

Abstract
We report a novel mouse model for the generation of sporadic tumors and show the efficiency of this approach by surveying Hedgehog (Hh)–related tumors. Up-regulation of the Hh pathway is achieved by conditionally regulated expression of an activated allele of Smoothened (R26-SmoM2) using either sporadic leakage or global postnatal induction of a ubiquitously expressed inducible Cre transgene (CAGGS-CreER). Following postnatal tamoxifen induction, CAGGS-CreER; R26-SmoM2 mice developed tumors with short latency and high penetrance. All mice exhibited rhabdomyosarcoma and basal cell carcinoma; 40% also developed medulloblastoma. In addition, mice showed a novel pancreatic lesion resembling low-grade mucinous cystic neoplasms in humans. In contrast, widespread activation of SmoM2 in the postnatal prostate epithelium results in no detectable morphologic outcome in 12-month-old mice. Comparison of gene expression profiles among diverse tumors identified several signature genes, including components of platelet-derived growth factor and insulin-like growth factor pathways, which may provide a common mechanistic link to the Hh-related malignancies. This experimental model provides a robust tool for exploring the process of Hh-dependent tumorigenesis and the treatment of such tumors. More generally, this approach provides a genetic platform for identifying tumorigenic potential in putative oncogenes and tumor suppressors and for more effective modeling of sporadic cancers in mice.
Introduction
Hedgehog (Hh) signaling plays many distinct roles in a variety of developmental processes (1). Hh proteins undergo autocleavage producing active lipid-modified signaling peptides that transduce signals through their interaction with a 12-pass transmembrane receptor, Patched1 (Ptch1). The binding of Hh to Ptch1 relieves inhibition of a seven transmembrane protein, Smoothened (Smo). Activated Smo signals through an intracellular signaling pathway to control the activities of three members of the Gli family of zinc finger transcriptional effectors, Gli1, Gli2, and Gli3. These Gli effectors regulate the transcription of downstream target genes, among which are Ptch1 and Gli1, negative and positive components of Hh pathway feedback systems, respectively (2).
The first evidence linking Hh pathway activity to human cancer was the identification of germ-line mutations of Ptch1 in Gorlin syndrome, a rare autosomal disease associated with an increased incidence of basal cell carcinoma (BCC), medulloblastoma, and rhabdomyosarcoma (3–5). Somatic mutations of several components of the Hh pathway, including Ptch1 and Smo, have also been detected in many sporadic BCCs and medulloblastomas (6–9). Further, recent studies have also implicated Hh pathway involvement in a wide range of tumors arising from organs of endodermal origin (8). These tumors include small cell lung cancer and carcinomas of the esophagus, stomach, pancreas, and prostate, none of which are typically associated with Gorlin syndrome. The use of cyclopamine and other small-molecule Hh pathway-specific antagonists has shown that Hh pathway activity is required for the growth of several cancers in mouse models and also a series of human cancer cell lines in vitro (8, 10, 11). However, the precise roles of the Hh pathway in tumor development, growth, and metastasis remain to be determined.
Currently, Ptch1+/− mice provide the major model for Hh-related tumorigenesis (12, 13). As in Gorlin’s patients, Ptch1+/− mice are predisposed to BCC and develop medulloblastoma and rhabdomyosarcoma but only at low penetrance (~10%; refs. 12, 13). Several mouse strains have been developed to attempt to model BCC and medulloblastoma by activating Hh signaling in either the skin or the brain (14–18); however, these models result in widespread up-regulation of Hh signaling in these tissues from embryonic stages and are frequently lethal at embryonic or early postnatal stages. In contrast, the majority of tumors in human patients occur sporadically and in a normal cellular context. Further, the tissue background itself can produce significant effects on tumor development and resulting phenotypes (19).
We have described a mouse strain, CAGGS-CreER, in which a Cre::ER fusion protein is ubiquitously expressed enabling tamoxifen-mediated control of Cre-mediated genetic modification (20). The Cre::ER fusion is inactive in most tissues, although rare sporadic leakage of Cre activity was observed. However, on injection of tamoxifen, high levels of recombination were observed in a wide range of adult tissues (20). We reasoned that the attributes of this model, low-level drug-independent sporadic recombination and precise drug-regulated high-level recombination, might form the basis of a strategy to explore the activity of putative oncogenes and tumor suppressors. To test this idea and at the same time to generate a robust mouse model that more faithfully mimics Hh-related sporadic tumors, we compounded the CAGGS-CreER transgene with a conditional allele of SmoM2 targeted into the ubiquitously expressed Rosa26 locus (21). SmoM2 encodes a mutant form of Smo previously identified in human BCC (15). In this allele, an activating mutation in the seventh transmembrane domain results in ligand-independent constitutive activation of Hh signaling in target tissues. We report a robust model for the generation of sporadic tumors, in which the frequency and latency of specific tumors are drug dependent. This model provides insights into novel aspects of the Hh-related tumorigenic program in the gastrointestinal tract. Further, transcriptional profiling of the diverse Hh-related tumors shows several common molecular links among distinct tumor types.
Materials and Methods
Mice
To generate CAGGS-CreER; R26-SmoM2 mice, the CAGGS-CreER transgenic line was crossed to R26-SmoM2 mice (mice were in a mixed genetic background, including 129/Sv and Swiss Webster as main components). The Ptch1+/− mice used in this study were maintained in a similar mixed background.
Tamoxifen induction in mice
Tamoxifen (Sigma, St. Louis, MO) was dissolved in corn oil (Sigma) at a concentration of 3 mg/mL. For both CAGGS-CreER; R26-SmoM2 and CAGGS-CreER; R26R studies, tamoxifen (1 mg/40 g body weight) was injected i.p. at postnatal day 10 (P10). Six weeks after tamoxifen injection, various organs were harvested from CAGGS-CreER; R26R mice and fixed in 4% paraformaldehyde. Frozen sections were cut at 14-μm intervals and subjected to standard X-gal staining.
Histology and immunohistochemistry
Adult CAGGS-CreER; R26-SmoM2 mice were cardiac perfused with 4% paraformaldehyde in PBS. Organs were harvested and further fixed in paraformaldehyde for 24 hours. All tissues were washed in 30% sucrose overnight and embedded in ornithine carbamyl transferase for cryosectioning at 14 μm. Tissues for paraffin sectioning were fixed, washed, dehydrated, and processed according to standard protocols in the Harvard Medical School Pathology Rodent Histopathology Core Facility (Boston, MA). Periodic acid-Schiff (PAS) and Alcian blue staining was carried out using standard protocols.
Immunohistochemistry was done on cryosections using the following primary antibodies: rabbit anti-GFP (1:1,000; Abcam, Cambridge, MA), rabbit anti-platelet-derived growth factor (PDGF) receptor α (PDGFRα; 1:1,200; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-Ki67 (1:1,000; NCL-Ki67-P, Novocastra, Newcastle, United Kingdom), mouse anti-desmin (1:100; Sigma), rabbit anti-Zic (gift of R. Segal Laboratory, Harvard Medical School, Boston, MA), mouse anti-NeuN (1:100; Chemicon, Temecula, CA), and mouse anti-myogenin (1:100; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA). 3,3′-Diaminobenzidine bright-field immunohistochemistry was done using the Envision + System (DAKO, Inc., Carpinteria, CA) according to the manufacturer’s instructions. Immunohistochemistry was done using a heat-based antigen retrieval protocol.
Affymetrix microarray and statistical analysis
Total RNA was purified from medulloblastoma and rhabdomyosarcoma tumor tissues and adjacent normal skeletal muscle and cerebellar tissues from three CAGGS-CreER; R26-SmoM2 mice in the tamoxifen postnatal injection group at 10 weeks of age. Total RNA was prepared from BCC excised from tail skin of three CAGGS-CreER; R26-SmoM2 mice and age-matched tail skin of three wild-type (WT; R26-SmoM2) littermates. Samples were prepared according to recommended protocol of Affymetrix. Probes were generated and hybridized to Mouse Expression Set 430 oligonucleotide arrays and scanned according to the manufacturer’s recommendations (Affymetrix, Santa Clara, CA). Resolver (Rosetta, Seattle, WA) software was used to identify those genes that showed statistically significant differences in each tumor type, relative to nontumorigenic tissues (fold change, >1.5; P < 0.001). Those genes that were significantly up-regulated or down-regulated in all three tumor types (analyzed independently) are tabulated in Supplementary Table S4.
Primary tumor cell culture and reverse transcription-PCR
Fresh medulloblastoma tissue was isolated at the core of a tumor mass in the brain of CAGGS-CreER; R26-SmoM2 mice. Tissue was thoroughly minced, digested with trypsin for 10 minutes, and then triturated into a near single-cell suspension. After filtering through a cell strainer, tumor cells were plated onto 35-mm gelatin-coated cell culture dishes in Neurobasal medium (Invitrogen, Carlsbad, CA) with N2 supplement (Invitrogen) with either KAAD-cyclopamine (3 μmol/L; Calbiochem, San Diego, CA) or vehicle. Three days after treatment, total cellular RNA was prepared using the Trizol reagent (Invitrogen). cDNA was synthesized from 1 μg total RNA using the SuperScript II kit (Invitrogen) and random hexamers. PCRs were done using 0.5 μL of undiluted and 4-fold serial dilutions of cDNA templates and the sequences of the amplification primers are the following: Ptch1 (5′-tgtctggcatcagtgaggag and 5′-gacaaggagccagagtccag), Gli1 (5′-atcacctgttggggatgctggat and 5′-ggcgtgaataggacttccgacag), Gli2 (5′-gagccaccccagcgtagaca and 5′-gccccaggtcgcactctag), Smo (5′-ttgtgctcatcaccttcagc and 5′-tgccaaacatggcaaataga), PDGFRα (5′-tgttggtgctgttggtgatt and 5′-tcccatctggagtcgtaagg), IGFBP4 (5′-agagcgaacatcccaacaac and 5′-acagtttggaatggggatga), IGFBP7 (5′-ggaaaatctggccattcaga and 5′-atttcatggagggcatcaac), MAP3K7 (5′-gggctgttcataatggcact and 5′-gagttgctctgcccttcatc), MAP4K4 (5′-catctccagggaaatcctca and 5′-taagtggcgtctgggttctc), and β-actin (5′-tcgtagatgggcacagtgtg and 5′-gttaccaactgggacgacatg).
Results
A somatic mouse model of Hh-related sporadic tumors
To characterize the CAGGS-CreER transgene as a potential driver line for modeling sporadic tumorigenesis in mouse, we first examined sporadic leakage and Cre-mediated activation of a conditional reporter allele R26R (22) in postnatal animals following injection of a single dose of tamoxifen (1 mg/40 g body weight) at P10. Major organs were harvested 6 weeks postinjection to assay the extent of drug-induced recombination. In the absence of tamoxifen injection, “leakiness” of CreER resulted in β-galactosidase activity in 0.1% to 5% of cells in the major organs examined; a broad contribution was observed within different cell populations in each organ with no obvious germ layer restriction. In contrast, 6 weeks after tamoxifen administration, a high rate of recombination was apparent in a wide variety of cell lineages within the major organs showing the effectiveness of a single pulse of tamoxifen in triggering widespread recombination in the postnatal mouse (Supplementary Fig. S1).
To determine the effectiveness of this sporadic/induced recombination model for the study of a tumor program, we linked this tool to recombination-mediated cell-autonomous up-regulation of the Hh signaling pathway (Fig. 1). A cDNA fragment encoding SmoM2 with a COOH-terminal YFP tag was targeted into the Rosa26 locus (R26) 3′ to a LoxP-flanked polyadenylation stop sequence cassette. When mice carrying this allele (R26-SmoM2) were crossed with a Wnt1-Cre line to enable widespread activation of SmoM2 in the dorsal neural tube and its neural crest derivative, typical Hh gain of function phenotypes were observed, showing the effectiveness of the R26-SmoM2 allele in Hh pathway activation (21). The R26-SmoM2 allele was crossed into a CAGGS-CreER background to examine the effects of spontaneous and drug-induced recombination-mediated activation of the R26-SmoM2 allele (Fig. 1).
Figure 1.
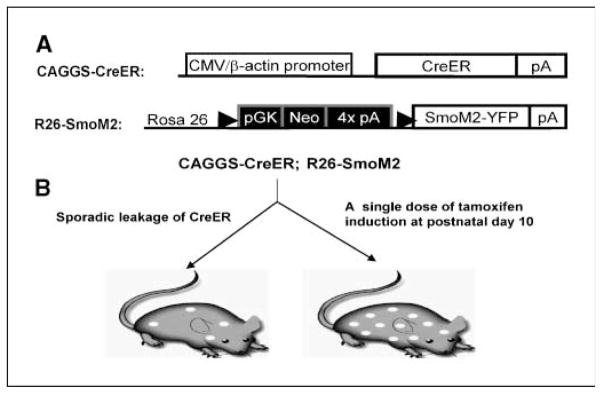
A somatic mouse model of Hh-related tumorigenesis. A, schematic representation of the CAGGS-CreER transgene and SmoM2 Rosa26 targeted alleles in the CAGGS-CreER; R26-SmoM2 model. B, recombination-mediated activation of SmoM2 by sporadic leakage and tamoxifen induction.
Tumorigenic spectrum and tumor incidence in CAGGS-CreER; R26-SmoM2 mice
To evaluate compound CAGGS-CreER; R26-SmoM2 mice as a somatic mouse model of Hh-related tumors, we analyzed a cohort of 20 Ptch1+/− mice, 33 CAGGS-CreER; R26-SmoM2 mice with sporadic CreER leakage, and 55 CAGGS-CreER;R26-SmoM2 mice that received tamoxifen as described above at P10. Animals were routinely monitored for the onset of malignancy.
The CAGGS-CreER; R26-SmoM2 model showed a significant difference in the tumor spectra compared with the Ptch1+/− model (Fig. 2A). Consistent with previous reports (13, 23, 24), no macroscopic BCC lesions were detected in Ptch1+/− mice and we only observed a low incidence of medulloblastomas and rhabdomyosarcomas, ~10% of mice at 18 weeks of age. In contrast, CAGGS-CreER; R26-SmoM2 mice showed a significantly enhanced tumorigenesis in the same time interval. In the absence of tamoxifen, all CAGGS-CreER; R26-SmoM2 mice exhibited rhabdomyosarcomas and 27% medulloblastomas (Fig. 2A). Tamoxifen administration increased the multiplicity of rhabdomyosarcoma present within a mouse (average number: −tamoxifen = 3; +tamoxifen = 7) and increased the incidence of medulloblastoma in this group (40%). In addition, tamoxifen-treated animals exhibited macroscopic BCC-like lesions from as early as 5 weeks of age, and all animals displayed BCC-like tumors by 8 weeks of age.
Figure 2.

Tumor fomation in CAGGS-CreER; R26-SmoM2 mice. A, distinct tumor spectra in CAGGS-CreER; R26-moM2 mice. Histograms show the fraction of Ptch1+/− (white), CAGGS-CreER; R26-SmoM2 sporadic leakage group (blue), and tamoxifen postnatal injection group (red) mice that developed the indicated tumors. B, survival curves of Ptch1+/−, sporadic leakage group of CAGGS-CreER; R26-SmoM2, and tamoxifen postnatal injection group showing the fraction of mice that survive up to 18 weeks. Genotypes are color coded.
Whereas the majority of mice in the sporadic leakage group survive to 18 weeks of age (all Ptch1+/− mice survive to this time), the overall survival of CAGGS-CreER; R26-SmoM2 mice was dramatically shortened following tamoxifen injection (Fig. 2B). By 18 weeks of age, because of heavy tumor burden and infection, all mice in this group were moribund, forcing compulsory compelling euthanasia according to institutional guidelines. In contrast, no tumor formation was observed in either CAGGS-CreER or R26-SmoM2 mice up to 12 months of age (data not shown).
Rhabdomyosarcomas in CAGGS-CreER; R26-SmoM2 mice
The CAGGS-CreER; R26-SmoM2 mice provide a particularly effective model to investigate the relationship between dysregulated Hh signaling and genesis of rhabdomyosarcoma, the most common soft-tissue sarcoma in children. Muscle tumors observed in CAGGS-CreER; R26-SmoM2 mice are histologically similar to those of Ptch1+/− mice and human embryonal rhabdomyosarcoma. These tumors ranged from 1 mm3 to 4,000 mm3 and were composed of a heterogeneous mixture of both round undifferentiated cells and elongated spindle-shape cells (Fig. 3A). Clear evidence of skeletal muscle differentiation (e.g., cross-striations) was apparent in some of the tumor cells (data not shown). Immunohistochemistry showed that both undifferentiated and differentiated tumor cells were positive for the muscle intermediate filament marker, desmin (Fig. 3A), indicating a likely origin from skeletal muscle progenitors. Analysis of Ki67 staining showed a relatively low proliferative index (Fig. 3A). Similar to the human embryonal rhabdomyosarcomas, transcriptional profiling of tumors in CAGGS-CreER; R26-SmoM2 mice showed a marked increase of MyoD, Myogenin, and Igf2 expression (Supplementary Table S1).
Figure 3.

Rhabdomyosarcoma (RBS), BCC, and medulloblastoma in CAGGS-CreER; R26-SmoM2 mice. A, characterization of rhabdomyosarcoma from a CAGGS-CreER; R26-SmoM2 mouse. The muscle tumor shows a mixture of round undifferentiated cells and elongated spindle-shape cells. Desmin is present in both undifferentiated and differentiated tumor cells. B, BCC in tail skin of mice in the postnatal tamoxifen injection group. C, sporadic and tamoxifen-induced SmoM2 expression in the cerebellum induces medulloblastoma. Typical histologic features of human classic medulloblastoma with small blue cells, numerous mitoses, and little histologic evidence of differentiation. Medulloblastomas in CAGGS-CreER; R26-SmoM2 mice expressed Zic1, an early marker of neuronal differentiation, and NeuN, a later marker of neuronal differentiation (C). Ki67 staining indicates a high mitotic index in tumors relative to the WT tissues (A, B, and C). Immunostaining using an anti-GFP antibody shows expression of SmoM2-YFP in tumor cells (A, B, and C).
Interestingly, those rhabdomyosarcomas that developed in the sporadic leakage group were mostly confined to the rear thigh and abdominal wall. In contrast, in the tamoxifen-treated group, rhabdomyosarcomas were also detected in skeletal muscle of the head, neck, tongue, and paratesticular regions (Supplementary Fig. S2). Further, tamoxifen injection at P10 also accelerated the mean age of muscle tumor onset from 9 weeks, for the first superficially visible tumors in the nontreated group, to 5 weeks following tamoxifen injection. Given that the tumorigenic program correlates with postnatal administration of tamoxifen, it seems likely that up-regulation of Hh signaling within the postnatal muscle lineage can lead to tumorigenesis, consistent with a cell autonomous action of SmoM2-YFP in a muscle progenitor. Muscle tumors that developed were predominantly YFP+ indicating expression of the SmoM2 allele (Fig. 3A).
To further analyze the effect of up-regulation of Hh signaling on postnatal muscle, we examined the normal-appearing muscle cells in CAGGS-CreER; R26-SmoM2 mice with postnatal tamoxifen injection at P10. Although SmoM2-expressing cells were detected, these muscle cells did not expressed Ki67 and myogenin, a marker for late myogenic progenitors (Supplementary Fig. S3). In contrast, Ki67 and myogenin were up-regulated in a subset of tumor cells in rhabdomyosarcoma (Supplementary Fig. S3). These results suggest that activation of SmoM2 in more differentiated muscle cells was not sufficient to generate a proliferative phenotype or to revert mature muscle into an immature cell type.
BCC in CAGGS-CreER; R26-SmoM2 mice
Although no obvious skin lesions were detected in 33 CAGGS-CreER; R26-SmoM2 mice in the sporadic leakage group up to 18 weeks of age, macrosopically detectable skin tumors were evident in the tail, ear, and foot regions as early as 5 weeks of age following tamoxifen administration. Such tumors include not only basaloid follicular hamartomas, as reported previously in a transgenic model (25), but also tumors with histologic features of human BCC, which consisted of multifocal nodular islands of atypical basal cells with peripheral palisading, frequent mitoses, and invasion of the underlying dermis (Fig. 3B). The majority of tumor cells were, as expected, SmoM2-YFP+ (Fig. 3B). Expression profiling showed that, like human BCC, the skin tumors in CAGGS-CreER; R26-SmoM2 mice expressed high levels of keratin 17 and Bcl-2 (Supplementary Table S2).
Medulloblastoma formation in CAGGS-CreER; R26-SmoM2 mice
CAGGS-CreER; R26-SmoM2 mice developed medulloblastomas within the cerebellum. The tumors were densely cellular and composed of sheets of small poorly differentiated cells with scant cytoplasm, displaying a high mitotic activity as visualized by Ki67 immunostaining (Fig. 3C). Immunohistochemistry showed that tumor cells were positive for early (Zic1) and late (NeuN) neuronal markers, which are characteristic of granule neuron specification (Fig. 3C). Expression of SmoM2-YFP in the majority of cells indicated that tumors were derived from SmoM2-expressing cells (Fig. 3C). In addition to the classic appearing tumors, nests of abnormal cells were frequently observed apart from the surface of the cerebellum consistent with similar preneoplastic lesions reported in Ptch1+/− mice (data not shown; ref. 26). Expression profiling revealed strong transcriptional up-regulation of characteristic markers of human medulloblastoma, such as Myc, Nmyc1, Foxm1, Ezh2, Otx2, and Sox18 (Supplementary Table S3).
Pancreatic and gastrointestinal lesions in CAGGS-CreER; R26-SmoM2 mice
Hh signaling has been linked to pancreatic cancers (8, 27). We detected a high rate of novel cystic metaplastic lesions in the pancreas of CAGGS-CreER; R26-SmoM2 mice. These lesions largely replaced the normal pancreatic architecture (Fig. 4). They are heterogeneous in size, lined by cuboidal epithelium with foci of columnar metaplasia, and are supported by proliferative ovarian-like stroma reminiscent of a mucinous cystic neoplasm (MCN). To further evaluate and characterize these epithelial changes as mucinous metaplasia, these lesions were evaluated by Alcian blue stain, which highlights intestinal-type mucins, and PAS stain, which highlights both gastric- and intestinal-type mucins. PAS-positive, Alcian blue-positive epithelium is not found in normal pancreatic parenchyma (Fig. 4) but is a feature of mucinous cystic tumors as well as adenocarcinoma of the pancreas. In these mice, the atypical epithelium of these cystic lesions of the pancreas was strongly positive for both PAS and Alcian blue stains, suggesting the presence of gastric intestinal mucins (Fig. 4). Mucin expression is particularly high in the smaller cysts (Fig. 4). Based on the architecture of the lesions, proliferative supporting stroma, and multiple foci of mucinous metaplasia, these cystic pancreatic lesions most closely resemble a spectrum of low-grade MCNs seen in human.
Figure 4.

Pancreatic mucinous cystic lesion in CAGGS-CreER; R26-SmoM2 mice. Alcian blue and PAS staining shows focal intestinal-type mucin expression (arrows) in the epithelium of the cysts, but not in normal pancreatic tissues. Note prominent mucin expression in smaller cysts.
We also noted diverticular harmartomatous lesions in both intestine and stomach (Supplementary Fig. S4), whereas the former was common (20% without tamoxifen; 80% with tamoxifen injection), the latter was rare (<5% of animals in the tamoxifen injection group). The epithelial cells within the diverticular lesion were disorganized and mildly hyperproliferative (Supplementary Fig. S4). However, no evidence of dysplasia was observed within the diverticular lesions or elsewhere in the gastrointestinal tract. Other endoderm-derived tissues, such as lung and prostate, both reported to cause Hh-dependent tumors, appeared normal.
Activation of SmoM2 is not sufficient to induce neoplastic transformation in mouse postnatal prostate epithelium
To extend our study using CAGGS-CreER;R26-SmoM2 as the model system and investigate the possibility that the tumors in these endoderm-derived tissues have a longer latency, extending beyond the maximum life span (18 weeks) of tamoxifen-treated CAGGS-CreER; R26-SmoM2 mice, we examined the potential involvement of Hh signaling in prostate tumorigenesis (28). SmoM2 was activated in prostate epithelium using Pb-Cre4, a Cre transgene specifically expressed in the postnatal prostate epithelium (29). Seven Pb-Cre4;R26-SmoM2 mice were observed to 12 months of age and additional eight mice to 9 months of age. However, we did not detect hyperproliferative lesions or neoplastic transformation of prostate epithelial cells in these mice (Fig. 5). Consistent with the results in the CAGGS-CreER;R26-SmoM2 model, these data suggest that activation of SmoM2 is not sufficient for tumor initiation in the mouse prostate.
Figure 5.

SmoM2 activation in postnatal prostate epithelium is not sufficient to induce neoplastic transformation. H&E staining was done on dorsal prostate from a WT R26-SmoM2 mouse and a Pb-Cre4; R26-SmoM2 mouse at 12 months of age. GFP antibody staining shows SmoM2-YFP expression in prostate epithelium. Ki67 staining of proliferative cells (white arrows) does not reveal a higher mitotic index in the prostate of Pb-Cre4; R26-SmoM2 mice compared with a WT control.
Expression profiles of Hh-related tumors
To identify common signature patterns within the transcriptional output of Hh tumors, we compared the expression profiles of the three principal tumor types: BCC, medulloblastoma, and rhabdomyosarcoma. Each tumor is likely to arise from a distinct cellular origin, ectodermal, neuroectodermal, and mesodermal, respectively. Tissues were collected at 10 weeks of age from mice in the tamoxifen injection group. The transcriptional profiles of medulloblastomas and hind limb rhabdomyosarcomas were compared with adjacent normal-appearing cerebellar and skeletal muscle tissues. Tamoxifen-induced BCC lesions in the tail skin of CAGGS-CreER; R26-SmoM2 mice were compared with tail skin from age-matched WT (R26-SmoM2) controls. Biostatistical analysis using the Resolver software generated a list of 157 genes with significantly altered expression in all three Hh-related tumors; 101 genes were up-regulated in all three tumors, whereas 56 genes were down-regulated (Supplementary Table S4).
As expected, well-validated transcriptional targets of the Hh pathway, such as Ptch1, Ptch2, Gli1, and Cyclin D1, were up-regulated in all three types of tumors. Surprisingly, Gli2, a key effector of the Hh transcriptional response but not one previously associated with Hh-mediated transcriptional regulation, was found to be significantly up-regulated in all tumor types.
PDGFRα and several components of the insulin-like growth factor (IGF) and the mitogen-activated protein kinase (MAPK) pathways, including IGFBP4 and IGFBP7 in the IGF pathway and MAP3K7 and MAP4K4 in the MAPK pathway, were significantly up-regulated in all three tumor sources (Supplementary Table S1). PDGFRα up-regulation in response to Hh pathway activation has been reported in BCC in human and Ptch1+/− mice (30); our new data suggest a possible broader involvement of this pathway. Interestingly, PDGFA, the ligand for PDGFRα, was also up-regulated in BCC and medulloblastoma but not in rhabdomyosarcoma (Supplementary Tables S1–S3). Further, high levels of PDGFRα expression were also confirmed by immunostaining of SmoM2-related rhabdomyosarcomas and medulloblastomas (Fig. 6A and B). Although we only detected up-regulation of IGF2 in rhabdomyosarcomas, a hallmark of embryonal type rhabdomyosarcoma, two IGF-binding proteins, IGFBP4 and IGFBP7, were highly up-regulated in all three tumors. Thus, the IGF regulatory system may be a common target in all Hh-related tumor types.
Figure 6.

Regulation of the Hh, PDGFRα, IGF, and MAPK pathways in Hh-related tumors. Immunohistochemistry using an anti-PDGFRα antibody identifies strong PDGFRα staining in rhabdomyosarcomas (A) and medulloblastomas (B), relative to control skeletal muscle and cerebellar tissues. C, semiquantitative RT-PCR analysis of Hh-regulated mRNA expression in cultured primary medulloblastoma cells from CAGGS-CreER; R26-SmoM2 mice in the presence or absence of the SmoM2 inhibitor, KAAD-cyclopamine. Treatments were done for 72 hours. Descending wedge, serial dilutions of cDNA templates.
To determine if expression of these genes requires ongoing Hh signaling, we used KAAD-cyclopamine, a derivative of the Hh pathway specific antagonist cyclopamine, to modulate SmoM2 activity (31) in primary cultures of cerebellar tumors. Freshly isolated medulloblastoma cells from 2-month-old CAGGS-CreER; R26-SmoM2 mice were treated 3 μmol/L KAAD-cyclopamine for 72 hours and gene expression was analyzed by semiquantitative reverse transcription-PCR (RT-PCR). All of the putative targets discussed above were down-regulated in response to KAAD-cyclopamine, whereas the levels of Smo and cytoplasmic actin appear to remain unaltered (Fig. 6C; data not shown). These data suggest an active dependence of Hh signaling to maintain elevated levels of PDGFRα, IGFBP4, IGFBP7, MAP3K7, and MAP4K4 transcripts.
Discussion
The CAGGS-CreER line as a Cre driver for modeling sporadic tumor in mice
Sporadic cancers result when initiating mutations acting at the single-cell level lead to neoplastic transformation in a genetically WT environment. However, in most current transgenic and conventional knockout models, an initiating oncogenic event is activated in all cells of a specific tissue (19). Other strategies have been developed to generate sporadic model systems. For example, intrachromosomal recombination and unequal sister chromatid exchange, which occur at the rate of 10−3 to 10−7 events per cell generation, have been used to activate K-ras, generating a mouse model that closely recapitulates spontaneous oncogene activation in somatic cells (32). With the increasing repertoire of conditional alleles of tumor suppressors and oncogenes, we reasoned that inducible Cre lines with broad expression and regulated recombination, such as CAGGS-CreER, may provide a more widely applicable and cost effective model of sporadic tumorigenesis.
In the work reported here, the CAGGS-CreER line combines sporadic leakage of CreER activity in single cells in a wide range of tissues with timed, titratable tamoxifen-mediated activation to produce a robust, regulated model of sporadic tumorigenesis. Although we have shown the effectiveness of this approach with a dominant-acting oncogene (SmoM2), the same general strategy is applicable to any Cre-dependent model that is configured appropriately for a specific genetic outcome. This may include as here “dominant” tumor models or where available conditional alleles for putative tumor suppressors. The latter strategy is likely to be particularly useful where germ-line mutation results in embryonic lethality as is the case for several key tumor regulators that include Apc, Pten, and Rb. The approach we have adopted establishes a basis for a global screening of tumor suppressor activity in the mouse. Further, the recent report of an ES cell line carrying the CAGGS-CreER transgene that is configured for easy detection of recombination at the Rosa26 locus should enable rapid tumor screening directly in founding chimeras (33).
Medulloblastoma, BCC, and rhabdomyosarcoma in CAGGS-CreER; R26-SmoM2 mice
The Ptch1+/− mouse model of Hh pathway-related tumors, a germ-line mutation of Ptch1, has a relatively low penetrance of the three principal tumor types associated with Gorlin syndrome: BCC, medulloblastoma, and rhabdomyosarcoma. In this model, only 10% of Ptch1+/− mice developed rhabdomyosarcoma and medulloblastoma (12, 13). Further, robust BCC formation requires UV irradiation and exhibits a long latency (23). The tumor incidence can be enhanced genetically through the additional loss of tumor suppressor function. For example, loss of p53 dramatically increased the frequency of medulloblastoma formation to 95% in Ptch1+/− mice (34). However, no alteration in the incidence or the time of onset of rhabdomyosarcoma and BCC was observed in this Ptch1+/−; P53−/− background (34). The CAGGS-CreER; R26-SmoM2 model has a significantly higher penetrance of all three signature tumors. Relative to existing models of Hh-mediated tumorigenesis (14–18), the short latency, high penetrance of tumor formation, and increased spectrum of tumor types within an individual mouse in the new model may facilitate preclinical screening of antitumor agents.
To date, much of the focus on Hh-related tumors has centered on medulloblastoma and BCC. Rhabdomyosarcoma, the most common childhood soft-tissue sarcoma, is also associated with Gorlin syndrome, although the frequency is low (3). Surprisingly, the CAGGS-CreER; R26-SmoM2 model developed multifocal rhabdomyosarcoma with 100% penetrance in both sporadic leakage and tamoxifen induction groups; tamoxifen injection enhanced the tumor frequency, increased the range of tumors, and accelerated the onset of tumor formation. Although it seems likely that rhabdomyosarcomas arise from within the skeletal muscle lineage, the underlying cellular origin of these tumors is unclear. Sonic Hh (Shh) signaling has been shown to play a critical role in both the specification of embryonic muscle progenitors and the subsequent differentiation of fast and slow twitch fibers (1). The high frequency of postnatal muscle tumors observed on Smo activation may point to a continuing role for Hh signaling in the development or repair of postnatal skeletal muscle. However, there are only few reports for an extended role of Hh signaling in the regulation of postnatal muscle cells (35). Our analysis on normal-appearing muscle of CAGGS-CreER; R26-SmoM2 mice suggests that SmoM2 activation in differentiated postnatal muscle cells is not sufficient to modify a normal differentiated skeletal muscle program. Rather, Hh signaling most likely acts within stem cells or progenitor cells to initiate rhabdomyosarcoma formation.
Hh signaling and tumors arising from endoderm derived tissues
Shh has been proposed recently to play an important role as a mediator of pancreatic carcinogenesis. Overexpression of the Shh ligand during the embryonic period leads to the development of pancreatic intraepithelial neoplastic lesions that are believed to be precursors of invasive pancreatic adenocarcinoma (27). Interestingly, we observed that activation of SmoM2 in the pancreas results in mucinous cystic metaplasia, a lesion that may correspond to the low-grade MCN seen in human. MCNs, although frequently benign, are thought to harbor a malignant potential (36). The current understanding of the progression from low-grade mucinous cystic neoplasia to high-grade dysplasia to pancreatic malignancy is still very limited, partly due to the lack of animal models. The mucinous pancreatic lesions seen in CAGGS-CreER, R26-SmoM2 mice most likely represent an early stage in the tumorigenic process and provide a new opportunity to explore events that control malignant transformation of the pancreas.
Tumorigenesis in several other endoderm-derived organs, including the lung, esophagus, stomach, and prostate, has also been linked to Hh signaling (8, 27, 28, 37–39). In some of these studies, a Hh ligand and concomitantly Hh signaling activity are up-regulated in human cancer lines, and continued signaling is required for growth of tumor cells in in vitro assays and xenograft models (27, 28, 37–39). Although we observed efficient CreER-mediated recombination in all these organs, no oncogenic transformation was observed even after tamoxifen induction. Further, our analysis of Pb-Cre4;R26-SmoM2 mice failed to detect neoplastic transformation in mouse prostate even up to 12 months of age. Thus, Hh signaling may not be sufficient to induce prostate tumorigenesis and may therefore have a restricted role to promote growth or metastasis.
A common Hh-regulated tumor program
A central question arising from these and other studies of Hh-related tumors is whether there are common mechanistic principles that underscore tumor development in diverse tissues. Transcriptional profiling of distinct Hh-dependent tumors has provided some insights. In addition to expected feedback components in the Hh pathway (Ptch1, Ptch2, and Gli1) and known cell cycle regulators (Cyclin D1) that provide a useful validation of the strategy, we identified several novel genes. We observed that Gli2, itself as a positive transcriptional regulator of Hh target genes (40), is up-regulated in all three tumors. Our results agree with several recent studies that have shown increased expression of Gli2 in medulloblastomas and BCC induced by Hh pathway activation (18, 41, 42). Further, overexpression of Gli2 in skin has been shown to induce BCC formation in a transgenic model (17), suggesting a role of Gli2 in Hh-induced tumorigenesis. Together, these findings suggest a possible common mechanism whereby tumor formation and growth may require the up-regulation of both Gli1 and Gli2 to maximize transcriptional output.
We also detected significant up-regulation of PDGFRα, IGF, and MAPK pathway components in all three Hh-related tumors, suggesting that Hh signaling may enable new signaling responses in tumor cells. Interestingly, PDGFRα is transcriptionally regulated by Gli1 in a multipotent mesenchymal cell line (10T1/2) and also in a Gli1-induced central nervous system hyperplasia (30, 43). Expression of PDGFRα has been reported to be significantly elevated in the Ptch1+/− mouse model of BCC and in human BCC (30). Activated PDGF signaling via the MAPK pathway has also been linked to human metastatic medulloblastoma (44). Interestingly, blocking PDGF and MAPK pathways inhibits growth and migration of BCC and medulloblastoma cells in vitro (30, 44). The results reported here also indicate a possible role of up-regulation of PDGFRα in rhabdomyosarcoma formation. The PDGF pathway may therefore serve as a common therapeutic target in all Hh-related tumors.
Previous studies have shown that IGF2 is indispensable for rhabdomyosarcoma and medulloblastoma development in Ptch1+/− mice (45). Further, Shh and IGF2 synergize in medulloblastoma formation (46) and IGF-induced activation of phosphatidylinositol 3-kinase signaling stabilizes the Shh transcriptional target N-myc in cerebellar granule neuron precursors (47). During fetal prostate development, Shh has been shown to regulate the expression of another IGF pathway regulator, IGFBP6 (48). IGFBP6 was also identified as a potential direct transcriptional target in Gli1-induced transformation of an epithelial cell line (49). The observed up-regulation of IGFBP4 and IGFBP7 in Hh-related tumors and their down-regulation on Hh pathway inhibition in tumor cells lend further support to a more general linkage between these two pathways.
The PDGF and IGF pathways can signal through intracellular cascades to regulate the activities of MAPKs, including the extra-cellular signal-regulated kinase and c-Jun NH2-terminal kinase (JNK) family kinases (30, 44). MAP4K4, a member of MAPK pathway, has been shown to act upstream of MAP3K7 in the MAPK cascade to activate the JNK family kinases (50). Interestingly, both MAP3K7 and MAP4K4 are up-regulated in all three Hh-related tumors and their expression in cultured tumor cells was also dependent on ongoing Hh pathway activity. Taken together, these data suggest a potential signaling network, such as Hh, PDGF, IGF, and MAPK pathways, in the Hh-dependent tumorigenic program.
Current strategies to develop more specific mechanism-based cancer therapy rely to a great extent on the identification of candidate targets. Animal studies of Hh pathway antagonists have shown considerable promise (8, 10, 11). However, the involvement of Hh signaling in development, maintenance, and regeneration of an organ may potentially complicate a treatment strategy based solely on direct pathway inhibition. The identification of other common components of Hh-associated malignancies could enable more effective combined therapeutic strategies for targeted treatment of Hh-related tumors.
Supplementary Material
Acknowledgments
Grant support: NIH grant R37-NS033642 (A.P. McMahon). J. Mao is a fellow of Damon Runyon Cancer Research Foundation and Charles H. Hood Foundation.
We thank Drs. Drucilla Roberts, Mari Mino-Kenudson, and Ben Stanger (Harvard Medical School) for analysis of specific pathologies and the members of McMahon laboratory and Rowitch laboratory for helpful discussion.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.McMahon AP, Ingham PW, Tabin CJ. Developmental roles and clinical significance of hedgehog signaling. Curr Top Dev Biol. 2003;53:1–114. doi: 10.1016/s0070-2153(03)53002-2. [DOI] [PubMed] [Google Scholar]
- 2.Lum L, Beachy PA. The Hedgehog response network: sensors, switches, and routers. Science. 2004;304:1755–9. doi: 10.1126/science.1098020. [DOI] [PubMed] [Google Scholar]
- 3.Gorlin RJ. Nevoid basal-cell carcinoma syndrome. Medicine (Baltimore) 1987;66:98–113. doi: 10.1097/00005792-198703000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Johnson RL, Rothman AL, Xie J, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–71. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- 5.Hahn H, Wicking C, Zaphiropoulous PG, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–51. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 6.Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature. 2001;411:349–54. doi: 10.1038/35077219. [DOI] [PubMed] [Google Scholar]
- 7.Wechsler-Reya R, Scott MP. The developmental biology of brain tumors. Annu Rev Neurosci. 2001;24:385–428. doi: 10.1146/annurev.neuro.24.1.385. [DOI] [PubMed] [Google Scholar]
- 8.Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature. 2004;432:324–31. doi: 10.1038/nature03100. [DOI] [PubMed] [Google Scholar]
- 9.Ruiz i Altaba A, Sanchez P, Dahmane N. Gli and hedgehog in cancer: tumours, embryos, and stem cells. Nat Rev Cancer. 2002;2:361–72. doi: 10.1038/nrc796. [DOI] [PubMed] [Google Scholar]
- 10.Romer J, Curran T. Targeting medulloblastoma: small-molecule inhibitors of the sonic hedgehog pathway as potential cancer therapeutics. Cancer Res. 2005;65:4975–8. doi: 10.1158/0008-5472.CAN-05-0481. [DOI] [PubMed] [Google Scholar]
- 11.Sanchez P, Clement V, Ruiz i Altaba A. Therapeutic targeting of the hedgehog-GLI pathway in prostate cancer. Cancer Res. 2005;65:2990–2. doi: 10.1158/0008-5472.CAN-05-0439. [DOI] [PubMed] [Google Scholar]
- 12.Hahn H, Wojnowski L, Miller G, Zimmer A. The patched signaling pathway in tumorigenesis and development: lessons from animal models. J Mol Med. 1999;77:459–68. doi: 10.1007/s001099900018. [DOI] [PubMed] [Google Scholar]
- 13.Corcoran RB, Scott MP. A mouse model for medulloblastoma and basal cell nevus syndrome. J Neurooncol. 2001;53:307–18. doi: 10.1023/a:1012260318979. [DOI] [PubMed] [Google Scholar]
- 14.Oro AE, Higgins KM, Hu Z, Bonifas JM, Epstein EH, Scott MP. Basal cell carcinomas in mice overexpressing sonic hedgehog. Science. 1997;276:817–21. doi: 10.1126/science.276.5313.817. [DOI] [PubMed] [Google Scholar]
- 15.Xie J, Murone M, Luoh SM, et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391:90–2. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- 16.Nilsson M, Unden AB, Krause D, et al. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc Natl Acad Sci U S A. 2000;97:3438–43. doi: 10.1073/pnas.050467397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grachtchouk M, Mo R, Yu S, et al. Basal cell carcinomas in mice overexpressing Gli2 in skin. Nat Genet. 2000;24:216–7. doi: 10.1038/73417. [DOI] [PubMed] [Google Scholar]
- 18.Hallahan AR, Pritchard JI, Hansen S, et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res. 2004;64:7794–800. doi: 10.1158/0008-5472.CAN-04-1813. [DOI] [PubMed] [Google Scholar]
- 19.Jonkers J, Berns A. Conditional mouse models of sporadic cancer. Nat Rev Cancer. 2002;2:251–65. doi: 10.1038/nrc777. [DOI] [PubMed] [Google Scholar]
- 20.Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–18. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- 21.Jeong J, Mao J, Tenzen T, Kottmann AH, McMahon AP. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. 2004;18:937–51. doi: 10.1101/gad.1190304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–1. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 23.Aszterbaum M, Epstein J, Oro A, et al. Ultraviolet and ionizing radiation enhance the growth of BCCs and trichoblastomas in patched heterozygous knockout mice. Nat Med. 1999;5:1285–91. doi: 10.1038/15242. [DOI] [PubMed] [Google Scholar]
- 24.Hahn H, Wojnowski L, Zimmer AM, Hall J, Miller G, Zimmer A. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nat Med. 1998;4:619–22. doi: 10.1038/nm0598-619. [DOI] [PubMed] [Google Scholar]
- 25.Grachtchouk V, Grachtchouk M, Lowe L, et al. The magnitude of hedgehog signaling activity defines skin tumor phenotype. EMBO J. 2003;22:2741–51. doi: 10.1093/emboj/cdg271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oliver TG, Read TA, Kessler JD, et al. Loss of patched and disruption of granule cell development in a preneoplastic stage of medulloblastoma. Development. 2005;132:2425–39. doi: 10.1242/dev.01793. [DOI] [PubMed] [Google Scholar]
- 27.Thayer SP, di Magliano MP, Heiser PW, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–6. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karhadkar SS, Bova GS, Abdallah N, et al. Hedgehog signalling in prostate regeneration, neoplasia, and metastasis. Nature. 2004;431:707–12. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- 29.Wu X, Wu J, Huang J, et al. Generation of a prostate epithelial cell-specific Cre transgenic mouse model for tissue-specific gene ablation. Mech Dev. 2001;101:61–9. doi: 10.1016/s0925-4773(00)00551-7. [DOI] [PubMed] [Google Scholar]
- 30.Xie J, Aszterbaum M, Zhang X, et al. A role of PDGFRα in basal cell carcinoma proliferation. Proc Natl Acad Sci U S A. 2001;98:9255–9. doi: 10.1073/pnas.151173398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taipale J, Chen JK, Cooper MK, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005–9. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- 32.Johnson L, Mercer K, Greenbaum D, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–6. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 33.Mao J, Barrow J, McMahon J, Vaughan J, McMahon A. An ES cell system for rapid, spatial, and temporal analysis of gene function in vitro and in vivo. Nucleic Acids Res. 2005;33:e155–9. doi: 10.1093/nar/gni146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wetmore C, Eberhart DE, Curran T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res. 2001;61:513–6. [PubMed] [Google Scholar]
- 35.Koleva M, Kappler R, Vogler M, Herwig A, Fulda S, Hahn H. Pleiotropic effects of sonic hedgehog on muscle satellite cells. Cell Mol Life Sci. 2005;62:1863–70. doi: 10.1007/s00018-005-5072-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brugge WR, Lauwers GY, Sahani D, Fernandez-del Castillo C, Warshaw AL. Cystic neoplasms of the pancreas. N Engl J Med. 2004;351:1218–26. doi: 10.1056/NEJMra031623. [DOI] [PubMed] [Google Scholar]
- 37.Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003;422:313–7. doi: 10.1038/nature01493. [DOI] [PubMed] [Google Scholar]
- 38.Berman DM, Karhadkar SS, Maitra A, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425:846–51. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- 39.Sanchez P, Hernandez AM, Stecca B, et al. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc Natl Acad Sci U S A. 2004;101:12561–6. doi: 10.1073/pnas.0404956101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development. 2002;129:4753–61. doi: 10.1242/dev.129.20.4753. [DOI] [PubMed] [Google Scholar]
- 41.Kimura H, Stephen D, Joyner A, Curran T. Gli1 is important for medulloblastoma formation in Ptc1+/− mice. Oncogene. 2005;24:4026–36. doi: 10.1038/sj.onc.1208567. [DOI] [PubMed] [Google Scholar]
- 42.Regl G, Neill GW, Eichberger T, et al. Human GLI2 and GLI1 are part of a positive feedback mechanism in basal cell carcinoma. Oncogene. 2002;21:5529–39. doi: 10.1038/sj.onc.1205748. [DOI] [PubMed] [Google Scholar]
- 43.Dahmane N, Sanchez P, Gitton Y, et al. The sonic hedgehog-Gli pathway regulates dorsal brain growth and tumorigenesis. Development. 2001;128:5201–12. doi: 10.1242/dev.128.24.5201. [DOI] [PubMed] [Google Scholar]
- 44.MacDonald TJ, Brown KM, LaFleur B, et al. Expression profiling of medulloblastoma: PDGFRA and the RAS/MAPK pathway as therapeutic targets for metastatic disease. Nat Genet. 2001;29:143–52. doi: 10.1038/ng731. [DOI] [PubMed] [Google Scholar]
- 45.Hahn H, Wojnowski L, Specht K, et al. Patched target Igf2 is indispensable for the formation of medulloblastoma and rhabdomyosarcoma. J Biol Chem. 2000;275:28341–4. doi: 10.1074/jbc.C000352200. [DOI] [PubMed] [Google Scholar]
- 46.Rao G, Pedone CA, Valle LD, Reiss K, Holland EC, Fults DW. Sonic hedgehog and insulin-like growth factor signaling synergize to induce medulloblastoma formation from nestin-expressing neural progenitors in mice. Oncogene. 2004;23:6156–62. doi: 10.1038/sj.onc.1207818. [DOI] [PubMed] [Google Scholar]
- 47.Kenney AM, Widlund HR, Rowitch DH. Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development. 2004;131:217–28. doi: 10.1242/dev.00891. [DOI] [PubMed] [Google Scholar]
- 48.Lipinski RJ, Cook CH, Barnett DH, Gipp JJ, Peterson RE, Bushman W. Sonic hedgehog signaling regulates the expression of insulin-like growth factor binding protein-6 during fetal prostate development. Dev Dyn. 2005;233:829–36. doi: 10.1002/dvdy.20414. [DOI] [PubMed] [Google Scholar]
- 49.Yoon JW, Kita Y, Frank DJ, et al. Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. J Biol Chem. 2002;277:5548–55. doi: 10.1074/jbc.M105708200. [DOI] [PubMed] [Google Scholar]
- 50.Yao Z, Zhou G, Wang XS, et al. A novel human STE20-related protein kinase, HGK, that specifically activates the c-Jun N-terminal kinase signaling pathway. J Biol Chem. 1999;274:2118–25. doi: 10.1074/jbc.274.4.2118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.