
| xtr | 2.6s | 60 | 48 | 10 | 2 | 41 | 9 | 14 |
| tni | 1.1f | 54 | 43 | 6 | 5 | 47 | 3 | 16 |
| tru | 1.0f | 42 | 41 | 1 | 0 | 23 | 2 | 7 |
| gac | 10.3s | 240 | 2 | 0 | 238 | 93 | 4 | 15 |
| ola | 1.2f | 3 | 3 | 0 | 0 | 3 | 2 | 0 |
| dre | 1.2f | 3180 | 3135 | 0 | 45 | 241 | 2 | 43 |
| cmi | 2.5c | 38 | 36 | 1 | 1 | 17 | 3 | 2 |
| pma | 2.0c | 344 | 194 | 125 | 25 | 224 | 1 | 74 |
| bfl | 1.1s | 14 | 14 | 0 | 0 | 3 | 1 | 3 |
| cin | 1.1f | 48 | 38 | 4 | 6 | 23 | 2 | 12 |
| csa | 1.1c | 272 | 236 | 22 | 14 | 133 | 2 | 7 |
| odi | 1.0s | 66 | 1 | 0 | 65 | 24 | 1 | 11 |
| sko | 1.0s | 1560 | 80 | 0 | 1480 | 1166 | 16 | 223 |
| dme | 1.0f | 215 | 98 | 0 | 117 | 24 | 2 | 44 |
| dsi | 1.0s | 14 | 12 | 0 | 2 | 3 | 1 | 4 |
| dse | 1.0s | 28 | 16 | 0 | 12 | 9 | 1 | 5 |
| der | 1.0f | 40 | 31 | 0 | 9 | 8 | 2 | 4 |
| dya | 1.0s | 23 | 21 | 0 | 2 | 4 | 1 | 4 |
| dan | 0.9s | 64 | 61 | 1 | 2 | 14 | 1 | 13 |
| dps | 1.1s | 50 | 50 | 0 | 0 | 5 | 1 | 4 |
| dpe | 1.0s | 60 | 46 | 7 | 6 | 21 | 2 | 18 |
| dwi | 0.9s | 11 | 11 | 0 | 0 | 4 | 1 | 1 |
| dvi | 1.8s | 69 | 68 | 0 | 1 | 6 | 2 | 12 |
| dmo | 0.9s | 61 | 43 | 0 | 18 | 12 | 2 | 6 |
| dgr | 1.2s | 53 | 53 | 0 | 0 | 9 | 1 | 7 |
| aga | 1.1s | 11 | 10 | 1 | 0 | 3 | 3 | 2 |
| aae | 0.7c | 143 | 138 | 3 | 2 | 23 | 1 | 3 |
| bmo | 1.1s | 64 | 58 | 0 | 6 | 39 | 3 | 10 |
| tca | 1.0c | 199 | 197 | 1 | 1 | 19 | 1 | 5 |
| ame | 1.0s | 53 | 48 | 0 | 5 | 24 | 5 | 10 |
| nvi | 0.9f | 34 | 30 | 4 | 0 | 17 | 3 | 3 |
| phu | 1.0c | 31 | 22 | 0 | 9 | 14 | 3 | 6 |
| api | 0.7s | 50 | 44 | 1 | 5 | 28 | 2 | 3 |
| dpu | 1.5s | 114 | 16 | 1 | 97 | 67 | 4 | 26 |
| isc | 1.3s | 7 | 2 | 5 | 0 | 7 | 3 | 1 |
| cre | 0.7c | 31 | 0 | 0 | 31 | 6 | 1 | 5 |
| cbr | 1.1f | 7 | 0 | 0 | 7 | 1 | 1 | 2 |
| cbe | 0.5c | 24 | 0 | 0 | 24 | 7 | 1 | 5 |
| cel | 0.9f | 13 | 0 | 0 | 13 | 2 | 1 | 1 |
| cja | 0.6c | 9 | 0 | 0 | 9 | 2 | 1 | 3 |
| hco | 0.2c | 212 | 0 | 0 | 212 | 38 | 3 | 22 |
| acn | 2.2c | 10 | 0 | 0 | 10 | 1 | 1 | 3 |
| ppa | 1.0c | 65 | 0 | 0 | 65 | 6 | 1 | 12 |
| min | 0.6c | 26 | 0 | 0 | 26 | 9 | 1 | 3 |
| mha | 0.8c | 12 | 4 | 1 | 7 | 11 | 2 | 2 |
| bma | 1.1c | 189 | 0 | 0 | 189 | 19 | 1 | 13 |
| tsp | 0.1c | 424 | 1 | 0 | 423 | 97 | 8 | 0 |
| sma | 0.7s | 32 | 22 | 5 | 5 | 23 | 7 | 4 |
| sja | 0.7c | 11 | 11 | 0 | 0 | 4 | 1 | 0 |
| sme | 0.8c | 66 | 63 | 2 | 1 | 15 | 1 | 13 |
| hro | 0.1c | 863 | 732 | 0 | 131 | 264 | 1 | 57 |
| cca | 0.1c | 1584 | 1493 | 0 | 91 | 410 | 1 | 20 |
| apo | 2.5c | 4 | 4 | 0 | 0 | 4 | 1 | 0 |
| lgi | 1.2s | 186 | 166 | 3 | 17 | 88 | 4 | 31 |
| aca | 2.6s | 11 | 10 | 1 | 0 | 6 | 1 | 1 |
| bgl | 4.6c | 17 | 16 | 0 | 1 | 7 | 1 | 0 |
| esc | 78c | 5 | 3 | 0 | 2 | 5 | 5 | 0 |
| apa | 25c | 36 | 0 | 0 | 36 | 22 | 3 | 1 |
| ami | 20c | 49 | 0 | 0 | 49 | 41 | 2 | 0 |
| nve | 0.6s | 708 | 625 | 7 | 76 | 345 | 3 | 68 |
| rsp | 0.1c | 177 | 162 | 10 | 5 | 55 | 2 | 10 |
| tad | 0.4s | 8 | 8 | 0 | 0 | 7 | 3 | 2 |
Abbreviatons: aae, Aedes aegypti; aca, Aplysia californica; acn, Ancylostoma caninum; aga, A. gambiae; ame, A. mellifera; ami, A. millepora; apa, A. palmata; api, Acyrthosiphon pisum; apo, Alvinella pompejana; bfl, Branchiostoma floridae; bgl, Biomphalaria glabrata; bma, Brugia malayi; bmo, Bombyx mori; bta, B. taurus; Blo., number of blocks (groups within an alignment; similar 5S rRNA copies built one block); cbe, Caenorhabditis brenneri; cbr, C. briggsae; cca, Capitella sp; cel, C. elegans; cfa, Canis familiaris; cho, Choloepus hoffmanni; cin, C. intestinalis; cja, C. japonica; cjc, Callithrix jacchus Marmoset; Clus., number of clusters of at least two 5S rRNA-coding regions within 10 000 nt; cmi, Callorhinchus mili; cpo, Cavia porcellus; cre, C. remanei; csa, C. savignyi; dan, Drosophila ananassae; der, D. erecta; dgr, D. grimshawi; dme, D. melanogaster; dmo, D. mojavensis; dno, Dasypus novemcinctus; dor, D. ordii; dpe, D. persimilis; dps, D. pseudoobscura; dpu, D. pulex; dre, D. rerio; dse, D. sechellia; dsi, D. simulans; dvi, D. virilis; Diff., number of different sequences; dwi, D. willistoni; dya, D. yakuba; eca, E. caballus; eeu, Erinaceus europaeus; esc, Euprymna scolopes; ete, Echinops telfairi; fca, Felis catus; gac, Gasterosteus aculeatus; gga, G. gallus; hco, Haemonchus contortus; hro, H. robusta; hsa, H. sapiens; isc, Ixodes scapularis; laf, L. africana; lgi, L. gigantea; mac, M. mulatta; mdo, Monodelphis domestica; meu, M. eugenii; mha, Meloidogyne hapla; min, M. incognita; mlu, Myotis lucifugus; mmu, Mus musculus; nve, Nematostella vectensis; nvi, Nasonia vitripennis; oan, Ornithorhynchus anatinus; ocu, Oryctolagus cuniculus; odi, O. dioica; oga, Otolemur garnettii; ola, O. latipes; opr, O. princeps; pca, P. capensis; phu, P. humanus; pma, P. marinus; ppa, Pristionchus pacificus; ppy, P. pygmaeus; pva, P. vampyrus; rno, R. norvegicus; rsp, Reniera sp; sar, Sorex araneus; sja, Schistosoma japonicum; sko, S. kowalevskii; sma, S. mansoni; sme, Schmidtea mediterranea; Spec., Species; spu, Strongylocentrotus purpuratus; ssc, Sus scrofa; str, Spermophilus tridecemlineatus; tad, T. adhaerens. tbe, Tupaia belangeri; tca, Tribolium castaneum; tgu, Taeniopygia guttata; tni, Tetraodon nigroviridis; tru, Takifugu rubripes; tsp, Trichinella spiralis; tsy, T. syrichta; ttr, T. truncatus; vpa, Vicugna pacos; xtr, X. tropicalis.
We distinguish the total number of candidates (Cop.) to be putatively functional (A), containing variations in sequence or structure (B), and questionable owing to variations in box motifs and secondary structure (Q). The second column depicts the state of each genome assembly: the number is calculated by the known genome size (Gregory, 2012) divided by the number of downloadable nucleotides in finished genomes (chromosomal status), scaffolds or contig.
If both sequence and structure showed several variations, compared with the rest of sequences of that species (for example, indels of at least 5 nt), then the candidate was defined as questionable (Table 1, Q-type), referring to possible pseudogenes.
Even more divergent candidates were deleted from our data sets and not further considered. In fact, we considered these genes to be not functional, to be pseudogenes.
We used the scoring step as a measure of the trustability of each candidate. All sequences, regardless of their score (A, B or Q), were considered in subsequent analyses.
For each organism, fasta, gff and stockholm alignment files are provided on the Supplemental Page.
Orthologous and paralogous 5S rRNA genes
For each taxon, we manually divided the stockholm alignment files into subgroups, defined here as ‘blocks' (Table 1).
For the identification of orthologous and paralogous 5S rRNA genes, we used consensus sequences of the blocks and analyzed them with the NeighborNet algorithm (Bryant and Moulton, 2004) and uncorrected p-distances in SplitsTree4 (Huson, 1998).
NTS analysis of clusters
Most 5S rRNA occur within 3000 nt. However, to detect a possible correlation of more distantly located 5S rRNAs, we defined 5S rRNA genes being part of one cluster, if and only if they were located on the same chromosome (scaffold or contig) within 10 000 nt independently of their orientation. We wrote postscript files for each taxon to display the genome-wide arrangement. PDF files are provided in the supplement.
The NTS regions <500 nt between two 5S rRNA candidates were aligned with clustalw. Fasta, gff and alignment files are available at the Supplemental Page.
Regulator analysis of 5S rRNA genes
Upstream promoter analysis
We selected the region comprising positions −35 to −25, upstream the 5S rRNA gene (Hallenberg and Frederiksen, 2001; Vizoso et al., 2011) and citations therein, and searched for conserved motifs with MEME (Bailey et al., 2009). We used parameters −minw 5 −maxw 8 to target a TATA box already described in the literature (see section below). The shuffling of sequences was performed with shuffle −0. Detailed results can be viewed on the Supplemental Page.
Internal promoter analysis
For each stockholm alignment, we created consensus sequences: (A) the most frequent nucleotide was represented in the consensus sequence, and (B) each nucleotide with a frequency >10% was part of the consensus sequence, following the IUPAC coding system.
Terminator analysis
For the terminator analysis, we analyzed 50 nt downstream of each 5S rRNA candidate and checked with rnabob descriptor the first occurrence of the pattern TTTT. Additionally, conserved motifs were identified by MEME (Bailey et al., 2009) (parameters: −minw 6 −maxw 20) within the 30 nt downstream. We used only unique sequences per species. Species with >80 different copies were neglected in this analysis, because of complexity reasons.
Linkage between 5S rRNA and other ncRNAs
We downloaded all known ncRNA classes from RFAM (Gardner et al., 2011), and in case of U1 snRNA and 5S rRNA, we included previous literature as mentioned above and searched them in the metazoan genomes with blast (Altschul et al., 1990). Additionally, we scanned the genomes for tRNAs with tRNAscan-SE (Lowe and Eddy, 1997).
As there is, to our knowledge, no established statistical model describing linkage between ncRNAs in a variety of species, we used a simple Gaussian mixture with a variable number of components.
Blast hits are not weighted, that is, hits with an E-value below a threshold of 10−4 are included, hits above the threshold are excluded. If genomic duplications due to possible assembly artefacts occur, the naive weight given to such a region could point towards linkage, where no real duplication was present. Therefore, we filtered the data: if exactly the identical number of nucleotides were observed between two linked genes, we assumed assembly artefacts (for example, multiple sequenced contigs) and used only one copy.
For each ncRNA gene copy, we test for linkage with 5S, we build a Gaussian mixture (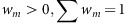 ):
):
Each Gaussian in the mixture describes the distance μ between a 5S rRNA gene copy and the other gene copy, while σ is the s.d. in this distance. As it is possible that either one 5S gene copy is linked with multiple copies of the other gene, or that multiple pairs of linked 5S rRNAs/other genes exist, we require a k-component mixture.
The number of components k is determined by increasing k from 1 up until no significant improvement in fit is possible. To prevent overfitting, a maximum of 10 Gaussians is allowed, less if the number of data points is lower than 40.
The parameter vector (μ1,…,μk), (σ1,…,σk) is fitted using expectation maximization (Hastie et al., 2001).
Results and Discussion
For the first time, we present here a complete overview of 5S rDNA in metazoans, including secondary structure prediction, genomic organization, sequence characteristics, putative regulatory motifs and linkage to other ncRNAs. Furthermore, we also found striking features in available mammalian genomes described below. Although this analysis shows many facts that depend on current genome assemblies, the reader should keep in mind that the assemblies of different organisms are extremely variable in terms of completeness and therefore are, at least for the number of copies, hardly comparable. Currently available metazoan genome assemblies very often lack multi-copy regions such as centromeres, telomeres and rRNA operons (Copeland et al., 2009; Dalloul et al., 2010; Alkan et al., 2011). Additionally, two identical gene copies located multiple times in the genome are often merged, or even completely removed (Marz et al., 2008; Alkan et al., 2011). According to Alkan et al. (2011), assemblies are in general 16.2% shorter than the reference genome, and 99.1% of validated duplicated sequences are missing from the assembled genome. However, in some assemblies we can find repeated sequences of the same locus, because at the contig or scaffold levels, some genomic regions are covered multiple times. In our analysis, we take these facts into account, and show—as a side effect—how much information we can obtain from genomic sequences when working with multiple-copy genes, regardless of genome assemblies. Available cytogenetic mapping data support our analysis as described in detail below.
Arrangement of 5S rRNA copies: number and evolutionary relationship
The overall summary of 5S rRNA copies in animals is depicted in Table 1. We discriminated between three different classes: (A) putative functional genes that passed all our filters, (B) those that showed slight variations in sequence or structure, and (Q) those that remained questionable and might even be possible pseudogenes.
Overall, we identified 12 766 5S rRNA sequences in 97 organisms, ranging from three sequences in the ricefish Oryzias latipes to 3180 sequences in the zebrafish Danio rerio. Both assemblies are in chromosomal stage. In both cases, real genomes are 1.2-fold larger than the assemblies (Gregory, 2012), see Table 1. The genome coverages of O. latipes and D. rerio is 10.6 × and ∼30 × , respectively. Owing to the assembly problems mentioned above, we assume the lower boundary for 5S rRNA copies in these fishes to be about 3180. In general, when the coverage of the genome is at least 8 × and the genome is sorted into chromosomes, it can be considered that the listed number of copies (Table 1) is a lower boundary. Cytogenetic mapping of the Squalius alburnoides being closely related to D. rerio showed several clusters on three chromosomes (Gromicho et al., 2006), in agreement with the 43 clusters on three chromosomes of the zebrafish in our study. Comparison of fish genomes bring in general difficulties due to polyploidy. The cytogenetic mapping of Gallus gallus showed one cluster on chromosome 9 (Cabral-de-Mello et al., 2011), which completely agrees with the one cluster we found also on chromosome 9.
The genome sequence of the most basal deuterostome acorn worm S. kowalevskii shows 1166 different copies. Protostomes seem to have, in general, a lower number of 5S rRNA copies. Although the genome of the polychaete worm Capitella capitata displays 1584 copies, we assume the real minimal number of 5S rRNA copies to be much smaller, because the genome is on contig stage, which is 10 times larger than the expected genome size (Gregory, 2012), see Supplemental Page. We found 410 different copies of 5S rRNA and we set this value as the minimal copy number in C. capitata. By cytogenetic mapping, Dichotomius have been shown to consist of a very strong characteristic cluster on chromosome 2 (Cabral-de Mello et al., 2010). The only coleoptera investigated in this manuscript (Tribolium) is not assembled on chromosomal level; however, it shows also one huge cluster of 151 5S rRNA copies. Previous reports have shown that copy number is very variable among metazoans: 1700–2000 copies (including pseudogenes) in humans (Sorensen and Frederiksen, 1991), 50–100 copies in Macaca fascicularis (Jensen and Frederiksen, 2000), 35–41 copies in the chicken (Daniels and Delany, 2003), 24 000–61 000 copies (including pseudogenes) in three amphibians (24 000 in Xenopus laevis (Hilder et al., 1983)) and only three copies in Plasmodium falciparum (Shippen-Lentz and Vezza, 1988). However, these estimates relied on the method used and on the ability to differentiate among functional and non-functional copies. Our results do not perfectly agree with these examples as we predicted, in general, a lower number of copies (18 in humans, 12 in M. mulatta, 6 in the chicken and 60 Xenopus tropicalis).
According to sequence and secondary structure features, we identified different 5S rRNA classes in some genomes as described below. Within species, alignments clearly unveiled disjunct sets, hereafter called ‘blocks', in 58 species, see Table 1. We aligned the consensus sequences of the 253 blocks retrieved. In the network obtained, we can distinguish four main 5S rRNA groups, see Figure 2—left.
Figure 2.

Evolutionary relationships among 5S rRNA. Left: orthologous 5S rRNAs consensus sequences of the 253 5S rRNA blocks (see Table 1). We can distinguish four 5S rRNA groups: vertebrates, basal deuterostomes and nematodes share high sequence similarity, whereas the sequences of other metazoans clustered into a distinct 5S rRNA group. Exceptions are marked with species names and ID of the block. Right: paralogous 5S rRNA genes in mammals. At least one 5S rRNA block consensus sequence of each mammalian genome analyzed is part of the here defined ‘basic 5S rRNA set'. The ‘core 5S rRNA set' comprises the sequences from the ‘basic set' except the one of Tarsius syrichta. These basic consensus sequences are highly conserved in sequence. Other 5S rRNA genes are highly divergent. The figure is drawn with the NeighborNet method of the SplitsTree package. All raw data and figures, including taxa names, are downloadable from the Supplemental Page.
Orthologous 5S rRNA genes
Vertebrate 5S rRNA sequences are clearly evolutionary separated from other metazoan sequences. Interestingly, basal deuterostomes (Hemichordata, Tunicata and Cephalochordata) and nematodes share high sequence similarity, whereas the sequences of other metazoans (Arthropoda, Lophotrochozoa, Cnidaria, Porifera and Placozoa) clustered into a distinct 5S rRNA group.
Paralogous 5S rRNA genes
When comparing consensus sequences of mammalian 5S rRNA blocks (Figure 2—right), we found, in contrast to non-mammalian sequences, a core 5S rRNA set that comprised at least one sequence of each mammalian species. Sequences within this core set were very similar (nearly no mutations), whereas consensus sequences of the other blocks were relatively divergent (some of them might even be non-functional, such as possibly Loxodonta africana 2, see Figure 2—left). No grouping or pattern can be observed in the divergent 5S rRNA set. 5S rRNA seems to have undergone two main evolutionary processes: on the one hand, the data suggest that the long-term evolution of the 5S rRNA genes in mammals is characterized by high selection pressure on housekeeping 5S rRNAs (for example, the 5S rRNA core set) and on the other hand gene diversification, which may provide adaptative potential to environmental change. In other words, we may be facing an evolutionary scenario in which strong purifying selection (and perhaps mechanisms involved in concerted evolution) maintains the integrity of housekeeping 5S rRNAs, whereas birth-and-death processes generate variation through duplications.
The distribution of some orthologous 5S sequences (Figure 2) might be explained by horizontal gene transfer of transposable elements similar to SPIN genes (Syvanen, 2012). However, this is, especially for housekeeping genes, under discussion.
5S rDNA clusters and NTS analysis
In order to study 5S rDNA sequences within species, we analyzed copies separated by less than 10 000 nt (that is, within a ‘cluster') in more detail. The number of clusters with at least two 5S rRNAs can be viewed in Table 1. The size of clusters depends on the genome and its assembly, and can be hardly compared.
In many species, we found clusters with differences in the length of their NTS. For example, in the honey bee Apis mellifera we found seven copies on contig GroupUn.750 with a constant spacer of 249 nt, whereas contig GroupUn.96 had five copies separated by a 711 nt spacer. Similarly, other species showed NTSs of different sizes in the same contig. This agrees with other species, as previously reported (for example, in molluscs (Vierna et al., 2011), arthropods (Perina et al., 2011) and chordates (Gornung et al., 2007)). In this work, we add to this list more chordate, annelid, arthropod, cephalochordate, placozoan, cnidarian and molluscan species. Sequence orientation and distances between 5S rRNA regions can be obtained for each organism on the Supplemental Page. In the following organisms, we have found 5S rRNA copies that displayed different orientations in the chromosome, a fact that is not in agreement with our expectations according to concerted evolution of repeats within the same cluster. In the cases in which distances among repeats were large (for example, in X. tropicalis, Drosophila melanogaster, D. virilis, D. mojavensis and D. willistoni), it is not unexpected that gene conversion was unable to homogenize the copies within the cluster. However, in other cases, distances between repeats were small (Petromyzon marinus, Pediculus humanus and Trichoplax adhaerens). This would indicate that the inversions are recent or that the unit of homogenization by gene conversion involves both repeats.
To determine the evolution of 5S rDNA in more detail, we cut and aligned the NTS regions <500 nt (alignment available on the Supplemental Page). As hypothezised by Vierna et al. (2011), a 5S rDNA sequence that is evolving concertedly within a given cluster can be transposed into another 5S rDNA cluster composed of repeats that are different to that one, but similar among them. After the occurrence of duplications involving both variants, it is possible to obtain an intermixed organization of 5S rDNA, in which NTSs located in the cluster are completely divergent. This is what we report here for some species (Daphnia pulex and D. rerio, see NTS alignment at Supplemental Page). These findings agree with the widespread idea that 5S rDNA repeats are transposed from one genome location to another (Rooney and Ward, 2005; Datson and Murray, 2006). Intermixed organization of NTS sequences was also found by Gornung et al. (2007); Perina et al. (2011); Vierna et al. (2011) in molluscs, crustacean and fishes species.
Totally unexpectedly, all NTS sequences (divided into four NTS types) retrieved from the mollusc Lottia gigantea and from the porifer Reniera sp. were almost identical. We blasted these NTSs against various nucleotide databases, but failed to find any similarities with previously reported sequences, such as bacterial/viral insertions. The same picture of very closely related NTS regions is given from the insects Anopheles gambiae and P. humanus, which is directly in contrast to closely related organisms sharing no related NTS features, such as Ciona intestinalis and C. savignyi or most of the drosophilids.
We have also retrieved putatively functional and non-functional 5S rRNA sequences within one cluster in many organisms. This has been reported for D. melanogaster before (Sharp et al., 1984).
In order to analyze the evolution of the NTS region at the species level, we selected the genus in which the most species were available (Drosophila, 12 species). We obtained the following results: (1) NTSs can be divided roughly into 10 different types, according to alignment clustering. In fact, NTS sequences that belong to the same type can be aligned because their degree of divergence is not high; (2) all species display only one type of NTS sequence in their genomes, except D. mojavensis and D. grimshawi, with two divergent NTS sequence types; (3) the drosophilids do not share their NTS type with their congeners; however, the very recent split species show similar NTS sequences (D. persimilis/D. pseudoobscura and D. simulans/D. sechellia); and (4) the different NTS types defined agree with the phylogeny of these 12 species (Drosophila 12 Genomes Consortium et al., 2007). Considering these results and the high degree of conservation of the 5S rRNA copies, we hypothesize an evolutionary scenario in which the long-term evolution of 5S rDNA in the genus Drosophila is driven by strong selection over the 5S rRNA copies, gene duplications and transpositions that generate new NTS loci, and homogenizing mechanisms within each array. The divergent NTSs retrieved from D. mojavensis and D. grimshawi could also point toward the occurrence of ancestral polymorphism. Birth-and-death evolution with a fast gene turnover, concerted evolution and mixed models combined with strong selection can explain these results.
Internal promoter analysis
The internal 5S rRNA boxes (Figure 1) are essential for transcription of 5S rRNA itself (Pieler et al., 1987; Bogenhagen, 1993; Hall, 2005). For instance, the C2H2 zinc finger protein TFIIIA binds Xenopus 5S rRNA internal promoters (named Box A, intermediate element and Box C (Pieler et al., 1987)), see Bogenhagen (1993) and Hall (2005). To analyze the occurrence of these essential boxes in our set of 5S rRNAs, we obtained consensus sequences for each block of 5S rRNAs taking into account the IUPAC coding system. In Figure 3a, consensus sequence of these consensus sequences is depicted. We found several conserved regions, which are shown highlighted. In the 5′-region of the 5S rRNA molecule, we found two strikingly conserved motifs: CAUAC (9–14 nt) and GAA (21–23 nt). Furthermore, we detected high conservation in the center of the structure, between positions 39 and 60. Finally, the 3′-end was highly conserved (GUG). By taking into account the secondary structure, the regions interacting with polymerase coincide. Our results confirm previous reports by Pieler et al. (1987); Sharp and Garcia (1988); Vizoso et al. (2011): the CAUAC covers the ICR-I, which was described for D. melanogaster by Pieler et al. (1987). The known ICR-II region (GUCCGAUC) is almost identical to our YCRUCYGAUC-motif, which occurs in all so far known 5S rRNAs and shows therewith very high conservation. For the ICR-III (as described in D. melanogaster: GAAAUUAAGCAGCG), we detected that the first U is often replaced by C, whereas the second U, which was reported for the molluscs as M (A or C), is highly conserved (Vizoso et al., 2011). In the case of ICR-IV, the known sequence for D. melanogaster (Pieler et al., 1987) does only match partially our consensus sequence.
Figure 3.

Consensus sequence and structure of metazoan 5S rRNA. Conserved motifs are highlighted. A full color version of this figure is available at the Heredity journal online.
Interestingly, most of our conserved motifs and structure can also be found in land plants, such as the CAUAC motif (Wicke et al., 2011). A comparison of the consensus structure of metazoans and land plants (Wicke et al., 2011) shows that they highly correspond with each other. There are slight differences concerning the length of hairpins, such as smaller internal loops in the 5′-end of the metazoan 5S, instead of two larger loops in plants (referred as B and C in Wicke et al. (2011)).
Potential regulatory upstream regions
We analyzed 40 nt upstream of each of our 5S rRNA sequences using MEME, and detected three conserved motifs. The motif WTAAA was retrieved in 35 species (1.7e–172, 370 sequences, see Supplemental Page). The TATA-box TATAAT was found in 13 genomes (5.2e−026, 71 sequences, see Supplemental Page). They were mainly protostomes, but we also found this motif in several sequences of the tunicate Oikopleura dioica. One copy per genome with this motif was found in three more deuterostomes. Interestingly, a TATA-like motif located at positions around −30 to −25 from the transcription start site is essential for efficient 5S rRNA transcription in vitro in Caenorhabditis elegans and C. briggsae (Nelson et al., 1998), Neurospora crassa (Tyler, 1987) and D. melanogaster (Sharp and Garcia, 1988). Upstream motif analysis has also detected TATA-like elements in several bivalve groups, with different degrees of conservation, at positions −30 to −25 (Vizoso et al., 2011), and in fishes (Martins and Wasko, 2004). Finally, we also found the motif TCTTGRGG (5.5e−077, 61 sequences) in 28 species (Supplemental Page). Among them, there were 25 mammals and three other species (the polychaete Capitella sp., the leech Helobdella robusta and the trematode Schistosoma mansoni). It is known that the mammalian 5S rRNA gene has a 12-nucleotide upstream promoter, the D-box (GGCTCTTGGGGC), which is important for efficient transcription in vitro and in vivo in humans (Hallenberg and Frederiksen, 2001). The D-box is located in positions −32 to −21 nt in humans and in M. fascicularis; in positions −33 to −22 nt in mouse and rat; and in positions −36 to −25 nt in hamster (Hallenberg and Frederiksen, 2001). However, we failed to find this motif in the remaining eight mammalian genomes (Rattus norvegicus, Dipodomys ordii, Ochotona princeps, Tursiops truncatus, Equus caballus, Echinops telfari, Procavia capensis and Macropus eugenii). Shuffling of all the sequences resulted in GC-rich motifs with large E-values (>360).
Terminator analysis
In Bogenhagen and Brown (1981); Huang and Maraia (2001) and Richard and Manley (2009), the transcription termination signal for 5S rDNA is described as one or more TTTT stretches. We analyzed 50 nt downstream of 5S rRNA-coding regions using MEME. We found three common patterns (Figure 4, for more specific patterns, see Supplement).
Figure 4.

Terminator pattern detected within the 30 nt downstream of 5S rRNA candidates, using MEME. Top: previously known poly-T region in 272 sequences with an E-value of 4.8 × 10−168. Middle: newly detected poly-A region in 99 sequences (1.6 × 10−270). Bottom: conserved region in 112 insect, one Sus scrofa and 17 bat P. vampyrus sequences (2.3 × 10−361) in agreement with Sharp et al. (1984) and Sharp and Garcia (1988).
As expected, we obtained 272 sequences with poly-T tail in 40 species. However, we also found 99 polyadenylated sequences in 26 species. We assume these are 5S rRNA copies that have been transcribed and inserted back into the genome as reported previously for mouse and rat (Drouin, 2000). These 99 sequences are found among all deuterostomes and are quite a large fraction compared with the 272 poly-T sequences. Sequence and structure of these 5S rRNA copies are completely conserved to putatively functional ones, suggesting that (a) these genes are transcribed (as reported for mouse and rat in Drouin (2000) and/or (b) the possible insertion by retroelements was very recent in terms of evolutionary time. For Drosophila only, we found 10 nucleotides located between the 5S rRNA-coding region and the poly-T tail. This is in agreement with Sharp and Garcia (1988), demonstrating that 135 nt are transcribed, whereas the mature transcript has only 120 nt.
This leads to the general question of how frequent longer intermediate transcripts are, compared with the size of the mature RNA. We tried to answer this question by plotting the distance from the RNA-coding region to the first TTTT motif, defined as terminator (Bogenhagen and Brown, 1981; Huang and Maraia, 2001; Richard and Manley, 2009). Figure 5 shows that, in general, deuterostomes have longer intermediate transcripts than protostomes. Human and fly share the feature of a 11-nt-longer preprocessed transcript. However, there are exceptions: in Dipodomys we found only 1 nt between RNA-coding region and TTTT, and in the most basal metazoan T. adhaerens, we found a distance of 7–13 nt.
Figure 5.

Evolutionary boxplot of distances between 5S rRNA-coding region and first occurrence of the downstream located TTTT motif. TTTT, number of sequences with TTTT within the first 30 nt; sum, number of terminator sequences analyzed; diff, number of different terminator sequences (50 nt analyzed); and uni, number of terminator sequences, which have at least one identical copy.
Martins and Wasko (2004) described for some fish species an additional conserved downstream motif (GAAACAA), but its function is not known. However, we only found this motif in very few of our conserved terminal regions, in unrelated species.
Finally, we detected an interesting feature in Acropora palmata, as analysis of terminal sequences, Figure 6, suggested a systematic insertion or deletion of thymines downstream of 5S rRNA-coding regions.
Figure 6.

A. palmata sequences downstream of 5S rRNA-coding regions, and systematic insertion or deletion of thymines.
5S rRNA and protein interactions
Scripture and Huber (1995) showed for X. laevis the interaction of protein eL5 to 5S rRNA helix III and loop C, as well as a adenine pairing in helix III. With our results, we were able to show that this might be a metazoan-wide feature, as these regions are conserved for all sequences (Figure 3). Interestingly, we were able to show that the interaction of transcription factor IIIA seems not to be consistent for all metazoans. This is in agreement with Lu et al. (2003), who showed that TFIIIA is interacting with helix V and helix II. Both regions were not conserved among our examined metazoan sequences. Although many data on 5S rRNA structure and interactions exists, its function is still not clearly elucidated (Smirnov et al., 2008).
Linkage between 5S rRNA and other ncRNAs
For 12 ncRNA genes out of all families of Rfam, we detected significant evolutionary linkage to 5S rDNA. For each of the ncRNAs, we provide a number of mixture model plots on the Supplemental Page.
Genomic linkage of 5S rDNA to major spliceosomal RNA genes has been reported for nematodes (Vahidi et al., 1988), crustaceans (U1, U5 (Pelliccia et al., 2001; Marz et al., 2008)), molluscs (U1, U2, (Cross and Rebordinos, 2005; Vierna et al., 2011)) and fishes (U1, U2, U5 (Manchado et al., 2006)). In this work, we detected a weak correlation to U1, U4 and U5 snRNA genes, and a slightly more evolutionary consistent linkage to U2. 5S rDNA was found in many organisms linked to U6 snRNA genes, probably owing to the high repetition of this snRNA, in addition to the copy number of 5S rDNA itself. Although we were able to identify linkages to U1 snRNA genes, a consistent linkage feature between organisms was not detected. To refuse the assumption that U1 snRNA genes were not detected correctly by blast, we analyzed this gene family the same way as 5S rDNAs (Figure 1). However, we obtained the same weak results on linkage.
The splice leader (SL RNA) is known to be linked to 5S rDNA in nematodes (Nilsen et al., 1989; Zeng et al., 1990) and protists (Keller et al., 1992). In this work, we found a strong linkage between SL RNA genes and 5S rDNA in all nematodes and platyhelminthes, which has never been reported before. Linkages were found to be sense and antisense (see Supplemental Page).
Furthermore, we were able to detect linkages to many tRNA genes, as already described by Freire et al. (2010) and Vizoso et al. (2011). Linkages to 5.8S rRNA genes, SRP RNA genes, Y RNA genes, Histone3 RNA and 7SK genes were detected rarely compared with all investigated organisms (see Supplemental Page). However, the linkage of 5S rRNAs and transposable elements, such as SRP RNA genes, seems to appear at least in primates and glires.
Additionally, we found linkages between 5S rDNA and one of eight miRNAs and two snoRNAs. One example is miRNA-562, which has never been described to be linked to 5S rDNA before (Figure 7). In this case, there are nine Gaussians that form the mixture. Evidence for possible linkage can be found in primates, mouse and bat, with most other species showing either no or no consistent information. We show four linkages consistent within primates at distances of around −1648, −1221, −440 and 714 nt. A fifth linkage at a distance of 3000 nt does not seem to be as well-supported by the data. Additionally, the linkages we found between mir-562 and 5S rDNA in mammal species can be traced back to eutherians.
Figure 7.

Evolutionary-conserved linkages of 5S rRNA and mir-562. Distance between any annotated mir-562 (denoted as '>' if sense; or '<' if antisense) and any 5S rRNA (adjusted at x=0, not displayed) in animal genomes (y axis, left). The y axis (right) denotes the probability (see curve functions) that these ncRNA gene linkages are of evolutionary importance. In primates, mir-562 is located conserved upstream (−1648 nt, antisense; −1221 nt, antisense; −440 nt; sense) and downstream (714 nt, sense) of 5S rRNA. In the bat P. vampyrus, the number of 5S rRNAs is constant (28 copies, see Table 1), but an expansion of mir-562 can be detected. Interestingly, each of the bat 5S rRNAs is linked (with variable distance) to at least one of the mir-562 copies.
In the bat P. vampyrus, the number of 5S rRNAs is constant to evolutionary related organisms (28 copies, see Table 1), but an expansion of mir-562 can be detected. Interestingly, each of the bat 5S rRNAs is linked (with variable distance) to at least one of the mir-562 copies.
Although a long-lasting stable linkage of 5S RNA is known for non-animals (for example, 5S–45S linkage in Bryophytes (Wicke et al., 2011)), we were not able to find a stable linkage within metazoans.
Unfortunately, the general sparsity (as, for example, in Figure 7) of the data requires manual intervention, which is starting the EM algorithm with multiple different start values for the parameters (μ, σ) or selection between two different proposed mixtures (different number k of peaks).
We are currently working on a more extensive statistical description of linkage between genes with the aim to automate the whole decision process regarding linkage analysis of a set of genomes that should greatly simplify analyses like the present one. Details are described in Marz and Höner zu Siederdissen (2013).
Conclusion
For the first time, a comprehensive set of putatively functional 5S rDNA sequences from current metazoan genome assemblies is published. This large amount of data allowed us to study metazoan 5S rDNA diversity in great detail, following a systematic approach and from an evolutionary perspective.
Among our main conclusions, we showed that 5S rRNA-coding regions in mammals are divided into two types that we name ‘housekeeping 5S rRNAs', which are very conserved, and ‘flexible 5S rRNAs', being much more variable. In addition, we found several paralog 5S rRNA-coding sequences in many species (58 out of 97 genomes).
We also reported a flexible genome organization of 5S rDNA, as it was found either (1) in clusters, linked to other ncRNAs, (2) in homogeneous clusters, with similar NTS sequences, (3) in heterogeneous clusters, with divergent NTS sequences, (4) in clusters in which coding regions displayed opposite orientations and (5) as dispersed copies. Interestingly, several species displayed more than one of these features.
The unexpected similarity found among NTS sequences of some distantly related taxa is unclear and intriguing. On the one hand, we might hypothesize that those sequences are unidentified elements that have a molecular role in the cell and therefore have been conserved along evolution. On the other hand, they might be the result of horizontal gene transfer events.
Remarkably, even though we found 5S rDNA to be linked to several ncRNAs in many species, we failed to detect a stable linkage throughout animal evolution.
As the biological meaning of various features that were found to characterize metazoan 5S rDNA are still to be elucidated, this work opens up very interesting possibilities for molecular biology research. For example, the meaning of different promoter and terminator sequences that have been found should be unveiled.
Dimarco et al. (2012) have recently reported that different 5S rRNA variants are transcribed in a sea urchin species. In the same way, whether transcription of paralog 5S rRNA sequences reported in the present work occurs equally in all tissues deserves with no doubts to be studied in detail.
Finally, our work supports the conclusions of various recent reports (Rooney and Ward, 2005; Kalendar et al., 2008; Vierna et al., 2009; Freire et al., 2010; Perina et al., 2011; Vierna et al., 2011; Vizoso et al., 2011), in which it was shown that the evolutionary patterns of 5S rDNA in animals are complex and cannot be explained only in the light of concerted evolution. Birth-and-death processes, selection, homogenizing mechanisms typically involved in concerted evolution and horizontal gene transfer events seem to be responsible of the diversity of this multigene family in metazoans.
Data archiving
All the data can be accessed via the following link: http://www.rna.uni-jena.de/supplements/5SRNA/index.html.
Acknowledgments
JV was funded by a ‘María Barbeito' fellowship and a travel grant from Xunta de Galicia and Universidade da Coruña (Spain). MM was funded by the Carl-Zeiss-Stiftung. This work was supported in part by DFG-Graduiertenkolleg 1384 ‘Enzymes and multienzyme complexes acting on nucleic acids', DFG project MA-5082/1 (SW and MM) and by the Austrian FWF, project ‘SFB F43 RNA regulation of the transcriptome' (CHzS).
The authors declare no conflict of interest.
References
- Alkan C, Sajjadian S., Eichler EE. Limitations of next generation genome sequence assembly. Nat Methods. 2011;8:61–65. doi: 10.1038/nmeth.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:202–208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzotti R, Pelliccia F, Rocchi A. Identification and characterization of U1 small nuclear RNA genes from two crustacean isopod species. Chromosome Res. 2003;11:365–373. doi: 10.1023/a:1024048108918. [DOI] [PubMed] [Google Scholar]
- Bogenhagen DF. Proteolytic footprinting of transcription factor TFIIIA reveals different tightly binding sites for 5S RNA and 5S DNA. Mol Cell Biol. 1993;13:5149–5158. doi: 10.1128/mcb.13.9.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogenhagen DF, Brown DD. Nucleotide sequences in Xenopus 5S DNA required for transcription termination. Cell. 1981;24:261–270. doi: 10.1016/0092-8674(81)90522-5. [DOI] [PubMed] [Google Scholar]
- Branlant C, Krol A, Ebel JP, Lazar E, Haendler B, Jacob M. U2 RNA shares a structural domain with U1, U4, and U5 RNAs. EMBO J. 1982;1:1259–1265. doi: 10.1002/j.1460-2075.1982.tb00022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant D, Moulton V. Neighbor-net: an agglomerative method for the construction of phylogenetic networks. Mol Biol Evol. 2004;21:255–265. doi: 10.1093/molbev/msh018. [DOI] [PubMed] [Google Scholar]
- Cabral-de Mello DC, Moura RC, Martins C. Chromosomal mapping of repetitive DNAs in the beetle Dichotomius geminates provides the first evidence for an association of 5S rRNA and histone H3 genes in insects, and repetitive DNA similarity between the B chromosome and A complement. Heredity (Edinb) 2010;104:393–400. doi: 10.1038/hdy.2009.126. [DOI] [PubMed] [Google Scholar]
- Cabral-de-Mello DC, Moura RC, Martins C. Cytogenetic mapping of rRNAs and histone H3 genes in 14 species of Dichotomius (Coleoptera, Scarabaeidae, Scarabaeinae) beetles. Cytogenet Genome Res. 2011;134:127–135. doi: 10.1159/000326803. [DOI] [PubMed] [Google Scholar]
- Cohen S, Agmon N, Sobol O, Segal D. Extrachromoso mal circles of satellite repeats and 5S ribosomal DNA in human cells. Mobile DNA. 2010;1:1–11. doi: 10.1186/1759-8753-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland CS, Marz M, Rose D, Hertel J, Brindley PJ, Santana CB, et al. Homology-based annotation of non-coding RNAs in the genomes of Schistosoma mansoni and Schistosoma japonicum. BMC Genomics. 2009;10:464–464. doi: 10.1186/1471-2164-10-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross I, Rebordinos L. 5S rDNA and U2 snRNA are linked in the genome of Crassostrea angulata and Crassostrea gigas oysters: does the (CT)n(GA)n microsatellite stabilize this novel linkage of large tandem arrays. Genome. 2005;48:1116–1119. doi: 10.1139/g05-075. [DOI] [PubMed] [Google Scholar]
- Dalloul RA, Long JA, Zimin AV, Aslam L, Beal K, Blomberg Le Ann, et al. Multi-platform next-generation sequencing 698 of the domestic turkey (Meleagris gal lopavo): genome assembly and analysis. PLoS Biol. 2010;8:e1000475. doi: 10.1371/journal.pbio.1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels LM, Delany ME. Molecular and cytogenetic organization of the 5S ribosomal DNA array in chicken (Gallus gallus) Chromosome Res. 2003;11:305–317. doi: 10.1023/a:1024008522122. [DOI] [PubMed] [Google Scholar]
- Datson PM, Murray BG. Ribosomal DNA locus evolution in Nemesia: transposition rather than structural rearrangement as the key mechanism. Chromosome Res. 2006;14:845–857. doi: 10.1007/s10577-006-1092-z. [DOI] [PubMed] [Google Scholar]
- Dimarco E, Cascone E, Bellavia D, Caradonna F. Functional variants of 5S rRNA in the ribosomes of common sea urchin Paracentrotus lividus. Gene. 2012;508:21–25. doi: 10.1016/j.gene.2012.07.067. [DOI] [PubMed] [Google Scholar]
- Drosophila 12 Genomes Consortium. Clark AG, Eisen MB, Smith DR, Bergman CM, Oliver B, Markow TA, et al. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- Drouin G. Expressed retrotransposed 5S rRNA genes in the mouse and rat genomes. Genome. 2000;43:213–215. doi: 10.1139/g99-100. [DOI] [PubMed] [Google Scholar]
- Drouin G, Moniz de Sá M. The concerted evolution of 5S ribosomal genes linked to the repeat units of other multigene families. Mol Biol Evol. 1995;12:481–493. doi: 10.1093/oxfordjournals.molbev.a040223. [DOI] [PubMed] [Google Scholar]
- Drouin G, Tsang C. 5S rRNA gene arrangements in protists: a case of nonadaptive evolution. J Mol Evol. 2012;74:342–351. doi: 10.1007/s00239-012-9512-5. [DOI] [PubMed] [Google Scholar]
- Eickbush TH, Eickbush DG. Finely orchestrated movements: evolution of the ribosomal RNA genes. Genetics. 2007;175:477–485. doi: 10.1534/genetics.107.071399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eirin-Lopez JM, Fernanda Ruiz M, Gonzalez-Tizon AM, Martinez A, Sanchez L, Mendez J. Molecular evolutionary characterization of the mussel Mytilus histone multigene family: first record of a tandemly repeated unit of five histone genes containing an H1 subtype with ”orphon” features. J Mol Evol. 2004;58:131–144. doi: 10.1007/s00239-003-2531-5. [DOI] [PubMed] [Google Scholar]
- Freire R, Arias A, Insua AM, Méndez J, Eiŕın-López JM. Evolutionary dynamics of the 5S rDNA gene family in the mussel Mytilus: mixed effects of birth-and-death and concerted evolution. J Mol Evol. 2010;70:413–426. doi: 10.1007/s00239-010-9341-3. [DOI] [PubMed] [Google Scholar]
- Fujiwara M, Inafuku J, Takeda A, Watanabe A, Fujiwara A, Kohno S, et al. Molecular organization of 5S rDNA in bitterlings (Cyprinidae) Genetica. 2009;135:355–365. doi: 10.1007/s10709-008-9294-2. [DOI] [PubMed] [Google Scholar]
- Gardner PP, Daub J, Tate J, Moore BL, Osuch IH, Griffiths-Jones S, et al. Rfam: Wikipedia, clans and the ”decimal” release. Nucleic Acids Res. 2011;39:141–145. doi: 10.1093/nar/gkq1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie JJ, Johnston JS, Cannone JJ, Gutell RR. Characteristics of the nuclear (18S, 5.8S, 28S and 5S) and mitochondrial (12S and 16S) rRNA genes of Apis mellifera (Insecta: Hymenoptera): structure, organization, and retrotransposable elements. Insect Mol Biol. 2006;15:657–686. doi: 10.1111/j.1365-2583.2006.00689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gongadze GM. 5S rRNA and ribosome. Biochemistry. 2011;76:1450–1464. doi: 10.1134/S0006297911130062. [DOI] [PubMed] [Google Scholar]
- Gornung E, Colangelo P, Annesi F. 5S ribosomal RNA genes in six species of Mediterranean grey mullets: genomic organization and phylogenetic inference. Genome. 2007;50:787–795. doi: 10.1139/g07-058. [DOI] [PubMed] [Google Scholar]
- Gregory T.2012. Animal Genome Size Database. ( . ( http://www.genomesize.com/ ).
- Griffiths-Jones S. RALEE–RNA ALignment editor in Emacs. Bioinformatics. 2005;21:257–259. doi: 10.1093/bioinformatics/bth489. [DOI] [PubMed] [Google Scholar]
- Gromicho M, Coutanceau JP, Ozouf-Costaz C, Collares-Pereira MJ. Contrast between extensive variation of 28S rDNA and stability of 5S rDNA and telomeric repeats in the diploid-polyploid Squalius alburnoides complex and in its maternal ancestor Squalius pyrenaicus (Teleostei, Cyprinidae) Chromosome Res. 2006;14:297–306. doi: 10.1007/s10577-006-1047-4. [DOI] [PubMed] [Google Scholar]
- Hastie T, Tibshirani R, Friedman J.(eds). (2001The Elements of Statistical Learning — Data Mining, Inference, and Prediction,2nd edn. Springer.
- Hall TM. Multiple modes of RNA recognition by zinc finger proteins. Curr Opin Struc Biol. 2005;15:367–373. doi: 10.1016/j.sbi.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Hallenberg C, Frederiksen S. Effect of mutations in the upstream promoter on the transcription of human 5S rRNA genes. Biochim Biophys Acta. 2001;1520:169–173. doi: 10.1016/s0167-4781(01)00264-0. [DOI] [PubMed] [Google Scholar]
- Hallenberg C, Frederiksen S. Effect of mutations in the up stream promoter on the transcription of human 5S rRNA genes. Biochim Biophys Acta. 2001;1520:169–173. doi: 10.1016/s0167-4781(01)00264-0. [DOI] [PubMed] [Google Scholar]
- Hilder VA, Dawson GA, Vlad MT. Ribosomal 5S genes in relation to C-value in amphibians. Nucleic Acids Res. 1983;11:2381–2390. doi: 10.1093/nar/11.8.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker IL. RNA consensus structure prediction with RNAalifold. Methods Mol Biol. 2007;395:527–544. doi: 10.1007/978-1-59745-514-5_33. [DOI] [PubMed] [Google Scholar]
- Huang Y, Maraia RJ. Comparison of the RNA polymerase III transcription machinery in Schizosaccharomyces pombe, Saccha769 romyces cerevisiae and human. Nucleic Acids Res. 2001;29:2675–2690. doi: 10.1093/nar/29.13.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH. SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics. 1998;14:68–73. doi: 10.1093/bioinformatics/14.1.68. [DOI] [PubMed] [Google Scholar]
- Jensen LR, Frederiksen S. The 5S rRNA genes in Macaca fascicularis are organized in two large tandem repeats. Biochim Bio phys Acta. 2000;1492:537–542. doi: 10.1016/s0167-4781(00)00139-1. [DOI] [PubMed] [Google Scholar]
- Kalendar R, Tanskanen J, Chang W, Antonius K, Sela H, Peleg O, et al. Cassandra retrotransposons carry indepen dently transcribed 5S RNA. Proc Natl Acad Sci USA. 2008;105:5833–5838. doi: 10.1073/pnas.0709698105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller M, Tessier LH, Chan RL, Weil JH, Imbault P. In Euglena, spliced-leader RNA (SL-RNA) and 5S rRNA genes are tandemly repeated. Nucleic Acids Res. 1992;20:1711–1715. doi: 10.1093/nar/20.7.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiya H, Kawakami M, Takemura S. Nucleotide sequence of 5S ribosomal RNA from the posterior silk glands of Bombyx mori. J Biochem. 1981;89:717–722. doi: 10.1093/oxfordjournals.jbchem.a133251. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Layat E, Sáez-Vásquez J, Tourmente S. Regulation of Pol I-transcribed 45S rDNA and Pol III-transcribed 5S rDNA in Arabidopsis. Plant Cell Physiol. 2012;53:267–276. doi: 10.1093/pcp/pcr177. [DOI] [PubMed] [Google Scholar]
- Little RD, Braaten BC. Genomic organization of human 5S rDNA and sequence of one tandem repeat. Genomics. 1989;4:376–383. doi: 10.1016/0888-7543(89)90345-5. [DOI] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Searles MA, Klug A. Crystal structure of a zinc finger-RNA complex reveals two modes of molecular recognition. Nature. 2003;426:96–100. doi: 10.1038/nature02088. [DOI] [PubMed] [Google Scholar]
- Manchado M, Zuasti E, Cross I, Merlo A, Infante C, Rebordinos L. Molecular characterization and chromosomal mapping of the 5S rRNA gene in Solea senegalensis: a new linkage to the U1, U2, and U5 small nuclear RNA genes. Genome. 2006;49:79–86. doi: 10.1139/g05-068. [DOI] [PubMed] [Google Scholar]
- Martins C, Wasko AP. Organization and Evolution of 5S Ribosomal DNA in the Fish Genome. Nova Science Publishers, Inc; 2004. [Google Scholar]
- Marz M, Höner zu Siederdissen C.2013Statistical approach for evolutionary gene linkage detection(in preparation).
- Marz M, Kirsten T, Stadler PF. Evolution of spliceosomal snRNA genes in metazoan animals. J Mol Evol. 2008;67:594–607. doi: 10.1007/s00239-008-9149-6. [DOI] [PubMed] [Google Scholar]
- Nelson DW, Linning RM, Davison PJ, Honda BM. 5′-flanking sequences required for efficient transcription in vitro of 5{S} {RNA} genes, in the related nematodes Caenorhabditis elegans} and Caenorhabditis briggsae. Gene. 1998;218:9–16. doi: 10.1016/s0378-1119(98)00392-8. [DOI] [PubMed] [Google Scholar]
- Nei M, Rooney AP. Concerted and birth-and-death evolution of multigene families. Annu Rev Genet. 2005;39:121–152. doi: 10.1146/annurev.genet.39.073003.112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW, Shambaugh J, Denker J, Chubb G, Faser C, Putnam L, et al. Characterization and expression of a spliced leader RNA in the parasitic nematode Ascaris lumbricoides var. suum. Mol Cell Biol. 1989;9:3543–3547. doi: 10.1128/mcb.9.8.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelliccia F, Barzotti R, Bucciarelli E, Rocchi A. 5S ribosomal and U1 small nuclear RNA genes: a new linkage type in the genome of a crustacean that has three different tandemly repeated units containing 5S ribosomal DNA sequences. Genome. 2001;44:331–335. doi: 10.1139/gen-44-3-331. [DOI] [PubMed] [Google Scholar]
- Perina A, Seoane D, González-Tizón AM, Rodŕıguez-Fariña F, Mart́ınez-Lage A. Molecular organization and phylogenetic analysis of 5S rDNA in crustaceans of the genus Pollicipes reveal birth- and death evolution and strong purifying selection. BMC Evol Biol. 2011;11:304–304. doi: 10.1186/1471-2148-11-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieler T, Hamm J, Roeder RG. The 5S gene internal control region is composed of three distinct sequence elements, organized as two functional domains with variable spacing. Cell. 1987;48:91–100. doi: 10.1016/0092-8674(87)90359-x. [DOI] [PubMed] [Google Scholar]
- Pieler T, Hamm J, Roeder RG. The 5S gene internal control region is composed of three distinct sequence elements, organized as two functional domains with variable spacing. Cell. 1987;48:91–100. doi: 10.1016/0092-8674(87)90359-x. [DOI] [PubMed] [Google Scholar]
- Query CC, Bentley RC, Keene JD. A specific 31-nucleotide domain of U1 RNA directly interacts with the 70K small nuclear ribonucleoprotein component. Mol Cell Biol. 1989;9:4872–4881. doi: 10.1128/mcb.9.11.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard P, Manley JL. Transcription termination by nuclear RNA polymerases. Gene Dev. 2009;23:1247–1269. doi: 10.1101/gad.1792809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney AP, Ward TJ. Evolution of a large ribosomal RNA multigene family in filamentous fungi: birth and death of a concerted evolution paradigm. Proc Natl Acad Sci USA. 2005;102:5084–5089. doi: 10.1073/pnas.0409689102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherly D, Boelens W, van Venrooij WJ, Dathan NA, Hamm J, Mattaj IW. Identification of the RNA binding segment of human U1 A protein and definition of its binding site on U1 snRNA. EMBO J. 1989;8:4163–4170. doi: 10.1002/j.1460-2075.1989.tb08601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnare MN, Gray MW. A candidate U1 small nuclear RNA for trypanosomatid protozoa. J Biol Chem. 1999;274:23691–23694. doi: 10.1074/jbc.274.34.23691. [DOI] [PubMed] [Google Scholar]
- Scripture JB, Huber PW. Analysis of the binding of Xenopus ribosomal protein L5 to oocyte 5 S rRNA. The major determinants of recognition are located in helix III-loop C. J Biol Chem. 1995;270:27358–27365. doi: 10.1074/jbc.270.45.27358. [DOI] [PubMed] [Google Scholar]
- Shambaugh JD, Hannon GE, Nilsen TW. The spliceosomal U small nuclear RNAs of Ascaris lumbricoides. Mol Biochem Parasit. 1994;64:349–352. doi: 10.1016/0166-6851(94)00040-9. [DOI] [PubMed] [Google Scholar]
- Sharp S, Garcia A, Cooley L, Söll D. Transcriptionally active and inactive gene repeats within the D. melanogaster 5S RNA gene cluster. Nucleic Acids Res. 1984;20:7617–7632. doi: 10.1093/nar/12.20.7617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp SJ, Garcia AD. Transcription of the Drosophila melanogaster 5S RNA gene requires an upstream promoter and four intragenic sequence elements. Mol Cell Biol. 1988;8:1266–1274. doi: 10.1128/mcb.8.3.1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp SJ, Garcia AD. Transcription of the Drosophila melanogaster 5S RNA gene requires an upstream promoter and four intragenic sequence elements. Mol Cell Biol. 1988;8:1266–1274. doi: 10.1128/mcb.8.3.1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shippen-Lentz DE, Vezza AC. The three 5S rRNA genes from the human malaria parasite Plasmodium falciparum are linked. Mol Biochem Parasit. 1988;27:263–273. doi: 10.1016/0166-6851(88)90046-1. [DOI] [PubMed] [Google Scholar]
- Smirnov AV, Entelis NS, Krasheninnikov IA, Martin R, Tarassov IA. Specific features of 5S rRNA structure - its interactions with macromolecules and possible functions. Biochemistry. 2008;73:1418–1437. doi: 10.1134/s000629790813004x. [DOI] [PubMed] [Google Scholar]
- Sorensen PD, Frederiksen S. Characterization of human 5S rRNA genes. Nucleic Acids Res. 1991;19:4147–4151. doi: 10.1093/nar/19.15.4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syvanen M. Evolutionary implications of horizontal gene transfer. Annu Rev Genet. 2012;46:341–358. doi: 10.1146/annurev-genet-110711-155529. [DOI] [PubMed] [Google Scholar]
- Thomas J, Lea K, Zucker-Aprison E, Blumenthal T. The spliceosomal snRNAs of Caenorhabditis elegans. Nucleic Acids Res. 1990;18:2633–2642. doi: 10.1093/nar/18.9.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler BM. Transcription of Neurospora crassa 5 S rRNA genes requires a TATA box and three internal elements. J Mol Biol. 1987;196:801–811. doi: 10.1016/0022-2836(87)90406-2. [DOI] [PubMed] [Google Scholar]
- Vahidi H, Curran J, Nelson DW, Webster JM, McClure MA, Honda BM. Unusual sequences, homologous to 5S RNA, in ribosomal DNA repeats of the nematode Meloidogyne arenaria. J Mol Evol. 1988;27:222–227. doi: 10.1007/BF02100078. [DOI] [PubMed] [Google Scholar]
- Vierna J, González-Tizón AM, Mart́ınez-Lage A. Long-term evolution of 5S ribosomal DNA seems to be driven by birth-and-death and selection in Ensis razor shells (Mollusca: Bivalvia) Biochem Genet. 2009;47:635–644. doi: 10.1007/s10528-009-9255-1. [DOI] [PubMed] [Google Scholar]
- Vierna J, Jensen KT, Mart́ınez-Lage A, González-Tizón AM. The linked units of 5S rDNA and U1 snDNA of razor shells (Mollusca: Bivalvia: Pharidae) Heredity. 2011;107:127–142. doi: 10.1038/hdy.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizoso M, Vierna J, González-Tizón AM, Mart́ınez-Lage A. The 5S rDNA gene family in mollusks: characterization of transcriptional regulatory regions, prediction of secondary structures, and long-term evolution, with special attention to Mytilidae mussels. J Hered. 2011;102:433–447. doi: 10.1093/jhered/esr046. [DOI] [PubMed] [Google Scholar]
- Wicke S, Costa A, Muñoz J, Quandt D. Restless 5S: the re-arrangement(s) and evolution of the nuclear ribosomal DNA in land plants. Mol Phylogenet Evol. 2011;61:321–332. doi: 10.1016/j.ympev.2011.06.023. [DOI] [PubMed] [Google Scholar]
- Zeng WL, Alarcon CM, Donelson JE. Many transcribed regions of the Onchocerca volvulus genome contain the spliced leader sequence of Caenorhabditis elegans. Mol Cell Biol. 1990;10:2765–2773. doi: 10.1128/mcb.10.6.2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Y, Weiner AM. A compensatory base change in U1 snRNA suppresses a 5' splice site mutation. Cell. 1986;46:827–835. doi: 10.1016/0092-8674(86)90064-4. [DOI] [PubMed] [Google Scholar]
- Úbeda-Manzanaro M., Merlo M. A., Palazón J. L., Sarasquete C., Rebordinos L. Sequence characterization and phylogenetic analysis of the 5S ribosomal DNA in species of the family Batrachoididae. Genome. 2010;53:723–730. doi: 10.1139/g10-048. [DOI] [PubMed] [Google Scholar]
- Ørum H, Nielsen H, Engberg J. Spliceosomal small nuclear RNAs of Tetrahymena thermophila and some possible snRNA-snRNA base-pairing interactions. J Mol Biol. 1991;222:219–232. doi: 10.1016/0022-2836(91)90208-n. [DOI] [PubMed] [Google Scholar]