

Abstract
The mechanisms regulating T helper 9 (TH9) cells and TH9-mediated diseases remain poorly defined. Here, we demonstrate that the receptor OX40 (Tnfrsf4) is a powerful inducer of TH9 cells in vitro and TH9-dependent airway inflammation in vivo. Under TGF-β based polarizing conditions, OX40 ligation eliminated production of induced regulatory T cells and TH17 cells, and divertedCD4+Foxp3− T cells to a TH9 phenotype. Mechanistically, OX40 activated the ubiquitin ligase TRAF6, which triggered the induction of NF-kB-inducing kinase (NIK) in CD4+ T cells and the non-canonical NF-kB pathway which subsequently lead toTH9 generation. Thus, our study identifies a previously unknown mechanism of TH9 induction and may have important clinical implications in allergic inflammation.
Following T cell receptor triggering the fate of the T cells is controlled, to a large extent, by signals from T cell costimulatory molecules and cytokines 1,2. In the case of naive CD4+ T cells, the cytokine milieu in which the cells are activated directsCD4+ T cells to further differentiate into functionally different subsets i.e., TH1, TH2, TH9, TH17, induced regulatory cells (iTregs) or T follicular helper (Tfh) cells, which in turn mediate different types of T cell immunity or immune-mediated diseases 3. For example, transforming growth factor-β (TGF-β)converts conventional CD4+ T cells to immunosuppressive iTregs by turning on the expression of Foxp3, while TGF-β in combination with the inflammatory cytokinesIL-1, IL-6 or TNF induces the transcription factors RORγt, RORα, and c-Rel that program the induction of TH17 cells 4. More recently, several studies suggest that together with IL-4 promote IL-9-producing ‘TH9 cells’ through induction of the transcription factors PU.1 and IRF4 5,6, although the exact nature of this response and the precise identity of TH9 cells remain somewhat contested. Additionally, activation of CD4+ T cells in an IL-12-rich environment (without TGF-β) stimulates T-bet expression, which programs the induction of TH1 cells, whereas an IL-4-rich environment supports TH2 cells via the induction of Gata3 7. This is of fundamental importance in immunology as individual subsets are often tailored to respond to specific pathogens. Thus, by inducing specific transcription factors, cytokines are critical in directing the differentiation of CD4+ T cell subsets.
OX40 (also known as a Tumor necrosis factor receptor super family 4, Tnfrsf4) is a T cell costimulatory molecule in the TNFR superfamily and traditionally signals through the NF-kB pathway 8. OX40 is rapidly induced upon T cell activation, most prominently on activated CD4+ T cells, and OX40 costimulation exerts a broad impact on the overall T cell immunity 9. In physiological immune responses, OX40 delivers a potent costimulatory signal to activated CD4+ T cells, supporting their survival and proliferation 9. OX40 also inhibits Foxp3+ Tregs 10,11, which may indirectly boost T effector cells by relieving them from Treg-mediated suppression. Furthermore, OX40 directly drives either a TH1 response or a TH2 response in various models, pointing to a key role for OX40 in the control of the TH cell differentiation process 12,13. OX40 is also involved in the development of CD4+ memory T cells, possibly by sustaining the expression of the survival factor Bcl-2 14, and in some models it is indispensible in the memory recall responses 15. However, OX40 can also contribute significantly to immune pathology in a variety of animal models. For example, over-expression of OX40 ligand (OX40L, also known as Tnfsf4) as a transgene in B6 mice induces spontaneous and systemic autoimmune diseases, characterized by heightened production of autoantibodies, severe inflammation in the gut, and extensive interstitial pneumonia in the lungs 16. Also, OX40 costimulation has been implicated in the pathogenesis of autoimmune colitis, experimental autoimmune encephalitis, arthritis, asthma, and transplant rejection 17. Such a diversity of responses suggests that OX40 mostly likely employs a variety of mechanisms to control immunity or immune pathology.
Considering the broad effects of OX40 on CD4+ T cell responses, it remains to be defined how OX40 intersects with cytokine signals in fine-tuning the differentiation of various TH subsets, and the molecular pathways involved, and whether OX40 favors a specific subset or broadly affects all TH subsets. In the present study, we addressed this question by assessing the induction of various CD4+ T cell subsets under TGF-based polarizing conditions. We found that OX40 is highly selective and potent in the induction of TH9 cells and operates via activation of the non-canonical NF-kB pathway.
RESULTS
OX40 favors the induction of TH9 cells in vitro
TGF-β, when combined withIL-2, IL-6, orIL-4, convertsCD4+ T conventional cells (Tconv) to iTregs, TH17 cells, and TH9 cells, respectively 4,18. To determine whether OX40 has any role in this process, we flow sorted naïve CD4+ Tconv (CD62L+CD44−Foxp3−) from wild type (WT) Foxp3gfp reporter mice, activated them in vitro under iTreg (TGF-β + IL-2), TH17 (TGF-β + IL-6), or TH9 (TGF-β + IL-4) polarizing conditions, and examined the induction of Foxp3, IL-17A, and IL-9 (the signature molecules for iTregs, TH17, TH9 cells respectively). To deliver OX40 signals to CD4+ Tconv in the culture, we used either OX40Ltransgenic (OX40Ltg) antigen presenting cells (APCs) or alternatively an agonist anti-OX40 mAb (OX86) to specifically engage OX40 on activated T cells 10,19. (10ng/ml) readily converted CD4+ Tconv to Foxp3+ iTregs upon activation with anti-CD3+ syngeneic APCs, and ~80% of CD4+ Tconv became Foxp3+ cells 3 days later (Fig. 1a). However, such iTreg induction was strongly inhibited when CD4+ Tconv were activated with anti-CD3+ OX40Ltg APCs. Similar inhibition of iTregs by OX40 was observed in an antigen-specific setting in which OT-II cells were activated with the cognate ovalbumin (OVA) antigen presented by either WTAPCs or OX40Ltg APCs (Fig. 1a). Conversion of CD4+ Tconv to TH17 cells by was also inhibited by OX40 ligation. As compared to the controls, TGF-β (3ng/ml) and IL-6 (10ng/ml) converted ~35% of the CD4+ Tconv to IL-17-producing cells 3 days after activation with anti-CD3+ WTAPCs, but OX40 ligation prevented the induction of IL-17-producing cells under identical TH17 polarizing conditions (Fig. 1b). We performed Therefore, OX40 can effectively shut down the reciprocal induction of both iTregs and TH17 cells.
Figure 1. Role of OX40 signaling in polarization of naive CD4+ T cells in vitro.
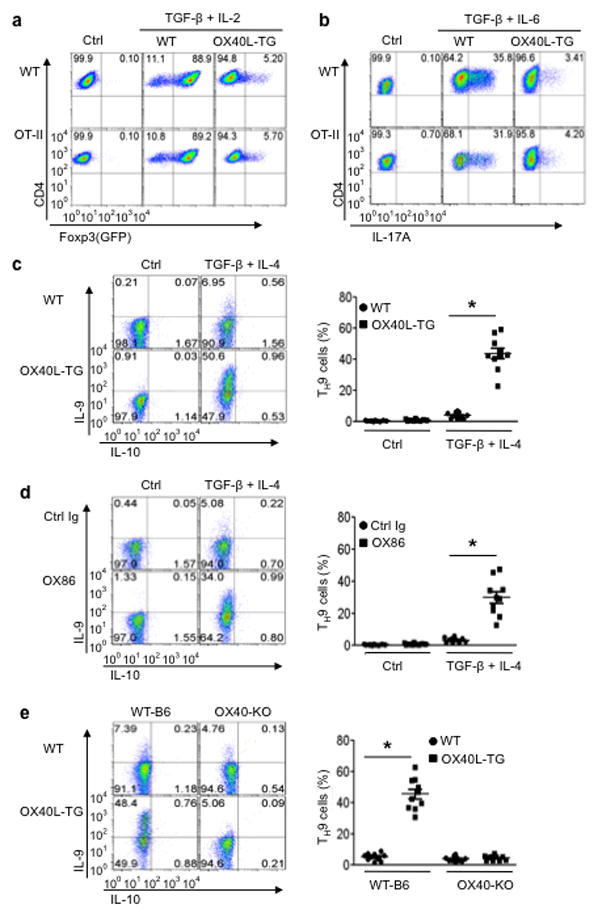
Naive CD4+ T cells sorted from WTB6, Ox40 KO, and OT-II Foxp3gfp reporter mice were cultured under iTreg, TH17, or TH9 polarizing conditions in vitro with or without engaging OX40 signaling, and induction of iTregs, TH17, and TH9 cells on day 3 was shown. (a) Induction of iTregs under polyclonal (anti-CD3) and antigen-specific settings (OVA323–339) in which WT APCs (WT) and OX40L-tg APCs (OX40L-TG) were used to activateCD4+ T cells. CD4+ T cells cultured without polarizing cytokines were included as controls (Ctrl). Numbers in quadrants denote the relative percentage of cells. The plot shown is a representative plot of 5 independent experiments. (b)Induction of TH17 cells under polyclonal and antigen-specific settings as described above. Numbers in quadrants denote the percentage of cells. The plot shown is a representative plot of 5 independent experiments. (c) Induction of TH9 cells from naïve B6 CD4+ T cells stimulated with anti-CD3+ WT APCs (WT) or OX40L-tg APCs (OX40L-TG). The panel on the right is a summary of all experiments performed and the dots denote individual experiments (n=10). (d)Naive CD4+ T cells were activated with anti-CD3plus WTAPCs with or without an agonist anti-OX40 mAb (OX86), and induction of TH9 cells was shown. The panel on the right is a summary of individual experiments (n=10). (e) Naïve B6 and Ox40 KO CD4+ T cells were activated with anti-CD3and APCs under TH9-polarizing conditions, and induction of IL-9 producing cells was shown. The panel on the rights is a summary of all experiments (n=10). * p <0.05.
We further examined whether OX40 also impacts TH9 induction by TGF-β and IL-4, and found that OX40 has a diametrically opposite effect on the induction of TH9 cells. TGF-β (3ng/ml) plus IL-4 (10ng/ml) converted ~7% of CD4+ Tconv to TH9 cells under conditions of anti-CD3plus WTAPC stimulation 3 days after the culture, which is within the published range of most studies 5,6,20 (Fig. 1c). Culturing CD4+ Tconv with anti-CD3+ OX40Ltg APCs drastically enhanced the induction of TH9 cells, and as much as 50% of the CD4+ Tconv became IL-9 producing cells. Additionally, instead of using OX40Ltg APCs, we used anti-CD3+ WTAPCs to activate CD4+ Tconv and included the agonist anti-OX40 mAb (OX86) to engage OX40 signaling. OX86 also markedly enhanced the induction of TH9 cells (Fig. 1d). The specificity of OX40 in TH9 induction was established by activating WTB6 and Ox40 knockout (KO) CD4+ Tconv under TH9 polarizing conditions, and induction of TH9 cells from Ox40 KO T cells remained similar regardless of activation with anti-CD3+ WTAPCs or anti-CD3+ OX40Ltg APCs (~5%), whereas WTCD4+ T cells were readily converted to TH9 cells by TGF-β + IL-4 upon anti-CD3+ OX40Ltg APC activation (~50%)(Fig. 1e). Furthermore, the induction of TH9 cells by OX40 is even more profound in an antigen-specific setting in which OT-II cells were activated with the cognate OVA antigen under TH9 polarizing conditions. Compared to the controls, ~80% of the OT-II cells were converted to TH9 cells by in the presence of OX40 ligation (Fig. 2a), which contrasts sharply to OT-II cells polarized without OX40 ligation (~9%). It should be noted that TH9 cells induced under conditions of OX40 ligation did not express other cytokines, as demonstrated by co-staining with IL-4, IL-5, IL-10, IL-13, IFN-γ, or IL-17 (Fig. 2b). In addition, quantitative Real-Time PCR and ELISA showed marked expression of IL-9 message and IL-9 protein by CD4+ Tconv polarized under TH9 conditions in the presence of OX40 ligation (Fig. 2c and Fig. 2d).
Figure 2. Role of OX40 ligation in polarization of OT-II T cell subsets in vitro.
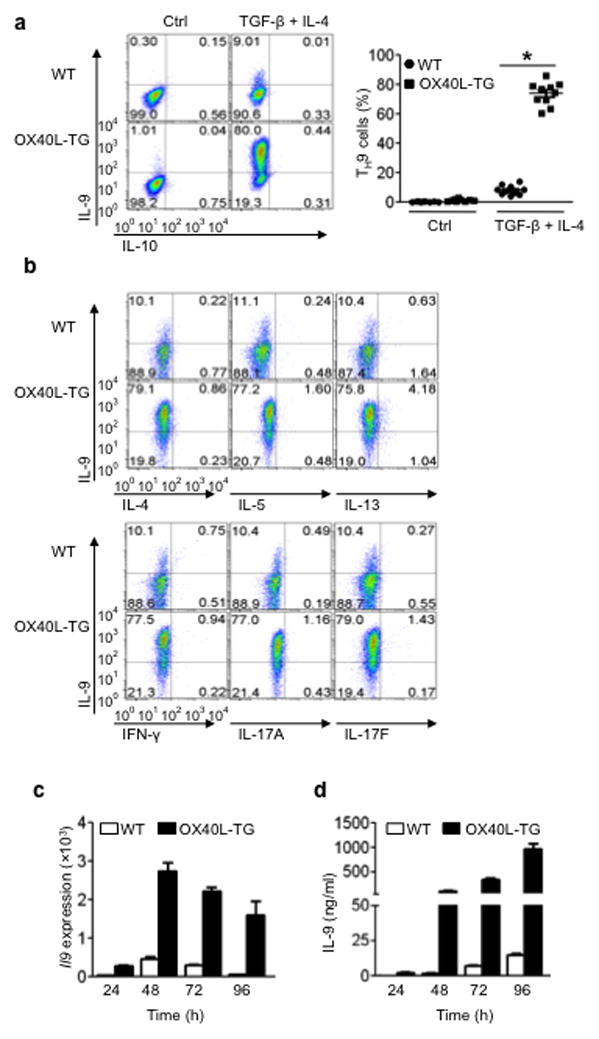
Naive OT-II CD4+ cells were stimulated with the cognate OVA antigen presented by either wt APCs (WT) or OX40L-tg APCs (OX40L-TG), with or without the TH9 polarizing cytokines, and induction of TH9 cells was assessed by flow cytometry, quantitative PCR, and ELISA. (a) Conversion of naive OT-II cells to TH9 cells with or without OX40 ligation on day 3; the data shown is a representative flow cytometryplot. The panel on the right represents all experiments, and dots denote individual experiments (n=10). (b) OT-II cells were polarized under TH9 conditions for 3 days, and IL-9 expression was assessed by co-staining for other cytokines. Data shown are representative plots of 4 individual experiments. (c) OT-II cells were activated with OVA presented by WTAPCs (WT) and OX40Ltg APCs (OX40L-TG) for various times, and IL-9 expression was quantified by Real-Time PCR. Data shown are mean ±SEM of 3 experiments. (d) OT-II cells were activated with OVA plus WTAPCs (WT) or OX40Ltg APCs (OX40L-TG) for various times, and production of IL-9 in the culture supernatants was assessed by ELISA. Data shown are mean ± SEM of 3experiments.
We repeated the TH9 induction experiments up to 10 times with consistent results. We also titrated the cytokines used in the cultures (0.5 to 3ng/ml for TGF-β and 10 to 30ng/ml for IL-4) as well as varied the time points (3 to 5 days of culture), OX40 consistently favored the polarization of TH9 cells (data not shown). Taken together, these experiments demonstrate the potency of OX40 costimulation in selective induction of TH9 cells.
PU.1 is dispensable for TH9induction by OX40
Recent studies in other settings indentified the transcription factor PU.1 (also known as Sfp1) as a key transcription factor for the induction of TH9 cells 6. The potency of OX40 in TH9 induction led us to examine whether Sfp1 is also involved in OX40-mediated induction of TH9 cells. To this end, we bred Sfp1 floxed mice with Cd4-cre mice to specifically delete Sfp1 in CD4+ T cells. Flow sorted CD4+ Tconv from Sfp1f/fCd4cre mice were stimulated with anti-CD3 plus either wt B6 or OX40Ltg APCs under TH9 polarizing conditions, and examined the induction of IL-9 producing cells by flow cytometry. Wt and Sfp1deleted CD4+ T cells expressed comparable levels of OX40 on the cell surface following activation (Fig. 3a), and both proliferation and survival were also similar between such two populations upon activation with anti-CD3+ wtAPCs (Fig. 3b). Thus, Sfp1 deletion in CD4+ T cells does not alter OX40 expression and cellular activation. When wt and Sfp1 deleted CD4+ Tconv were subjected to the same TH9 polarizing conditions (activated by anti-CD3+ wtAPCs), ~6% of wt CD4+ T cells and ~3% of Sfp1 deleted CD4+ T cells were converted to IL-9 producing cells, thus representing a ~50% reduction (Fig. 3c), which is consistent with a role for PU.1 in TH9 induction under certain conditions 6. When OX40 was engaged in the cultures (activated by anti-CD3+ OX40Ltg APCs), induction of TH9 cells by TGF-β+ IL-4 was essentially the same between wt and Sfp1 deleted CD4+ Tconv, and in either case, ~40% of the CD4+ Tconv were converted to IL-9-producing cells (Fig. 3c). Similar findings were observed in cultures in which wt and Sfp1 deleted CD4+ T cells were activated with anti-CD3+ wt APCs and OX86 was added to stimulate OX40 signaling (data not shown). These findings suggest that PU.1 is dispensable in OX40-mediated induction of TH9 cells.
Figure 3. Role of PU.1 and cytokine signaling in OX40-mediated induction of TH9 cells.
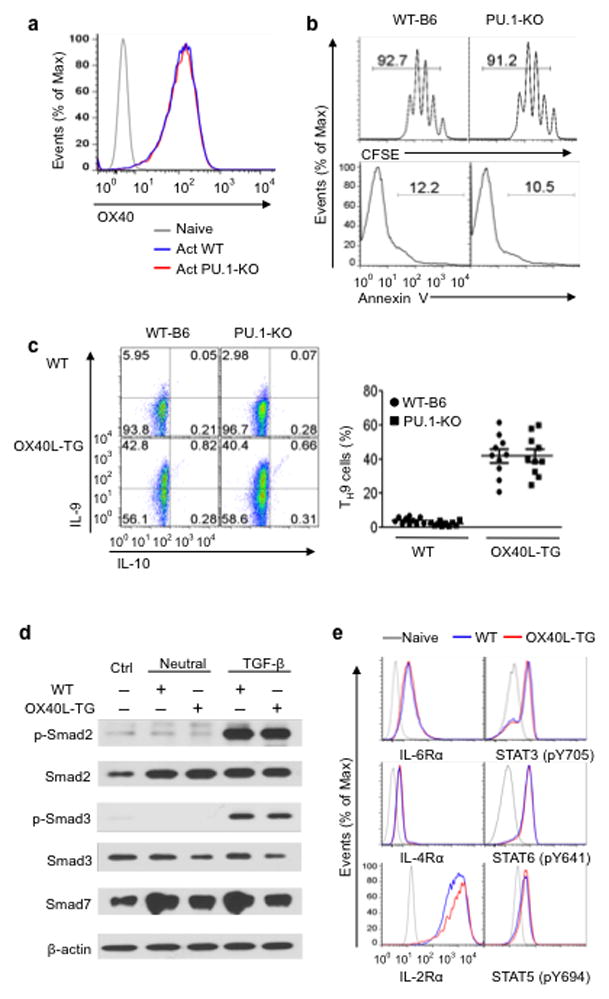
(a) Naive CD4+ T cells sorted from wt B6 and PU.1 (Sfp1) deleted mice were activated with anti-CD3and APCs for 3 days and OX40 expression was assessed by flow cytometry and shown. (b) Proliferation and survival of WT-B6 and Sfp1 KOCD4+ T cells as determined by CFSE dilutions and Annexin V staining 3 days after activation with anti-CD3and WT APC. (c) Naive CD4+ T cells from WTB6 and Sfp1 KO mice were activated with anti-CD3 plus wt APCs (WT) or OX40Ltg APCs (OX40L-TG) under TH9-polarizing conditions for 3 days, and induction of TH9 cells was shown. Numbers in quadrants indicate the percentage of in each section, and the panel on the right summaries all experiments with dots denoting individual experiments. (d) Immunoblotting analysis of Smad2, Smad3, Smad7 in CD4+ Tconv activated with anti-CD3 plus WT APCs (WT) or OX40Ltg APCs (OX40L-TG) with or without additional TGF-β. The plot shown represents 1 of 4 experiments 24 hrs after T cell activation. (e) Expression of IL-2Rα, IL-4Rα, and IL-6Rα as well as activation of Stat5, Stat6, and Stat3, respectively, in CD4+ Tconv activated with anti-CD3 plus either WTAPCs (WT) or OX40Ltg APCs (OX40L-TG). The analysis was done 24 hrs after T cell activation and unstimulated naive T cell were included in the analysis as controls. Data are representative of 1 of 3 independent experiments.
Wt CD4+ Tconv cells showed no alterations in TGF-β signaling upon activation with or without OX40 ligation, as assessed by immunoblotting for Smad expression and phosphorylation (Smad2, 3, 7) (Fig. 3d), which is compatible with the need for TGF-β in TH9 induction by OX40. Furthermore, expression of IL-2Rα, IL-4Rα, and IL-6Rα as well as phosphorylation of Stat5, Stat6, and Stat3 were virtually identical between CD4+ Tconv activated in the presence or absence of OX40 ligation (Fig. 3e). Thus, OX40 seems to employ a mechanism specifically favoring the induction of TH9 cells.
OX40 activates canonical and non-canonical NF-kB pathways
As a member in the TNFR superfamily, OX40 signals primarily through the activation of NF-kB, which consists of both the canonical (p50-RelA) and the non-canonical (p52-RelB) pathways 21. We performed a series of experiments to simultaneously assess the relative expression of such pathways during T cell activation. We activated wt B6 CD4+ Tconv in vitro for various times with or without OX40 ligation, followed by extraction of both cytosolic and nucleus proteins from the activated cells. Induction of key molecules involved in the activation of both NF-kB pathways was determined by immunoblotting and then compared. At 36 hrs following T cell activation, p105 (the precursor of p50) was strongly induced in the cytosol; generation of p50 was similarly induced in CD4+ Tconv activated with or without OX40 ligation (Fig. 4a). p100 (the precursor of p52) was also strongly induced in the cytosol, but only under conditions of OX40 ligation was p100 processed to generate p52 (Fig. 4a). Nuclear accumulation of p50 and RelA (i.e. canonical pathway) was prominent in CD4+ Tconv activated with either anti-CD3+ wt APCs or anti-CD3+ OX40Ltg APCs. In contrast, nuclear accumulation of p52 and RelB (i.e. non-canonical pathway) was observed only in CD4+ Tconv activated in the presence of OX40 ligation (Fig. 4b). These data suggest that canonical NF-kB (p50-RelA) is a default pathway in activated T cells, and OX40 ligation additionally activates the non-canonical (p52-RelB) pathway in CD4+ T cells.
Figure 4. Role of OX40 ligation in the activation of both canonical and non-canonical NF-kB pathways.
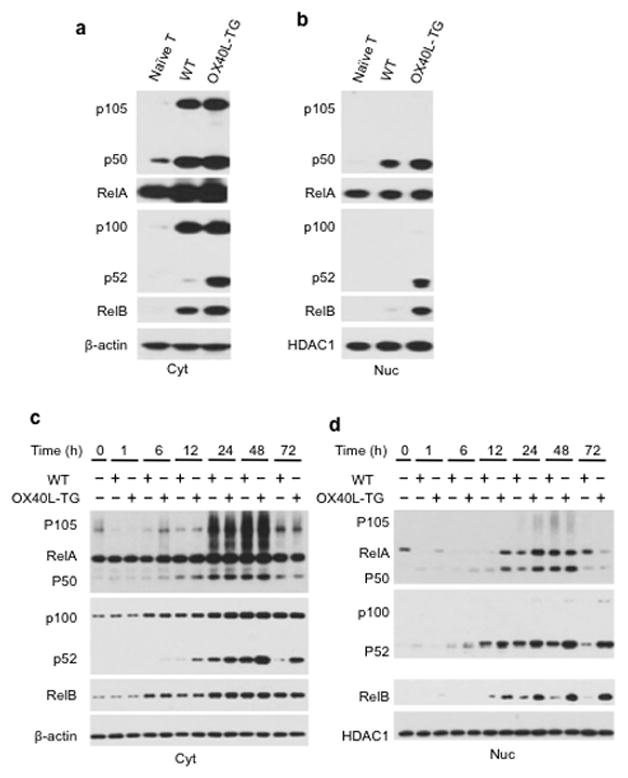
Naive CD4+ Tconv were activated with anti-CD3plus WT APCs (WT) or OX40Ltg APCs (OX40L-TG), and at different time points, cytosolic (Cyt) and nucleus (Nuc) proteins were extracted and examined for NF-kB activation by immunoblotting. Naive unstimulated CD4+ T cells were used as controls. (a, b) Induction of the canonical (p50-RelA) and non-canonical (p52-RelB) NF-kB pathways in the cytosol (a) and nucleus (b) 36 hrs after T cell activation is shown. (c, d) Time-dependent assessment of p50-RelA and p52-RelB pathways (from 1 hr to 72 hrs following T cell activation) in the cytosol (Cyt) and nucleus (Nuc). β-actin and HDAC1 (histone deacetylase 1) were used as loading controls. Data shown are representative of 1 of 3 independent experiments.
Kinetic analyses revealed striking differences in the tempo of canonical and non-canonical NF-kB activation by OX40. Induction of p105 and p50 in the cytosol by OX40 as well as their nuclear translocation were rapid and transient, occurring predominantly in the first 24 and 48 hrs after T cell activation (Fig. 4c and Fig. 4d). In contrast, induction of p52 and RelB by OX40 and their nuclear accumulation were slow but sustained, and 3 days after T cell activation when p50 and RelA were barely detectable in the nucleus, nuclear expression of p52 and RelB remained very strong in OX40 ligated T cells (Fig. 4d). These data suggest that OX40 triggers the activation of both NF-kB pathways; while the canonical pathway is transient, activation of the non-canonical NF-kB pathway is sustained for a much longer period of time.
OX40 requires the non-canonical pathway in TH9 induction
To dissect the relative roles of the canonical versus non-canonical NF-kB pathway in TH9 induction by OX40, we used both loss-of-function and gain-of-function approaches. We bred dominant negative IkBα-tg (IkBαΔN-tg) mice in which a mutated form of IkBα, which is resistant to phosphorylation and degradation, is over-expressed as a transgene (driven by the lck promoter), and therefore, the canonical NF-kB pathway is constitutively repressed 22. We crossed these mice with Foxp3gfp reporter mice, and flow sorted naive CD4+ Tconv from such mice. The sorted CD4+ Tconv cells were stimulated with anti-CD3 plus either wt APC or OX40Ltg APCs under TH9 polarizing conditions, and induction of IL-9 producing cells was determined by flow cytometry. We confirmed that in IkBαΔN-tg CD4+ T cells, the canonical pathway (p50-RelA) activation was strongly inhibited, but induction of non-canonical pathway (p52-RelB) by OX40 ligation and their nuclear translocation were similar to those in wt CD4+ T cells (Fig. 5a and Fig. 5b). We also confirmed that IkBαΔN-tg T cells expressed similar levels of OX40 as wt T cells upon activation in vitro; they also proliferated vigorously upon anti-CD3+ wt APC stimulation and were not apoptotic (Supplementary Fig. 1). Induction of TH9 cells by OX40 was comparable between wt B6 and IkBαΔN-tg CD4+ T cells, and in either case, ~40% of CD4+ Tconv were converted to IL-9 producing cells 3 days after the culture (Fig. 5c).
Figure 5. Role of the canonical NF-kB pathway in OX40-mediated induction of TH9 cells.
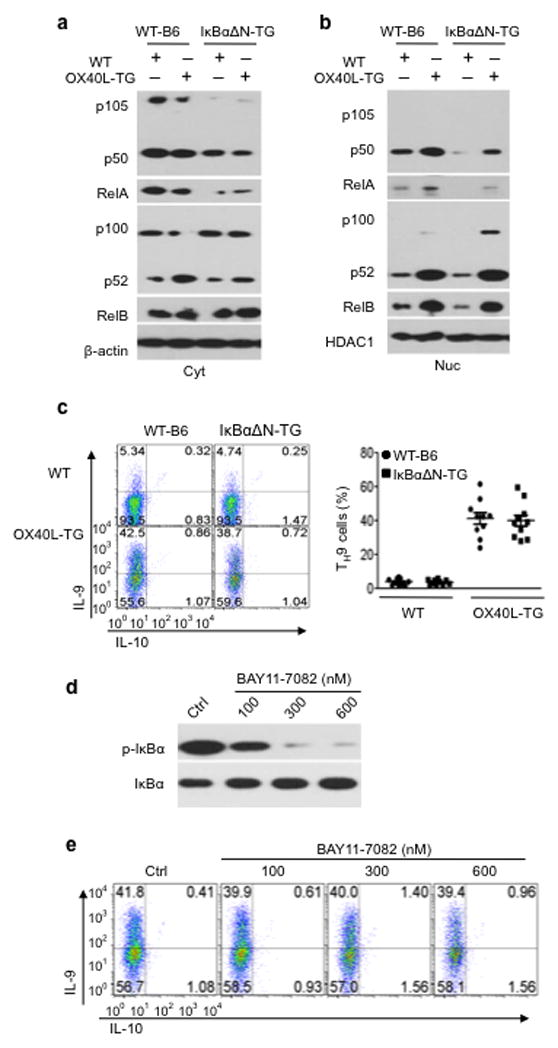
(a, b) WT-B6 and IkBαΔN-TGCD4+ cells were activated for 24 hrs with anti-CD3 plus WT APCs (WT) or OX40Ltg APCs (OX40L-TG), and induction of p50-RelA and p52-RelB in the cytosol (Cyt) and nucleus (Nuc) was assessed by immunoblotting. The plot shown is representative data of 1 of 3 experiments. (c)Naive WT-B6 and IkBαΔN-TGCD4+ T cells were activated with anti-CD3plus WT APCs (WT) or OX40Ltg APCs (OX40L-TG) under TH9 polarizing conditions, and the plots shown are IL-9-producing cells on day 3, as determined by flow cytometry. Numbers in quadrants indicate relative percentage of cells in each section. The panel on the right is a summaryofTH9 cells and the dots represent individual experiments (n=10). (d) Phosphorylation of IkB in CD4+ T cells with or without the addition of different doses of an IkBα inhibitor, BAY11-7082, two days after stimulation. (e) WTB6 CD4+ T cells were stimulated with anti-CD3and OX40Ltg APCs under TH9 polarizing conditions, and BAY11-7082 was added as indicated. Induction of IL-9 producing cells was examined 3 days later. Data are representative of 1 of 3 independent experiments.
In another set of experiments, we stimulated wt B6 CD4+ Tconv with anti-CD3+ OX40Ltg APCs under TH9 polarizing conditions, and in these cultures, we added a chemical inhibitor of canonical NF-kB activation (BAY11-7082), which selectively inhibits IkB phosphorylation 23, and then assessed TH9 induction at different time points. The doses of BAY11-7082 used were shown to potently inhibit IkB activation (Fig. 5d). As shown in Fig. 5e, we observed that TH9 induction by OX40 was not affected by the addition of Bay11-7082 despite strong inhibition of IkB, and ~40% of the CD4+ Tconv became TH9 cells regardless the presence or absence of BAY11-7082. Thus, the canonical NF-kB pathway appears dispensable in OX40-mediated induction of TH9 cells.
To determine whether the non-canonical pathway plays any roles in TH9 induction, we bred p52 KO mice in which activation of the non-canonical (p52-RelB) pathway is prevented because of the lack of p52 generation 24,25. We again sorted CD4+ Tconv from p52 KO mice, activated them under TH9 polarizing conditions with or without OX40 ligation, and examined the induction of TH9 cells 3 days later. First of all, we showed that wt B6 and p52 KO CD4+ Tcells are comparable in OX40 expression, proliferation (shown by CFSE dilutions), and survival (shown by Annexin V staining) upon anti-CD3+ APC activation in vitro (Supplementary Fig. 2). Approximately 40% of wt CD4+ Tconv were converted to IL-9 producing cells by TGF-β + IL-4 upon anti-CD3and OX40Ltg APC activation (Fig. 6a). However, p52 KO CD4+ Tconv could not be converted to TH9 cells under identical culture conditions (Fig. 6a). Thus, the non-canonical p52-RelB pathway plays a key role in OX40-mediated induction of TH9 cells.
Figure 6. Role of the non-canonical NF-kB pathway in OX40-mediated induction of TH9 cells.
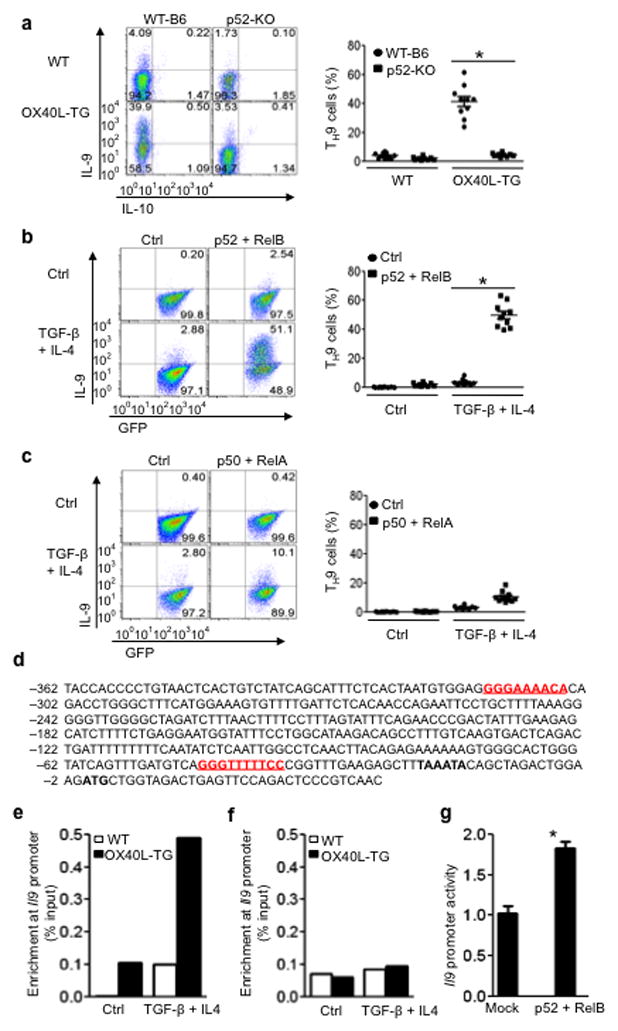
(a) WT-B6 and p52-KO CD4+ T cells were activated with anti-CD3 plus WT APCs (WT) or OX40Ltg APCs (OX40L-TG) under TH9 polarizing conditions for 3 days, and induction TH9cells was shown. The panel on the right is the percentage of TH9 cells among naïve CD4+ T cells and the dots denote individual experiments (n=10). (b) Retroviral transduced WTCD4+ Tconv expressing RelB and p52 were cultured with or without TH9 polarizing cytokines for 3 days, and induction of TH9 cells was shown. The panel on the right depicts the results of all experiments (n=10). (c) WT-B6 CD4+ T cells were transduced with retroviral vectors encoding p50 and RelA, and IL-9 expression by the transduced T cells under TH9-polarizing conditions on day 3 was shown. The right panel depicts the results of all experiments (n=10). (d) Putative NF-kB binding sites in the IL-9 promoter region. The red color depicts the, and the TATA box is shown in bold font. (e) WTCD4+ T cells were activated with anti-CD3 plus either WT APCs (WT) or OX40Ltg APCs (OX40L-TG) under either neutral (Ctrl) or TH9 inducing conditions (TGF-β + IL-4), and enrichment of RelB at the IL-9 promoter region was determined by ChIP and shown. Data shown are representative of 1 of 3 independent experiments. (f) WTCD4+ T cells were cultured under either neutral or TH9 polarizing conditions, and ChIP of RelA binding to the IL-9 promoter region was shown. One of 3independent experiments is shown. (g) 293T cells were transfected with IL-9 promoter-luciferase construct with or without expression vectors encoding full-length RelB and p52. The promoter activity is presented as fold increase over cells transfected with the empty vector alone. Data are 1 of 3 independent experiments (mean ± SEM), * p<0.05.
We then used a gain-of-function approach to ascertain the role of the p52-RelB pathway in TH9 induction. We used a retroviral mediated gene transfer approach to over-express p52-RelB in wt CD4+ T cells, and then examined the induction of TH9 cells by TGF-β + IL-4 only (without OX40 ligation). When the successfully transduced wt CD4+ Tconv (marked by the expression of GFP) were cultured under TH9 polarizing conditions, as much as 50% of the p52-RelB transduced T cells became IL-9 producing cells, whereas the control vector transduced cells showed minimal IL-9 expression (<5%)under identical culture conditions (Fig. 6b). In stark contrast, wt CD4+ T cells transduced with p50-RelA did not show marked induction of IL-9 under the same polarizing conditions (Fig. 6c), thus establishing the specificity of the p52-RelB pathway in TH9 induction by OX40. These findings complement the data obtained using the p52 KO CD4+ T cells, and together they demonstrate an important role for the p52-RelB pathway in TH9 induction by OX40.
Sequence analysis of the IL-9 promoter region revealed two putative NF-kB binding sites (Fig. 6d), and chromatin immunoprecipitation (ChIP) showed that p52-RelB could directly bind to and preferentially enriched at the IL-9 promoter region in TH9 cells polarized under conditions of OX40 ligation (Fig. 6e). However, ChIP ofp50-RelA in similarly polarized TH9 cells did not show its selective enrichment in the IL-9 promoter region (Fig. 6f). These data suggest that p52-RelB may have the potential to directly drive IL-9 transcription. To test this possibility, we cloned ~1000 bp of the IL-9 promoter region upstream to a luciferase reporter gene, and transfected this construct into 293T cells. We then determined the luciferase activities with or without co-transfection of p52-RelB expression constructs, and found that a significant increase in IL-9 promoter activities could be demonstrated in the presence of p52-RelB (Fig. 6g).
TRAF6 is critical to the induction of p52-RelB pathway
The requirement for p52-RelB in TH9 induction prompted us to examine how OX40 triggers the activation of the non-canonical NF-kB pathway. A key step in NF-kB activation by TNFR superfamily members is the recruitment of TNFR-associated factors (TRAF), the adaptor molecules required for downstream signaling 26. RT-PCR based array analysis for all known TRAFs in activated CD4+ Tconv (48 hrs after T cell activation) showed that TRAF6 mRNA was preferentially induced upon OX40 ligation, while expression of other TRAFs was not markedly different with or without OX40 ligation(Fig. 7a). The increased expression of TRAF6 protein in CD4+ Tconv following OX40 stimulation was further confirmed by immunoblotting, and the difference in TRAF6 expression by activated CD4+ T cells with or without OX40 ligation was striking (Fig. 7b).
Figure 7. Analysis of TRAF6 in OX40 triggered activation of non-canonical NF-kB pathway.
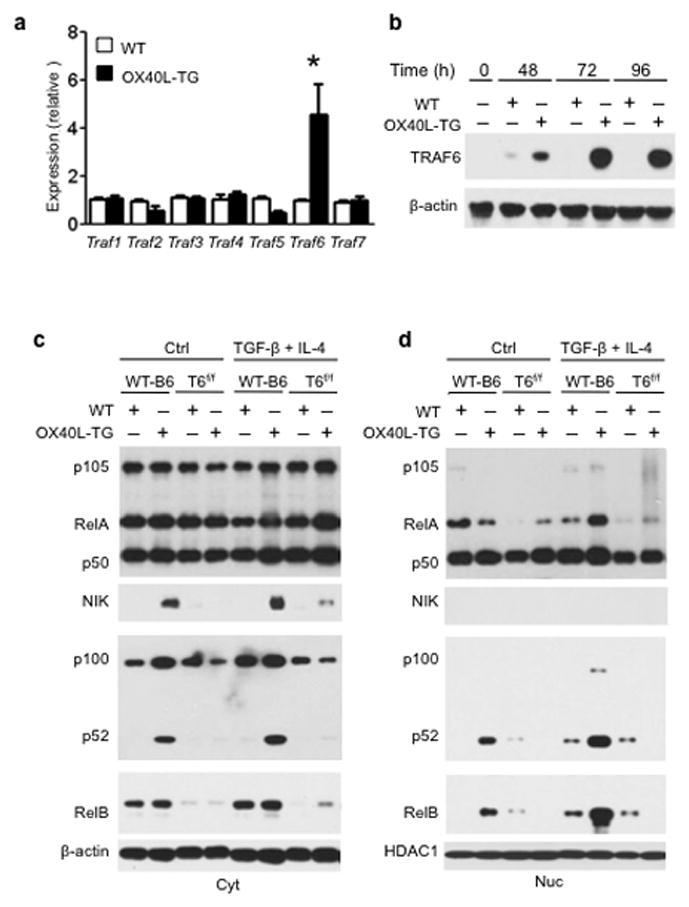
(a) Naive WTCD4+ T cells were stimulated with anti-CD3 plus either WTAPCs (WT) or OX40Ltg APCs (OX40L-TG) for 48 hrs, and expression of Traf1 to Traf7 was quantitated by real-time PCR. (b) Immunoblotting showing strikingly different TRAF6 expression in activated CD4+ T cells with or without OX40 ligation at various time points. (c, d) Naive WT-B6and TRAF6 deleted (Traf6f/fCd4cre) CD4+ T cells (T6f/f) were activated with wt APCs (WT) or OX40Ltg APCs (OX40L-TG) under either neutral or TH9-polarzing conditions for 48 hrs. Cytosolic (Cyt) and nucleus (Nuc) proteins were extracted and expression of p105, p100, RelA, RelB, p52, p50, and NIK was assessed simultaneously by immunoblotting and compared. Data were representative of 3 independent experiments. * p<0.05.
Besides being an adaptor molecule, TRAF6 also functions as an ubiquitin ligase 27. The exact role of TRAF6 in regulating OX40 signaling in T cells has not been examined. We then crossed Traf6 floxed mice with Cd4-Cre mice to specifically delete Traf6 in CD4+ T cells (Traf6f/fCd4cre) 28. We sorted CD4+ Tconv from wt B6 and Traf6f/fCd4cre mice, activated them with anti-CD3 plus either wt APCs or OX40Ltg APCs, and then examined the activation of NF-kB pathways by immunoblotting. OX40 ligation on activated CD4+ T cells triggered strong activation of NF-kB-inducing kinase (NIK) under either neutral or TH9 polarizing conditions, which corroborates a recent report in a different model (Fig. 7c) 29. NIK activation mediates the processing of p100 to p52, and this is further correlated very well with prominent nuclear accumulation of p52 and RelB (Fig. 7c and Fig. 7d). Deletion of Traf6 in CD4+ Tconv prevented the induction of NIK by OX40 and the subsequent generation of p52 from p100. The defective p52 generation in Traf6deleted CD4+ T cells inhibited the nuclear accumulation of p52-RelB, thus blocking activation of the non-canonical NF-kB pathway (Fig. 7c and Fig. 7d). TRAF6 therefore plays a pivotal role in the induction of NIK upon OX40 ligation, and the subsequent activation of the non-canonical NF-kB pathway.
OX40 fails to convert Traf6f/fCd4cre cells to TH9 cells
To determine the role of TRAF6 in TH9 induction, we again flow sorted CD4+ Tconv from wt B6 and Traf6f/fCd4cre mice, stimulated them with anti-CD3 plus either wt or OX40Ltg APCs under TH9 polarizing conditions, and then examined the induction of TH9 cells at different time points. Wt B6 and cells showed similar expression of OX40 upon activation (Fig. 8a); they also showed comparable proliferation (CFSE dilutions) and survival (Annexin V staining) upon anti-CD3+ APC stimulation in vitro (Fig. 8b). OX40 ligation converted a large fraction (~40%)of wt CD4+ Tconv to TH9 cells (Fig. 8c). In sharp contrast, OX40 ligation failed to convert Traf6f/fCd4cre CD4+ T cells to TH9 cells (<3%). This inhibition of TH9 induction is very similar to that observed in p52 KO cells (Fig. 6a). In both p52 KO and Traf6 deleted CD4+ T cells, PU.1, IRF4, and Gata3mRNA and protein expression did not show marked differences upon T cell activation when compared to those in wt B6 CD4+ T cells (Supplementary Fig 3).
Figure 8. Conditional deletion of Traf6 in CD4+ T cells prevents TH9 induction by OX40.

(a) OX40 expression by activated WTB6 (Act WT) and TRAF6-deleted CD4+ T cells (Act T6f/f) 3 days after activation. One of 3 experiments is shown. (b) Proliferation and survival of wt B6 and Traf6-deleted CD4+ T cells (T6f/f) 3days after anti-CD3and APC stimulation. One of 3 experiments is shown. (c) Naive WT-B6and Traf6f/fCd4cre CD4+ T cells (T6f/f) were activated with anti-CD3plus either WT APCs (WT) or OX40Ltg APCs (OX40L-TG) under TH9 polarizing conditions for 3 days, and induction of IL-9 producing cells was determined by flow cytometry. The panel on the right shows the summary of TH9 cell frequency induced in all experiments; the dots denote individual experiments (n=10). (d) Activated CD4+ cells from Traf6f/fCd4cre mice were transduced with retroviral vectors encoding p52 and RelB or control vectors. The transduced T cells were cultured under TH9 polarizing conditions (without OX40 ligation) for 3 days, and induction of IL-9 producing cells among the successfully transduced T cells (marked by GFP expression) was determined by flow cytometry. The right panel indicates the relative frequency of TH9 cells induced in all experiments (n=10). (e) Activated CD4+ T cells from Traf6 deleted mice were transduced with retroviral vectors encoding p50 and RelA, and further cultured under TH9 polarizing conditions for 3 days. IL-9 expression was assessed by flow cytometry and shown. The right panel shows the summary of all experiments (n=10). * p<0.05.
To ascertain that the lack ofTH9 induction in Traf6f/fCd4creCD4+ T cells is truly due to the impaired p52-RelB activation, we transduced Traf6f/fCd4cre CD4+ T cells with retroviral vectors encoding RelB and p52, and the successfully transduced T cells were marked by the expression of GFP. Such GFP positive Traf6f/fCd4cre CD4+ T cells were further cultured under TH9 polarizing conditions for 3 days (without OX40 ligation), and induction of IL-9 producing cells within the GFP positive population was determined by flow cytometry. Control vector transduced Traf6f/fCd4cre T cells did not show detectable levels of IL-9, however Traf6f/fCd4cre T cells transduced with p52-RelB readily became IL-9 producing cells under TH9 polarizing conditions (Fig. 8d) Conversely, CD4+ T cells transduced with p50-RelA failed to produce IL-9 under identical culture conditions (Fig. 8e), further demonstrating the importance of the p52-RelB pathway in TH9 induction. Together, these data demonstrate an indispensable role for TRAF6 and p52-RelB in OX40-mediated induction of TH9 cells.
OX40 triggers TH9-mediated airway inflammation in vivo
To examine the role of the OX40-TH9 axis in vivo, we first compared wt B6 and OX40L-tg mice for signs of tissue inflammation in vivo by histopathology. Consistent with previous reports 16, OX40L-tg mice developed signs of extensive autoimmune diseases (data not shown). Histologically, the lung was prominently involved, with signs of severe interstitial and airway inflammation 19. Masson’s trichrome (MTC) staining showed epithelial hyperplasia and sub-mucosal fibrosis, and PAS&AB staining revealed hyper-proliferation of mucin-producing cells in the airway epithelium in OX40L-tg mice (Fig. 9a). Quantitative assessment showed that as much as 30% of airway epithelial cells were mucin-producing cells in OX40L-tgmice, and this is reminiscent of that seen in IL-9 transgenic mice in the lung airways 30. Indeed, Real-Time PCR analysis for a panel of pro-inflammatory cytokines in the lung tissues revealed high expression of IL-9and IL-13in OX40L-tg, but not in wt, mice (Fig. 9b). This was further confirmed by flow cytometry, which identified an expanded population of CD4+CD44hi T effector/memory cells that spontaneously produce IL-9 in situ (Fig. 9c).
Figure 9. The in vivo effect ofOX40 in TH9 induction and airway inflammation.
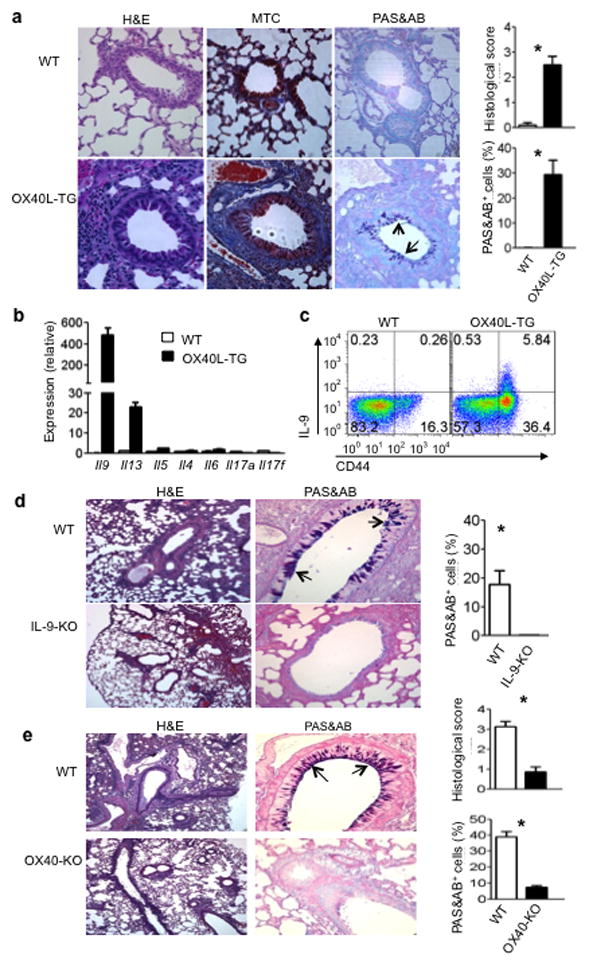
(a) Tissue pathology in the lungs of wt B6 and OX40Ltg mice (20 wks of age), as assessed by H&E, MTC, and PAS&AB staining. The pictures shown are representative of 5 animals in each group. The arrows denote mucin-producing cells in the airway epithelium; the histograms show quantitative histology scores and % of PAS&AB+ cells among airway epithelial cells. Magnifications, 400X. (b) Quantitative RT-PCR analysis of a panel of cytokines in the lung samples from wt B6 and OX40Ltg mice. Data shown are mean ±SEM of 3 animals. (c) Tissue infiltrating cells in the lungs of WTB6 and OX40Ltg mice were isolated and examined for IL-9 expression by flow cytometry. Cells were labeled with anti-CD4 and anti-CD44, and IL-9 expression in the CD4+ population was shown. One of 10 experiments is shown. (d) WT and IL-9-KO mice were briefly treated with an agonist anti-OX40 mAb, as described in the Methods section, and changes in the lung airways were assessed by H&E (100X) and PAS&AB staining (400X). The arrows denote mucin-producing cells in the airway epithelium. The pictures shown are representative of 5 animals in each group. The histogram shows the % of mucin-producing cells relative to airway epithelial cells. (e) WTB6 mice and OX40-KO mice were firstly sensitized and then re-challenged with OVA, and the lung tissue pathology as well as airway inflammation were shown. The arrows show mucing-producing cells, and the histograms depict quantitative histology scores and mucin-producing cells. Each group included 3 animals. * p < 0.05.
In a different set of experiments, we injected an agonist anti-OX40 mAb (OX86) into wt and Il9 KO mice, and then assessed changes in the lungs. A brief treatment with OX86 induced prominent bronchial epithelial hyperplasia and hyper-proliferation of mucin-producing cells in the wild type mice, as shown by PAS&AB staining (Fig. 9d). In contrast, these changes were completely absent in similarly treated Il-9 KO mice (Fig. 9d). Furthermore, in a standard asthma model involving OVA sensitization and re-challenge, hyper-proliferation of mucin-producing cells was similarly induced in wt B6 mice, but not in Ox40 KO mice (Fig. 9e). ELISA revealed markedly reduced IL-9 in the bronchial alveolar lavage (BAL) in OVA challenged Ox40 KO mice (0.08 ± 0.02 ng/ml, n=3) as compared to that in wt B6 mice (3.83 ± 0.43 ng/ml, n=3 (p<0.05). Additionally, TH9 cells derived from OT-II cells under conditions of OX40 ligation induced marked proliferation of mucin-producing cells upon adoptive transferring to Rag1−/− mice, and this effect was markedly inhibited by neutralizing IL-9 (Supplementary Fig. 4). Together, these data demonstrate a direct role of OX40 in TH9 induction and airway inflammation in vivo.
Discussion
The recently identified IL-9 producing ‘TH9 cells’ are a new addition to the family of TH subsets 31,32. However, the precise identity of TH9 cells and the exact mechanisms that regulate TH9 induction remain somewhat contested 18. Here, we demonstrate a significant mechanism of TH9 cell induction. First, the costimulatory molecule OX40 is unexpectedly potent in favoring TH9 induction. OX40 ligation together with TGF-β and IL-4 can covert as much as 80% of CD4+ Tconv to TH9 cells, demonstrating a remarkable potency of this costimulatory pathway in the induction of TH9 cells. Second, we identified a new molecular mechanism by which OX40 favors TH9 cells. OX40 ligation activates the ubiquitin ligase TRAF6, which mediates the activation of NIK; NIK activation results in the processing of p100 to generate the p52subunit, and together with RelB, they form the transcriptionally active p52-RelB complex. This non-canonical NF-kB pathway is essential forTH9 induction by OX40. Third, TH9 cells induced by OX40 appear to be distinct from other published TH9 cells in that they do not express other cytokines 31–33, suggesting that TH9 cells generated under conditions of OX40 costimulation may represent authentic TH9 cells. This new finding further solidifies TH9 cells as a distinct TH subset and may facilitate further mechanistic studies of TH9 cells as well as the role of TH9 cells in various diseases. Finally, we provide in vivo evidence that OX40 is critically important in driving hyper-proliferation of mucin-producing cells in the airway epithelium, demonstrating the in vivo importance of TH9 cells.
In most studies, TH9 cells are induced most effectively by TGF-β and IL-4 34, but cells committed to IL-9 production are often at very low levels (<10%), especially under conditions in which APCs are involved in activation of CD4+ T cells 35. This low frequency of TH9 cells makes functional and mechanistic studies of such cells difficult, and also casts considerable uncertainty on whether TH9 cells are truly a separate lineage. In fact, TH9 cells induced by TGF-β and IL-4 co-express other cytokines including IL-4 or IL-10 31,32. The recent findings that TH17 cells, TREGS, or even innate immune cells can also express IL-9 suggest that IL-9 may be a shared cytokine among multiple cellular subsets 33,36,37. This is a particular concern considering that the transcription factors PU.1 and IRF4, though closely associated with TH9, are not specific to TH9 cells, and they are also involved in the induction of other cell types including TH2 cells 38. In the current studies, we demonstrate that ligation of OX40, together with TGF-β and IL-4, induce high levels of TH9 cells, and such TH9 cells don’t express cytokines commonly seen in TH9 cells induced under other conditions 18. Previous studies have shown that OX40 potently shuts down the induction of IL-10 in activated T cells 39. These data, together with our findings that OX40 also turns off the induction of TH17 cells and iTregs, suggest that TH9 cells induced under conditions of OX40 stimulation are different cells from those induced without OX40 ligation. Those induced by TGF-β and IL-4, possibly through the action of PU.1 and IRF4, may be meta stable or intermediaries, as they express features of other TH subsets, whereas additional OX40 signaling may promote further differentiation of TH9 cells by turning off the expression of other cytokines. However, whether OX40 signaling transiently modulates IL-9 expression while suppressing other cytokines or stably re-programs the cytokine locus in differentiated TH9 cells remains to be defined. This is a significant area that deserves further investigation.
Despite thepotency of OX40 in TH9 induction, OX40 does not seem to operate through the well-described TH9-associated transcription factors PU.1 and IRF4 5,6. Instead, OX40 ligation triggers sustained activation of the non-canonical NF-kB pathway in CD4+ T cells that is critically involved in the induction of TH9 cells. OX40 maybe unique in this regard, because stimulation via GITR, another member in the TNFR superfamily that is also expressed on activated CD4+ T cells 8, fails to support TH9 induction (our unpublished observation). Thus, there appears to be a certain degree of selectivity among TNFR superfamily members in the activation of the non-canonical NF-kB pathway, but the exact mechanisms as to why OX40 preferentially engages the p52-RelB pathway remain to be clearly defined. The promoter region of Il9 contains multiple NF-kB binding sites, as does the conserved non-coding region of Il9 18. Indeed, p52-RelB, but not p50-RelA, can directly bind to the Il9 promoter region and triggers IL-9 transcriptional activity under TH9 polarizing conditions. The involvement of the non-canonical p52-RelB pathway does not exclude the importance of other factors in TH9 induction. In fact, TGF-β and IL-4 are mandatory for optimal TH9 induction by OX40, since in the absence of TGF-β and IL-4, OX40 instead promotes a strong TH1 response 9. Thus, the non-canonical p52-RelB pathway most likely collaborates with other factors downstream of such cytokine signaling that collectively coordinate the induction of TH9 cells, although the factors activated by the TH9 polarizing cytokines are not known and are not addressed in this study. Some emerging data suggest the possibility of T-bet inhibition by such cytokines 40. Nonetheless, the striking effect of OX40 in TH9 induction suggests that the p52-RelB pathway mostly likely predominates this response, and other factors downstream of the polarizing cytokines may simply create a permissive environment for p52-RelB to act during TH9 differentiation. In fact, two recent studies demonstrating the involvement of NF-kB in IL-9 expression in different settings and different cell types seem to support this notion 41,42. In addition, the effect of OX40 ligation also involves the repression of TH17 cells and iTregs, but the mechanisms responsible for such an effect await further identification. Studies in this area may uncover additional insights on the role of OX40 in the control of T cell responses.
In summary, our study identified OX40 as a powerful inducer of TH9 cells and uncovered a previously unknown mechanism triggered by OX40 in selective induction of TH9 cells. TH9 cells induced by OX40 are different in that they express remarkably high levels of IL-9 without other TH2 or TH17 cytokines. Such TH9 cells are stable, can be induced in vivo and exhibit a potent effect on airway epithelial cells and mucin-producing cells. These new findings may have important clinical implications 43.
ONLINE METHODS
Animals
OT-II and Cd4-Cre mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Tnfrsf4 (OX40) KO, Tnfsf4 (OX40L-tg), p52 KO, Traf6 floxed (Traf6f/f), and Foxp3gfp reporter mice have been described previously 4,25,28,44,45. Some strains were crossed with the Foxp3gfp mice to genetically mark Foxp3+ TREGS for cell sorting. Il9 KO mice were provided by A. McKenzie (MRC Laboratory of Molecular Biology); IkBαΔN-tg mice were obtained from M. Alegre (University of Chicago). Sfp1 (PU.1) floxed mice (Sfp1 f/f) were provided by D. Tenen (Harvard Medical School). Animal use and care conformed to the guidelines established by the Animal Care Committee at Harvard Medical School in Boston.
Polarization of naïve CD4+ T Cells in vitro
Naive CD4+ T cells (CD62L+CD44−Foxp3−) were prepared by flow sorting; the sorted cells (1×105 cells/well) were activated with anti-CD3 (2ug/ml; clone 2C11, eBioscience) plus equal numbers of syngeneic APCs in 96 well tissue culture plates (Sigma-Aldrich). In some experiments, OT-II CD4+ T cells were stimulated with OVA323–339 (1 μg/ml) presented by syngeneic APCs. APCs were prepared by depleting T cells from total spleen cells with anti-CD3-PE and anti-PE microbeads (Miltenyi Biotec), followed by a brief treatment with mitomycin C (50 μg/ml, Sigma-Aldrich) before each experiment. Induction of iTregs, TH17 cells, and TH9 cells was performed as previously reported 4,32. Briefly, cells were activated in the presence of 3ng/ml hTGF-β1 and 10ng/ml IL-2for iTreg induction, and for TH17 polarization, 3ng/ml hTGF-β1, 10ng/ml IL-6 were used. Furthermore, 3ng/ml hTGF-β1 and 10ng/ml IL-4 were used to polarize CD4+ T cells to TH9 cells. All recombinant cytokines were purchased from R&D systems. CD4+ T cells cultured under different polarizing conditions (for 1 to 4 days) were harvested and cell polarization assessed by intracellular staining and quantitative Real-Time PCR. Cytokines in the culture supernatants was determined by ELISA.
Intracellular staining
For cytokine staining, CD4+ T cells activated under different polarizing conditions were re-stimulated with PMA (50 ng/ml) and ionomycin (550 ng/ml; Sigma-Aldrich) for 4 hours in the presence of Golgi Stop (BD PharMingen). Cells were fixed and permeabilized with Cytofix/Cytoperm solution (BD PharMingen) and stained with fluorochrome-conjugated anti-IL-9 (RM9A4, BioLegend), anti-IL-10 (JES5-16E3; BD PharMingen), anti-IL-13 (JES10-5A2; BD Pharmingen), anti-IL-4 (11B11; BD Pharmingen), anti-IL-5 (TRFk5; eBioscience), anti-IFN-γ (XMG1.2; BioLegend), anti-IL-17F (9D3.1C8; BioLegend) and anti-IL-17A (TC11; BioLegend). In some experiments, iTregs was determined by the expression of GFP; Stat3, Stat5, and Stat6 in activated CD4+ T cells was assessed by intracellular staining with phospho-specific mAbs (BD PharMingen). Antibodies against PU.1 (9G7; Cell Signaling Technology), IRF4 (3E4; eBioscience), and Gata3 (L50-823; BD PharMingen) were used to determine their expression in naïve and activated CD4+ T cells according to manufacturer’s instructions. All samples were acquired using LSRII flow cytometer (Beckton Dickinson) and data analysis was performed using Flowjo 7.6 software (Tree Star) 46.
Quantitative PCR and ELISA
Cellular RNA was extracted using the RNeasy mini kit (Qiagen) and reverse transcribed into cDNA using the ABI PRISM TaqMan reverse transcription method. Expression of genes of interest and of GAPDH control was assessed in simplex RT-PCR reactions with FAM and VIC probes (Applied BioSystems). All primer and probe sets were purchased from Applied BioSystems. Transcript levels of target genes were calculated according to the 2−ddCT method as described by the manufacturer (ABI PRISM 7700 user bulletin; Applied BioSystems) and expressed as arbitrary units as previously reported 10. IL-9 levels in culture supernatants were measured with the ELISA kit (eBioscience) according to the manufacturer’s instructions.
Immunoblotting
Protein extracts were resolved by SDS-PAGE, transferred onto an immunobilon membrane, and blotted with specific antibodies. The following antibodies were used for immunoblotting: anti-β-actin (Sigma-Aldrich), anti-p105/p50, and anti-RelA (Abcam), anti-p100/p52, anti-RelB, anti-NIK, anti-HDAC1, anti-IkB, anti-Smads and other secondary antibodies (Cell Signaling Technology). Expression of target molecules was revealed by chemoilluminant method as previously reported 47.
Chromatin immunoprecipitation (ChIP)
Chromatins were extracted from naive CD4+Foxp3− T cells polarized under various conditions for 48 to 72 hrs (15×106 cells) after fixation with formaldehyde. Immunoprecipitation of chromatin was performed with anti-RelB, anti-RelA, and isotype control antibody (Abcam) using EZ ChIP kit (Millipore) according to the manufacturer’s instructions. Immunoprecipitated DNA was reverse cross-linked, extracted with phenol/chloroform, and further precipitated with ethanol. ChIP DNA was then analyzed by real-time PCR using the following primer sets (Rel B: 5′-CTTTTCCTTTAGTATTTCAGAACCCGA-3′ and 5′-TCAGTCTACCAGCATCTTCCAGT-3′; RelA: 5′ AATGAGTGAAAGGCAAGTCTTGCTTT-3′ and 5′ TCGGGTTCTGAAATACTAAAGGAAAAG-3′). The data are presented as the relative binding based on normalization to input DNA, as previously reported 5.
Promoter luciferase assay
A luciferase reporter assay was performed as previously reported 48. Briefly, the IL-9 promoter region, 1kb in size upstream of the transcription start site, was cloned into pGL3 luciferase reporter plasmid (Promega). HEK293 T cells were transfected with the reporter constructs, together with expression plasmids carrying murine p50, p52, RelB, RelA cDNA using Lipofectamine 2000 method (Invitrogen). After 48 h, the luciferase activities were analyzed using the dual-luciferase reporter assay system (Promega). Co-transfection with the Renilla-luciferase expression vector pRL-TK was performed in all reporter assays. For all samples, the assays were repeated for at least three times and the data were normalized for transfection efficiency by dividing firefly luciferase activity by that of the Renilla luciferase.
Retroviral mediated gene expression
The cDNA fragments encoding RelA, p50, RelB, and p52 were amplified by PCR and further cloned into pMYs-IRES-EGFP retroviral vector (Cell Biolabs). The retrovirus particles were prepared by transfecting the packaging Plat-E cells based on the manufacturer’s recommendations. To transduce T cells, naive CD4+ T cells were first activated with the Dynabeads Mouse CD3/CD28 T Cell Expander (Invitrogen) for 24 hrs, followed by incubating with freshly prepared retroviral particles by centrifugation at 780g and 32°C in the presence of 10μg/ml polybrene (Sigma-Aldrich) for 2hrs. After the centrifugation, cells were further cultured at 32°C for 6 hrs, followed by culturing the transduced T cells under various polarizing conditions for additional 3 days in complete 1640 medium at 37°C. Polarization of CD4+ T cells to TH9 cells was assessed by intracellular cytokine staining as described above.
Tissue histopathology
Tissue samples were prepared from animals, embedded in paraffin, sectioned and stained with hematoxylin and eosin (H&E). Periodic acid schiff/alcian blue (PAS&AB) and Masson’s trichrome staining (MTC) were also performed to assess mucin-producing cells and tissue damage 30. To quantify tissue histopathology, peri-bronchial and peri-vascular cellular infiltration as well as fibrosis were graded using a semi-quantitative score system (0 to 4 and 0 denotes no inflammation) as previously reported 5. For each slide, five randomly chosen areas were scored. For quantifying PAS/AB staining, mucus production was assessed by determining the percentage of epithelial cells that stained strongly positive 49. Individual slides were examined independently and representative sections were shown.
Induction of lung inflammation
Groups of WTB6 and OX40 KO mice (8 weeks of age) were sensitized i.p. with 50 μg OVA dissolved in aluminum hydroxide (50mg in 100ml alum, Sigma). Mice were challenged with 5% OVA aerosol 4 weeks later for 2 days (30 min each), before analyses. In other experiments, OT-II cells were polarized to TH9 cells by TGF-β and IL-4 upon activation with OVA and OX40L-tg APCs for 4 days (IL-9+ cells >80%). TH9 cells (1×106) were injected into syngeneic Rag1−/− hosts via the tail vein. OT-II cells activated without the polarizing cytokines were used as controls. The host mice were challenged with aerosolized OVA (5% in saline) via the airway 30 min daily for 2 days. Groups of mice were also treated with a neutralizing anti-IL-9 (150 μg, clone 222622, R&D) or control IgG before OVA challenge. Mice were killed 1 day after the OVA challenge; bronchial alveolar lavage (BAL) was performed, and cells in BAL fluid were determined using cytospin and modified Wright-Giemsa staining. Tissue samples were prepared and stained with H&E, PAS&AB for analysis. Each group included 5 mice.
Statistical analysis
The Mann-Whitney test was used for histology score analysis. Other measurements were performed using unpaired two-tailed Student’s t test. A p value < 0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank N. Ishii (Tohoku University) for the Ox40l-tg mice;M. Alegre (University of Chicago) for IkBαΔN-tg mice; D. Tenen (Harvard Medical School) for Sfp1 floxed mice (Sfp1f/f). This work was supported by the National Institutes of Health and the Juvenile Diabetic Research Foundation International.
Footnotes
AUTHOR CONTRIBUTIONS
XX, SB, WL, XC, HW designed and performed various experiments, EJT, YXF, YC, MCW provided key reagents and animal models, and XCL initiated the project, supervised the study, and wrote the manuscript.
COMPETING INTERESTS STATEMENT
The authors have no conflict of interests to declare.
References
- 1.Janeway CA, Bottomly K. Signals and signs for lymphocyte responses. Cell. 1994;76:275–285. doi: 10.1016/0092-8674(94)90335-2. [DOI] [PubMed] [Google Scholar]
- 2.Li XC, Rothstein DM, Sayegh MH. Costimulatory pathways in transplantation: challenges and new developments. Immunological Reviews. 2009;229:271–293. doi: 10.1111/j.1600-065X.2009.00781.x. [DOI] [PubMed] [Google Scholar]
- 3.Dong C. Diversification of T helper cell lineages: finding the family root of IL-17-producing cells. Nat Rev Immunol. 2006;6:329–334. doi: 10.1038/nri1807. [DOI] [PubMed] [Google Scholar]
- 4.Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector Th17 and regulatory cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 5.Staudt V, et al. Interferon-regulatory factor 4 is essential for the developmental program of T helper 9 cells. Immunity. 2010;33:192–202. doi: 10.1016/j.immuni.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 6.Chang HC, et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nature Immunology. 2010;11:527–534. doi: 10.1038/ni.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 8.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 9.Croft M. Control of immunity by the TNFR-related molecule OX40 (CD134) Annu Rev Immunol. 2010;28:57–78. doi: 10.1146/annurev-immunol-030409-101243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vu MD, et al. OX40 costimulation turns off Foxp3+ Tregs. Blood. 2007;110:2501–2510. doi: 10.1182/blood-2007-01-070748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med. 2008;205:825–839. doi: 10.1084/jem.20071341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Demirci G, Li XC. Novel roles of OX40 in the allograft response. Curr Opin Organ Transplantation. 2008;13:26–30. doi: 10.1097/MOT.0b013e3282f3def3. [DOI] [PubMed] [Google Scholar]
- 13.Ito T, et al. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med. 2005;202:1213–1223. doi: 10.1084/jem.20051135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity. 2001;15:445–455. doi: 10.1016/s1074-7613(01)00191-1. [DOI] [PubMed] [Google Scholar]
- 15.Vu MD, et al. Critical, but Conditional, Role of OX40 in Memory T Cell-Mediated Rejection. J Immunol. 2006;176:1394–1401. doi: 10.4049/jimmunol.176.3.1394. [DOI] [PubMed] [Google Scholar]
- 16.Murata K, et al. Constitutive OX40/OX40 Ligand Interaction Induces Autoimmune-Like Diseases. J Immunol. 2002;169:4628–4636. doi: 10.4049/jimmunol.169.8.4628. [DOI] [PubMed] [Google Scholar]
- 17.Sugamura K, Ishii N, Weinberg AD. Therapeutic targeting of the effector T cell costimulatory molecule OX40. Nature Rev Immunol. 2004;4:420–431. doi: 10.1038/nri1371. [DOI] [PubMed] [Google Scholar]
- 18.Perumal NB, Kaplan MH. Regulation of IL-9 transcription in T helper cells. Trends Immunol. 2011;32:146–150. doi: 10.1016/j.it.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao X, et al. New insights on OX40 in the control of T cell immunity and immune tolerance in vivo. J Immunol. 2012;188:892–901. doi: 10.4049/jimmunol.1101373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noelle RJ, Nowak EC. Cellular sources and immune functions of interleukin-9. Nature Reviews Immunology. 2010;10:683–687. doi: 10.1038/nri2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vallabhapurapu S, Karin M. Regulation and function of NF-kB transcription factors in the immune system. Ann Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 22.Zhou P, et al. Transplantation Tolerance in NF-{kappa}B-Impaired Mice Is Not Due to Regulation but Is Prevented by Transgenic Expression of Bcl-xL. J Immunol. 2005;174:3447–3453. doi: 10.4049/jimmunol.174.6.3447. [DOI] [PubMed] [Google Scholar]
- 23.Zhan Y, et al. Glucocorticoid-induced TNF receptor expression by T cells is reciprocally regulated by NF-kappaB and NFAT. J Immunol. 2008;181:5405–5413. doi: 10.4049/jimmunol.181.8.5405. [DOI] [PubMed] [Google Scholar]
- 24.Franzoso G, et al. Mice deficient in nuclear factor (NF)-kappa B/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J Exp Med. 1998;187:147–159. doi: 10.1084/jem.187.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lo JC, et al. Coordination between NF-kB family members p50 and p52 is essential for mediating LTbR signals in the development and organization of secondary lymphoid tissues. Blood. 2006;107:1048–1055. doi: 10.1182/blood-2005-06-2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hildebrand JMYZ, Buchta CM, Poovassery J, Stunz LL, Bishop GA. Roles of tumor necrosis factor receptor associated factor 3 (TRAF3) and TRAF5 in immune cell functions. Immunological Reviews. 2011;244:55–74. doi: 10.1111/j.1600-065X.2011.01055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paul PK, et al. The E3 ubiquitin ligase TRAF6 intercedes in starvation-induced skeletal muscle atrophy through multiple mechanisms. Mol Cell Biol. 2012;32:1248–1259. doi: 10.1128/MCB.06351-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.King CG, et al. TRAF6 is a T cell-intrinsic negative regulator required for the maintenance of immune homeostasis. Nature Medicine. 2006;12:1088–1092. doi: 10.1038/nm1449. [DOI] [PubMed] [Google Scholar]
- 29.Murray SE, et al. NF-κB inducing kinase plays an essential T cell intrinsic role in graft-versus-host disease and lethal autoimmunity in mice. J Clin Invest. 2011;121:4775–4786. doi: 10.1172/JCI44943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Temann UA, Geba GP, Rankin JA, Flavell RA. Expression of interleukin 9 in the lungs of transgenic mice causes airway inflammation, mast cell hyperplasia, and bronchial hyperresponsiveness. J Exp Med. 1998;188:1307–1320. doi: 10.1084/jem.188.7.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Veldhoen M, et al. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nature Immunology. 2008;9:1341–1146. doi: 10.1038/ni.1659. [DOI] [PubMed] [Google Scholar]
- 32.Dardalhon V, et al. Interleukin 4 inhibits TGF-β-induced-Foxp3+T cells and generates, in combination with TGF-β, Foxp3− effector T cells that produce interleukins 9 and 10. Nature Immunology. 2008;9:1347–1355. doi: 10.1038/ni.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nowak EC, et al. IL-9 as a mediator of Th17-driven inflammatory disease. J Exp Med. 2009;206:1653–1660. doi: 10.1084/jem.20090246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmitt E, et al. IL-9 production of naive CD4+ T cells depends on IL-2, is synergistically enhanced by a combination of TGF-β and IL-4, and is inhibited by IFN-γ. Journal of Immunology. 1994;153:3989–3995. [PubMed] [Google Scholar]
- 35.Tan C, Gery I. The unique features of Th9 cells and their products. Crit Rev Immunol. 2012;32:1–10. doi: 10.1615/critrevimmunol.v32.i1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilhelm C, et al. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nature Immunology. 2012;12:1071–1077. doi: 10.1038/ni.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu LF, et al. Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature. 2006;442:997–1002. doi: 10.1038/nature05010. [DOI] [PubMed] [Google Scholar]
- 38.Goswami R, Kaplan MH. A brief history of IL-9. Journal of Immunology. 2011;186:3283–3288. doi: 10.4049/jimmunol.1003049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ito T, et al. OX40 ligand shuts down IL-10-producing regulatory T cells. PNAS. 2006;103:13138–13143. doi: 10.1073/pnas.0603107103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Das J, et al. Transforming growth factor beta is dispensable for the molecular orchestration of Th17 cell differentiation. The Journal of Experimental Medicine. 2009;206:2407–2416. doi: 10.1084/jem.20082286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jash A, et al. Nuclear Factor of Activated T Cells 1 (NFAT1)-induced Permissive Chromatin Modification Facilitates Nuclear Factor-κB (NF-κB)-mediated Interleukin-9 (IL-9) Transactivation. Journal of Biological Chemistry. 2012;287:15445–15457. doi: 10.1074/jbc.M112.340356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stassen M, et al. IL-9 and IL-13 production by activated mast cells is strongly enhanced in the presence of lipopolysaccharide: NF-kappa B is decisively involved in the expression of IL-9. J Immunol. 2001;166:4391–4398. doi: 10.4049/jimmunol.166.7.4391. [DOI] [PubMed] [Google Scholar]
- 43.Kaur D, Brightling C. OX40/OX40 ligand interactions in T cell regulation and asthma. Chest. 2012;141:494–499. doi: 10.1378/chest.11-1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pippig SD, et al. Robust B Cell Immunity but Impaired T Cell Proliferation in the Absence of CD134 (OX40) J Immunol. 1999;163:6520–6529. [PubMed] [Google Scholar]
- 45.Sato T, et al. Consequences of OX40-OX40 ligand interactions in langerhans cell function: enhanced contact hypersensitivity responses in OX40L-transgenic mice. Eur J Immunol. 2002;32:3326–3335. doi: 10.1002/1521-4141(200211)32:11<3326::AID-IMMU3326>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 46.Xiao X, et al. OX40/OX40L Costimulation Affects Induction of Foxp3+ Regulatory T Cells in Part by Expanding Memory T Cells In Vivo. J Immunol. 2008;181:3193–3201. doi: 10.4049/jimmunol.181.5.3193. [DOI] [PubMed] [Google Scholar]
- 47.Hleb M, et al. Evidence for cyclin D3 as a novel target of rapamycin in human T lymphocytes. J Biol Chem. 2004;279:31948–31955. doi: 10.1074/jbc.M400638200. [DOI] [PubMed] [Google Scholar]
- 48.Maruyama T, et al. Control of the differentiation of regulatory T cells and TH17 cells by the DNA-binding inhibitor Id3. Nature Immunology. 2011;12:86–95. doi: 10.1038/ni.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jember AGH, Zuberi R, Liu FT, Croft M. Development of Allergic Inflammation in a Murine Model of Asthma Is Dependent on the Costimulatory Receptor OX40. J Exp Med. 2001;193:387–392. doi: 10.1084/jem.193.3.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.