

Abstract
Small-molecule inhibitors that block the MDM2-p53 protein-protein interaction (MDM2 inhibitors) are being intensely pursued as a new therapeutic strategy for cancer treatment. We previously published a series of spirooxindole-containing compounds as a new class of MDM2 small-molecule inhibitors. We report herein a reversible ring opening-cyclization reaction for some of these spirooxindoles, which affords four diastereomers from a single compound. Our biochemical binding data showed that the stereo-chemistry in this class of compounds has a major effect on their binding affinities to MDM2; with >100-fold difference between the most potent and the least potent stereoisomers. Our study has led to the identification of a set of highly potent MDM2 inhibitors with a stereochemistry that is different from that of our previously reported compounds. The most potent compound (MI-888) binds to MDM2 with a Ki value of 0.44 nM and achieves complete and long-lasting tumor regression in an animal model of human cancer.
INTRODUCTION
Compounds containing a spirocyclic oxindole-pyrrolidine ring (spirooxindole) system have recently attracted significant attention because of their unique structure and broad biological activities (Figure 1).1 For example, the natural product spirotryprostatin A is a cell cycle regulator,2 while pteropodine is a modulator of muscarinic M1 and 5-HT2 receptors.3 Additionally, spirooxindole-containing compounds have been reported as actin polymerization inhibitors, 4 and as inhibitors of tubulin polymerization.5
Figure 1.

Examples of Spirooxindole-Containing Compounds with Interesting Biological Activities
We previously reported sprirooxindole-containing compounds that are potent and specific small-molecule inhibitors that block the MDM2-p53 protein-protein interaction.6 For example, compound MI-219 (Figure 1) binds to MDM2 with low nanomolar affinity, effectively blocks the MDM2-p53 protein-protein interaction in cells, reactivates p53 in tumor cells with wild-type p53 in vitro and in vivo, and strongly inhibits tumor growth in animal models of human cancer.6b Our data thus suggested that modifications of spirooxindole-containing compounds may lead to a highly promising MDM2 inhibitor for the treatment of human cancer by reactivation of the powerful tumor suppressor function of p53.
Our spirooxindole-containing MDM2 inhibitors were synthesized using an efficient 1,3-dipolar cycloaddition reaction (Scheme 1).6d Compound 1a was obtained in high purity and was found to be relatively stable in aprotic solvents such as CH2Cl2, THF, EtOAc, and DMSO. Stereochemistry was assigned to compound 1a and its analogues, including MI-219, using the X-ray crystal structures of the key intermediate I and of compound II (Figure 1 and Scheme 1).7 However, when a pure sample of 1a was treated with MeOH or CH3CN/H2O, up to four peaks, all with the same molecular weight, were observed by analytical HPLC (Supporting Information - SI). Isolation and purification of the fractions from compound 1a and subsequent analysis by 1H, DEPT 135, and 13C NMR showed that individual components possess the same number of protons, CH, CH2, CH3, and quaternary carbons. The data suggested that these individual peaks represent different isomers or stereoisomers.
Scheme 1.

Synthesis and Chemical Structures of Our Previously Reported MDM2 Inhibitors
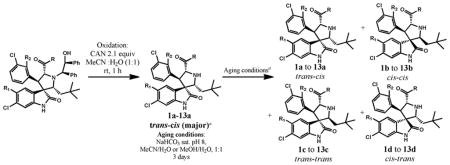
Based upon these initial observations and on our subsequent investigations, we hypothesized that reversible ring opening and cyclization of the pyrrolidine ring takes place in some of these compounds. A cascade reaction of this type can generate four possible diastereomers from one single stereoisomer, as shown in Table 1. Although reversible ring opening-cyclization has been previously observed for certain spirooxindole-containing alkaloids,8 no detailed investigation has been reported.
Table 1.
Isomerization of Selected Spirooxindole-Containing Compoundsa
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Compounds (a–d) | R1 | R2 | R | Yieldb(%) | Ratio after Oxidation a:b: (c+d)c | Ratio after Aging a:b: (c+d)c,d |
| 1 | 1 | H | H | NMe2 | 50e | 95:2:3 | 6:71:24 |
| 2 | 2 | H | H | NHMe | 61 | 87:5:8 | 3:71:26 |
| 3 | 3 | H | F | NMe2 | 57 | 75:19:6 | 35:46:19 |
| 4 | 4 | H | F | NHMe | 84 | 67:2:31 | 9:79:12 |
| 5 | 5 | H | F |
|
86 | 88:2:10 | 1:80:19 |
| 6 | 6 | H | F |
|
79 | 89:10:1 | 12:61:27 |
| 7 | 7 | H | F | OEt | 74 | 89:6:3 | 44:50:6 |
| 8 | 8f | H | F | OH | 25g | — | 41:34:25h |
| 9a | 9 | H | F | NH2 | 56 | 0:44:56 | 0:96:4 |
| 10 | 10 | F | H | NMe2 | 60 | 56:25:19 | 10:71:19i 2:88:8 |
| 11a | 11 | F | H | NHMe | 87 | 19:52:27 | 1:74:25 |
| 12 | 12 | F | H |
|
81 | 30:32:38 | 3:58:37 |
| 13a | 13 | F | H |
|
50 | 15:67:18 | 2:74:24 |
In cases of 9, 11, 12 and 13, the cis-cis isomers were obtained as the predominant products of ceric ammonium nitrate oxidation without aging (equilibrium in CH3CN and H2O at pH 8 for 3 days)
Isolated yield after flash column chromatography
Ratio determined by HPLC analysis; assignments were not made for c and d;
The combined yield of a to d in the aging step was determined from the yield in the oxidation step;
Yield after HPLC separation;
Compound 8a-8d was obtained from the hydrolysis by LiOH-THF-H2O of 7a;
Hydrolysis yield;
Starting from pure 8a;
Ratio after crude product was allowed to stand in MeOH for 2 h.
We herein report our detailed investigation of the isomerization of selected spirooxindole-containing compounds. Our biochemical binding data showed that the stereochemistry in this class of compounds has a major effect on their binding affinities to MDM2; with >100-fold difference between the most potent and the least potent stereoisomers. Importantly, our investigation has led to the identification of a set of highly potent MDM2 inhibitors with a stereochemistry that is different from that of our previously reported compounds.6 One such compound is capable of achieving complete and long-lasting tumor regression in an animal model of human cancer.
RESULTS AND DISCUSSION
We first investigated the isomerization reaction for selected spirooxindole-containing compounds (entries 1–13, Table 1). The trans-cis isomers, 1a-7a, were obtained as major products (67–95%) after ceric ammonium nitrate (CAN) oxidation (Table 1, entries 1–7).
However, treatment of the oxidation reaction products (entries 1–7) with CH3CN and H2O at pH 8 for 3 days (aging) produced a mixture of four isomers with the cis-cis isomers (1b to 7b) as the major components. Hydrolysis of 7a yielded a mixture of four isomers 8a-8d. Interestingly, for entry 9, after CAN oxidation, a mixture of 9b-9d was obtained without detectable amount of 9a. Although 10a constituted 56% after oxidation, this isomer rapidly decreased to 10% within 2 h in MeOH. When R1 = F (Table 1, entries 11 and 13), the cis-cis isomers (11b and 13b) were found to be the predominant product after oxidation with a small amount of the other three isomers. For entry 12, a mixture of four isomers was obtained after oxidation with a similar amount of isomers 12a and 12b. Thus, substitution of an F at the 5-position of the oxindole ring or removal of a methyl group from the amide nitrogen seemed to favor isomer b. These data show that for these compounds, isomerization occurs in CH3CN and H2O or in MeOH.
To investigate the isomerization in more detail, we obtained four individual isomers 5a-5d (entry 5) to high purity. Isomer 5a, 5c or 5d each was gradually converted into a mixture of four isomers over a period of up to four days in CH3CN-H2O (Figure 2). Isomer 5b, on the other hand, was quite stable in CH3CN-H2O, only 4% being converted into a mixture of three other isomers after 12 days.
Figure 2.

Interconversion of 5a-5d in MeCN-H2O. Panels A to D show isomerization of compounds 5a-5d. The proportion of compounds is on the Y axis and reaction time is on the X axis. Conditions: Trifluoroacetate salts (0.1 mg) of the amine were placed in 0.25 mL of 50% aqueous MeCN. Purity was analyzed at the indicated time points by analytical reverse phase HPLC.
To determine the absolute stereochemistry of isomer b, we obtained the crystal structure of 13b, which revealed the three hydrophobic groups in a cis-cis substitution pattern on the pyrrolidine ring (Figure 3). The crystal structure of an intermediate after the ring opening (14), however, showed that the substitution pattern of the three hydrophobic groups on the pyrrolidine ring was trans-cis (Figure 3). Hence, these data indicate that initial product after oxidative removal of the chiral auxiliary has a trans-cis configuration on the pyrrolidine ring, and upon cleavage of the chiral auxiliary, the isomerization occurs when the final products are exposed to H2O and MeCN prior to the work-up.
Figure 3.

Absolute Stereochemistry from X-ray Crystallography
Based on the X-ray crystallographic data from 13b and 14, we propose the reaction mechanism shown in Scheme 2 for the observed isomerization. The trans-cis isomers 1a-13a (Table 1) are the initial products from CAN oxidation. In aqueous solution, the trans-cis isomers 1a-13a undergo a reversible retro-Mannich reaction affording the respective ring-opened transition state (a-TS) in which the pyrrolidine ring has been opened. Recyclization of a-TS affords four stereoisomers.9 We did not attempt to determine the crystal structures of stereoisomers c and d because very limited amounts of the compounds could be obtained, due to their facile conversion to other isomers. Stereochemistry of isomers c and d were assigned based on the stability profiles of 5c and 5d in Figure 2 and the stereochemistry for isomers a and b.
Scheme 2.

Proposed Isomerization Mechanism of Spirooxindole-Containing Compounds 1a-13a
We next investigated binding kinetics of isomers 5a-5d to MDM2 in the binding assay medium using our fluorescence- polarization (FP) assay (Figure 4). DMSO stock solutions of 5a-5d were freshly prepared from lyophilized powder of 5a-5d right before the experiments. Then, aqueous incubation solutions of 5a-5d were prepared by diluting the fresh DMSO stock solutions in FP binding assay buffer and final DMSO concentration was 5%. These incubation solutions, in which isomerization of 5a-5d was taking place, were stored at room temperature for the whole time range of experiments. Aliquots of the incubation solutions of 5a-5d were tested at different time points to determine MDM2 binding affinities. Since isomerization of 5a-5d is sensitive to the incubation time, total time used for binding assay plate preparation and incubation was precisely controlled so that assay plates were read exactly 10 min after aliquots were pipetted from the incubation solution.
Figure 4.

Binding Kinetics of 5a, 5b, 5c and 5d. The IC50 of MDM2 binding affinity is on the Y axis and incubation time is on the X axis. See experimental section for binding assay conditions.
In the first time-point, isomer 5b has the highest binding affinity, followed by 5c, 5a, and 5d (Figure 4). The stereochemistry has a significant effect on their binding affinity to MDM2; isomer 5b is >300-times more potent than isomer 5d and is 18-times more potent than 5a. However, after 100 h, the binding affinities for these four isomers converged (Figure 4).
To understand the binding kinetics of 5a-5d, we investigated the isomerization kinetics of 5a-5d in binding assay medium by direct HPLC analysis of a solution of the individual compound in binding medium at indicated time points (Figure 5). At the 100 h time-point, there was a similar distribution for each individual isomer, regardless which isomer was used at the beginning, and isomer 5b and 5a accounted for approximately 50% and 40%, respectively. Isomer 5b has a slow isomerization kinetic and was the most stable, followed by isomer 5d, 5a, and 5c. Isomer 5c had very fast isomerization kinetics and approximately 20% of it was already isomerized into 5b within 30 min. Hence, the high binding affinity for isomer 5c in the beginning (Figure 4) should be interpreted with caution since there was a significant amount of 5d in the binding assay medium. Therefore, the binding kinetics of 5a-5d (Figure 4) is consistent with the observed isomerization kinetics (Figure 5).
Figure 5.

Isomerization of 5a-5d in Binding Assay Medium. Panels A to D correspond to isomerization studies of isomers 5a-5d. The proportion of compounds is on the Y axis and reaction time is on the X axis. Conditions: the individual sample 5a, 5b, 5c or 5b (0.1 mg) was dissolved in binding medium (0.25 mL) and the samples were incubated at room temperature. The solution of corresponding isomer in binding medium was directly analyzed in analytical reverse phase HPLC at the indicated time points.
We also investigated the isomerization kinetics for 1a, 1b, 6a, 6b, 12a, and 12b in the binding assay medium. Our data showed that in general, isomers a isomerized relatively fast, whereas isomers b were fairly stable (SI). For instance, after 12 days in the medium, 93.9% of 1b remained when started with 1b but 53% of 1a converted into 1b and only 17.6% of 1a remained when started with 1a.
We purified a sufficient amount of cis-cis isomers 1b-13b and trans-cis isomer 1a-8a and 12a and determined their binding affinities to MDM2 (Table 2). Based upon the isomerization kinetics observed for 5a/5b, 1a/1b, 6a/6b and 12a/12b, there was minimal isomerization for these isomers in the binding assay medium in the initial 20 min. Therefore, the incubation time in all binding experiments was controlled precisely at 15 min, resulting in less than 20 min experimental time in total, including plate preparation and incubation, to minimize the influence of the isomerization on MDM2 binding affinity. Our binding data showed that in general, the cis-cis isomers b have better binding affinities to MDM2 than the corresponding trans-cis isomers a. For example, 4b and 5b are >15-times more potent than their respective isomer 4a and 5a. Interestingly, 12a and 12b have very similar binding affinities.
Table 2.
Binding Affinity of Selected Spirooxindole Inhibitors to MDM2 Protein
| Entry | Compounda,b | IC50c (nM) | Kid (nM) | Compounda,b | IC50c (nM) | Kid (nM) |
|---|---|---|---|---|---|---|
| 1 | 1a | 10700±1300 | 1500±180 | 1b | 126±13 | 16.1±1.7 |
| 2 | 2a | 565±57 | 76.2±7.7 | 2b | 30.9±2.9 | 3.1±0.3 |
| 3 | 3ab | 2285±183 | 311±24 | 3b | 137±11 | 17.6±1.0 |
| 4 | 4a | 387±39 | 51.8±4.2 | 4b | 25.9±1.5 | 2.4±0.2 |
| 5 | 5a | 141±11 | 18.1±1.0 | 5b | 11.3±1.2 | 1.0 |
| 6 | 6ab | 410±39 | 54.9±4.2 | 6b | 24.6±7.0 | 2.2±0.5 |
| 7 | 7a | 19500±4000 | 2700±500 | 7b | 721±79 | 97.5±10.7 |
| 8 | 8a | 498±44 | 67.0±5.9 | 8b | 55.1±5.2 | 6.4±0.6 |
| 9 | 9a | n.d.e | 9b | 22.5±2.2 | 1.9±0.2 | |
| 10 | 10a | n.d.e | 10b | 835±121 | 113±16 | |
| 11 | 11a | n.d.e | 11b | 112±12 | 14.2±1.1 | |
| 12 | 12ab | 180±24 | 23.5±2.1 | 12b | 104±16 | 13.1±1.0 |
| 13 | 13a | n.d.e | 13b | 66.8±7.4 | 8.0±1.0 | |
| 14 | 1d | 181000±31000 | 25000±18000 | |||
| 15 | 5c | 24.1±2.3 | 2.2±0.2 | 5d | 3100±460 | 430±62 |
Purity was >95% unless otherwise stated;
Purity of 4a was 94%; of 6a, 93.5% and of 12a, 82%;
See experimental section for binding assay procedures;
Calculation of Ki based on IC50;10
n.d. not determined.
Our binding data also provided initial structure-activity relationship for isomers b. Substitution of F at the 2- position of the phenyl ring of 1b and 2b has no effect on the MDM2 binding affinity (compare 1b versus 3b and 2b versus 4b). However, substitution of F at the 5-position of the oxindole ring of 1b and 2b was detrimental to binding, as shown by the binding affinity difference between 1b and 10b, and between 2b and 11b. Removal of one methyl from the amide improves the binding affinity of 1b, 3b, 4b and 10b (compare 1b versus 2b, 3b versus 4b, 4b versus 9b and 10b versus 11b). Changing the amide of 9b to either an ester (7b) or an acid (8b) decreased the binding affinity to MDM2.
We investigated the cell growth inhibitory activity of isomers 5a and 5b in the SJSA-1 osteosarcoma cell line, which has an amplified MDM2 gene and has been extensively used for evaluation of MDM2 inhibitors (Table 3).6
Table 3.
Cell Growth Inhibitory Activity of 5a and 5b with different treatment time
| Time | IC50 of 5a (μM) | IC50 of 5b (μM) |
|---|---|---|
| Day 1 | 30.32±6.23 | 18.06±2.40 |
| Day 2 | 0.95±0.16 | 0.63±0.04 |
| Day 3 | 0.18±0.02 | 0.18±0.01 |
| Day 4 | 0.15±0.03 | 0.14±0.05 |
Surprisingly, our data showed that despite the difference in their binding affinities to MDM2, there is no significant difference in their activity against cellular growth of the SJSA-1 cell line. To investigate this discrepancy between binding affinities and cellular activities of these two compounds, we monitored the isomerization of 5a and 5b in cell culture medium by HPLC analysis of MeCN extracts of cell growth medium containing 5a or 5b at indicated time points (Figure 6). Both 5a and 5b were found to isomerize in cell growth medium (Figure 6, panels 1 and 2). Starting from either 5a or 5b, the distributions of four isomers were very similar after incubation for one day in the cell culture medium. After incubation for 2 days, the most potent stereoisomer 5b in the binding assay accounts for approximately 50% of all 4 isomers, either starting from 5a or from 5b. Hence, the relatively rapid isomerization of both 5a and 5b in the cell culture medium explains why these two isomers show similar activity in the cell growth inhibitory assay.
Figure 6.

Isomerization Studies of 5a and 5b in Cell Culture Medium. Panels A and B depict the isomerization studies of compounds 5a and 5b, respectively. The proportion of compounds is on the Y axis and reaction time is on the X axis. See SI for conditions of the isomerization study in cell growth inhibition assay medium.
We next investigated the in vivo activity of 5a and 5b in a pharmacodynamic (PD) experiment using SJSA-1 osteosarcoma tumor xeonografts in severe combined immunodeficient (SCID) mice. SCID mice bearing SJSA-1 tumors were treated with a single dose of 5a or 5b at 200 mg/kg via oral gavage and the tumor tissues were harvested at indicated time points for Western blotting analysis of p53 activation. Western blotting showed that both 5a and 5b effectively activated p53 in tumor tissue in a time-dependent manner, as shown by the accumulation of p53 protein itself, and MDM2 and p21 proteins, two p53 targeted gene products (Figure 7).
Figure 7.

Western Blot Analysis of p53 Activation Induced by 5a and 5b in SJSA-1 Osteosarcoma Tumor Tissue with a Single, Oral Dose of Either Compound at 200 mg/kg. FL-PARP, full-length PAPR; Cl-PARP, cleaved PARP; Pro-Cas3, pro-caspase-3; Cl-Cas3, cleaved caspase-3.
Isomer 5b was clearly more potent than 5a in activating p53 in tumor tissues. At 24 h, 5b resulted in stronger apoptosis induction than 5a based upon cleavage of poly(ADPribose) polymerase (PARP) and activation of caspase-3, two biochemical markers of apoptosis. These PD data in tumor-bearing mice demonstrate that isomer 5b is more potent than isomer 5a in vivo in activation of p53 and induction of apoptosis, in contrast to the cell growth data observed in these two isomers (Table 3).
To further understand the clear difference between isomers 5a and 5b in the in vivo PD experiment in SJSA-1 tumor tissue, we investigated the isomerization of 5a and 5b in vivo in both plasma and tumor tissue in SCID mice bearing SJSA-1 tumors. SCID mice were orally administered with a single, oral dose of 5a or 5b at 100 mg/kg, and samples of plasma and tumor tissue were collected at 1 h, 6 h, and 24 h and analyzed using the LC-MS/MS method. The data are summarized in Table 4.
Table 4.
Concentration of 5a and 5b in Plasma and Tumor Tissue in SCID Mice Bearing SJSA-1 Tumors when Dosed with Either 5a or 5b.
| Dosing | Plasma Concentration (ng/ml)a | |||
|---|---|---|---|---|
| Isomer | 1 h | 6 h | 24 h | |
| 5b | 5b | 16800±1931 | 2577±815 | 5.9±0.8 |
| 5a | 3343±1385 | 47±20 | BLQb | |
| 5a | 5b | 2380±448 | 210±141 | BLQ |
| 5a | 617±97 | 75±24 | BLQ | |
| Tumor Concentration(ng/g)a | ||||
| 1 h | 6 h | 24 h | ||
| 5b | 5b | 19133±3775 | 1963±683 | BLQ |
| 5a | 1122±297 | 65±24 | BLQ | |
| 5a | 5b | 1797±527 | 120±84 | BLQ |
| 5a | 2973±1036 | 27±18 | BLQ | |
Three mice per time point.
BLQ: below the lower limit of quantification.
In plasma, the concentration of 5b in mice dosed with 5b was 7-fold higher than that in mice dosed with 5a at 1 h and was 12-fold at 6 h. In tumor tissue, the concentration of 5b in mice dosed with 5b was 11-fold higher than that in mice dosed with 5a at 1 h, and was 16-fold at 6 h. Hence, the much stronger induction of p53 activation and of PARP cleavage by 5b than 5a in SJSA-1 tumor tissue (Figure 7) is consistent with the data that much higher concentrations of 5b were achieved when dosed with 5b than dosed with 5a in tumor tissue, and 5b is 18-times more potent than 5a in terms of binding to MDM2.
To further analyze isomerization of compound 5 in vivo, we determined the ratio of four isomers (5a-5d) when dosed with either 5a or 5b based upon the HPLC spectra from analysis of the extracts of plasma and tumor tissue homogenate (Figure 8). Our data showed that when dosed with 5b, 5b remained the dominant isomer at 1 h and 6 h time-points and there was only a small percentage of the other three isomers (<10% when combined). In contrast, when dosed with 5a, there was rapid isomerization and isomer 5b became the dominant form at the 6 h time-point. These data show that 5b isomerizes slowly in vivo whereas 5a isomerizes very fast.
Figure 8.

Isomerization of 5a and 5b in tumor tissue or plasma. Panels A and B depict the isomerization of 5a and panels C and D depict the isomerization of 5b in plasma and tumor tissue accordingly. The proportion of compounds is on the Y axis and the time when samples were harvested is on the X axis. Bars indicate the standard error.
We determined the antitumor activity of 5a and 5b in the SJSA-1 xenograft model. SCID mice bearing established SJSA-1 tumors were treated with 5a at 200 mg/kg or 5b at 100 mg/kg and 200 mg/kg daily via oral gavage for 14 days (Figure 9). Isomer 5a at 200 mg/kg daily dosing inhibited tumor growth by 41% as compared to vehicle control (p < 0.0001) at the end of the treatment (day 26). In comparison, 5b at 100 mg/kg inhibited tumor growth by 67% (p <0.0001) but shrank tumors at 200 mg/kg by an average of 91% at the end of treatment (day 26, see SI). There was no significant weight loss (SI) or other signs of toxicity for either compound. The efficacy data show that isomer 5b has much better in vivo antitumor activity than isomer 5a, consistent with the much stronger p53 activation and apoptosis induction by 5b than by 5a in tumor tissues (Figure 7).
Figure 9.
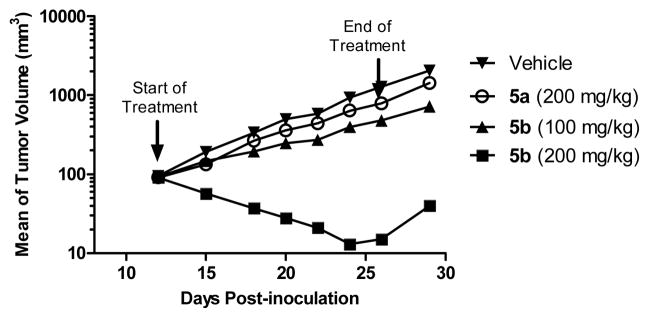
Antitumor Activity of 5a and 5b in the SJSA-1 Osteosarcoma Tumor Xenograft Model.
Although 5b had a strong antitumor activity and shrank tumors by 91% in the SJSA-1 xenograft model, it failed to achieve complete tumor regression. We hypothesized that the failure of complete tumor regression by 5b may be due to its less than ideal pharmacokinetic (PK) properties. To this end, we have performed extensive modifications to spirooxindol-containing compounds to further improve their in vivo PK profile, which ultimately led to identification of compound 15b (MI-888) (Figure 10).12 Compound 15b binds to MDM2 with a Ki value of 0.44 nM. In comparison, the corresponding cis-trans isomer 15a has a Ki value of 4.5 nM in binding to MDM2, being 10-times less potent than 15b.
Figure 10.
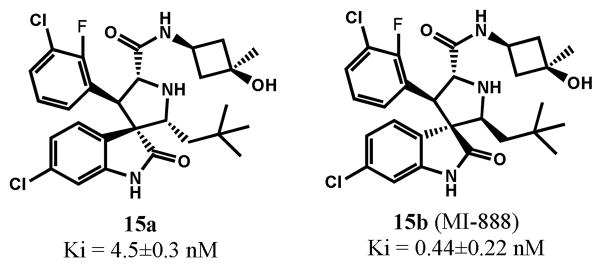
Structures and MDM2 binding affinity of 15a and 15b
To test the therapeutic potential and the potential difference in their antitumor activity between 15a and 15b, both compounds were tested in the SJSA-1 tumor xenograft model. In this experiment, SCID mice bearing established SJSA-1 tumors were treated with 15a at 100 mg/kg or 15b at 100 mg/kg via oral gavage for 14 days (Figure 11).
Figure 11.

Antitumor Activity of 15a and 15b in the SJSA-1 Osteosarcoma Tumor Xenograft Model
In contrast to 5b, and other previously reported potent MDM2 inhibitors, such as MI-219, Nutlin-3a and AM-8553,6b,13 15b was capable of achieving rapid and complete tumor regression. After treatments with 15b for 10 days at 100 mg/kg, all tumors of the treated mice completely regressed. Importantly, the complete regression was longlasting; all mice treated with 15b remained completely tumor free for an additional 64 days. In comparison, while 15a at 100 mg/kg daily dosing inhibited tumor growth by 78% as compared to vehicle control (p < 0.0001) at the end of the treatment (day 24), it failed to achieve tumor regression. There was no significant weight loss or other signs of toxicity for either compound (SI). The efficacy data thus clearly demonstrate that isomer 15b is much more efficacious than isomer 15a.
CONCLUSIONS
In the present study, we have performed detailed investigations on the reversible ring opening-recyclization reaction of selected spirooxindole-containing MDM2 inhibitors. This reaction afforded four diastereoisomers from one single stereoisomer. The stable isomers (isomers b) for these compounds have a cis-cis configuration and bind to MDM2 with better affinities than the other three isomers. Importantly, our investigation has led to the identification of a set of highly potent MDM2 inhibitors with a low nanomolar binding affinity. In particular, compound 15b (MI-888) has a Ki value of 0.44 nM to MDM2 and is capable of achieving complete and long-lasting tumor regression in an animal model of human cancer. Our data clearly suggest that MI-888 is a highly promising MDM2 inhibitor and warrants extensive preclinical investigation as a potential clinical development candidate.
EXPERIMENTAL SECTION
1. Chemistry
All reactions were conducted in round-bottomed flasks with a Teflon-coated magnet stirring bar. Experiments involving moisture and/or air sensitive components were performed under an N2 atmosphere. Commercial reagents and anhydrous solvents were used without further purification. Crude reaction products were purified by flash column chromatography on silica gel. Further purification was performed on a semipreparative HPLC (Waters Delta 600) with SunFire C18 reverse phase column (19 mm × 150 mm). The mobile phase was a gradient flow of solvent A (water, 0.1% of TFA) and solvent B (CH3CN, 0.1% of TFA or MeOH, 0.1% of TFA) at a flow rate of 10 mL/min. Proton nuclear magnetic resonance (1H NMR) and carbon nuclear magnetic resonance (13C NMR) spectroscopy were performed on a Bruker Advance 300 NMR spectrometer. Low resolution ESI mass spectra analyses were determined on a Thermo-Scientific LCQ Fleet mass spectrometer. The analytical reverse phase HPLC (rp-HPLC) model was Waters 2795 Separation (UV detection at 230 nm and 254 nm wave lengths). All final compounds were purified to ≥95% purity as determined by analytical HPLC analysis using reverse phase column (SunFire, C18-5 μm, 4.6 × 150 mm) unless otherwise stated.
The procedures for synthesis of compounds 1a-6a, 12a, and 15a were adapted from our previously reported methods6d,7 shown in Scheme 1 with some modifications.6a The trans-cis isomers 1a-6a and 15a were obtained as major products after ceric ammonium nitrate (CAN) oxidative cleavage of chiral auxiliary. The trans-cis isomers 9a, 11a, and 13a were minor isomers after CAN oxidation and efforts made to obtain 9a-13a in high purity proved futile.
Detailed procedures for the synthesis of 7a and 8a via different synthetic routes are in the Supporting Information (SI).
(2′R,3S,4′R,5′R)-6-chloro-4′-(3-chlorophenyl)-N,N-dimethyl-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (1a, TFA salt)
1H NMR (300 MHz, CD3OD): 7.40-7.32 (m, 4H), 6.87 (s, 1H), 6.76 (d, J = 8.22 Hz, 1H), 6.55 (d, J = 8.17 Hz, 1H), 5.47 (d, J = 7.88 Hz, 1H), 4.28 (d, J = 6.38 Hz, 1H), 4.17 (d, J = 7.88 Hz, 1H), 2.97 (s, 3H), 2.78 (s, 3H), 2.05 (dd, J = 15.31, 7.63, Hz, 1H), 1.16 (d, J = 15.31 Hz, 1H), 0.84 (s, 9H) 13C (75 MHz, CD3OD): 180.46, 167.42, 145.49, 139.42, 136.92, 136.38, 131.97, 130.50, 130.23, 128.37, 128.30, 124.57, 123.26, 112.09, 63.35, 63.27, 62.84, 55.37, 42.47, 38.03, 36.96, 30.99, 29.59 ESI-MS calculated for C25H3035Cl2N3O2 [M+H]+ = 474.17, Found: 474.50. [α]D25 = − 40.4° (c = 0.0113, MeCN)
(2′R,3S,4′R,5′R)-6-chloro-4′-(3-chlorophenyl)-N-methyl-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (2a, TFA salt)
1H NMR (300 MHz, CD3OD): 7.28-7.16 (m, 3H), 7.16-7.06 (m, 1H), 6.92-6.82 (m, 2H), 6.80-6.76 (m, 1H), 4.92 (d, J =10.23, 4.20-4.10 (m, 2H), 2.73 (s, 3H), 1.99 (s, J = 15.32, 6.75 Hz, 1H), 1.47 (dd, J = 15.54, 3.49 Hz, 1H), 0.80 (s, 9H) 13C (75 MHz, CD3OD): 180.04, 167.26, 144.79, 137.00, 136.58, 135.84, 131.40, 129.99, 129.84, 128.18, 128.11, 126.71, 123.42, 111.88, 64.23, 64.23, 62.35, 57.08, 42.71, 30.95, 29.58, 27.00. ESI-MS calculated for C24H2835Cl2N3O2 [M+H]+ = 460.16, Found: 460.52
(2′R,3S,4′S,5′R)-6-chloro-4′-(3-chloro-2-fluorophenyl)-N,Ndimethyl-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (3a, TFA salt)
1H NMR (300 MHz, CD3OD): 7.80-7.70 (m, 1H), 7.50-7.40 (m, 1H), 7.32-7.22 (m, 1H), 6.92-6.88 (m, 1H), 6.74 (dd, J = 8.15, 1.75 Hz, 1H), 6.48 (d, J = 8.11 Hz, 1H), 5.58 (d, J = 7.94 Hz, 1H), 4.38 (d, J = 7.94 Hz, 1H), 4.26 (d, J = 7.55 Hz, 1H), 2.99 (s, 3H), 2.84 (s, 3H), 2.06 (dd, J = 15.42, 7.72 Hz, 1H), 1.13 (d, J = 15.37 Hz, 1H), 0.89 (s, 9H) 13C (75 MHz, CD3OD): 180.07, 166.93, 157.51 (d, JC-F = 243.98 Hz), 145.59, 137.11, 132.54, 128.77, 127.66, 126.74 (d, JC-F = 4.55 Hz), 125.89 (d, JC-F =13.40 Hz), 123.68, 122.85 (d, JC-F = 10.19 Hz), 123.10 (d, JC-F = 18.37 Hz), 112.07, 63.03, 62.05, 61.76, 42.09, 37.70, 36.89, 30.77, 29.34 ESI-MS calculated for C25H2935Cl2FN3O2 [M+H]+ = 492.16, Found: 492.44
(2′R,3S,4′S,5′R)-6-chloro-4′-(3-chloro-2-fluorophenyl)-Nmethyl-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (4a, TFA salt, 94% purity)
1H NMR (300 MHz, CD3OD): 7.66-7.54 (m, 1H), 7.38-7.28 (m, 1H), 7.22-7.10 (m, 1H), 6.87 (s, 1H), 6.82-6.72 (m, 2H), 5.21 (d, J = 10.00 Hz, 1H), 4.50 (d, J = 9.93 Hz, 1H), 4.30-4.24 (m, 1H), 2.75 (s, 3H), 2.07 (dd, J = 15.35, 7.35 Hz, 1H), 1.8 (d, J = 14.57 Hz, 1H), 0.80 (s, 9H) 13C (75 MHz, CD3OD): 180.31, 167.24, 157.81 (d, JC-F = 247.78 Hz), 145.44, 136.94, 132.11, 128.66, 127.94, 126.47 (d, JC-F = 4.45 Hz), 125.29, 125.01 (d, JC-F = 13.96 Hz), 123.37, 122.64 (d, JC-F = 18.03 Hz), 122.04, 64.35, 63.69, 61.74, 49.51, 42.34, 30.92, 29.55, 27.10. ESI-MS calculated for C24H2735Cl2FN3O2 [M+H]+ = 478.15, Found: 478.92
(2′R,3S,4′S,5′R)-6-chloro-4′-(3-chloro-2-fluorophenyl)-N-((S)-3,4-dihydroxybutyl)-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (5a, TFA salt)
1H NMR (300 MHz, CD3OD): 7.63-7.53 (m, 1H), 7.40-7.30 (m, 1H), 7.23-7.13 (m, 1H), 6.87 (d, J = 1.36 Hz, 1H), 6.86-6.74 (m, 2H), 5.19 (d, J = 10.22 Hz, 1H), 4.49 (d, J = 10.22 Hz,1H), 4.25 (dd, J = 7.35, 2.71 Hz, 1H), 3.56-3.43 (m, 1H), 3.42-3.30 (m, 4H), 2.07 (dd, J = 15.43, 7.44 Hz,1 H), 1.72-1.58 (m, 1H), 1.56-1.40 (m, 1H), 1.30 (dd, J = 15.43, 2.62 Hz, 1H), 0.80 (s, 9H). 13C (75 MHz, CD3OD): 180.26, 166.78, 157.81 (d, JC-F = 247.93 Hz), 145.43, 136.90, 132.16, 128.82, 127.96, 126.48 (d, JC-F = 4.49 Hz), 125.43, 124.94 (d, JC-F = 14.00 Hz), 123.37, 122.63 (d, JC-F = 18.23 Hz), 112.00, 71.01, 67.25, 64.38, 63.80, 61.69, 50.06, 42.36, 38.30, 33.80, 30.94, 29.55. ESI-MS calculated for C27H3335Cl2FN3O4 [M+H]+ = 552.18, Found: 552.92; [α]D25 = − 40.1° (c = 0.0106, MeOH)
(2′R,3S,4′S,5′R)-6-chloro-4′-(3-chloro-2-fluorophenyl)-N-(2-morpholinoethyl)-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (6a, TFA salt, 93.5% purity)
1H NMR (300 MHz, CD3OD): 7.59 (t, J = 6.93 Hz, 1H), 7.41 (td, J = 7.55, 0.97 Hz, 1H), 7.22 (t, J = 7.90 Hz, 1H), 6.90 (d, J = 1.79 Hz, 1H), 6.77 (dd, J = 8.14, 1.79 Hz, 1H), 6.48 (d, J = 8.15 Hz, 1H), 5.21 (d, J = 8.22 Hz, 1H), 4.50 (d, J = 8.19 Hz, 1H), 4.17 (dd, J = 7.63, 2.23 Hz, 1H), 4.10-3.80 (m, 4H), 3.75-3.45 (m, 4H), 3.40-3.00 (m, 4H), 2.08-1.96 (m, 1H), 1.19 (dd, J = 15.32, 2.23 Hz, 1H), 0.81 (s, 9H). 13C (75 MHz, CD3OD): 180.28, 168.91, 157.75 (d, JC-F = 248.38 Hz), 145.50, 137.13, 132.29, 128.63, 127.67, 126.63 (d, JC-F = 4.50 Hz), 125.74 (d, JC-F = 13.69 Hz), 124.72, 123.44, 122.83 (d, JC-F = 18.25 Hz), 112.20, 65.16, 64.25, 63.84, 62.22, 58.01, 53.84, 50.05, 42.39, 35.72, 30.95, 29.52. ESI-MS calculated for C29H3635Cl2FN4O3 [M+H]+ = 577.21, Found: 577.48. [α]D25 = − 43.3° (c = 0.0139, MeCN).
(2′R,3S,4′S,5′R)-ethyl 6-chloro-4′-(3-chloro-2-fluorophenyl)-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxylate (7a, TFA salt)
1H NMR (300 MHz, CD3OD): 7.50-7.40 (m, 2H), 7.28-7.18 (m, 1H), 6.91 (d, J = 1.81 Hz, 1H), 6.78(dd, J = 8.13, 1.87 Hz, 1H), 6.46 (d, J = 8.14 Hz, 1H), 5.30 (d, J = 8.52 Hz, 1H), 4.43 (d, J = 8.52 Hz, 1H), 4.40-4.20 (m, 2H), 4.08 (dd, J = 7.47, 2.43 Hz, 1H), 2.00 (dd, J = 15.32, 7.53 Hz, 1H), 1.22 (t, J = 7.12 Hz, 3H), 1.19 (dd, J = 15.32, 2.55 Hz, 1H), 0.81 (s, 9H). 13C (75 MHz, CD3Cl, note free amine): 181.33, 171.91, 156.54 (d, JC-F = 248.02 Hz), 142.85, 134.15, 129.91, 128.37 (d, JC-F = 14.61 Hz), 127.19 (d, JC-F = 3.19 Hz), 125.94, 124.94, 124.63 (d, JC-F = 4.50 Hz), 122.10, 121.69 (d, JC-F = 18.50 Hz), 110.72, 67.16, 65.70, 63.19, 61.74, 51.17, 43.45, 30.33, 29.97, 14.32. ESI-MS calculated for C25H2835Cl2FN2O3 [M+H]+ = 493.15, Found: 493.44
(2′R,3S,4′S,5′R)-6-chloro-4′-(3-chloro-2-fluorophenyl)-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxylic acid (8a, TFA salt)
1H NMR (300 MHz, CD3OD): 7.56-7.46 (m, 1H), 7.46-7.36 (m, 1H), 7.26-7.18 (m, 1H), 6.91 (d, J = 1.61 Hz, 1H), 6.76 (dd, J = 8.10, 1.50 Hz, 1H), 6.47 (d, J = 8.14 Hz, 1H), 5.29 (d, J = 8.67 Hz, 1H), 4.43 (d, J = 8.67 Hz, 1H), 4.11 (d, J = 7.47, 2.13 Hz, 1H), 2.02 (dd, J = 15.36, 7.50 Hz, 1H), 1.16 (dd, J = 15.36, 2.24 Hz, 1H), 0.81 (s, 9H). ESI-MS calculated for C23H2435Cl2FN2O3 [M+H]+ = 465.11, Found: 465.42;
(2′R,3S,4′R,5′R)-6-chloro-4′-(3-chlorophenyl)-N-((S)-3,4-dihydroxybutyl)-5-fluoro-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (12a, TFA salt, 82% purity)
1H NMR (300 MHz, CD3OD): 7.32-7.22 (m, 3H), 7.18-7.10 (m, 1H), 7.06 (d, J = 8.74 Hz, 1H), 6.88 (d, J = 6.08 Hz, 1H), 4.97 (d, J = 10.79 Hz, 1H), 4.22 (d, J = 10.79 Hz, 1H), 4.26-4.18 (m, 1H), 3.50-3.20 (m, 5H), 2.03 (dd, J = 15.35, 6.56 Hz, 1H), 1.68-1.52 (m, 2H), 1.50-1.40 (m, 1H), 0.84 (s, 9H). 13C (75 MHz, CD3OD): 179.64, 166.47, 140.40 (d, JC-F = 2.94 Hz), 136.54, 136.06, 131.64, 130.16, 130.02, 128.14, 127.27 (d, JC-F = 40.14 Hz), 123.05 (d, JC-F = 19.44 Hz), 115.94 (d, JC-F = 25.54 Hz), 113.09, 70.91, 67.24, 64.19, 64.10, 62.73, 57.52, 42.66, 38.14, 33.85, 30.99, 29.57. ESI-MS calculated for C27H3335Cl2FN3O4 [M+H]+ = 552.18, Found: 552.75
(2′R,3S,4′R,5′R)-6-chloro-4′-(3-chlorophenyl)-N-((1s,3S)-3-hydroxy-3-methylcyclobutyl)-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (15a, TFA salt)
1H NMR (300 MHz, MeOD-d4): 7.40-7.50 (m, 1H), 7.40-7.30 (m, 1H), 7.20-7.10 (m, 1H), 6.85 (s, 1H), 6.83-6.75 (m, 2H), 5.11 (d, J = 10.11 Hz, 1H), 4.47 (d, J = 10.09 Hz, 1H), 4.22 (dd, J = 7.32, 2.72 Hz, 1H), 4.00-3.80 (m, 1H), 2.50-2.30 (m, 2H), 2.15-1.95 (m, 2H), 1.95-1.80 (m, 1H), 1.40-1.20 (m, 1H), 1.29 (s, 3H), 0.80 (s, 9H).13C NMR (75 MHz, MeOD-d4): 180.0, 165.9, 157.6 (d, JC-F = 248 Hz), 145.3, 136.7, 131.9, 128.6, 127.7, 126.2 (d, JC-F = 4.76 Hz), 125.2, 124.8 (d, JC-F = 13.8 Hz), 123.1, 122.4 (d, JC-F = 18.1 Hz), 111.8, 64.2, 63.5, 61.4, 49.7, 45.5, 45.4, 42.2, 38.4, 30.7, 29.4, 27.4. ESI-MS calculated for C28H3335Cl2FN3O3 [M+H]+: 548.17, Found: 548.25. [α]D25 = −30.6° (c = 0.0154 g/mL in MeOH);
The cis-cis isomers 1b-7b, 10b, 12b, and 15b were obtained as major products after aging the materials purified by flash column chromatography in appropriate solvents for 3–4 days. In cases of 9b, 11b, and 13b, the cis-cis isomers were obtained as major products after the CAN oxidation without aging. Generally, the side isomers c and d were observed in small amounts after oxidation or after aging.
Standard procedure for aging: 30 mg material obtained from CAN oxidation (prepurified by flash column chromatography) was placed in a round bottomed flask with a magnetic stirring bar. MeCN (2.4 mL) was added to dissolve the product. H2O (2.0 mL) was added to this MeCN solution followed by 0.4 mL NaHCO3 saturated solution to adjust the pH to 8. This solution was allowed to stir at room temperature for 3–4 days. The percentage of isomer 2 and other isomers was determined using analytical HPLC. Further purification was performed by semi-preparative or preparative HPLC. Pure MeOH can also be used as aging solvent.
(2′S,3R,4′R,5′R)-6-chloro-4′-(3-chlorophenyl)-N,N-dimethyl-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (1b, TFA salt)
1H NMR (300 MHz, CD3OD): 7.58 (d, J = 8.08 Hz, 1H), 7.32-7.10 (m, 5H), 6.78 (d, J = 1.54 Hz, 1H), 5.69 (d, J = 10.03 Hz, 1H), 4.41 (d, J = 6.93 Hz, 1H), 4.14 (d, J = 10.03 Hz, 1H), 2.96 (s, 3H), 2.76 (s, 3H), 1.90 (dd, J = 15.41, 8.26 Hz, 1H), 1.15 (d, J = 15.41 Hz, 1H), 0.89 (s, 9H). 13C (75 MHz, CD3OD): 178.04, 168.67, 145.47, 137.24, 135.99, 135.36, 131.69, 130.50, 129.79, 128.44, 126.49, 124.27, 123.84, 112.31, 65.29, 64.52, 60.50, 57.35, 42.59, 38.06, 37.00, 31.03, 29.66 ESI-MS calculated for C25H3035Cl2N3O2 [M+H]+ = 474.17, Found: 474.68. [α]D25 = 32.1° (c = 0.0126, MeCN)
(2′R,3R,4′R,5′R)-6-chloro-4′-(3-chlorophenyl)-N,N-dimethyl-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (1d, TFA salt)
1H NMR (300 MHz, CD3OD): 7.72 (d, J = 8.08 Hz, 1H), 7.28-7.22 (m, 1H), 7.20-7.12 (m, 2H), 7.12-7.08 (m, 1H), 7.04 (d, J = 7.65 Hz, 1H), 6.81 (d, J = 1.79 Hz, 1H), 5.54 (d, J = 10.87 Hz, 1H), 4.37 (dd, J = 6.54, 4.52 Hz, 1H), 4.15 (d, J = 10.85 Hz, 1H), 2.98 (s, 3H), 2.81 (s, 3H), 1.70 (dd, J = 15.24, 6.67 Hz, 1H), 1.20 (dd, J = 15.26, 4.43 Hz, 1H), 0.91 (s, 9H). 13C (75 MHz, CD3OD): 177.03, 169.16, 144.95, 136.94, 135.68, 135.34, 131.25, 130.20, 130.10, 128.88, 128.33, 124.24, 123.43, 112.43, 65.98, 65.14, 60.60, 60.24, 44.23, 38.07, 37.04, 30.97, 29.77 ESI-MS calculated for C25H3035Cl2N3O2 [M+H]+ = 474.17, Found: 474.50. [α]D25 = − 132.9° (c = 0.0031, MeCN)
(2′S,3R,4′R,5′R)-6-chloro-4′-(3-chlorophenyl)-N-methyl-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (2b, TFA salt)
1H NMR (300 MHz, CD3OD): 7.63 (d, J = 8.06 Hz, 1H), 7.30-7.14 (m, 4H), 7.12-7.00 (m, 1H), 6.82-6.76 (m, 1H), 5.29 (d, J = 11.24 Hz, 1H), 4.47 (d, J = 6.68 Hz, 1H), 4.16 (d, J = 11.22 Hz, 1H), 2.73 (s, 3H), 1.92 (dd, J = 15.40, 8.39 Hz, 1H), 1.17 (d, J = 16.85 Hz, 1H), 0.90 (s, 9H). 13C (75 MHz, CD3OD): 177.78, 168.54, 145.38, 137.07, 135.77, 134.60, 131.40, 130.29, 129.48, 128.29, 126.32, 124.24, 124.16, 112.18, 65.11, 64.18, 62.85, 56.95, 43.42, 30.97, 29.66, 26.98 ESI-MS calculated for C24H2835Cl2N3O2 [M+H]+ = 460.16, Found: 460.48.
(2′S,3R,4′S,5′R)-6-chloro-4′-(3-chloro-2-fluorophenyl)-N,Ndimethyl-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (3b, TFA salt)
1H NMR (300 MHz, CD3OD): 7.68-7.57 (m, 2H), 7.45-7.35 (m, 1H), 7.22-7.10 (m, 2H), 6.84-6.77 (m 1H), 5.68 (d, J = 10.24 Hz, 1H), 4.64 (d, J = 10.24 Hz, 1H), 4.48 (dd, J = 8.20, 1.70 Hz, 1H), 2.98 (s, 3H), 2.80 (s, 3H), 1.89 (dd, J = 15.48, 8.26 Hz, 1H), 1.14 (dd, J = 15.48, 1.67 Hz, 1H), 0.89 (s, 9H). 13C (75 MHz, CD3OD): 177.96, 168.37, 152.99 (d, JC-F = 253.32 Hz), 145.26, 137.39, 132.82, 128.82, 126.95, 126.70 (d, JC-F = 4.79 Hz), 124.28, 122.09 (d, JC-F = 13.13 Hz), 118.90, 115.11, 112.24, 64.69, 59.97, 42.64, 37.96, 37.03, 31.03, 29.62 ESI-MS calculated for C25H2935Cl2FN3O2 [M+H]+ = 492.16, Found: 492.46.
(2′S,3R,4′S,5′R)-6-chloro-4′-(3-chloro-2-fluorophenyl)-Nmethyl-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (4b, TFA salt)
1H NMR (300 MHz, CD3OD): 7.65-7.55 (m, 2H), 7.40-7.34 (m, 1H), 7.20-7.10 (m, 2H), 6.84-6.80 (m, 1H), 5.26 (d, J = 11.15 Hz, 1H), 4.64 (d, J = 11.19 Hz, 1H), 4.45 (d, J = 7.86 Hz, 1H), 2.74 (s, 3H), 1.87 (dd, J = 15.19, 8.58 Hz, 1H), 1.11 (d, J = 15.21 Hz, 1H), 0.90 (s, 9H). 13C (75 MHz, CD3OD): 177.75, 168.20, 159.53 (d, JC-F = 248.25 Hz), 145.22, 137.24, 132.59, 128.58, 126.63 (d, JC-F = 5.25 Hz), 124.16, 123.43, 126.66, 122.59 (d, JC-F = 4.05 Hz), 121.54 (d, JCF = 12.75 Hz), 112.13, 64.49, 64.40, 62.73, 55.34, 43.33, 31.01, 29.58, 27.06 ESI-MS calculated for C24H2735Cl2FN3O2 [M+H]+ = 478.15, Found: 478.25.
(2′S,3R,4′S,5′R)-6-chloro-4′-(3-chloro-2-fluorophenyl)-N-((S)-3,4-dihydroxybutyl)-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (5b, TFA salt)
1H NMR (300 MHz, CD3OD): 7.60 (d, J = 8.15 Hz, 1H), 7.55 (t, J = 7.12 Hz, 1H), 7.38 (t, J = 7.57 Hz, 1H), 7.20-7.10 (m, 2H), 6.78 (d, J = 1.70 Hz, 1H), 5.27 (d, J = 11.40 Hz, 1H), 4.62 (d, J = 11.40 Hz, 1H), 4.52 (dd, J = 8.33, 1.29 Hz, 1H), 3.45-3.25 (m, 5H), 1.90 (dd, J = 15.46, 8.38 Hz, 1H), 1.64-1.48 (m, 1H), 1.46-1.32 (m, 1H), 1.14 (dd, J = 15.46, 1.34 Hz, 1H), 0.87 (s, 9H). 13C (75 MHz, CD3OD): 177.77, 167.67, 157.90 (d, JC-F = 251.2 Hz), 145.21, 137.26, 132.67, 128.64, 126.89 (d, JC-F = 1.88 Hz), 126.65 (d, JC-F = 4.89 Hz), 124.17, 123.43, 122.60 (d, JC-F = 19.0 Hz), 121.52 (d, JC-F = 13.0 Hz), 112.14, 70.99, 67.25, 64.45, 62.873, 43.39, 38.26, 33.78, 31.02, 29.57. ESI-MS calculated for C27H3335Cl2FN3O4 [M+H]+ = 552.18, Found: 552.42. [α]D25 = − 36.2° (c = 0.0147, MeOH)
(2′S,3S,4′S,5′R)-6-chloro-4′-(3-chloro-2-fluorophenyl)-N-((S)-3,4-dihydroxybutyl)-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (5c, TFA salt)
1H NMR (300 MHz, CD3OD): 8.37 (s, broad, NH), 7.62 (d, J = 8.11 Hz, 1H), 7.50-7.34 (m, 2H), 7.36-7.10 (m, 2H), 6.83 (d, J = 1.83 Hz, 1H), 4.32 (d, J = 12.09 Hz, 1H), 4.24-4.14 (m, 1H), 3.50-3.10 (m, 5H), 1.86-1.72 (m, 2H), 1.58-1.44 (m, 1H), 1.44-1.26 (m, 1H), 0.80 (s, 9H). ESI-MS calculated for C27H3335Cl2FN3O4 [M+H]+ = 552.18, Found: 552.42.
(2′R,3R,4′S,5′R)-6-chloro-4′-(3-chloro-2-fluorophenyl)-N-((S)-3,4-dihydroxybutyl)-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (5d, TFA salt)
1H NMR (300 MHz, CD3OD): 7.49 (d, J = 8.02 Hz, 1H), 7.36-7.24 (m, 1H), 7.11 (dd, J = 8.09, 1.89 Hz, 1H), 7.06-6.96 (m, 1H), 6.96-6.88 (m, 1H), 6.80 (d, J = 1.88 Hz, 1H), 4.89 (d, J = 11.57 Hz, 1H), 4.46 (d, J = 11.57 Hz, 1H), 4.35-4.25 (m, 1H), 3.56-3.40 (m, 1H), 3.40-3.20 (m, 4H), 1.70-1.40 (m, 3H), 1.10 (dd, J = 14.59, 3.06 Hz, 1H), 0.91 (s, 9H). ESI-MS calculated for C27H3335Cl2FN3O4 [M+H]+= 552.18, Found: 552.40.
(2′S,3R,4′S,5′R)-6-chloro-4′-(3-chloro-2-fluorophenyl)-N-(2-morpholinoethyl)-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (6b, TFA salt)
1H NMR (300 MHz, CD3OD): 7.66-7.48 (m, 2H), 7.42-7.32 (m, 1H), 7.20-7.10 (m, 2H), 6.78 (d, J = 1.68 Hz, 1H), 5.38 (d, J = 11.48 Hz, 1H), 4.65 (d, J = 11.48 Hz, 1H), 4.52 (d, J = 7.59 Hz, 1H), 4.00-3.70 (m, 4H), 3.70-3.60 (m, 2H), 3.50-3.40 (m, 2H), 3.40-3.10 (m, 4H), 1.97 (dd, J = 15.40, 8.62 Hz, 1H), 1.12 (d, J = 15.40 Hz, 1H), 0.88 (s, 9H). 13C (75 MHz, CD3OD): 177.73, 169.20, 145.19, 137.15, 132.54, 128.69, 126.84, 126.64 (d, JC-F = 4.80 Hz), 124.11, 123.72, 122.57 (d, JC-F = 13.19 Hz), 121.77 (d, JC-F = 13.19 Hz), 112.06, 65.08, 64.59, 64.32, 62.69, 57.41, 53.70, 48.47, 43.55, 35.61, 31.06, 29.56. ESI-MS calculated for C29H3635Cl2FN4O3 [M+H]+ = 577.21, Found: 577.48. [α]D25 = − 24.7° (c = 0.0171, MeOH)
(2′S,3R,4′S,5′R)-ethyl 6-chloro-4′-(3-chloro-2-fluorophenyl)-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxylate (7b, TFA salt)
1H NMR (300 MHz, CD3OD): 7.59 (d, J = 7.41 Hz, 1H), 7.50-7.42 (m, 1H), 7.40-7.32 (m, 1H), 7.17-7.07 (m, 2H), 6.78 (d, J = 1.65 Hz, 1H), 5.61 (d, J = 12.25 Hz, 1H), 4.56 (d, J = 12.25 Hz, 1H), 4.47 (dd, J = 8.50, 1.50 Hz, 1H), 4.25 (dq, J = 10.77, 7.12 Hz, 1H), 4.13 (dq, J = 10.77, 7.12 Hz, 1H), 1.93 (dd, J = 15.39, 8.67 Hz, 1H), 1.34 (dd, J = 15.39, 1.57 Hz, 1H), 1.10 (t, J =7.12 Hz, 3H), 0.87 (s, 9H). ESI-MS calculated for C25H2835Cl2FN2O3 [M+H]+ = 493.15, Found: 493.44.
(2′S,3R,4′S,5′R)-6-chloro-4′-(3-chloro-2-fluorophenyl)-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxylic acid (8b, TFA salt)
1H NMR (300 MHz, CD3OD): 7.58-7.42 (m, 2H), 7.36-7.26 (m, 1H), 7.14-7.02 (m, 1H), 6.75 (s, 1H), 5.43 (d, J = 12.00 Hz, 1H), 4.58 (d, J = 11.97 Hz, 1H), 4.35 (d, J = 8.53 Hz, 1H), 1.87 (dd, J = 15.08, 9.31 Hz, 1H), 1.09 (d, J = 15.31 Hz, 1H), 0.85 (s, 9H). ESI-MS calculated for C23H2435Cl2FN2O3 [M+H]+ = 465.11, Found: 465.38.
(2′S,3R,4′S,5′R)-6-chloro-4′-(3-chloro-2-fluorophenyl)-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (9b, TFA salt)
1H NMR (300 MHz, CD3OD): 7.54-7.44 (m, 2H), 7.36-7.26 (m, 1H), 7.14-7.00 (m, 2H), 6.70 (d, J = 1.76 Hz, 1H), 5.22 (d, J = 11.36 Hz, 1H), 4.50 (d, J = 11.36 Hz, 1H), 4.41 (d, J = 8.23, 1.85 Hz, 1H), 1.81 (dd, J = 15.46, 8.31 Hz, 1H), 1.06 (dd, J = 15.46, 1.90 Hz, 1H), 0.78 (s, 9H). ESI-MS calculated for C23H2535Cl2FN3O2 [M+H]+ = 464.13, Found: 464.60.
(2′S,3R,4′R,5′R)-6-chloro-4′-(3-chlorophenyl)-5-fluoro-N,Ndimethyl-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (10b, TFA salt)
1H NMR (300 MHz, CD3OD): 7.66 (d, J = 8.49 HZ, 1H), 7.35-7.12 (m, 4H), 6.86 (d, J = 6.02 Hz, 1H), 5.67 (d, J = 10.03 Hz, 1H), 4.41 (dd, J = 8.20, 1.79 Hz, 1H), 4.13 (d, J = 10.03 Hz, 1H), 2.97 (s, 3H), 2.74 (s, 3H), 1.91 (dd, J = 15.46, 8,20 Hz, 1H) 1.17 (dd, J = 15.46, 1.82 Hz, 1H), 0.91 (s, 9H). 13C (75 MHz, CD3OD):177.80, 168.54, 156.04 (d, JC-F = 243.74 Hz), 141.04, 136.12, 135.16, 131.81, 130.66, 129.73, 128.52, 125.50 (d, JC-F =7.37 Hz), 123.70 (d, JC-F = 19.58 Hz), 114.43 (d, JC-F =25.50 Hz), 113.62, 65.81, 64.45, 60.58, 57.31, 42.59, 38.04, 37.00, 31.04, 29.63. ESI-MS calculated for C25H2935Cl2FN3O2 [M+H]+ = 492.16, Found: 492.50.
(2′S,3R,4′R,5′R)-6-chloro-4′-(3-chlorophenyl)-5-fluoro-Nmethyl-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (11b, TFA salt)
1H NMR (300 MHz, CD3OD): 7.68 (d, J = 8.47 Hz, 1H), 7.32-7.16 (m, 3H), 7.04 (d, J = 7.63 Hz, 1H), 6.85 (d, J = 6.01 Hz, 1H), 5.23 (d, J = 11.21 Hz, 1H), 4.46 (dd, J = 8.23, 1.76 Hz, 1H), 4.13 (d, J = 11.21 Hz, 1H), 2.71 (S, 3H), 1.90 (dd, J = 15.46, 8.23 Hz, 1H), 1.17 (dd, J = 15.48, 1.75 Hz, 1H), 0.89 (s, 9H). 13C (75 MHz, CD3OD): 177.57, 168.31, 159.98 (d, JC-F = 244.73 Hz), 140.90, 135.98, 134.31, 131.59, 130.56, 129.45, 128.47, 125.84 (d, JC-F = 7.48 Hz), 123.61 (d, JC-F = 19.45 Hz), 114.33 (d, JC-F = 25.29 Hz), 113.57, 65.62, 64.15, 62.93, 57.04, 63.42, 31.01, 29.60, 27.02. ESI-MS calculated for C24H2735Cl2FN3O2 [M+H]+ = 478.15, Found: 478.46.
(2′S,3R,4′R,5′R)-6-chloro-4′-(3-chlorophenyl)-N-((S)-3,4-dihydroxybutyl)-5-fluoro-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (12b, TFA salt)
1H NMR (300 MHz, CD3OD): 7.68 (d, J = 8.47 Hz, 1H), 7.32-7.17 (m, 3H), 7.12-7.02 (m, 1H), 6.85 (d, J = 6.00 Hz, 1H), 5.23 (d, J = 11.28 Hz, 1H), 4.46 (dd, J = 8.41, 1.91 Hz, 1H), 4.10 (d, J = 11.28 Hz, 1H), 3.50-3.20 (m, 5H), 1.90 (dd, J = 15.18, 8.62 Hz, 1H), 1.65-1.47 (m, 1H), 1.47-1.33 (m,1H), 1.18 (d, J = 15.18 Hz,1 H), 0.89 (s, 9H). 13C (75 MHz, CD3OD): 177.56, 167.81, 155.99 (d, JC-F = 243.27 Hz), 140.87, 135.98, 134.28, 131.63, 130.60, 129.51, 128.60, 125.82 (d, JC-F = 7.39 Hz), 123.58 (d, JCF = 19.68 Hz), 114.30 (d, JC-F = 25.32 Hz), 113.55, 70.86, 67.23, 65.53, 64.16, 63.07, 57.35, 43.43, 38.15, 33.78, 31.01, 29.57. ESI-MS calculated for C27H3335Cl2FN3O4 [M+H]+= 552.18, Found: 552.38; [α]D25 = 1.1° (c = 0.0334, free amine in MeOH).
(2′S,3R,4′R,5′R)-6-chloro-4′-(3-chlorophenyl)-5-fluoro-N-((1r,4R)-4-hydroxycyclohexyl)-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (13b, TFA salt)
1H NMR (300 MHz, CD3OD): 7.66 (d, J = 8.45 Hz, 1H), 7.30-7.16 (m, 3H), 7.10-7.03 (m, 1H), 6.85 (d, J = 5.98 Hz, 1H), 5.22 (d, J = 11.37 Hz, 1H), 4.46 (dd, J = 8.31, 1.68 Hz, 1H), 4.09 (d, J = 11.37 Hz, 1H), 3.70-3.50 (m, 1H), 3.50-3.39 (m, 1H), 2.00-1.84 (m, 3H), 1.82-1.70 (m, 1H), 1.58-1.46 (m, 1H), 1.40-1.12 (m, 4H), 1.08-0.90 (m, 1H), 0.88 (s, 9H). 13C (75 MHz, CD3OD): 177.42, 166.80, 155.82 (d, JC-F = 243.61 Hz), 140.67 (d, JC-F = 2.77 Hz), 135.73, 134.18, 131.36, 130.33, 129.44, 128.37, 125.77 (d, JC-F = 7.38 Hz), 123.38 (d, JC-F = 19.54 Hz), 114.06 (d, JC-F = 25.18 Hz), 113.39, 69.97, 65.12, 64.06, 62.94, 57.41, 49.88, 43.37, 34.38, 34.29, 30.98, 30.82, 29.39. ESI-MS calculated for C29H3535Cl2FN3O3 [M+H]+ = 562.20, Found: 562.36.
(2′S,3R,4′S,5′R)-6-Chloro-4′-(3-chloro-2-fluorophenyl)-N-(cis-3-hydroxy-3-ethylcyclobutyl)-2′-neopentyl-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (15b, MI-888, TFA salt)
1H NMR (300 MHz, MeOD-d4): 7.62-7.53 (m, 2H), 7.45-7.35 (m, 1H), 7.20-7.10 (m, 2H), 6.80-6.85 (m, 1H), 5.11 (d, J = 11.07 Hz, 1H), 4.57 (d, J = 11.07 Hz, 1H), 4.40 (d, J = 7.39 Hz, 1H), 4.00-3.80 (m, 1H), 2.50-2.35 (m, 1H), 2.35-2.20 (m, 1H), 2.10-1.90 (m, 1H), 1.90-1.60 (m, 2H), 1.30 (s, 3H), 1.20-1.05 (m, 1H), 0.88 (s, 9H). 13C NMR (75 MHz, MeOD-d4): 177.85, 167.96, 157.67 (d, JC-F = 249.51 Hz), 144.96, 136.85, 132.22, 128.53, 126.58, 126.34 (d, JC-F = 4.76), 123.91, 123.69, 122.30 (d, JC-F = 18.97 Hz), 122.02 (d, JC-F = 13.10 Hz), 111.84, 67.14, 64.67, 64.53, 62.91, 45.53, 45.47, 43.38, 38.08, 30.84, 29.50, 27.39. ESI-MS calculated for C28H3335Cl2FN3O3 [M+H]+: 548.19, Found: 548.25. [α]D25 = − 16.3° (MeOH, c = 0.0284 g/mL)
2. FP-based Protein Binding Assay
The binding affinity of MDM2 inhibitors was determined by an optimized, sensitive and quantitative fluorescence polarization-based (FP-based) binding assay, using a recombinant human His-tagged MDM2 protein (residues 1–118) and a FAM tagged p53-based peptide as the fluorescent probe.
The design of the fluorescent probe was based upon a previously reported high affinity p53-based peptidomimetic compound (5-FAM-βAla-βAla-Phe-Met-Aib-pTyr-(6-Cl-LTrp)-Glu-Ac3c-Leu-Asn-NH2),11 termed PMDM6-F. The Kd value of PMDM6-F with the MDM2 protein was determined to be 1.4 ± 0.3 nM by monitoring the total fluorescence polarization of mixtures composed with the fluorescent probe at a fixed concentration and the MDM2 protein with increasing concentrations up to full saturation. Fluorescence polarization values were measured using the Infinite M-1000 plate reader (Tecan U.S., Research Triangle Park, NC) in Microfluor 2 96-well, black, round-bottomed plates (Thermo Scientific).
In the saturation experiments, 1 nM of PMDM6-F and increasing concentrations of proteins were added to each well to a final volume of 125 μl in the assay buffer (100 mM potassium phosphate, pH 7.5, 100 μg/ml bovine γ-globulin, 0.02% sodium azide (Invitrogen), with 0.01% Triton X-100 and 4% DMSO). Plates were incubated at room temperature for 15–30 minutes with gentle shaking to ensure equilibrium. The polarization values in millipolarization units (mP) were measured at an excitation wavelength of 485 nm and an emission wave-length of 530 nm. Equilibrium dissociation constants (Kd) were then calculated by fitting the sigmoidal dose-dependent FP increases as a function of protein concentrations using Graphpad Prism 5.0 software (Graphpad Software, San Diego, CA).
Ki values of tested compounds were determined in a dose-dependent competitive binding experiment. Mixtures of 5 μl of a solution of the tested compound in different concentrations in DMSO and 120 μl of preincubated protein/fluorescent probe complex with fixed concentrations in the assay buffer (100 mM potassium phosphate, pH 7.5, 100 μg/ml bovine γ-globulin, 0.02% sodium azide, with 0.01% Triton X-100) were added to assay plates and incubated at room temperature with gentle shaking. The incubation time was controlled precisely at 15 minutes to minimize the influence of the initial isomerization to the binding affinities. Final concentrations of the protein and fluorescent probe in the competitive assays were 10 nM and 1 nM, respectively, and final DMSO concentration was 4%. Negative controls containing protein/fluorescent probe complex only (equivalent to 0% inhibition), and positive controls containing free fluorescent probe only (equivalent to 100% inhibition), were included in each assay plate. FP values were measured as described above. IC50 values were determined by nonlinear regression fitting of the sigmoidal dose-dependent FP decreases as a function of total compound concentrations using Graphpad Prism 5.0 software (Graphpad Software, San Diego, CA). Ki values of tested compounds to the MDM2 protein were calculated using the measured IC50 values, the Kd value of the fluorescent probe to the protein, and the concentrations of the protein and fluorescent probe in the competitive assays.10
3. Binding Kinetics Studies of 5a, 5b, 5c and 5d and Isomerization Studies in Binding Medium
Aliquots of freshly prepared DMSO stock solutions of compounds 5a-5d were diluted in FP binding assay medium to prepare the aqueous compound incubation solutions in which compound isomerization was taking place. Final compound concentration in the incubation solution was 25 μM, and 5% of DMSO was added to enhance the solubility. These solutions were stored at room temperature for the whole time range of the experiment. 80 μL aliquots of compound solutions were mixed with 20 μL of freshly prepared MDM2/PMDM6-F mixture in the assay plates at different time points. Final concentrations of the protein, fluorescent probe, and DMSO are the same as those in the competitive assays described above. Negative and positive controls were also included in each assay plate. Experimental time was precisely controlled so that each plate was read exactly 10 min after compound solution was pipetted from the incubation solution stock, which typically includes 5 min for plate preparation and 5 min of incubation at room temperature with gentle shaking. mP values were measured and IC50 values were determined as described above. Thus, it should be noted that all IC50 values presented are values actually obtained 10 min after the incubation times presented.
4. Cell Growth Inhibition Assay
The SJSA-1 osteosarcoma tumor cell line was purchased from the American Type Culture Collection. Cells were seeded in 96-well flat bottom cell culture plates at a density of (3–4)×103 cells/well and grown overnight, then incubated with compounds at different concentrations. The rate of cell growth inhibition after treatment with different concentrations of a compound was determined by assaying with a lactate dehydrogenase-based WST-8 assay (WST-8; Dojindo Molecular Technologies Inc., Gaithersburg, MD). WST-8 solution was added to each well to a final concentration of 10%, and then the plates were incubated at 37 °C for 2–3 h. The absorbance of the samples was measured at 450 nm using a TECAN ULTRA reader. The concentrations of the compounds that inhibited cell growth by 50% (IC50) were calculated by comparing absorbance of the untreated cells with the treated cells.
5. Pharmacodynamic (PD) Study
The SJSA-1 xenograft tumor model was used for PD studies. To develop xenograft tumors, 5 × 106 SJSA-1 cancer cells with 50% Matrigel were injected subcutaneously on the dorsal side of SCID mice (Charles River), one tumor per mouse. When xenograft tumors reached a mean of ~100 mm3 (70–130 mm3), two mice were treated with vehicle control (10% PEG400: 3% Cremophor: 87% PBS), 16 mice were treated with a single oral dose of compound 5a at 200 mg/kg and another 16 mice were treated with a single oral dose of compound 5b at 200 mg/kg. Mice treated with vehicle control were sacrificed at 6 h and the tumor tissue was harvested for Western blot analysis. Two mice treated with 5b were sacrificed each time at 1, 3, 6, and 24 h and tumor tissue was harvested. The same schedule was performed on mice treated with 5a.
6. Western Blotting
For western blot analysis, tumor cells or tumor tissues were lyzed in ice-cold RIPA buffer: 20 mM Tris- HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM b-glycerophosphate, 1 mM sodium orthovanadate and 1μg/ml leupeptin. The expressions of indicated proteins in the whole cell lysates were detected by western blot analysis using the following antibodies: anti-p53 (OP43; Calbiochem), anti-MDM2 (sc-965; Santa Cruz), anti-p21 (556431; BD Biosciences), anti-PARP (9542, Cell Signaling Technology), anti-caspase-3 (AAP-113, Stressgen Bioreagents) and HRP-conjugated anti-GAPDH (sc- 5778; Santa Cruz).
7. In vivo Experiment
For in vivo studies, the SJSA-1 xenograft tumor model was used. To develop xenograft tumors, 5 × 106 SJSA-1 cancer cells with 50% Matrigel were injected subcutaneously on the dorsal side of SCID mice (Charles River), one tumor per mouse. When xenograft tumors reached a mean of ~100 mm3 (70–130 mm3) the mice were randomized into groups of 8 mice. Vehicle Control (10% PEG400: 3% Cremophor: 87% PBS) was administered p.o. once per day for 14 days. Compound 5a was administered orally once per day for 14 days at 200 mg/kg and compound 5b was administered orally once per day for 14 days at 100 mg/kg and 200 mg/kg. Tumor sizes and animal weights were measured three times per week. Data are represented as mean tumor volumes. Tumor volume (mm3) = (AxB2)/2 where A and B are the tumor length and width (in mm), respectively. Statistical analyses were done by two-way ANOVA and unpaired two-tailed t test, using Prism (version 4.0, GraphPad). P < 0.05 was considered statistically significant. The same protocol was applied to compounds 15a and 15b.
8. Pharmacokinetics of 5a and 5b in tumor bearing SCID mice
The SJSA-1 xenograft tumor model was used for PK studies. To develop xenograft tumors, 5 × 106 SJSA-1 cancer cells with 50% Matrigel were injected subcutaneously on the dorsal side of SCID mice (Charles River), one tumor per mouse. When xenograft tumors reached a mean of ~100 mm3 (70–130 mm3), three mice were treated with vehicle control (10% PEG400: 3% Cremophor: 87% PBS), nine mice were treated with a single oral dose of compound 5a at 100 mg/kg and another nine mice were treated with a single oral dose of compound 5b at 100 mg/kg. Three mice treated with 5a were sacrificed each time at 1, 6, and 24 h, and the plasma and the tumor tissue were harvested and frozen at −78 °C immediately. The same schedule was performed on mice treated with 5b.
The quantitative LC-MS/MS analysis were conducted using an Agilent 1200 HPLC system coupled to an API 3200 mass spectrometer (Applied Biosystems, MDS Sciex Toronto, Canada) equipped with an API electrospray ionization (ESI) source. Aliquots (5 μL) were injected into a reversed-phase column (5 cm × 4.6 mm I.D., packed with 3.5 μm Xbridge C18, Waters). The mobile phase consisted of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). The gradient of mobile phase was as follows: 5% B for 0.5 min, 5% to 60% B over 7.5 min, 60% B for an additional 7.5 min, and then immediately set back to 5% B for re-equilibration. The flow rate of the mobile phase was 1.0 mL/min.
Internal standard (150 μL, 100 ng/mL in acetonitrile) was added to 50 μL of previously collected mice plasma or tumor tissue homogenate to precipitate proteins. 5 μL of supernatant was injected into the LC-MS/MS instrument. The mass spectrometer was operated at ESI positive ion mode and the detection of the ions was performed in the multiple reaction monitoring (MRM) mode, monitoring the transition of m/z 552.2→m/z 320.1 for 5a and 5b. Analyte concentrations in plasma and tumor tissue homogenate were obtained from a calibration curve constructed from the plot of peak area ratio versus concentration. The data was accepted based on the performance of quality control samples (QCs) prepared using SCID mice blank plasma (QCs at three concentrations in duplicate). The calibration range for 5b was established from 1.0 to 1000 ng/ml in blank SCID mice plasma, and 25 ng/g to 10000 ng/g in blank SCID mice tumor tissue homogenate. The calibration range for 5a was established from 2.0 to 2000 ng/ml in blank SCID mice plasma, and 25.0 ng/g to 10000 ng/g in blank SCID mice tumor homogenate.
Supplementary Material
Acknowledgments
We are grateful for financial support from the National Cancer Institute, National Institutes of Health (Grants R01CA121279, P50CA06956, and P50CA097248), the University of Michigan Cancer Center (Core Grant P30CA046592), and Ascenta Therapeutics, Inc. and Sanofi S.A.
Footnotes
Supporting Information (SI). The SI contains experimental procedures and characterization data for all new compounds, experimental procedures for isomerization studies, protein binding test, cytotoxicity studies, tumor size measurement, and statistic data of efficacy studies. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Trost BM, Brennan MK. Synthesis. 2009;18:3003–3025. [Google Scholar]; (b) Galliford CV, Scheidt KA. Angew Chem Int Ed. 2007;46:8748–8758. doi: 10.1002/anie.200701342. [DOI] [PubMed] [Google Scholar]; (c) Marti C, Carreira EM. Eur J Org Chem. 2003:2209–2219. [Google Scholar]; (d) Bartkovitz DJ, Chu X-J, Ding Q, Graves BJ, Jiang N, Zhang J, Zhang Z. PCT patent application WO 2011/067185. 2011; (e) Chen XH, Wei Q, Luo SW, Xiao H, Gong LZ. J Am Chem Soc. 2009;131:13819–13825. doi: 10.1021/ja905302f. [DOI] [PubMed] [Google Scholar]; (f) Sebahar PR, Williams RM. J Am Chem Soc. 2000;122:5666–5667. [Google Scholar]; (g) Nussbaum F, Danishefsky SJ. Angew Chem Int Ed. 2000;39:2175–2178. doi: 10.1002/1521-3773(20000616)39:12<2175::aid-anie2175>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 2.Edmondson S, Danishefsky SJ, Sepp-Lorenzino L, Rosen N. J Am Chem Soc. 1999;121:2147–2155. [Google Scholar]
- 3.Kang TH, Matsumoto K, Tohda M, Murakami Y, Takayama H, Kitajima M, Aimi N, Watanabe H. Eur J Pharmacol. 2002;444:38–45. doi: 10.1016/s0014-2999(02)01608-4. [DOI] [PubMed] [Google Scholar]
- 4.Lo MMC, Neumann CS, Nagayama S, Perlstein EO, Schreiber SL. J Am Chem Soc. 2004;126:16077–16086. doi: 10.1021/ja045089d. [DOI] [PubMed] [Google Scholar]
- 5.Antonchick AP, Gerding-Reimers C, Catarinella M, Schürmann M, Preut H, Ziegler S, Rauh D, Waldmann H. Nature Chem. 2010;2:735–740. doi: 10.1038/nchem.730. [DOI] [PubMed] [Google Scholar]
- 6.(a) Yu S, Qin D, Shangary S, Chen J, Wang G, Ding K, McEachern D, Qiu S, Nikolovska-Coleska Z, Miller R, Kang S, Yang D, Wang S. J Med Chem. 2009;52:7970–7973. doi: 10.1021/jm901400z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, Nikolovaska-Coleska Z, Ding K, Wang G, Chen J, Bernard D, Zhang J, Lu Y, Gu Q, Shah RB, Pienta KJ, Ling X, Kang S, Guo M, Sun Y, Yang D, Wang S. Proc Natl Acad Sci. 2008;105:3933–3938. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ding K, Lu Y, Nikolovska-Coleska Z, Wang G, Qiu S, Shangary S, Gao W, Qin D, Stuckey J, Krajewski K, Roller PP, Wang S. J Med Chem. 2006;49:3432–3435. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]; (d) Ding K, Lu Y, Nikolovska-Coleska Z, Qiu S, Ding Y, Gao W, Stuckey J, Krajewski K, Roller PP, Tomit Y, Parrish DA, Deschamps JR, Wang S. J Am Chem Soc. 2005;127:10130–10131. doi: 10.1021/ja051147z. [DOI] [PubMed] [Google Scholar]
- 7.Ding K, Wang G, Deschamps JR, Rarrish DA, Wang S. Tetrahedron Lett. 2005;46:5949–5951. [Google Scholar]
- 8.(a) Marti C, Carreira EM. Eur J Org Chem. 2003:2209–2219. [Google Scholar]; (b) Laus G, Brössner D, Senn G, Wurst K. J Chem Soc, Perkin Trans 2. 1996:1931–1936. [Google Scholar]; (c) Finch N, Taylor WI. J Am Chem Soc. 1962;84:3871–3877. [Google Scholar]
- 9.(a) Wenkert E, Udelhofen JH, Bhattacharyya NK. J Am Chem Soc. 1959;81:3763–3768. [Google Scholar]; (b) van Tamelen EE, Yardley JP, Miyano M, Hinshaw WB., Jr J Am Chem Soc. 1969;91:7333–7341. [Google Scholar]; (c) Seaton JC, Nair MD, Edwards OE, Marion L. Can J Chem. 1960;40:1035–1042. [Google Scholar]
- 10.Nikolovska-Coleska Z, Wang R, Fang X, Pan H, Tomita Y, Li P, Roller PP, Krajewski K, Saito NG, Stuckey JA, Wang S. Anal Biochem. 2004;332:261–73. doi: 10.1016/j.ab.2004.05.055. [DOI] [PubMed] [Google Scholar]
- 11.García-Echeverría C, Chène P, Blommers MJJ, Furet P. J Med Chem. 2000;43:3205–3208. doi: 10.1021/jm990966p. [DOI] [PubMed] [Google Scholar]
- 12.Zhao Y, Yu S, Sun W, Liu L, Kumar S, Lu J, McEachern D, Li X, Sun D, Wang S. J Med Chem. (in review) [Google Scholar]
- 13.(a) Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]; (b) Rew Y, Sun D, Turiso FGLD, Bartberger MD, Beck HP, Canon J, Chen A, Chow D, Deignan J, Fox BM, Huang X, Jiang M, Jiao X, Jin L, Kayser F, Kopecky D, Li Y, Lo MC, Long A, Michelsen K, Oliner JD, Osgood T, Ragains M, Saiki AY, Schneider S, Toteva M, Yakowec P, Yan X, Ye Q, Yu D, Zhao X, Zhou J, Medina JC, Olson S. J Med Chem. 2012;55:4936–4954. doi: 10.1021/jm300354j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.