

Abstract
Detailed studies of the genetics of speciation have focused on a few model systems, particularly Drosophila. The copepod Tigriopus californicus offers an alternative that differs from standard animal models in that it lacks heteromorphic chromosomes (instead, sex determination is polygenic) and has reduced opportunities for sexual conflict, because females mate only once. Quantitative trait loci (QTL) mapping was conducted on reciprocal F2 hybrids between two strongly differentiated populations, using a saturated linkage map spanning all 12 autosomes and the mitochondrion. By comparing sexes, a possible sex ratio distorter was found but no sex chromosomes. Although studies of standard models often find an excess of hybrid male sterility factors, we found no QTL for sterility and multiple QTL for hybrid viability (indicated by non-Mendelian adult ratios) and other characters. Viability problems were found to be stronger in males, but the usual explanations for weaker hybrid males (sex chromosomes, sensitivity of spermatogenesis, sexual selection) cannot fully account for these male viability problems. Instead, higher metabolic rates may amplify deleterious effects in males. Although many studies of standard speciation models find the strongest genetic incompatibilities to be nuclear–nuclear (specifically X chromosome–autosome), we found the strongest deleterious interaction in this system was mito–nuclear. Consistent with the snowball theory of incompatibility accumulation, we found that trigenic interactions in this highly divergent cross were substantially more frequent (>6 × ) than digenic interactions. This alternative system thus allows important comparisons to studies of the genetics of reproductive isolation in more standard model systems.
Keywords: QTL, reproductive isolation, speciation, copepod, nuclear–mitochondrial interaction
Introduction
The most accepted explanation for the evolution of postzygotic isolation is the accumulation of Dobzhansky–Muller Incompatibilities (Coyne and Orr, 2004). Between isolated populations, different alleles arise through mutation or migration and go to fixation by drift or selection. Although these alleles are neutral or adaptive in their own genetic background, epistatic interactions within a hybrid background may be deleterious (Coyne and Orr, 2004). As populations accumulate more fixed differences, the number of potential digenic interactions between novel alleles increases as the square of the number of substitutions—the so-called snowball effect (Orr, 1995). The number of higher-order interactions is expected to increase even faster (Orr, 1995), although higher-order epistasis contributing to the onset of reproductive isolation—as opposed to accumulating after isolation is already established—has very rarely been tested (Coyne and Orr, 2004; but see Rieseberg et al., 1996).
Because selection can lead to rapid fixation of different alleles, genes involved in processes resulting in the most differentiation due to selection seem to have a disproportionate role in hybrid breakdown. For example, mechanisms underlying hybrid breakdown are often attributed to genetic conflicts and their resolution (Arnqvist et al., 2000; Presgraves, 2010). These conflicts are largely determined by the details of the particular breeding and sex determination systems. The speciation literature in animals has focused almost exclusively on heterogametic species. In fact, three often cited ‘rules' of speciation involve conflict between heteromorphic sex chromosomes and autosomes. The first, Haldane's Rule, is the observation that the heterogametic sex is more likely to suffer sterility or inviability in hybrids. This pattern holds for a broad range of taxa, including both XY and ZW systems (Coyne and Orr, 1989; Schilthuizen et al., 2011). The second ‘rule' of speciation, the Large X effect, postulates that genes underlying speciation tend to localize disproportionately to the X (or Z) chromosome (Coyne and Orr, 1989, 2004; Turelli and Moyle, 2007). A third ‘rule', sometimes known as Darwin's Corollary to Haldane's Rule (Turelli and Moyle, 2007), is that hybrid crosses frequently result in asymmetric incompatibility. This pattern could be driven by interactions involving the X (or Z) chromosome, as well as a number of other uniparentally inherited factors, including mitochondria and plastids (Turelli and Moyle, 2007; Burton and Barreto, 2012).
Importantly, a great number of taxa—from plants to many invertebrates to widespread representatives of many vertebrate groups, such as fish, amphibians and reptiles—have non-heterogametic mechanisms of sex determination. For these taxa, two of the major ‘rules' of speciation, Haldane's Rule and the Large X effect, cannot apply. Such taxa might therefore be predicted to show slower accumulation of incompatibilities, particularly those causing sterility, with greater symmetry between sexes (Phillips and Edmands, 2012). It is just beginning to be possible to test these predictions with the recent development of models lacking heteromorphic sex chromosomes, such as Helianthus, Mimulus, Solanum, Microbotryum and Saccharomyces (for example, Lowry et al., 2008; Rieseberg and Blackman, 2010; Giraud and Gourbière, 2012). Although firm conclusions would be premature, studies in some of these alternative model systems have shown comparatively modest numbers of incompatibilities (Lowry et al., 2008) and little disparity in the evolution of male vs female sterility (Moyle and Nakazato, 2008).
Another alternative model is Tigriopus californicus, a copepod that inhabits supralittoral splash pools from Alaska to Baja California. T. californicus has extremely limited gene flow, with extensive genetic divergence between even geographically close populations (for example, Burton and Lee, 1994). Interpopulation hybridization typically results in mild hybrid vigor in the F1 generation, across all levels of genetic divergence, and hybrid breakdown in the F2 and backcross generations, which increases at higher levels of divergence (for example, Edmands, 1999). T. californicus does not have heteromorphic sex chromosomes (Ar-Rushdi, 1963)—instead, sex determination appears to be polygenic (Voordouw and Anholt, 2002). That is, multiple unlinked loci are believed to contribute to sex determination within populations. Although the number of loci contributing to sex determination is unknown, it is thought to be on the order of 5–8 per population (B Anholt, personal communication), and different populations may be fixed or segregating for sex determination alleles at alternate loci. Similarly, the degree of dominance and epistasis for sex determination is as yet uncharacterized in this system.
T. californicus also differs from more commonly studied speciation models in that it has reduced opportunities for intra- and inter-sexual conflict. In T. californicus, males clasp and guard immature females until the females are ready to mate, and females only mate a single time, producing multiple clutches of eggs from stored sperm (Burton, 1985). This potentially reduces other selective pressures, which may drive reproductive isolation. Intrasexual conflict (such as sperm competition) is limited, reducing opportunities for faster male evolution (Wu and Davis, 1993). The scope for post-mating intersexual conflict is also limited, as both sexes share an interest in maximizing the number of viable offspring produced from each mating, thus eliminating a subset of reasons for arms races between the sexes. Consistent with these limits on sexual conflict, as well as the absence of heteromorphic sex chromosomes, a previous study of the species reported a lack of elevated hybrid male sterility, relative to hybrid female sterility (Willett, 2008).
In the absence of sex chromosome–autosome conflicts and with limited potential for sexual conflict, another potential driver of hybrid breakdown in T. californicus is mitochondrial–nuclear conflict (reviewed in Burton and Barreto, 2012). Given a higher mutation rate than the nuclear genome (particularly in T. californicus; Willett, 2012) and smaller effective population size, mitochondrial genomes can rapidly diverge due to drift. Nuclear genes interacting with the mitochondrial genome are expected to evolve rapidly as well, to compensate for incompatibilities, potentially leading to the accumulation of Dobzhansky–Muller Incompatibilities (Burton and Barreto, 2012). Some interactions between candidate nuclear genes and the mitochondria have been found (Burton and Barreto, 2012), but the overall contribution of mitonuclear to hybrid breakdown has not been tested at a genome-wide level.
One pair of Californian populations, Santa Cruz (SC) and San Diego (SD), has been the subject of concerted efforts to characterize these patterns. Extensive genomic resources have been developed using these populations, including a published transcriptome (Barreto et al., 2011) and a saturated F2 linkage map (Foley et al., 2011), containing several genes of interest, including nuclear genes proposed to interact with the mitochondria (for example, Burton and Barreto, 2012). A previous study (Pritchard et al., 2011) assessed genotypic ratios in SC × SD F2 males from a unidirectional cross (that is, a single mitochondrial background), using an incomplete linkage map, and found several regions with significant distortions.
The current study differs from Pritchard et al. (2011) in that it includes both reciprocal crosses and both sexes. It maps not just Mendelian ratio distortions but also sterility, developmental and morphological traits. It also uses a much larger number of individuals and a new, saturated linkage map (Foley et al., 2011). By using genome-wide markers and a large number of individuals, we compare patterns of two- vs three-locus epistasis in segregation distortion, something which has never been done in Tigriopus and has rarely been done in any other system. By comparing reciprocal crosses, we provide the first genome-wide screen for nuclear–mitochondrial interactions impacting hybrid breakdown in this species. By comparing males and females, we offer the first genomic assessment of sex-specific hybridization effects in this species, addressing how gender differences manifest themselves in the absence of sex chromosomes. By mapping regions underlying a series of traits, we assess whether T. californicus shows a pattern found in other taxa, in which genetic conflicts driving sterility accumulate faster and are more sex-specific than those underlying other characters. In these ways the current study provides the first comprehensive assessment of how the genetic architecture of postzygotic isolation in T. californicus compares with that in more standard model organisms.
Materials and methods
Quantitative trait loci (QTL) mapping was done using adult F2 hybrids from reciprocal crosses between the SC and SD populations. A previous study used F2 larvae from one of these two reciprocal crosses to construct a linkage map (Foley et al., 2011). Details of the laboratory culture conditions, crossing protocol and marker development are described in Foley et al. (2011) and will be summarized briefly here.
Crossing protocol and phenotypic measurements, including survivorship and sex ratio
In order to create the F2 mapping population, we first set up reciprocal crosses between SD and SC isofemale lines. Before crosses began, isofemale lines were maintained in mass culture for 2.5 years, in a standard seawater, Tetramin (fish food) and Spirulina (cyanobacterium) medium at 20 °C. Reciprocal crosses between the lines were conducted in polyethylene containers (Gladware) filled with 400 ml of culture medium, each with 50 males and 50 virgin females. The males and virgin females were identified and collected by separating pre-copulatory clasped pairs. Parental controls, as a baseline for mortality and sex-ratio estimates, were conducted in the same fashion. In the absence of heteromorphic sex chromosomes, the primary differential contribution to these reciprocal crosses is expected to be cytoplasmic—in particular the mitochondria. Reciprocal crosses are denoted as SCmit and SDmit for SC and SD mitochondrial backgrounds, respectively.
Males were removed after 1 week, and females were removed after ∼2 weeks, when copepodids (juveniles) were observed. These F1 progeny were allowed to mature and, as clasped pairs were observed, they were dissected apart and paired with a new partner of the same cross type, descended from a different container (to avoid inbreeding). One male and one virgin female were placed in each Petri plate (60 × 15 mm2, 12 ml culture medium). F1 × F1 crosses were generated in two blocks, with 17 days in between. Males were removed once an eggsac was observed.
For the first 40 F1 females from each reciprocal, the first clutch of F2 offspring was used to assess the total number of live larvae, 30-day larval mortality and the sex ratio of progeny. Similar data were collected from parental and F1 control crosses at the same time. We tested for differences among crosses and strains in survivorship and sex ratio using a χ2 test in R, (chi square Test (stats)). In each case, we tested the counts of the smaller class, using the proportions of the numerically larger sample as the expectation. We considered results to be significantly different at P<0.05.
On the day of hatching, 250 nauplii (larvae) from the first clutches of F2 offspring were collected for genotyping and the construction of the genetic linkage map (Foley et al., 2011). F1 females were removed to fresh plates as subsequent clutches of F2s hatched. The second clutch of F2 offspring was used to obtain individual genotypes and phenotypes for QTL mapping. Male development time (Dev) was taken as the date (from hatching) at which a male clasped a female. On the same day clasping was recorded, clasped pairs were separated. To test fertility (Fert) each male was placed in a 12-ml petri plate with three SD virgin females, and each female in a petri plate with three SD males. Fertility tests were done only with SD because preliminary assays showed no hybrid breakdown when reciprocal F2 were backcrossed to SD. Male fertility was scored as a binomial variable as the production, or not, of offspring. Female development time was scored as her age when her first eggsac was produced, and her fertility was scored as a binomial variable as her production, or not, of offspring.
After fertility was established, morphometric measurements were taken. We took photographs of individuals with a Sony CCD-IRIS/RGB video camera (Sony Corporation, Park Ridge, NJ, USA) mounted on a Leica MZ12 dissection scope (Leica Microsystems, Wetzlar, Germany) at × 32 magnification. We scored body size metrics cephalothorax length and width for both sexes. For females, we also scored eggsac length and area (ESA). For males, we also scored the width of the clasper (Clasper), a specialized enlarged structure at the end of male antennules. Body size metrics were scored by the ImageJ freeware (National Institutes of Health, Bethesda, MD, USA; Abramoff et al., 2004).
Significant block effects in F2 individuals were found for all traits measured (data not shown). All morphological and life history traits for block 1 individuals were therefore mean-corrected, within sex and mitochondrial background, before any subsequent analyses were performed. Ultimately, genotype and phenotype data were collected from 1514 individuals: 375 SCmit females (SCmitF), 362 SCmit males (SCmitM), 394 SDmit females (SDmitF), and 383 SDmit males (SDmitM). For brevity, only the major, relevant, results of the morphological and developmental analysis are presented below, and the detailed results are reported in Supplementary Materials.
Genotyping
After each individual was photographed, they were preserved for future DNA extraction. Individuals were rinsed briefly in diH2O and blotted dry on filter paper. Eggsacs were removed from females to prevent contamination by paternal DNA. We have previously established that the DNA in stored sperm is not sufficient to contaminate female samples (Pritchard and Edmands, 2013). Individuals were stored at −80 °C in 200-μl PCR tubes until all samples were collected.
To each vial, 50 μl of lysis buffer (10 mM Tris pH 8.3, 50 mM KCl, 0.5% Tween 20, and 200 μg proteinase K per ml buffer was added. The vials were incubated at 65 °C for 1 h, followed by 100 °C for 15 min. Samples were split into 20- and 30-μl aliquots. The 20-μl aliquots were dried down at 65 °C for 4 h and then sent to the Roswell Park Cancer Institute for genotyping. The 30-μl aliquots were archived at −80 °C in case some individuals needed to be re-genotyped.
In total, 73 nuclear single-nucleotide polymorphism markers for QTL mapping (Figure 1) were selected from among the 190 mapped markers of Foley et al. (2011). These markers were selected at the ends of chromosomes, and at relatively even distances between. A particular effort was made to retain markers developed within candidate genes known to interact with the mitochondrial genome (Willett and Burton, 2004; Burton and Barreto, 2012). Primers and extension sequences were developed by Jeffrey Conroy using the iPlex Gold software (Sequenom, San Diego, CA, USA). Sequences were automatically scored using the iPlex Gold Assay on the MassARRAY Compact (Sequenom) mass spectrometer. All F2 individuals used for QTL mapping were scored. Additionally, eight males and eight females from each of the parental populations and reciprocal F1 crosses were simultaneously genotyped to ensure marker classes were being scored accurately.
Figure 1.

Linkage map used in QTL analysis of T. californicus. Five candidate genes for nuclear–mitochondrial interactions are underlined: TcCYC (cytochrome C), RPOL (mitochondrial RNA polymerase), TcTFB1 (mitochondrial transcription factor B1), TcRISP (mitochondrial rieske iron-sulfur protein) and TcTFAM (transcription factor A, mitochondrial). Marker distances were taken from Foley et al. (2011).
Sex and mitochondrial effects on non-Mendelian ratios (NMR)
Analyses were conducted in R 2.12.0 (R Development Core Team, 2010), unless otherwise noted. At each marker, we tested for the significance of the effects of sex and mitochondrial background on departure from a 1:2:1 genotypic ratio (SC-homozygote:heterozygote:SD-homozygote). We examined these in a categorical regression framework in R vglm (VGAM), with the model family specification ‘multinomial' appropriate for a non-ordered nominal response variable. Model selection was performed among the models of sex, cross, sex+cross and sex+cross+sex × cross, using Wilks' Generalized Likelihood Ratio Test. The simplest, best-fit model was retained. To correct for multiple testing, we used a Bonferroni threshold of P<6.85E-4, assuming 73 independent tests.
Mapping NMR
NMR loci were mapped in SAS 9.2 (SAS Institute Inc, Carey, NC, USA) package QTL (Hu and Xu, 2009). It is not possible to conduct a permutation analysis for determining significance thresholds for ratio distortion of marker classes without simulating QTL genotypes for the ‘missing' individuals. We therefore used the mathematical approach of Piepho (2001). Briefly, this method scales the significance threshold to the sum of the absolute changes in the Likelihood Ratio Statistic scores. The more variance in the Likelihood Ratio Statistic between markers across the genome, the higher the threshold will be. We calculated significance thresholds in two ways. We estimated a genome-wide threshold, and—because this might be unduly biased by an extremely high peak on a single chromosome, which might disguise more modest effects elsewhere—we also calculated chromosome-specific thresholds.
To interpret the direction of the QTL effect, we plotted the proportion of SC homozygotes relative to SD homozygotes at each single-nucleotide polymorphism—log scaled—as well as the heterozygote. We also plotted the expected fitness of the heterozygote as the average of the two homozygote classes (this is the additive expectation), and assessed departures from additivity using a χ2 test. A two LOD (logarithm of the odds) drop-off from the QTL maximum was taken to delineate the 95% confidence interval of the QTL (Lynch and Walsh, 1998). Where there are two, non-overlapping, 95% confidence intervals we may infer two potentially separate QTL on the same chromosome.
Developmental, reproductive and morphometric QTL
All phenotype data were mean-corrected for block effects, and Box-Cox transformed for normality in the boxcox (Mass) package in R, unless the 95% confidence interval around the suggested power parameter, lambda, did not overlap with 1 (Venables and Ripley, 2002). All phenotypes, including sex, were mapped using the Multiple QTL Mapping function (Arends et al., 2010) in R, package qtl (Broman et al., 2003) after testing for sex (where appropriate) and cross-specific effects as well as epistasis in scantwo (qtl). Like composite interval mapping, Multiple QTL Mapping incorporates marker covariates into the Haley–Knott regression framework to reduce spurious QTL (Arends et al., 2010). Genotypes were imputed using a Kosambi map function at 1-cM intervals. We mapped QTL using a dominance model, with significance thresholds determined by 1000 permutations. We mapped cephalothorax length, cephalothorax width, fertility and development in both sexes; eggsac length and eggsac area in females; clasper size in males; and sex (Supplementary Table S1). We calculated correlation (Pearson's) between these traits, within sex and mitochondrial background. We attempted dimension reduction and tested the performance of principal components (data not shown), as well as a composite ‘body size' estimator. However, we found no increase in our ability to identify QTL using this approach, and we therefore report the direct trait measurements.
Locus-specific mortality estimates
We estimated minimum locus-specific probability of survivorship at each of our NMR QTL following Luo and Xu (2003), modified slightly, to account for three marker classes per locus as opposed to four, as we did not distinguish between alleles according to maternal or paternal origin. We considered only the single-most distorted locus per chromosome, apart from on chromosome 1 where there is evidence for two independent distortion loci. Briefly, we know from the larval data (Foley et al., 2011) that genotypic ratios at hatching do not differ from Mendelian (1:2:1) expectations. We report the larval genotype frequencies from Foley et al. (2011) in the top panel of Figure 2 for the subset of markers used in the current study to illustrate the shift in genotype frequencies from larvae to adults. We used this Mendelian expectation as our pre-mortality baseline estimate of genotypic class frequencies.
Figure 2.

Bar plots of the relative frequencies of SD homozygous (light gray), SC homozygous (dark gray) and heterozygous (black) individuals at each marker locus across the genome; for the linkage-mapping population of nauplii (from Foley et al., 2011) and by sex and mitochondrial background for the F2 QTL population. *0.05<P>0.01 after Bonferroni correction; **0.01<P>0.0001 after Bonferroni correction; ***P<0.0001 after Bonferroni correction, asterisks vertical over bars.
For each distortion locus, we identified which of the three genotype classes was most
overabundant given the Mendelian expectation. To estimate the marginal minimum
pre-mortality population size, for each locus we assumed zero mortality for the most
overabundant class. From the number of individuals of this class, we estimated the
predicted numbers of the other two classes necessary to obtain the undistorted 1:2:1
ratio. The sum of these was our locus-specific estimate of pre-mortality population
size. Survivorship 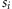 , due
to genotype at locus
, due
to genotype at locus 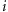 ,
was the estimated pre-mortality population size divided by the observed population
size.
,
was the estimated pre-mortality population size divided by the observed population
size.
Epistasis in NMR
To test for epistasis in NMR for pairs of marker loci, we calculated the expectation of two-locus genotypes conditioned on the frequency of genotypic classes at each individual locus. We opted for this expectation as it is conservative and unlikely to confound interlocus effects with intralocus patterns of NMR. We then tested whether this expectation deviated from the actual occurrence of genotypes using χ2. Only between-chromosome comparisons were considered. We retained the most significant P-value for each chromosome pair.
Higher-order epistasis in NMR
We estimated the significance of three-way epistasis for NMR as above, using the table(base) function in R. We conservatively disregarded any three-way interactions, which included chromosomal pairs previously detected as significant for two-way epistasis, as it would not be clear whether the significance was being driven by the strength of the two-way interaction or by additional, more complex epistasis. We could not estimate three-way epistasis among the four sex × marker classes individually, because too many cells in the contingency tables were represented by too few individuals, so we tested instead for three-way interactions among all individuals, among males and females separately and for the mitochondrial backgrounds separately. As we were particularly interested in mitochondrial–nuclear interactions a priori, we examined mitochondrial × two-way nuclear interactions separately from three-way nuclear interactions.
In order to evaluate the significance of these results, we utilized Bonferroni correction for multiple testing. When testing multiple linked loci, the number of markers can greatly overestimate the number of independent tests. In the case of higher degrees of epistasis, this overestimation is exponential. We used the method of Cheverud (2000) to account for multiple testing among linked markers and assumed two independent tests per chromosome for the purposes of determining our threshold. As our longest chromosome is 34.8-cM long, this was a conservative estimate. We disregarded all comparisons that involve markers on the same chromosome, which were necessarily in disequilibrium.
Results
Survivorship and sex ratio
Survivorship was low in the F2 (SCmit: 0.26, SE=0.05; SDmit: 0.29, SE=0.04) and moderate in the F1 (SCmit: 0.48, SE=0.08; SDmit: 0.53, SE=0.07) and parentals (SC: 0.51, SE=0.07; SD: 0.42, SE=0.08). The F2 values were significantly lower than all other cohorts. The proportion of females was high in the F2 (SCmit: 0.66, SE=0.06; SDmit 0.64, SE=0.06) and one F1 cohort (SDmit: 0.62, SE=0.07), moderate in parental SD (0.50, SE=0.07) and one F1 cohort (SCmit: 0.49, SE=0.07) and low in parental SC (0.26, SE=0.06). Proportions were significantly lower in F1 SCmit compared with both F2 cohorts, in SD parentals compared with one F2 cohort (SDmit) and in SC parentals compared with all other cohorts. If we assume a 1:1 ‘genetic' sex ratio at hatching, our empirical estimates of larval survival and adult sex ratio suggest sex-specific survival rates of 0.34 for females and 0.17 for males for SCmit and 0.37 and 0.21 for females and males, respectively, for SDmit.
Sex and mitochondrial effects on NMR
As previously reported (Foley et al., 2011 and Figure 2), no evidence of NMR was found at any of the 190 loci screened in 250 newly hatched nauplii (larvae) used to construct the T. californicus linkage map. This rules out pre-hatching mortality, differential gametic fertilization success and meiotic drive as potential mechanisms for NMR. Because we were unable to phenotypically or genetically determine the sex of nauplii, we could not measure sex-ratio distortion at hatching. Any NMR measured in the adults across the sexes must have been due to genetic mortality, but adult single-sex NMR may have been due to a combination of sex-ratio distortion and larval mortality. In adults, NMR was evident overall (Figure 2). Some combination of sex and cross (mitochondrial background) interactions for NMR were significant at 9 out of 12 chromosomes (Table 1). Because of the significance of sex and mitochondrial effects, we divided our mapping population into four classes on the basis of sex and mitochondrial background for subsequent QTL mapping.
Table 1. Significance of sex, cross, and the full model, including sex, cross and their interaction term on patterns of non-Mendelian ratios in T. californicus genetic markers.
| Chr | Marker position (cM) | Model | Δ log likelihood | df | P |
|---|---|---|---|---|---|
| 1a | 23.767 | Full | 97.5652 | 6 | 8.08E-19 |
| 1 | 9.872 | Sex+cross | 206.8571 | 4 | 1.26E-43 |
| 2 | 0 | Full | 88.51938 | 6 | 6.15E-17 |
| 3 | 18.01 | Sex | 25.23805 | 2 | 3.31E-06 |
| 7 | 12.25 | Sex | 22.81742 | 2 | 1.11E-05 |
| 8 | 7.203 | Sex | 24.06168 | 2 | 5.96E-06 |
| 9 | 19.359 | Full | 52.20212 | 6 | 1.7E-09 |
| 10 | 26.627 | Sex | 194.7377 | 2 | 5.17E-43 |
| 11 | 0 | Sex | 26.37187 | 2 | 1.88E-06 |
| 12 | 0 | Sex | 40.33378 | 2 | 1.74E-09 |
The results for the most significant marker, among linked markers with concordant effects, are reported. The chromosome (Chr), marker position (taken from Foley et al., 2011) and best-fit model are shown, as well as the difference in log-likelihood between the best-fit model and the minimal model, and its significance (χ2).
Only a single marker supported this model. All other reported results are concordant across multiple linked markers.
Mapping NMR
All chromosomes had at least one significant NMR QTL above the chromosomal threshold (Supplementary Table S2, Figure 3) in at least one class. Taking the more stringent threshold, calculated across the entire genome for each class separately, there was no evidence of NMR on chromosomes 5 and 12. There were two QTL with non-overlapping 95% confidence intervals on chromosome 1 for SCmit females and males and SDmit males. There was also evidence in SDmit males for two non-overlapping QTL on chromosome 2. LOD scores for females were overall lower than for males, and LOD scores for the SDmit cross were much lower than for the SCmit cross.
Figure 3.

QTL for NMR in T. californicus F2. For each sex and mitochondrial background, the LOD plots are shown, with LOD maxima indicated by open circles, and the two LOD confidence interval shown by the solid black line. Genome-wide significance thresholds are indicated by the solid gray line and chromosome-specific thresholds by the dotted gray line. The additional horizontal gray line in the SCmit M LOD plot indicates a break in the axis. Transparent gray boxes indicate intervals containing a candidate gene (see Figure 1). Effect plots are shown below the LOD plot, with the log relative survival of the SC homozygote relative to the SD homozygote in black, the heterozygote in gray and the additive expectation of the heterozygote as dotted gray. Significant deviations from the additive expectation are indicated by asterisks.
The two most distorted chromosomes were chromosomes 1 and 10. In all the cases, for chromosome 1, the SC homozygotes were much more likely to survive to adulthood than the SD homozygotes, and in three of the four classes (apart from SDmitF), there was a significant excess in heterozygotes. For chromosome 10, the proportion of heterozygotes in each class did not deviate from expected (Figure 3); however, for females there was a deficit of SC homozygotes on chromosome 10, and for males there was an excess of SC homozygotes. Beyond chromosome 1, there were several examples of heterozygote advantage but only in males. For females, there was strong heterozygote disadvantage on chromosome 8 (Figure 3).
For both SCmit sexes, SC alleles were favored at all QTL—with the exception of the marginal QTL on chromosomes 3 and 7 in females, and chromosome 10 with its pattern of opposing sex-specific effects. Within the SD mitochondrial background, there was no clear trend in favor of alleles from either population. In this background, again chromosome 10 had a striking symmetric sex bias. Another seven QTL favored SC alleles, five favored SD alleles (three in males, two in females) and one QTL favored heterozygotes.
There was little evidence for NMR at any of the five a priori candidate genes for nuclear–mitochondrial interactions, chosen based on a large body of work on mitonuclear interactions in T. californicus (reviewed in Burton and Barreto, 2012). These five candidate genes are cytochrome c, mitochondrial RNA polymerase, mitochondrial transcription factors A and B1 and mitochondrial rieske iron-sulfur protein (highlighted in gray on Figure 3). In SCmit males, one NMR peak above the stringent genome-wide threshold, on chromosome 7, lay within a marker interval surrounding a candidate gene. However, the heterozygote, not the SC homozygote, had a significant advantage. In SDmit females, the broad confidence interval on chromosome 6 overlapped a candidate gene. In this case, the SC allele was favored, not the SD allele.
Developmental, reproductive and morphometric QTL
In addition to NMR QTL, a number of QTL were found for developmental and morphometric traits. These QTL explained a significant proportion of the variation in these traits. However, no QTL were found for fertility (Table 2, Supplementary Table S1). Sex ratio mapped to several loci in each cross (Figure 4). The largest effect in both crosses was on chromosome 10, and it was concordant between the crosses. Chromosomes 1 and 2 also had effects on sex ratio in both crosses. For chromosome 1, more females than males were SD homozygotes and a higher proportion of males than females were heterozygous. For chromosome 2, the effects differed between the crosses. Several other loci were associated more weakly with sex ratio in a single cross.
Table 2. Number of significant QTL for NMR for each class of T. californicus F2 hybrids.
| NMR | CTW | CTL | Dev | Fert | ESL | ESA | Sex | |
|---|---|---|---|---|---|---|---|---|
| SCmitF | 10 | 5 (20.4%) | 3 (9.9%) | 3 (12.3%) | 0 (0%) | 2 (13.0%) | 3 (12.9%) | 5 (23.3%) |
| SCmitM | 10 | 5 (26.9%) | 4 (22.4%) | 1 (5.9%) | 0 (0%) | — | — | |
| SDmitF | 7 | 1 (4.6%) | 4 (13.1%) | 1 (7.1%) | 0 (0%) | 5 (24.8%) | 4 (18.5%) | 5 (24.8%) |
| SDmitM | 11 | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0%) | 0 (0.0%) | — |
Abbreviations: CTL, cephalothorax length; CTW, cephalothorax width; Dev, development time; ESA, eggsac area; ESL, eggsac length; Fert, fertility; NMR, non-Mendelian ratios; QTL, quantitative trait loci.
For CTW, CTL, Dev, Fert, ESL, ESA and sex, the number of QTL is shown with the percentage of variance explained in brackets.
Figure 4.

LOD plots and effect plots for sex in an F2 mapping population of T. californicus by mitochondrial background. In the LOD plot, the horizontal black line represents the significance threshold. In the effect plot, the black line is the additive effect of the SD allele and the gray line is the dominance effect; female is scored as 0 and male as 1. An asterisk indicates a significant dominance effect.
Locus-specific mortality estimates
We estimated locus-specific mortality effects in our mapping population within sex and cross, as well as across sex. The large interactions between sex and genotype for sex-ratio distortion suggest that at least some of the apparent mortality might be driven by sex-ratio distorters. Disregarding sex as a term in the mortality estimates would be one way to both minimize the effects of sex ratio distorters on mortality estimates and to determine at which loci these effects were strongest. When we measured across sex, our estimates of survivorship at each locus were higher than if we considered the sexes separately (Table 3). NMR even became nonsignificant at several loci. Considering the sexes separately, the locus-specific probability of survival was estimated to be as low as 0.61 at chromosome 10 for SCmit females.
Table 3. Minimum estimated locus-specific probability of survival s i of an F2 individual due to genotype-specific mortality, based on observed patterns of non-Mendelian ratios, with significance of distortion estimated by χ2.
| QTL | SCmitF s i | SCmitM s i | SCmit s i | SDmitF s i | SDmitM s i | SDmit s i | ||
|---|---|---|---|---|---|---|---|---|
| 1a | 0.748 | 0.674 | 0.772 | 0.722 | 0.738 | 0.840 | ||
| 1b | 0.899 | 0.645 | 0.738 | 0.779 | 0.838 | |||
| 2 | 0.734 | 0.867 | 0.778 | 0.927 | ||||
| 3 | 0.780 | 0.825 | 0.937 | ns | ||||
| 4 | 0.824 | 0.875 | 0.888 | 0.826 | 0.874 | |||
| 5 | 0.896 | 0.905 | ||||||
| 6 | 0.899 | 0.890 | 0.919 | 0.971 | 0.946 | ns | ||
| 7 | 0.921 | 0.775 | 0.860 | 0.839 | 0.916 | |||
| 8 | 0.778 | 0.886 | 0.783 | 0.745 | 0.853 | 0.860 | ||
| 9 | 0.888 | 0.752 | 0.825 | 0.745 | 0.851 | |||
| 10 | 0.610 | 0.679 | 0.889 | 0.646 | 0.686 | 0.888 | ns | |
| 11 | 0.845 | 0.773 | 0.801 | 0.817 | 0.778 | 0.824 | ||
| 12 | 0.923 | 0.965 | ns | |||||
Abbreviations: ns, not significant; QTL, quantitative trait locus.
Bold values represent loci at which survival is higher in the combined cross than in either sex individually.
There were two QTL with non-overlapping two LOD (logarithm of the odds) confidence intervals on chromosome 1 (1a and 1b) but only one non-overlapping QTL on the remaining chromosomes.
Epistasis in NMR
We tested for bi-locus Dobzhansky–Muller incompatibilities within all individuals combined, males and females separately, SCmit and SDmit separately, and all four classes individually (Table 4). The strongest deviation from Mendelian ratios overall, and within each sex, was when we jointly considered chromosome 1 and the mitochondrial background. Several other interactions between mitochondrial background and chromosomes 2 and 9 were found but only in males. Between autosomes, the strongest effect we found was for an interaction between chromosomes 4 and 7 with a deficit of SC chromosome 7 homozygotes with SD chromosome 4 homozygotes. The same interaction was seen in males and females separately and in both mitochondrial backgrounds considered separately. The interaction between chromosomes 4 and 7 was not significant in any of the 4 sex × mit classes when they were analyzed separately. For the SDmit cross, and particularly the males, there was also a significant interaction between chromosomes 4 and 6.
Table 4. Significant inter-chromosomal epistasis for non-Mendelian ratios in F2 hybrid T. californicus, showing proportions of two-locus allelic classes relative to expected frequency for combinations of sex and mitochondrial background, after accounting for the main effect of single locus genotype frequency.
| Chr1 | Chr2 | M1 | M2 | A1A2 | A1B2 | A1H2 | B1A2 | B1B2 | B1H2 | H1A2 | H1B2 | H1H2 | χ 2 | P= | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All | 1 | Mit | 2 | — | 1.15 | 0.85 | — | 0.23 | 1.76 | — | 1.12 | 0.89 | — | 134.55 | 3 × 10−27 |
| 4 | 7 | 5 | 1 | 1.05 | 1.05 | 0.95 | 0.092 | 1.22 | 1.25 | 1.28 | 0.90 | 0.93 | 74.34 | 7 × 10−13 | |
| F | 1 | Mit | 2 | — | 1.16 | 0.84 | — | 0.26 | 1.74 | — | 1.19 | 0.81 | — | 99.10 | 8 × 10−20 |
| 4 | 7 | 5 | 1 | 0.98 | 1.00 | 1.00 | 0.14 | 1.21 | 1.28 | 1.31 | 0.91 | 0.90 | 38.90 | 5 × 10−6 | |
| M | 1 | Mit | 2 | — | 1.14 | 0.86 | — | 0.15 | 1.83 | — | 1.03 | 0.97 | — | 42.20 | 5 × 10−8 |
| 2 | Mit | 2 | — | 1.27 | 0.70 | — | 0.64 | 1.37 | — | 1.04 | 0.95 | — | 32.72 | 4 × 10−6 | |
| 9 | Mit | 3 | — | 1.30 | 0.66 | — | 0.80 | 1.20 | — | 0.90 | 1.10 | — | 25.54 | 0.0001 | |
| 4 | 7 | 4 | 5 | 1.13 | 1.08 | 0.93 | 0.04 | 1.25 | 1.22 | 1.26 | 0.88 | 0.95 | 36.58 | 1 × 10−5 | |
| 4 | 6 | 1 | 1 | 1.56 | 0.77 | 0.74 | 0.84 | 1.24 | 0.98 | 0.84 | 0.98 | 1.09 | 27.97 | 0.00048 | |
| SCmit | 4 | 7 | 5 | 1 | 1.16 | 0.97 | 0.95 | 0.17 | 1.22 | 1.22 | 1.23 | 0.93 | 0.94 | 32.22 | 8.5 × 10−5 |
| SDmit | 4 | 7 | 5 | 1 | 0.93 | 1.13 | 0.96 | 0.04 | 1.22 | 1.28 | 1.34 | 0.86 | 0.91 | 44.96 | 4 × 10−7 |
| 4 | 6 | 1 | 1 | 1.62 | 0.85 | 0.72 | 0.80 | 1.14 | 1.02 | 0.78 | 0.99 | 1.10 | 32.40 | 7.9 × 10−5 | |
| SCmitF | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — |
| SCmitM | 6 | 8 | 2 | 2 | 0.98 | 0.32 | 1.10 | 0.91 | 2.08 | 0.63 | 1.03 | 0.85 | 1.02 | 30.87 | 0.00015 |
| SDmitF | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — |
| SDmitM | 4 | 6 | 1 | 1 | 2.04 | 0.76 | 0.56 | 0.78 | 1.15 | 1.01 | 0.61 | 1.02 | 1.16 | 41.00 | 2 × 10−6 |
Significantly interacting chromosomes are shown, with the most significantly interacting pair of markers M1 and M2 (numbered by position from 0 cM on each chromosomal linkage map). Here A equals the SC homozygote, B equals the SD homozygote and H equals the heterozygote at locus 1 or 2. The proportion of the observed frequency of each marker class relative to the random expectation is shown, with model χ2 and P-value. The Bonferroni threshold is P<1.74 × 10−4, assuming 288 independent tests for All, F and M; P<1.89 × 10−4 assuming 264 tests for all other groups where there are no mitochondrial interactions.
Higher-order epistasis in NMR
Accounting for these simplest incompatibilities, we then tested for additional complex epistasis for NMR. There were abundant higher-order interactions above the Bonferroni threshold (Supplementary Table S3A). Some of these were nuclear × nuclear interactions with the mitochondrial background. Across all individuals pooled, we detected five mitochondrial × two-way interactions. When we conducted the analysis on males alone, there was one detectable interaction, and there were no interactions detectable when we analyzed females alone. The analysis of purely nuclear three-way epistasis indicated there were many more significant three-way interactions than two-way interactions. Across the pool of all individuals there were 15 three-way interactions. We detected the most three-way epistasis in the analysis of males alone, more than within females, and more than in the pool of all individuals, despite the reduction in sample size. In males, we detected 42 interactions, and in females we detected 2. We detected more three-way interactions in the SDmit reciprocal of the cross than in the SCmit when we analyzed them separately: 15 three-way interactions compared with 4. In sum, these 78 detectable interactions were 6.5 × the number (12) of comparable digenic interactions we were able to identify.
We might predict that combinations of coadapted alleles would be more common than combinations of alleles from different genetic backgrounds. However, among the significant two-way interactions, there was no detectable excess of individuals homozygous at all loci for alleles from the same population of origin (data not shown). All chromosomes were involved in multiple significant three-way interactions. Chromosomes 8 and 9 were involved in the fewest, and chromosomes 2 and 11 were involved in the most (Supplementary Table S3B).
Associations among traits
Body size, fertility and eggsac size (in females) were positively correlated with each other and negatively correlated with development time (Supplementary Table S4). However, the presence of QTL for NMR on a chromosome did not significantly predict QTL for any other trait (Fisher's Exact Test, not shown). We likewise did not find strong evidence that mitonuclear compatibility was associated with increased body size, fertility, development rate or eggsac size. Instead, in the SC background, we found that SC alleles were associated with larger size or faster development in 25 of the 26 morphometric and developmental QTL. In the SD background, 10 loci followed this same pattern, while 5 showed effects in the opposite direction (Supplementary Table S1).
Discussion
Overview
The current study compares the genetics of postzygotic isolation in T. californicus to that of more standard model organisms, which typically have heteromorphic sex chromosomes and greater opportunities for sexual conflict, testing specific predictions about the role of mitochondrial effects, sex-specific effects and different phenotypic consequences of hybridization, such as sterility vs inviability. A previous study of the same pair of populations (Pritchard et al., 2011) found gene regions contributing to hybrid inviability in many of the same general locations (for example, significant NMR on chromosomes 1, 2, 4, 9, 10 and 11, but not on 3, 5, 8 and 12, with some disagreement for chromosomes 6 and 7). However, they could not address mitochondrial effects, sex-specific effects or different phenotypic consequences because they assessed only one mitochondrial background, one sex (males) and one phenotype (genotypic ratios in survivors). Here, by comparing reciprocal crosses, we find that the strongest genetic conflicts are nuclear–mitochondrial, rather than nuclear–nuclear. By comparing sexes, we find a possible sex ratio distorter (but no sex chromosome) and stronger hybrid incompatibilities in males, despite the absence of heteromorphic sex chromosomes. By assessing a series of phenotypic traits, we find multiple QTL for viability and other traits in both sexes, but no QTL for sterility, a sharp contrast to the common pattern of hybrid male sterility factors being the first to accumulate. Finally, by using a saturated linkage map and a much larger number of individuals than the previous study (Pritchard et al., 2011), we were able to assess complex epistasis, finding many more three-locus interactions than two-locus interactions, thereby supporting one prediction of the ‘snowball' hypothesis (Orr, 1995).
Mitochondrial effects
In the absence of rapidly accumulating X-autosome conflicts, other types of gene interactions can be expected to have a greater role in postzygotic isolation. Mitonuclear interactions are major candidates, as the mitochondrial genome typically evolves more quickly than the nuclear genome due to higher substitution rates (in many taxa) and fourfold lower effective population sizes. Mitochondrial conflicts may be especially likely in Tigriopus due to its particularly elevated rate of mitochondrial evolution (synonymous site substitution rate ∼55-fold higher for mtDNA relative to nucDNA: Willett, 2012) and geographically isolated populations with small effective population sizes (evidenced by fluctuating census population sizes and low genetic variation; Burton et al., 2006) prone to fixing different deleterious mtDNA mutations (evidenced by maladapted nuclear–mitochondrial complexes in natural populations; Edmands et al., 2009). Indeed, the strongest epistatic interaction we detected in this study was between the mitochondrion and chromosome 1, with additional, milder mitochondrial interactions with chromosomes 2 and 9 in males. None of these nuclear QTL overlapped with the five a priori candidate genes identified through extensive work on mitonuclear conflicts in T. californicus (for example, Burton and Barreto, 2012). This illustrates the value of the agnostic QTL approach but is not entirely surprising given that upwards of 1000 nuclear-encoded proteins are estimated to function in the mitochondrion (Reichert and Neupert, 2004), providing a large number of potential candidates for mitonuclear coadaptation. It is important to note that this study used isofemale lines, rather than outbred populations, so we do not know if identified reproductive QTL are fixed within populations, an important issue for understanding the evolution of reproductive isolation (for example, Cutter, 2012). We do know, however, that there is extremely low genetic variation within T. californicus populations (for example, Burton and Lee, 1994) and that none of the markers used in this study were found to be polymorphic within the SC or SD populations (Foley et al., 2011), making it unlikely that the gene interactions detected were the result of rare variants.
Chromosome 1 was significantly distorted in all four cohorts, although the degree of distortion depended on mitochondrial background. Some of the complexity of this pattern might reflect multiple linked loci with sex and cross-specific effects. Non-overlapping confidence intervals in SCmit females and males, as well as in SDmit males indicate at least two independent loci on chromosome 1 contributing to NMR. The SCmit cross was more distorted overall—with more NMR QTL, and more QTL with higher LOD thresholds and a much higher predicted mortality—than the SDmit cross. This suggests pervasive nuclear–mitochondrial interactions, consistent with the importance of the role of cytosolic elements in Darwin's Corollary to Haldane's Rule (Turelli and Moyle, 2007). Interestingly, however, instead of simple mitonuclear coadaptation, we found that SC alleles were overrepresented in both hybrid backgrounds, in both sexes: strongly in the SCmit cross, less so in the SDmit cross. One possible explanation for the excess of SC alleles may be that SC and SD are fixed for alternative deleterious recessives at multiple loci but that SD has a higher burden of these alleles. This may be consistent with the overall association between SC alleles and larger size and faster development in both crosses.
Possible sex ratio distorter but no sex chromosome
Sex-specific NMR was found across much of the genome. However, the most striking differences were for one NMR QTL on chromosome 10. Examining the bar plots of genotype frequencies (Figure 2), we can see that for chromosome 10 SD homozygotes are much more likely to be female, and SC homozygotes are more likely to be male, in both mitochondrial backgrounds.
Because it is impossible to morphologically determine naupliar sex, we were unable to distinguish NMR due to mortality vs sex determination. Several lines of evidence together suggest the ratios on chromosome 10 are not a product of sexually antagonistic effects on survival. If there were sex specific mortality in the homozygotes, we would expect that combining the sexes would produce a large excess of heterozygotes. Instead, combining the sexes neatly eliminates any significant NMR in SDmit (and very nearly in SCmit). Based on that evidence alone, we cannot definitively rule out an additive effect of the locus on sexually antagonistic survival. However, after over 4.5 years of free mating (∼50 generations) in an experimental hybrid swarm between SC and SD, chromosome 10 is the only region of the genome that maintains a high degree of heterozygosity (BC Phillips, VL Pritchard and SEdmands, unpublished data), suggesting balancing selection. In this swarm, approximately half the females are heterozygous, whereas only 1% of the males are, and the rest of the individuals are homozygous for the SC haplotype. Although we still cannot rule out the hypothesis of sexually antagonistic effects on survival, the maintenance of this level of variation over so many generations suggests instead that there is a sex ratio distorter on chromosome 10, with the SD allele contributing to an increased chance of being female relative to the background genotype.
Chromosome 10 is unlikely to be acting as a hitherto unrecognized sex chromosome in nature within populations across the range of T. californicus, and in particular within either SC or SD. Chromosome 10 has been associated with adult sex-ratio in some, but not all, interpopulation hybridizations (Pritchard and Edmands, 2013). The pattern we observe instead suggests that SC is fixed for a ‘maleness' allele and SD for a ‘femaleness' allele (relative to the hybrid genetic background) at chromosome 10. If the putative sex-ratio distorter on chromosome 10 were a large-effect allele segregating in both populations, we would see a very different pattern in our reciprocal crosses. The SC males would transmit the large effect ‘maleness' allele in one direction of the cross, while in the other direction the SD males would contribute the allele. We would then predict to see the SC allele overrepresented in females in the SCmit cross, and the SD allele overrepresented in females in the SDmit cross. Instead, we find the SD allele overrepresented in females in both crosses.
There must instead be a number of other sex-ratio distorters segregating within our SC and SD populations, and by extension in our crosses. Other loci in the F2 population certainly contribute to sex determination. The significant QTL for sex ratio in both crosses on chromosomes 1 and 2 are potential candidates. Simulations of Fisherian selection in small populations with migration suggest that between 5 and 8 sex-determining alleles are expected to be segregating within any given population of T. californicus (B Anholt, personal communication). Sex-ratio distortion, then, was likely responsible for at least some of the inflated mortality estimates in Table 3.
Sex-specific effects
We found no hybrid sterility in either sex and elevated hybrid inviability in males. Relative to females, males had more single locus QTL and nuclear–mitochondrial interactions for NMR and lower predicted locus-specific survival rates. Female-biased sex ratios were also observed in both crosses, suggesting higher male mortality.
The explanations for weaker hybrid males in Tigriopus are likely different from those cited for more commonly studied model systems, especially Diptera (for example, Wu and Davis, 1993; Presgraves and Orr, 1998; Coyne and Orr, 2004; Musters et al., 2006). A primary explanation for the pattern, male heterogamety, does not apply. A second possible explanation, intensified sexual selection in males (Wu and Davis, 1993), may a have a role but is likely to be less important in Tigriopus, where sexual selection in males is expected to be reduced as females mate only once, thereby limiting specific opportunities for both intra- and inter-sexual conflict. Further, heightened sexual selection in males is expected to speed the evolution of hybrid male sterility, not viability, because viability loci are thought to be largely shared between the sexes (Hollocher and Wu, 1996). However, neither the current study nor previous work on T. californicus (Willett, 2008) show evidence of higher male than female infertility due to hybrid breakdown.
A third possible explanation for the excess of male viability problems is specific to mitonuclear interactions. Because male fitness is irrelevant to the fitness of maternally inherited mitochondria, mutations in the mitochondria that are neutral or favorable to females are expected to be positively selected, and male nuclear genes will have to evolve to compensate (Innocenti et al., 2011)—the so called ‘sex-specific selective sieve'. Moreover, if mitochondria are able to bias primary sex ratios towards females, mitochondria may drive intragenomic arms races of the kind believed to underlie a majority of cases of hybrid breakdown in male fertility in plants (Rieseberg et al., 1996; Rieseberg and Blackman, 2010). The sex-selective sieve hypothesis may not, however, explain the results of the current study, because, like the sexual selection hypothesis, it is expected to impact male fertility more than survival (Innocenti et al., 2011).
A fourth possible explanation for the observed pattern of elevated male inviability is that hybrid incompatibilities may be aggravated in the sex with a higher metabolic rate (usually, but not always, males). This would be akin to mitonuclear mismatches having more deleterious effects in more metabolically active tissues, a pattern observed in organisms ranging from plants to mammals (Lane, 2011). In T. californicus, males are known to be more susceptible to thermal stress (Willett, 2010), suggesting that males are more metabolically sensitive overall.
Complex epistasis
Complex, higher-order interactions are expected to accumulate as populations diverge and to account for an exponentially larger proportion of hybrid breakdown as divergence progresses (Orr, 1995). Overall, we found many more three-way than two-way interactions for NMRs in our lines. We should emphasize here that our Bonferroni threshold was conservative with respect to linkage and is increasingly conservative for higher-order interactions. The patterns of epistasis we found for NMR varied with both sex and mitochondrial background. As predicted by the ‘snowball-effect' model (Orr, 1995), two-way (and higher order) interactions should tend to accumulate over time by the exponent of the order of their interaction. At a certain point in divergence, three-way interactions (and higher) will contribute increasingly more to hybrid breakdown than two-way interactions. Our results lend support to this idea. Moreover, our cumulative estimates of mortality for single locus and multi-locus interaction NMR were unrealistically high, suggesting pervasive, complex epistasis beyond what we were able to model.
Complex epistasis may cause the majority of hybrid breakdown in wide crosses, but these effects have rarely been characterized within species (but see Rieseberg et al., 1996). The genetics of incipient hybrid breakdown has instead usually been studied between species pairs long after the establishment of complete isolation (Coyne and Orr, 2004). The population structure of T. californicus, characterized by many isolated populations spanning a broad range of genetic divergence (Burton and Lee, 1994), may be particularly suitable for studying crosses at varying degrees of genetic distance and isolation.
Our results are consistent with the predictions of the Dobzhansky–Muller model, under which hybrid incompatibilities are epistatic (Orr, 1995), and likely to have a lower additive component. We found very little epistasis for morphological and developmental traits, as opposed to the pervasive epistasis we found for NMR.
Summary
Patterns of hybrid breakdown in T. californicus largely conform to expectations of a system with limited antagonistic coevolution between the sexes and no degenerate sex chromosome. No sterility was evident, allowing us to demonstrate the role of higher-order epistatic interactions to a degree unprecedented in an animal model. Consistent with the snowball model, there were many more detectable three-way interactions than pairwise. There were pervasive mitonuclear interaction effects on NMR and development, but these did not correspond to candidate genes. Sex-specific effects were also pervasive, with males generally showing lower hybrid viability, but the explanations for these weaker males likely differ from those in systems with heterogamety and high levels of sexual conflict. In sum, the genetics of postzygotic isolation in T. californicus was found to be quite different from that in more commonly studied systems in plants and animals, demonstrating the necessity of expanding studies of speciation genetics to alternative model systems with a diversity of life history strategies.
Data archiving
Data deposited in the Dryad repository: doi:10.5061/dryad.g763q
Acknowledgments
We thank Shiven Chaudhry, Grace Jang, Tara Matsudo, Helen Truong and Valerie Velasco for laboratory assistance. We thank Jeffrey Conroy at the Roswell Park Cancer Institute for design and implementation of the SNP genotyping assays. We thank Barret Phillips, Michael Bruford and several anonymous reviewers for comments on the manuscript. This work was funded by the US National Science Foundation Grant DEB-0743472 to SE.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Heredity website (http://www.nature.com/hdy)
Supplementary Material
References
- Abramoff M, Magalhães PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- Ar-Rushdi AH. The cytology of achiasmatic meiosis in the female Tigriopus (Copepoda) Chromosoma. 1963;13:526. [Google Scholar]
- Arends D, Prins P, Jansen RC, Broman KW. R/qtl: high-throughput multiple QTL mapping. Bioinformatics. 2010;26:2990–2992. doi: 10.1093/bioinformatics/btq565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnqvist G, Edvardsson M, Friberg U, Nilsson T. Sexual conflict promotes speciation in insects. Proc Natl Acad Sci USA. 2000;97:10460–10464. doi: 10.1073/pnas.97.19.10460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreto FS, Moy GW, Burton RS. Interpopulation patterns of divergence and selection across the transcriptome of the copepod Tigriopus californicus. Mol Ecol. 2011;20:560–572. doi: 10.1111/j.1365-294X.2010.04963.x. [DOI] [PubMed] [Google Scholar]
- Broman KW, Wu H, Sen S, Churchill GA. R/qtl: QTL mapping in experimental crosses. Bioinformatics. 2003;19:889–890. doi: 10.1093/bioinformatics/btg112. [DOI] [PubMed] [Google Scholar]
- Burton RS. Mating system of the intertidal copepod Tigriopus californicus. Mar Biol. 1985;86:247–252. [Google Scholar]
- Burton RS, Barreto FS. A disproportionate role for mtDNA in Dobzhansky-Muller incompatibilities. Mol Ecol. 2012;20:4942–4957. doi: 10.1111/mec.12006. [DOI] [PubMed] [Google Scholar]
- Burton RS, Ellison CK, Harrison JS. The sorry state of F2 hybrids: consequences of rapid mitochondrial evolution in allopatric populations. Am Nat. 2006;168:S14–S24. doi: 10.1086/509046. [DOI] [PubMed] [Google Scholar]
- Burton RS, Lee BN. Nuclear and mitochondrial gene genealogies and allozyme polymorphism across a major phylogeographic break in the copepod Tigriopus californicus. Proc Natl Acad Sci USA. 1994;91:5197–5201. doi: 10.1073/pnas.91.11.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheverud JM.2000Detecting epistasis among quantitative trait lociIn: Wolf JB, Brodie ED III, Wade MJ (eds).Epistasis and the Evolutionary Process Oxford University Press: Oxford, UK; 58–81. [Google Scholar]
- Coyne J, Orr HA.1989Two rules of speciationIn: Otte D, Endler J (eds). Speciation and its Consequences Sinauer Associates: Sunderland, MA, USA; 180–206. [Google Scholar]
- Coyne J, Orr HA. Speciation. Sinauer Associates: Sunderland, MA, USA; 2004. [Google Scholar]
- Cutter AD. The polymorphic prelude to Bateson-Dobzhansky-Muller incompatibilities. Trends Ecol Evol. 2012;27:209–218. doi: 10.1016/j.tree.2011.11.004. [DOI] [PubMed] [Google Scholar]
- Edmands S. Heterosis and outbreeding depression in interpopulation crosses spanning a wide range of divergence. Evolution. 1999;53:1757–1768. doi: 10.1111/j.1558-5646.1999.tb04560.x. [DOI] [PubMed] [Google Scholar]
- Edmands S, Northrup SL, Hwang AS. Maladapted gene complexes within populations of the intertidal copepod Tigriopus californicus. Evolution. 2009;63:2184–2192. doi: 10.1111/j.1558-5646.2009.00689.x. [DOI] [PubMed] [Google Scholar]
- Foley BR, Rose CG, Rundle DE, Leong W, Moy GW, Burton RS, et al. A gene-based SNP resource and linkage map for the copepod Tigriopus californicus. BMC Genomics. 2011;12:568. doi: 10.1186/1471-2164-12-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraud T, Gourbière S. The tempo and modes of evolution of reproductive isolation in fungi. Heredity. 2012;109:204–214. doi: 10.1038/hdy.2012.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollocher H, Wu C-I. The genetics of reproductive isolation in the Drosophila simulans clade: X vs autosomal effects and male vs female effects. Genetics. 1996;143:1243–1255. doi: 10.1093/genetics/143.3.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Xu S. PROC QTL—A SAS procedure for mapping quantitative trait loci. Int J Plant Genomics. 2009;2009:141234. doi: 10.1155/2009/141234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innocenti P, Morrow EH, Dowling DK. Experimental evidence supports a sex-specific selective sieve in mitochondrial genome evolution. Science. 2011;332:845–848. doi: 10.1126/science.1201157. [DOI] [PubMed] [Google Scholar]
- Lane N. Mitonuclear match: optimizing fitness and fertility over generations drives ageing within generations. Bioessays. 2011;33:860–869. doi: 10.1002/bies.201100051. [DOI] [PubMed] [Google Scholar]
- Lowry DB, Modliszewski JL, Wright KM, Wu CA, Willis JH. The strength and genetic basis of reproductive isolating barriers in flowering plants. Phil Trans Roy Soc B. 2008;363:3009–3021. doi: 10.1098/rstb.2008.0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Xu S. Mapping viability loci using molecular markers. Heredity. 2003;90:459–467. doi: 10.1038/sj.hdy.6800264. [DOI] [PubMed] [Google Scholar]
- Lynch M, Walsh B. Genetic Analysis of Quantitative Traits. Sinauer Associates Inc: Sunderland, MA, USA; 1998. pp. 448–449. [Google Scholar]
- Moyle LC, Nakazato T. Comparative genetics of hybrid in compatibility: sterility in two Solanum species crosses. Genetics. 2008;179:1437–1453. doi: 10.1534/genetics.107.083618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musters H, Huntley MA, Singh RS. A genomic comparison of faster-sex, faster-x and faster-male evolution between Drosophila melanogaster and Drosophila pseudoobscura. J Mol Evol. 2006;62:693–700. doi: 10.1007/s00239-005-0165-5. [DOI] [PubMed] [Google Scholar]
- Orr HA. The population genetics of speciation: the evolution of hybrid incompatibilities. Genetics. 1995;139:1805–1813. doi: 10.1093/genetics/139.4.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips BC, Edmands S. Does the speciation clock tick more slowly in the absence of heteromorphic sex chromosomes. Bioessays. 2012;34:166–169. doi: 10.1002/bies.201100164. [DOI] [PubMed] [Google Scholar]
- Piepho HP. A quick method for computing approximate thresholds for quantitative trait loci detection. Genetics. 2001;157:425–432. doi: 10.1093/genetics/157.1.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presgraves DC. The molecular evolutionary basis of species formation. Nature Rev. Genetics. 2010;11:175–180. doi: 10.1038/nrg2718. [DOI] [PubMed] [Google Scholar]
- Presgraves DC, Orr HA. Haldane's rule is obeyed in taxa lacking a hemizygous sex. Science. 1998;282:952–954. doi: 10.1126/science.282.5390.952. [DOI] [PubMed] [Google Scholar]
- Pritchard VL, Dimond L, JHarrison JS, Velázquez CCS, Zieba JT, Burton RS, et al. Interpopulation hybridization results in widespread viability selection across the genome in Tigriopus californicus. BMC Genet. 2011;12:54. doi: 10.1186/1471-2156-12-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard VL, Edmands S. The genomic trajectory of hybrid swarms: outcomes of repeated crosses between populations of Tigriopus californicus. Evolution. 2013;67:774–791. doi: 10.1111/j.1558-5646.2012.01814.x. [DOI] [PubMed] [Google Scholar]
- R Development Core Team . R: A language and environment for statistical computing. R Foundation for Statistical Computing: Vienna, Austria; 2010. [Google Scholar]
- Reichert AS, Neupert W. Mitochondriomics or what makes us breathe. Trends Genet. 2004;20:555–562. doi: 10.1016/j.tig.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Blackman BK. Speciation genes in plants. Ann Bot. 2010;106:439–455. doi: 10.1093/aob/mcq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg LH, Sinervo B, Linder CR, Ungerer MC, Arias DM. Role of gene interactions in hybrid speciation: evidence from ancient and experimental hybrids. Science. 1996;272:741–745. doi: 10.1126/science.272.5262.741. [DOI] [PubMed] [Google Scholar]
- Schilthuizen M, Giesbers MCWG, Beukeboom LW. Haldane's rule in the 21st century. Heredity. 2011;107:95–102. doi: 10.1038/hdy.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli M, Moyle LC. Asymmetric postmating isolation: Darwin's corollary to Haldane's rule. Genetics. 2007;176:1059–1088. doi: 10.1534/genetics.106.065979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables WN, Ripley BD.2002Modern Applied Statistics with S4th edn.Springer: New York, NY, USA [Google Scholar]
- Voordouw MJ, Anholt BR. Heritability of sex tendency in a harpacticoid copepod, Tigriopus californicus. Evolution. 2002;56:1754–1763. doi: 10.1111/j.0014-3820.2002.tb00189.x. [DOI] [PubMed] [Google Scholar]
- Willett CS. No evidence for faster male hybrid sterility in population crosses of an intertidal copepod (Tigriopus californicus) Genetica. 2008;133:129–136. doi: 10.1007/s10709-007-9191-0. [DOI] [PubMed] [Google Scholar]
- Willett CS. Potential fitness trade-offs for thermal tolerance in the intertidal copepod Tigriopus californicus. Evolution. 2010;69:2521–2534. doi: 10.1111/j.1558-5646.2010.01008.x. [DOI] [PubMed] [Google Scholar]
- Willett CS. Quantifying the elevation of mitochondrial DNA evolutionary substitution rates over nuclear rates in the intertidal copepod Tigriopus californicus. J Mol Evol. 2012;74:310–318. doi: 10.1007/s00239-012-9508-1. [DOI] [PubMed] [Google Scholar]
- Willett CS, Burton RS. Evolution of interacting proteins in the mitochondrial electron transport system in a marine copepod. Mol Biol Evol. 2004;21:443–453. doi: 10.1093/molbev/msh031. [DOI] [PubMed] [Google Scholar]
- Wu CI, Davis AW. Evolution of postmating reproductive isolation: the composite nature of Haldane's rule and its genetic bases. Am Nat. 1993;142:187–212. doi: 10.1086/285534. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.