

Abstract
Our previously reported Bcl-2/Bcl-xL inhibitor, 4, effectively inhibited tumor growth but failed to achieve complete regression in vivo. We have now performed extensive modifications on its pyrrole core structure, which has culminated in the discovery of 32 (BM-1074). Compound 32 binds to Bcl-2 and Bcl-xL proteins with Ki values of < 1 nM and inhibits cancer cell growth with IC50 values of 1-2 nM in four small-cell lung cancer cell lines sensitive to potent and specific Bcl-2/Bcl-xL inhibitors. Compound 32 is capable of achieving rapid, complete and durable tumor regression in vivo at a well-tolerated dose-schedule. Compound 32 is the most potent and efficacious Bcl-2/Bcl-xL inhibitor reported to date.
INTRODUCTION
A common feature in many different types of human tumors is overexpression of the prosurvival Bcl-2 family members Bcl-2 and Bcl-xL,1-4 which make tumor cells resistant to conventional cancer therapeutic agents. Therefore, it has been proposed that small-molecule inhibitors of Bcl-2 and Bcl-xL may have a promising therapeutic potential for the treatment of human cancer.3
Compounds 15 and 26 represent two highly potent and specific Bcl-2/Bcl-xL inhibitors. Preclinical studies have shown that 1 and 2 are effective as single agents against lymphomas, chronic lymphoid leukemia (CLL) and a subset of small-cell lung cancer (SCLC) models, and can enhance the antitumor activity of conventional anticancer drugs and γ-irradiation in preclinical models of diverse tumor types.3 Compound 2 is currently in Phase I/II clinical trials, where it has shown promising single-agent activity in patients with CLL and B-cell lymphomas.
Because design of Bcl-2 and Bcl-xL inhibitors involves targeting the interaction of Bcl-2/Bcl-xL proteins with their pro-apoptotic binding partners such as BAD and BIM proteins, a challenging task in drug discovery, very few new, potent, specific and bona fide small-molecule inhibitors of this interaction have been reported, even after the discovery of 1 and 2. Recently, we reported the structure-based design of a family of new, highly potent and specific Bcl-2/Bcl-xL inhibitors (Figure 1).7-9 Our initial lead compound 3 binds to Bcl-2 and Bcl-xL with high affinities and potently inhibits cell growth in cancer cell lines that are sensitive to 1 and 2, but it lacks chemical stability and fails to achieve significant in vivo antitumor activity.7 Subsequent structure-based design and optimization of 3 led to compounds 4 and 5, which have excellent chemical stability, bind to Bcl-2 and Bcl-xL with Ki values of <1 nM and inhibit cancer cell growth with low nanomolar activity.8 While 5 effectively inhibits tumor growth and in fact induces tumor regression in the H146 small-cell lung cancer model at its maximum tolerated dose (MTD), the tumor regression it caused was transient,8 suggesting further optimization was needed toward our goal of developing a new class of Bcl-2/Bcl-xL inhibitors for cancer treatment. Very recently, we have reported further structure-based optimization of compound 5, with a focus on two regions in the molecule, which led to the successful discovery of a superior compound, 6 (BM-957).9 Compound 6 binds to Bcl-2 and Bcl-xL with Ki values < 1 nM and inhibits tumor cell growth with IC50 values of 21-22 nM against H146 and H1417 small-cell cancer cell lines.9 Significantly, 6 achieved tumor regression in an animal model of human cancer.9
Figure 1.

Chemical structures of 1 (ABT-737)5, 2 (ABT-263)6 and our recently reported potent and specific Bcl-2/Bcl-xL inhibitors.
In the previous study, which yielded compound 6, we focused our modifications on the nitro group and the soluble “tail” containing the N,N-dimethylamino group in compound 4. In the present study, we report our further optimization of 4, with a focus on its 1H-pyrrole-3-carboxylic acid core structure. Our efforts have culminated in the discovery of 32 (BM-1074), which, based upon its cellular activity and in vivo efficacy, is arguably the most potent and efficacious Bcl-2/Bcl-xL inhibitor discovered to date.
Results and Discussion
Previously, we have shown that removal of the acid group from the pyrrole carboxylic acid of 4, yielding compound 7, resulted in a >50-fold decrease in binding affinity to Bcl-2 and a modest decrease in binding affinity to Bcl-xL.8 Compound 7, at concentrations as high as 10 μM, was found to be completely inactive in inhibition of cell growth in the H146 cancer cell line (Table 1), suggesting that very high binding affinity to Bcl-2/Bcl-xL is clearly needed in order for small-molecule inhibitors to effectively inhibit cancer cell growth.8 Converting this acid group into a methylamide (compound 8) has a modest negative effect on binding to Bcl-2 but has no effect on binding to Bcl-xL (Table 1). Interestingly, compound 8 has an IC50 value of 36 nM in the H146 cell line (Table 1), and is thus slightly more potent than compound 4, suggesting that compound 8 has superior cell permeability compared to compound 4. These binding and cellular data showed that modifications of the acid group of 4 can have a significant negative or positive effect on binding to Bcl-2/Bcl-xL and on cellular activity. Accordingly, we have made additional modifications at this position in order to further explore the structure-activity relationships and to identify promising new compounds. All the designed and synthesized new compounds were tested with our standard fluorescence-polarization (FP) assays7 for their binding affinities to Bcl-2 and Bcl-xL proteins and for their cell growth inhibitory activity in the H146 small-cell lung cancer cell line, which is sensitive to potent and bona fide Bcl-2/Bcl-xL inhibitors such as compounds 1-6,7 and the results are presented in Table 1.
Table 1.
Structure-activity relationship studies on the pyrrole ring of lead compound 4
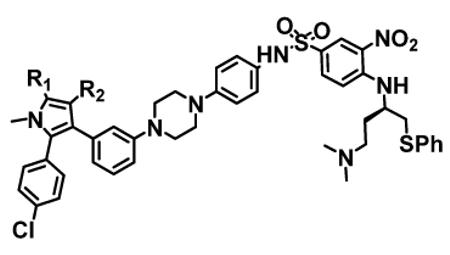
| Compound ID | R1 | R2 | Binding Affinities (IC50 ± SD, nM) |
Cell growth inhibition (IC50 ± SD, nM) |
|
|---|---|---|---|---|---|
|
| |||||
| Bcl-2 | Bcl-XL | H-146 | |||
| 4 | −CH3 | −CO2H | 1.3 ± 0.2 | 6 ± 1 | 61 ± 39 |
| 7 | −CH3 | −H | 99 ±5 | 11 ± 6 | >1,000 |
| 8 | −CH3 | −CONHCH3 | 5 ± 1 | 6 ± 3 | 36 ± 26 |
| 9 | −CH3 | −CONH2 | 19 ± 5 | 14 ± 5 | 245 ± 17 |
| 10 | −CH3 |
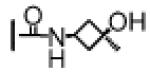
|
7.5 ± 1.2 | 15 ± 1 | 12 ± 2 |
| 11 | −CH3 |
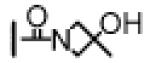
|
5.6 ± 0.6 | 9.4 ± 0.6 | 66 ± 4 |
| 12 | −CH3 | −CONHSO2CH3 | 0.9 ± 0.2 | 5 ± 1 | 38 ± 22 |
| 13 | −CH3 | −Cl | 260 ± 93 | 12 ± 8 | >1,000 |
| 14 | −CH3 | −CF3 | 271 ± 74 | 13 ± 2 | >1,000 |
| 15 | −CH3 | −CN | 17 ± 6 | 4.5 ± 1.3 | 491 ± 160 |
| 16 | −CF3 | −CO2H | 1.1 ± 0.6 | 6 ± 2 | 496 ± 59 |
| 17 | −Cl | −CO2H | 1.5 ± 0.9 | 4.7 ± 1.5 | 100 ± 17 |
First, changing the N-methylamide in compound 8 to an amide group resulted in compound 9, which is 4- and 2-times less potent than 8, respectively, in binding to Bcl-2 and Bcl-xL. Compound 9 has an IC50 value of 245 nM in the H146 cell line and is thus 6-times less potent than 8 in this cell line. Replacement of the methyl group in the N-methylamide of compound 8 with a cis-3-hydroxy-3-methylcyclobutyl group or a 3-methylazetidine-3-ol group resulted in compounds 10 and 11, respectively. Compounds 10 and 11 bind to Bcl-2 with affinities similar to that of compound 8 but both compounds are slightly less potent than 8 in binding to Bcl-xL. While compounds 10 and 11 have very similar affinities to Bcl-2 and Bcl-xL proteins, their activity in inhibition of cell growth in the H146 cell line differs by a factor of 6, further indicating that modifications of this site can have a significantly different effect on binding affinities to Bcl-2/Bcl-xL and cellular activity for this class of compounds.
In the co-crystal structure of 4 in complex with Bcl-xL (Figure 2), the acid group in 4 forms a hydrogen bond/salt bridge with Arg 132 in Bcl-xL,8 and we synthesized compound 12 to test if the methylsulfonylamide, a bioisostere of the carboxylic acid, can replace the acid group in 4 and achieve high binding affinities to Bcl-2 and Bcl-xL and potent cellular activity in the H146 cell line. Compound 12 is as potent as 4 in binding to both Bcl-2 and Bcl-xL proteins, and has an IC50 value of 38 nM in the H146 cell line, thus twice as potent as 4. These results show that the methylsulfonylamide group can indeed effectively replace the acid group, achieving not only high binding affinities to Bcl-2/Bcl-xL, but also potent cellular activity.
Figure 2.

Crystal structure of 4 (yellow) in a complex with Bcl-xL (gray) (PDB entry: 3SP7).8 The red oval highlights space available for further modifications around the pyrrole ring. The hydrogen bond/salt bridge of the acid group to Arg 132 is indicated by a dashed cyan line.
In addition to their ability to form hydrogen bonds and salt bridges, acid and methylsulfonylamide groups have strong electron withdrawing properties. To further define the contributions of these groups for binding to Bcl-2 and Bcl-xL and cellular activity in compounds 4 and 12, we synthesized compounds 13-15 to investigate if other electron withdrawing groups, such as Cl, CF3 and CN, can effectively replace the acid group in compound 4 to achieve high binding affinities to Bcl-2 and Bcl-xL and potent cellular activity. Although compound 13 with Cl and compound 14 with CF3 have similar potencies to Bcl-xL as compared to 4, both compounds are 100-times less potent than 4 in binding to Bcl-2. Compounds 13 and 14 are also >100-times less potent than 4 in inhibition of cell growth in the H146 cancer cell line. Compound 15, in which the acid group in 4 has been replaced by a nitrile, is >10-times less potent than 4 in binding to Bcl-2 but has a similar binding affinity to Bcl-xL, as compared to 4. Compound 15 has an IC50 value of 496 nM in the H146 cell line, and is thus 8 times less potent than 4.
In our co-crystal structure of 4 in complex with Bcl-xL (Figure 2), the methyl group in 4 (R1 = CH3) is in close contact with the hydrophobic portion of the Glu129 side chain in Bcl-xL. We thus synthesized compounds 16 and 17 to determine whether the methyl group can be replaced by other small hydrophobic groups such as CF3 and Cl groups to achieve high binding affinities to Bcl-2/Bcl-xL. As suggested by the co-crystal structure for compound 4, both 16 and 17 bind to Bcl-2 and Bcl-xL with the same high affinities as compared to compound 4 (Table 1). In the H146 cell line, compound 17 is slightly less potent than 4, but 16 is 8-times less potent than 4.
In all the synthesized compounds described here, the pyrrole was retained as the core structure. We next designed and synthesized 5 classes of compounds to investigate if other 5-membered heteroaromatic groups can effectively replace the pyrrole moiety in compound 4 and achieve high binding affinities to Bcl-2/Bcl-xL and potent cellular activity. Since our data showed that removal of the carboxylic acid in 4 greatly decreases its binding to Bcl-2 and cellular activity while replacement of the carboxylic acid with a methylsulfonylamide group can effectively maintain both high binding affinities to Bcl-2/Bcl-xL and potent cellular activity, we have synthesized several new compounds possessing or lacking an acid or a methylsulfonylamide group in each class of compounds. The binding and cellular data for these compounds are summarized in Table 2.
Table 2.
Structure-activity relationships for compounds in which other 5-membered hetero-aromatic rings were used to replace the pyrrole in the initial lead compound 4
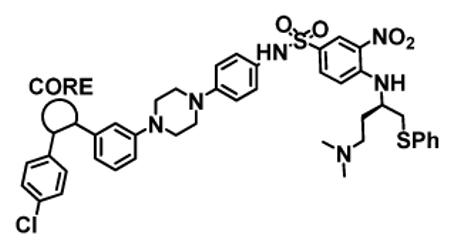
| Compound ID |
Core | Binding Affinities (IC50 ± SD, nM) |
Cell growth inhibition (IC50 ± SD, nM) |
Compound ID |
Core | Binding Affinities (IC50 ± SD, nM) |
Cell growth inhibition (IC50 ± SD, nM) |
||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| Bcl-2 | Bcl-XL | H-146 | Bcl-2 | Bcl-XL | H-146 | ||||
| 7 |

|
99 ± 5 | 11 ± 6 | >10,000 | 4 |
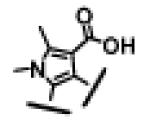
|
1.3 ± 0.2 | 6 ± 1 | 61 ± 39 |
| 12 |

|
0.9 ± 0.2 | 5 ± 1 | 38 ± 22 | |||||
| 18 |
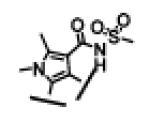
|
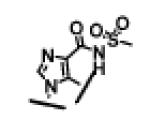
46 ± 10 | 18 ± 1 | >10,000 | 23 |
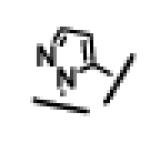
|
3.8 ± 1.1 | 8.3 ± 0.7 | >1,000 |
| 19 |
|
110 ± 4 | 32 ± 9 | >10,000 | 24 |
|
5.4 ± 1.1 | 5 ± 2 | >1,000 |
| 20 |
|
210 ± 28 | 15 ± 1 | >10,000 | 25 |
|
7.1 ± 1.6 | 6.1 ± 2.2 | >1,000 |
| 21 |
|
140 ± 53 | 26 ± 3 | >10,000 | 26 |
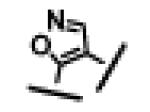
|
3.7 ± 0.6 | 5.9 ± 1.7 | 541 ± 76 |
| 22 |
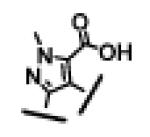
|
350 ± 146 | 22 ± 7 | >10,000 | 27 |
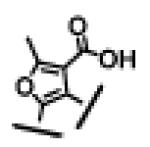
|
3.8 ± 1.0 | 4.7 ± 0.2 | 199 ± 46 |
For each of these five different classes, compounds 23-27, containing either an acid or a methylsulfonylamide group, show high binding affinities to both Bcl-2 and Bcl-xL proteins with IC50 values of 3-7 nM. However, these compounds have a significantly weaker activity in cellular assays than compounds 4 and 12. While compounds 26 and 27 have IC50 values of 541 nM and 199 nM, respectively, compounds 23, 24 and 25 have minimal activity at 1 μM.
In comparison, analogues (18-22) without either an acid or a methylsulfonylamide group show at least a 10-fold weaker binding affinity to Bcl-2 than their corresponding analogues 23-27. Compounds 18-22 are also 5-10 times less potent in their binding to Bcl-xL than compounds 23-27. Consistent with their weaker affinities to both Bcl-2 and Bcl-xL, compounds 18-22 have IC50 values of >10 μM in the H146 cell line in the cell growth inhibition assay, further emphasizing that very high binding affinities are needed in order for small molecule inhibitors of Bcl-2 and Bcl-xL to achieve potent cellular activity.
Judged by the binding affinities to Bcl-2 and Bcl-xL and the cellular activity in the H146 cell line, compounds 4 and 12 are two of the most potent and promising compounds and, accordingly, we focused on these two compounds in our further optimization.
Based upon the co-crystal structure of compound 4 complexed with Bcl-xL (Figure 2), the N-methyl group in 4 inserts into a hydrophobic pocket in Bcl-xL, which can, however, accommodate a hydrophobic group larger than methyl. Indeed, our previous study showed that replacement of the N-methyl group in 4 (R2 = CH3, Table 3) with either an N-ethyl or N-isopropyl group yielded analogues 5 and 28 with high binding affinities to Bcl-2/Bcl-xL and significantly improved cellular activity (Table 3).8 In fact, compounds 5 and 28 are 8- and 20-times more potent than compound 4 in inhibition of cell growth in the H146 cell line.
Table 3.
Structure-activity relationship of analogues of compounds 4 and 12
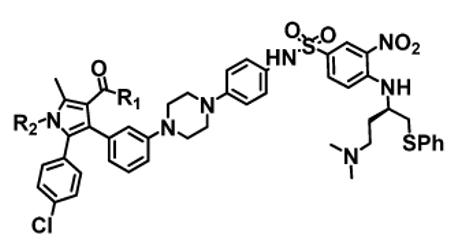
| COMPOUND | R1 | R2 | Binding Affinities (IC50 ± SD, nM) | Cell Growth Inhibition (IC50 ± SD, nM) |
||
|---|---|---|---|---|---|---|
|
| ||||||
| Bcl-2 | Bcl-XL | Mcl-1 | H-146 | |||
| 5 | −OH | Et- | 2 ± 1.6 | 6.6 ± 2.3 | NT | 8.1 ± 3.5 |
| 28 | −OH | iPr- | 1.4 ± 0.5 | 4.8 ± 0.1 | NT | 3 ± 2.4 |
| 29 | −OH | Pr- | 10 ±4 | 8.2 ± 2.2 | NT | 42 ± 5 |
| 30 | −OH | Bu- | 23 ± 2 | 16 ± 2 | NT | 67 ± 5 |
| 31(BM-1075) | −NHSO2CH3 | Et- | 1.8 ± 0.3 | 7.0 ± 1.8 | > 2000 | 4.8 ± 0.9 |
| 32 (BM-1074) | −NHSO2CH3 | iPr- | 1.8 ± 0.2 | 6.9 ± 1.8 | > 2000 | 1.3 ± 0.3 |
NT = not tested
To probe this site further and to identify the optimal group for binding to Bcl-2/Bcl-xL and cellular activity, we designed and synthesized compounds 29 and 30 in which the N-methyl group in compound 4 was replaced by either an N-n-propyl or N-n-butyl group, respectively. Although both 29 and 30 have high binding affinities to Bcl-2 and Bcl-xL, they are several times less potent than compounds 5 and 28 in their binding to Bcl-2. Consistent with their weaker affinities to Bcl-2, compounds 29 and 30 are >5- and >10-times less potent than compounds 5 and 28, respectively, in inhibition of cell growth in the H146 cell line. Our data therefore showed that the N-ethyl and N-isopropyl groups at this site in compounds 5 and 28 are most optimal for achieving high binding affinities to Bcl-2 and Bcl-xL and potent cell growth inhibition of the H146 cell line.
Since the methylsulfonylamide group can effectively replace the acid group in compound 4 and retain high binding affinities to Bcl-2/Bcl-xL and potent cellular activity, we next synthesized compounds 31 and 32 in which the acid group in compounds 5 and 28 was replaced by a methylsulfonylamide group (Table 3). As shown in Table 3, compounds 31 and 32 show high binding affinities to both Bcl-2 and Bcl-xL proteins and are very potent in inhibition of cell growth in the H146 cell line, and achieve IC50 values of 4.8 nM and 1.3 nM, respectively. To assess their binding specificity, we tested compounds 31 and 32 for their binding affinity to Mcl-1 in our optimized FP assay (Table 3).7 Our data showed that compounds 31 and 32 have no appreciable binding to Mcl-1 at concentrations as high as 2 μM. Hence, compounds 31 and 32 are potent and specific Bcl-2 and Bcl-xL inhibitors.
Based upon their high binding affinities to Bcl-2 and Bcl-xL and potent cell growth inhibitory activity in the H146 cell line, 31 and 32 represent promising new lead compounds for further in vivo evaluation for their therapeutic potential.
First, we evaluated the maximum tolerated dose (MTD) of both compounds in severe combined immunodeficient (SCID) mice. Both compounds administered intravenously (i.v.) in mice at 15 mg/kg, daily, 5 days a week for 2 weeks, were found to be well tolerated and did not cause significant weight loss (<5%) or other signs of toxicity during and after the treatment. However, at 25 mg/kg, both compounds caused significant weight loss (~10%) and, therefore, we concluded that 15 mg/kg dosed intravenously is the MTD for both drugs in SCID mice.
We next tested compounds 31 and 32 for their ability to induce apoptosis at the MTD in H146 xenograft tumors in SCID mice. A single dose of either compound at 15 mg/kg was administered i.v. to SCID mice bearing the H146 xenograft tumors. Animals were sacrificed at 3 h, 6 h and 24 h time points, and tumor tissues were removed for western blot analysis for cleavage of Poly ADP ribose polymerase (PARP) and caspase-3, two critical biochemical apoptosis markers. The data in Figure 3 showed that both compounds induce robust cleavage of PARP and caspase-3 at both 3 and 6-hr time-points in H146 tumor tissues, indicative of strong apoptosis induction in vivo.
Figure 3.

Western blot analysis of tumor tissues for cleavage of PARP and caspase-3. Mice were treated with a single dose of compound 31 or 32 (15 mg/kg, i.v.), were sacrificed at 3, 6 and 24-h time points and tumors were removed for western blot analysis. Cleavage of Cl PARP, cleaved PARP; Cl Cas-3, cleaved caspase-3.
Based upon the strong apoptosis induction in vivo observed for both 31 and 32, we evaluated their antitumor efficacy in the H-146 xenograft tumor model, and the results are shown in Figure 4. Both compounds 31 and 32 show strong antitumor activity. While compound 31 achieves only partial tumor regression, 32 is capable of achieving rapid, complete and persistent tumor regression. At day 62, 18 days after the treatment was ended, none of the 8 mice treated with 32 had measurable tumors, and at day 117, 74 days after the end of the treatment with 32, four mice (50%) did not have measurable tumors. The average tumor size for the 8 mice was 47 mm3 at day 117, as compared to 130 mm3 at the start of the treatment (day 33). Furthermore, all the mice treated with compound 32 suffered no significant weight loss (<5%) and did not show other signs of toxicity during or after the treatment. In our previous study, we had tested compound 5 for its antitumor activity in the H146 xenograft model and found that while 5 effectively inhibited tumor growth at 25 mg/kg, i.v., 5 days per week for 2 weeks, it failed to achieve long-lasting tumor regression.8 Hence, compound 32 at 15 mg/kg is considerably more effective than compound 5 at 25 mg/kg in inducing complete and persistent tumor regression in the H146 xenograft model.
Figure 4.


Antitumor activity of compounds 31 and 32 in the H146 small-cell lung cancer xenograft model in SCID mice. Tumors were grown to an average size of 126 mm3 and compound 31 or 32 was administered at 15 mg/kg intravenously, daily, 5 days a week for 2 weeks. (a). Tumor growth. (b). Animal body weight.
To further define their anticancer activity, we next tested compounds 31 and 32 in three additional small-cell lung cancer cell lines, known to be sensitive to 1 and 2, and the data are summarized in Table 4. While all four compounds effectively inhibit cell growth in these three cancer cell lines, compound 32 is the most potent of the compounds. Compound 32 has IC50 values of 1.0 nM, 1.4 nM and 2.3 nM in these three cancer cell lines and is >10-times more potent than 2, and >50-times more potent than 1, against each of these three cell lines, based upon their IC50 values.
Table 4.
Cell growth inhibitory activity of compounds 31, 32, 1 and 2 in three small-cell lung cancer cell lines
| Cell Lines | IC50 ± SD (nM) | |||
|---|---|---|---|---|
|
| ||||
| 1 | 2 | 31 | 32 | |
| H1963 | 54.0±28.2 | 26.6±7.9 | 8.2±4.9 | 1.0±0.5 |
| H187 | 137.7±71.3 | 38.4±26.8 | 7.9±3.9 | 1.4±1.3 |
| H1417 | 173.4±122.1 | 54.2±11.1 | 11.1±2.0 | 2.3±0.2 |
Chemistry
Initially, we employed a convergent synthesis7 (Method A) for the preparation of the target molecules. The piperazine 38, embedded in our target molecules, was prepared in three steps starting with the reaction of the aniline (33) and 4-fluoro-3-nitrobenzene-1-sulfonyl chloride (34), forming the sulfonamide (35).7 Displacement of the activated fluorine in 35 with (R)-N1,N1-dimethyl-4-(phenylthio)butane-1,3-diamine (36) and acid removal of the Boc protecting group generated 38. The second fragment was designed as a variable scaffold, in which diversity could be built upon to develop compounds for SAR. These scaffolds consisted of various 5-membered hetero-aromatic rings with a 4-chlorophenyl and as a synthetic handle, an adjacent 3-iodophenyl group, to which 38 could be attached. Accordingly, in this methodology, 38 was subjected to Buchwald-Hartwig coupling7, 10 with the synthesized scaffolds 9a, and 18a-21a furnishing 9 and 18-21, respectively. However, this synthetic route was complicated by poor yields of the target molecules, especially when the scaffolds contained a carboxylic acid or bioisosteres and, ultimately, this precluded further use of this methodology for further studies.
Consequently, a stepwise synthesis7, 8 (Method B), applicable to all scaffolds was carried out. In this methodology, (4-nitrophenyl)piperazine (39) was subjected to Ullman coupling11 with the synthetic scaffolds 4b, 5b, 12a, 13a, 14a, 15a, 16b, 17a, 22a, 23b, 24b, 27a, 28b, 29a, 30a, 31a, and 32a producing intermediates 4c, 5c, 12b, 13b, 14b, 15b, 16c, 17b, 22b, 23c, 24c, 27b, 28c, 29b, 30b, 31b, and 32b, respectively. Intermediates 25a and 26a were obtained using the method described in Scheme 6. Base hydrolysis of ethyl ester intermediates 17b and 27b, or low temperature acid hydrolysis of the tert-butyl ester intermediates 29b and 30b, gave the corresponding carboxylic acids. Reduction of the nitro groups in these intermediates (17b-carboxylic acid, 27b-carboxylic acid, 29b-carboxylic acid, 30b-carboxylic acid, 4c, 5c, 12b, 13b, 14b, 15b, 16c, 22b, 23c, 24c, 25a, 26a, 28c, 31b, and 32b) by catalytic hydrogenation led to aniline intermediates that were subjected to the same synthetic strategy used for the preparation of 37, generating the target compounds 4, 5, 12-17 and 22-32. Decarboxylation of 4 by acid treatment generated target compound 7. Target compounds 8, 10, and 11 were obtained by EDCI-mediated coupling to carboxylic acid 4 of methylamine, (1s,3s)-3-amino-1-methylcyclobutanol, and 3-methylazetidine-3-ol, respectively.
Scheme 6.

Synthesis of pyrazole acid intermediates 25a and 26a
Preparation of the 2-methylfuran scaffold 27a, and the variously substituted 1H-pyrrole scaffolds 4b, 5b, 9a, 12a, 13a, 14a, 15a, 16b, 28b, 29a, 30a, 31a, 32a is outlined in Scheme 2. Intermediates 44a-d were prepared using an established synthetic strategy8 in which condensation of the β-ketoesters 40a-c with 3-iodobenzaldehyde (41a) produced intermediates 42a-c, respectively, and the bromo-analogue (42d) was prepared by the condensation of β-ketoester (40a) with 3-bromobenzaldehyde (41b). Compounds 42a-d were subjected to a Stetter reaction with 4-chlorobenzaldehyde furnishing 44a-d.8, 12 The furan intermediate (27a) was prepared by heating 44a with HCl, and AcOH in EtOH. The 2-trifluoromethyl-1H-pyrrole intermediate (16a) was prepared by condensation of intermediate (44b) with methylamine. Paal-Knorr cyclization of 44a with methylamine, of 44d with methylamine, and 44c with ethylamine, n-propylamine, n-butylamine, or iso-propylamine furnished pyrroles 4a, 46, 5a, 28a, 29a, and 30a, respectively.13 Base hydrolysis of the ethyl ester intermediates 4a, 16a or low temperature acid hydrolysis of the tert-butyl ester intermediates 5a, 28a furnished the corresponding 1H-pyrrole-3-carboxylic acid scaffolds 4b, 16b and 5b, 28b, respectively.8
Scheme 2.

Synthesis of variously substituted 1H-pyrroles and 2-methyl furan scaffolds
EDCI-catalyzed coupling of ammonia to the carboxylic acid 4b furnished the 1H-pyrrole-3-carboxamide scaffold 9a, but the same conditions failed to produce the N-(methylsulfonyl)-carboxamide (12a) which was instead prepared by formation of the acid chloride of 4b and heating it with excess methanesulfonamide. In a similar manner used for the preparation of 12a, 31a and 32a were prepared from 5b and 28b, respectively. Scaffold 13a was prepared in two steps starting with the acid-mediated decarboxylation of 4b followed by chlorination at the 3-position with N-chlorosuccinamide (NCS). The 3-trifluoromethyl-1H-pyrrole scaffold 14a was prepared from 46 in three steps. Acid-mediated decarboxylation of 46 generated intermediate 47 which was iodinated at the 3-position with N-iodosuccinamide (NIS), producing 48. A trifluoromethyl group was selectively installed at the 3-position of the iodopyrrole by Cu(I) catalyzed coupling with methyl 2,2-difluoro-2-(fluorosulfonyl)acetate, producing 14a.14 The 1H-pyrrole-3-carbonitrile scaffold 15a was prepared by dehydration of pyrrole-3-carboxamide 9a.15
Imidazole scaffolds 18a and 23b were prepared as outlined in Scheme 3. The imidazole 18a was synthesized by a two-step 1,3-dipolar addition of toluenesulfonylmethyl isocyanide (TosMIC) with the imine 50.16 Compound 50 was prepared by reaction of 4-chloroaniline (49) with 41a under Dean-Stark conditions.16 The imidazole (23b) was prepared using a procedure described by Yang, et al., for the construction of imidazol[1,5-a][1,4]benzodiazapines.17 EDCI coupling of 49 with 3-iodobenzoic acid (51) generated the amide 52, which was treated with diethyl chlorophosphate to generate the unstable iminophosphate which was condensed under basic conditions with ethyl isocyanoacetate to produce the imidazole 23a.17 Base hydrolysis of the ethyl ester (23a) produced its corresponding acid that was converted to its acid chloride and reacted with methanesulfonamide to generate the imidazole scaffold (23b).
Scheme 3.

Synthesis of imidazole scaffolds 18a and 23b
The pyrazole scaffolds (19a and 24b) were prepared using a previously established method.18 Treatment of 53a or 53b with N,N-dimethylformamide dimethylacetal (54) generated the enaminones (55a and 55b) which, condensed with 4-chlorophenylhydrazine (56), produced the pyrazoles (19a and 24a), respectively.18 Base hydrolysis of the ethyl ester (24a) produced the corresponding carboxylic acid that was converted to its acid chloride and reacted with methanesulfonamide to produce the pyrazole scaffold 24b.
The pyrazoles 20a and 21a and the isoxazole 22a were prepared from a common intermediate, the enaminone (59). 3-Iodophenylacetic acid (57) was converted to its acid chloride by refluxing in thionyl chloride. Friedel-Craft acylation of chlorobenzene with 3-iodophenylacetyl chloride, using a standard method19, resulted in removal of the iodine atom from 58 and, to avoid this problem, the reaction was diluted and carried out at 0°C, producing compound 58 as the major product. Treatment of ketone 58 with 54 produced the enaminone (59). Condensation of methyl hydrazine with 59 produced a mixture of 2 regio-isomers that were separated to give the pyrazoles (20a and 21a).20 Condensation of 59 with hydroxylamine produced only one isoxazole (22a).21
The 2-chloropyrrole (17a) was prepared in four steps starting from intermediate 58. Compound 58 was treated with Br2 in AcOH to produce the α-bromoketone (60), reacted with ethyl 2-cyanoacetate under basic conditions to produce compound 61,22 treatment of which with 4M HCl in 1,4-dioxane formed the 2-chloropyrrole (62), which was methylated with CH3I under basic conditions to give the ethyl ester (17a).22
The pyrazole acids (25a and 26a) were synthesized following the procedure outlined in Scheme 6. Condensation of 63 with methyl hydrazine produced two regio-isomers, (64a and 64b),23 which were separated and were treated separately with NIS, producing 65a and 65b.24 In parallel, the pinacol boronic ester (68) was prepared by displacement of the activated fluorine in 4-fluoronitrobenzene (67) with the piperazine (66). This boronic ester was subjected to Suzuki coupling with 65a or 65b.24 The Suzuki coupling condition also resulted in hydrolysis of the methyl ester and furnished the pyrazole acids 25a and 26a.
Summary
In the present study, we have optimized compound 4, which is a potent Bcl-2/Bcl-xL inhibitor but was unable to achieve tumor regression in animal models of human cancer. In contrast to our previous study9, in which our modifications were focused on the nitro group and the soluble thiophenyl-containing “tail” group in compound 4, the present study was centered on the pyrrole core structure, which has a significant effect on binding affinities to Bcl-2/Bcl-xL and cellular activity in cancer cells. Our optimization efforts have yielded compound 32 which binds to Bcl-2 and Bcl-xL with Ki values <1 nM. Similar to other initial lead compounds, compound 32 does not show any appreciable binding to Mcl-1 protein at concentrations as high as 2 μM. Compound 32 potently inhibits cancer cell growth in the H146 small-cell lung cancer cell line and achieves an IC50 value of 1.3 nM. Compound 32 also potently inhibits cell growth inhibition, with IC50 values of 1.0-2.3 nM, in three other small-cell lung cancer cell lines which were known to be sensitive to 1 and 2. In direct comparison, compound 32 is >10- and >50-times more potent than 2 and 1, respectively, against these cancer cell lines. Significantly, compound 32 achieves rapid, complete and persistent tumor regression in the H146 xenograft tumor model in mice at a well-tolerated dose-schedule. Taken together, these data show that compound 32 is arguably the most potent and efficacious Bcl-2/Bcl-xL inhibitor reported to date and warrants extensive evaluation as a potential clinical development candidate for the treatment of human cancer.
Experimental
General Information
Unless otherwise stated, all reactions were performed under a nitrogen atmosphere in dry solvents under anhydrous conditions and all commercial reagents were used as supplied without further purification. NMR spectra were obtained on a Bruker 300 UltraShield spectrometer at a 1H frequency of 300 MHz and 13C frequency of 75 MHz. Chemical shifts (δ) are reported in parts per million (ppm) relative to an internal standard. The final products were purified by a C18 reverse phase semi-preparative HPLC column with solvent A (0.1% of TFA in water) and solvent B (0.1% of TFA in CH3CN) as eluents. All final compounds have purity ≥ 95% as determined by Waters ACQUITY UPLC. Synthesis of compounds 4, 5, 7, 8, 28 and their intermediates were reported previously.8
Ethyl 2-acetyl-4-(4-chlorophenyl)-3-(3-iodophenyl)-4-oxobutanoate (44a)8
Ethyl acetoacetate 44a (2.27g, 17.4 mmol), 3-iodobenzaldehyde 41a (4.04g, 17.4 mmol), piperidine (70 μL), and acetic acid (200 μL) were dissolved in toluene (10 ml) and refluxed with azeotropic removal of water using a Dean-Stark apparatus. After reaction overnight, the solution was cooled, diluted with EtOAc, washed sequentially with 1.0M HCl, saturated sodium bicarbonate, and brine, then dried over sodium sulfate. Purification by column chromatography (9:1 hexane:EtOAc) provided 5.3 g of the product 42a as a mixture of isomers. Triethylamine (1.55 mL) was added to a slurry of 42a (5.3 g, 15.4 mmol), 4-chlorobenzaldehyde 43 (2.16 g, 15.4 mmol), and 3-ethyl-5-(2-hydroxyethyl)-4-methylthiazolium bromide (0.583 g, 2.3 mmol) and the mixture was stirred with heating at 70 °C until 42a was consumed. After cooling to room temperature, the mixture was diluted with EtOAc, washed sequentially with 1.0M HCl, saturated sodium bicarbonate, and brine, and dried over sodium sulfate. The EtOAc was removed in vacuo and provided 7.5 g of crude 44a which was used without further purification.
Ethyl 5-(4-chlorophenyl)-4-(3-iodophenyl)-1,2-dimethyl-1H-pyrrole-3-carboxylate (4a)8
A 2.0M solution of methylamine in MeOH (35 mL, 70 mmol) was added to compound 44a (7.5 g, 15.4 mmol) dissolved in 5 mL of MeOH. After 24 hours the solution was acidified with 1.0 M HCl and the compound was extracted with EtOAc. The EtOAc solution was washed with brine, dried over sodium sulfate and concentrated in vacuo to provide crude 4a. Purification by column chromatography (5:1 hexane:EtOAc) provided 4.9 g (59 % after 3 steps) of 4a.
Ethyl 5-(4-chlorophenyl)-4-(3-iodophenyl)-2-methylfuran-3-carboxylate (27a)
Concentrated HCl (25 mL), and glacial acetic acid (5 mL) were added to a solution of 44a (1.0 g, 2.06 mmol) in EtOH ( 25 mL) and the mixture was heated to 70 °C. After 4 hours, the reaction was quenched with saturated sodium bicarbonate, extracted with EtOAc, washed with brine, dried over sodium sulfate, filtered, and the EtOAc was removed in vacuo to produce crude 27a. Purification by column chromatography provided 834 mg (96% yield) of 27a. 1H-NMR (300 MHz, CDCl3) δ ppm 7.74-7.65 (m, 2H), 7.29-7.17 (m, 5H), 7.11 (t, J = 7.74 Hz, 1H), 4.09 (q, J = 7.13 Hz, 2H), 2.68 (s, 3H), 1.06 (t, J = 7.15 Hz, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 163.83, 159.06, 146.84, 139.22, 136.78, 135.95, 133.65, 130.11, 129.63, 128.88(2C), 128.59, 126.93(2C), 121.23, 115.78, 94.03, 60.22, 14.39, 14.08.
Ethyl 2-(2-(4-chlorophenyl)-1-(3-iodophenyl)-2-oxoethyl)-4,4,4-trifluoro-3-oxobutanoate (44b)
Starting with the ethyl trifluoroacetoacetate (40b), compound 44b was prepared according to the procedure described for the preparation of compound 44a.
Ethyl 5-(4-chlorophenyl)-4-(3-iodophenyl)-1-methyl-2-(trifluoromethyl)-1H-pyrrole-3-carboxylate (16a)
Starting with 44b, compound 16a was prepared in a similar manner described for the preparation of compound 4a. 1H-NMR (300 MHz, CDCl3) δ ppm 7.54-7.45 (m, 2H), 7.35 (d, J = 8.51 Hz, 2H), 7.10 (d, J = 8.50 Hz, 2H), 7.01-6.85 (m, 2H), 4.22 (q, J = 7.14 Hz, 2H), 3.58 (s, 3H), 1.18 (t, J = 7.13 Hz, 3H); ESI-MS m/z 533.83 (M+H)+.
tert-Butyl 2-acetyl-4-(4-chlorophenyl)-3-(3-iodophenyl)-4-oxobutanoate (44c)
Starting with tert-butyl acetoacetate (40c), compound 44c was prepared according to the procedure described for the preparation of compound 44a.
tert-Butyl 5-(4-chlorophenyl)-1-ethyl-4-(3-iodophenyl)-2-methyl-1H-pyrrole-3-carboxylate (5a)
Ethylamine (11.7 mL, 2.0 M in MeOH, 23.40 mmol) was added to a solution of compound 44c (3.0 g, 5.85 mmol) in MeOH (20 mL). After 24 hours, the solution was acidified with 1.0 M HCl, stirred briefly and the compound was extracted with EtOAc. The EtOAc solution was washed sequentially with saturated NaHCO3, brine, then dried over Na2SO4 and concentrated in vacuo to provide crude 5a. Purification by column chromatography (using a gradient of hexane and DCM, the product eluted at 100% DCM) provided 1.30 g (43% yield) of 5a. 1H-NMR (300 MHz, CDCl3) δ ppm 7.53 (t, J = 1.63 Hz, 1H), 7.44 (dt, J = 1.39, 7.76 Hz, 1H), 7.26 (d, J = 8.50 Hz, 2H), 7.07 (d, J = 8.49 Hz, 2H), 6.93 (dt, J = 1.34, 7.66 Hz, 1H), 6.84 (t, J = 7.72 Hz, 1H), 3.80 (q, J = 7.18 Hz, 2H), 2.61 (s, 3H), 1.27 (s, 9H), 1.14 (t, J = 7.18 Hz, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 165.26, 140.07, 138.85, 135.37, 134.70, 134.12, 132.78(2C), 130.50, 129.73, 129.66, 129.16, 128.75(2C), 122.64, 113.05, 93.37, 79.83, 39.18, 28.26(3C), 16.25, 11.47.
tert-Butyl 5-(4-chlorophenyl)-4-(3-iodophenyl)-1-isopropyl-2-methyl-1H-pyrrole-3-carboxylate (28a)
Starting with iso-propylamine and 44c, compound 28a was prepared according to the procedure described for the preparation of compound 5a. In this case the reaction was heated in a sealed tube. 1H-NMR (300 MHz, CDCl3) δ ppm 7.50 (t, J =1.39 Hz, 1H), 7.42 (dt, J = 1.30, 7.67 Hz, 1H), 7.25 (d, J = 8.41 Hz, 2H), 7.05 (d, J = 8.39 Hz, 2H), 6.92-6.87 (m, 1H), 6.82 (t, J = 7.69 Hz, 1H), 4.33 (sept, J = 7.07 Hz, 1H), 2.69 (s, 3H), 1.41 (d, J = 7.09 Hz, 6H), 1.25 (s, 9H); 13C-NMR (75 MHz, CDCl3) δ ppm 165.35, 139.97, 139.05, 135.17, 134.57, 134.14, 133.16(2C), 131.21, 130.26, 129.53, 129.11, 128.56(2C), 122.48, 113.91, 93.34, 79.83, 48.81, 28.18(3C), 22.48(2C), 13.06.
tert-Butyl 5-(4-chlorophenyl)-4-(3-iodophenyl)-2-methyl-1-propyl-1H-pyrrole-3-carboxylate (29a)
Starting with n-propylamine and 44c, compound 29a was prepared according to the procedure described for the preparation of compound 5a. 1H-NMR (300 MHz, CDCl3) δ ppm 7.53 (t, J = 1.56 Hz, 1H), 7.46-7.41 (m, 1H), 7.25 (d, J = 8.48 Hz, 2H), 7.05 (d, J = 8.49 Hz, 2H), 6.94-6.89 (m, 1H), 6.84 (t, J = 7.69 Hz, 1H), 3.75-3.66 (m, 2H), 2.59 (s, 3H), 1.51 (sex, J = 7.69 Hz, 2H), 1.27 (s, 9H), 0.75 (t, J = 7.42 Hz, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 165.28, 140.07, 138.86, 135.71, 134.66, 133.99, 132.76(2C), 130.55, 130.00, 129.65, 129.15, 128.71(2C), 122.53, 112.94, 93.37, 79.81, 45.96, 28.23(3C), 24.24, 11.67, 11.28.
tert-Butyl 1-butyl-5-(4-chlorophenyl)-4-(3-iodophenyl)-2-methyl-1H-pyrrole-3-carboxylate (30a)
Starting with n-butylamine and 44c, compound 30a was prepared according to the procedure described for the preparation of compound 5a. 1H-NMR (300 MHz, CDCl3) δ ppm 7.53 (s, 1H), 7.43 (d, J = 7.75 Hz, 1H), 7.25 (d, J = 8.38 Hz, 2H), 7.05 (d, J = 8.40 Hz, 2H), 6.92 (d, J = 7.60 Hz, 1H), 6.84 (t, J = 7.70 Hz, 1H), 3.79-3.69 (m, 2H), 2.60 (s, 3H), 1.47 (quin, J = 7.68 Hz, 2H), 1.27 (s, 9H), 1.14 (sex, J = 7.28 Hz, 2H), 0.78 (t, J = 7.31 Hz, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 165.27, 140.07, 138.87, 135.66. 134.65, 133.98, 132.78(2C), 130.53, 129.97, 129.65, 129.13, 128.68(2C), 122.53, 112.95, 93.35, 79.79, 44.15, 32.98, 28.23(3C), 19.97, 13.71, 11.66.
Ethyl 2-acetyl-3-(3-bromophenyl)-4-(4-chlorophenyl)-4-oxobutanoate (44d)
Starting with 3-bromobenzaldehyde (41b) and ethyl acetoacetate (40a), compound 44d was prepared according to the procedure described for the preparation of compound 44a.
Ethyl 4-(3-bromophenyl)-5-(4-chlorophenyl)-1,2-dimethyl-1H-pyrrole-3-carboxylate (46)
Starting with 44d, compound 46 was prepared in the manner described for the preparation of compound 4a. 1H-NMR (300 MHz, CDCl3) δ ppm 7.31 (s, 1H), 7.29-7.20 (m, 3H), 7.07-6.90 (m, 4H), 4.07 (q, J = 7.1 Hz, 2H), 3.39 (s, 3H), 2.61 (s, 3H), 1.03 (t, J = 7.1 Hz, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 165.55, 138.25, 136.80, 134.06, 133.84, 132.45(2C), 130.55, 129.96, 129.39, 128.93, 128.80, 128.68(2C), 122.51, 121.17, 111.21, 59.39, 31.92, 14.02, 11.87.
5-(4-Chlorophenyl)-4-(3-iodophenyl)-1,2-dimethyl-1H-pyrrole-3-carboxylic acid (4b)8
NaOH (8.2 g, 204.3 mmol) was added to a solution of 4a (4.9 g, 10.2 mmol) in 300 ml of 1:1:1 dioxane, EtOH, and H2O, and the solution was heated at reflux until no compound 4a was observed by TLC. After cooling, the reaction was slowly neutralized with 1M HCl and the compound was extracted with EtOAc. The EtOAc solution was washed with brine, dried over Na2SO4 and concentrated in vacuo to produce compound 4b as a pale solid which was not further purified.
5-(4-Chlorophenyl)-4-(3-iodophenyl)-1-methyl-2-(trifluoromethyl)-1H-pyrrole-3-carboxylic acid (16b)
Compound 16b was prepared according to the procedure described for the preparation of 4b. 1H-NMR (300 MHz, CDCl3) δ ppm 7.55-7.41 (m, 2H), 7.35 (d, J = 8.46 Hz, 2H), 7.09 (d, J = 8.42 Hz, 2H), 7.06-6.99 (m, 1H), 6.92 (t, J = 7.76 Hz, 1H), 3.59 (s, 3H). ESI-MS m/z 505.83 (M+H)+.
5-(4-Chlorophenyl)-4-(3-iodophenyl)-1,2-dimethyl-1H-pyrrole-3-carboxamide (9a)
Ammonia (1.5 mL, 0.5M in 1,4-dioxane, 0.73 mmol) was added to a solution of 4b (164 mg, 0.36 mmol), EDCI (104 mg, 0.54 mmol), HOBt (70 mg, 0.54 mmol), and DIEA (188 μL, 1.08 mmol) in 4 mL of DCM. After stirring overnight, the solvent was removed in vacuo and the crude was purified by column chromatography (the compound eluted at a 5:1 to 1:1 ratio of DCM:EtOAc) to give 145 mg ( 90 % yield) of 9a. 1H-NMR (300 MHz, CDCl3) δ ppm 7.60-7.51 (m, 2H), 7.27 (d, J = 8.49 Hz, 2H), 7.13-7.07 (m, 1H), 7.03 (d, J = 8.50 Hz, 2H), 6.96 (t, J = 7.72 Hz, 1H), 5.40-4.91 (m, 2H), 3.40 (s, 3H), 2.63 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 167.64, 139.85, 137.42, 136.19, 135.65, 134.10, 132.47(2C), 130.37, 130.24, 130.20, 129.98, 128.84(2C), 120.07, 113.60, 94.38, 31.89, 11.82. ESI-MS m/z 451.25 (M+H)+.
5-(4-Chlorophenyl)-4-(3-iodophenyl)-1,2-dimethyl-N-(methylsulfonyl)-1H-pyrrole-3-carboxamide (12a)
Oxalyl chloride (40 μL, 0.44 mmol) followed by DMF (catalytic) was added to a solution of the carboxylic acid 4b (100 mg, 0.22 mmol) in DCM (4 mL) and heated under reflux for 30 min. After cooling, DCM and excess oxalyl chloride were removed in vacuo to produce the corresponding acid chloride. The resulting solid was re-dissolved in 1,2-DCE (5 mL), then methanesulfonamide (104 mg, 1.1 mmol) and DMAP (13 mg, 0.11 mmol) were added, and the solution heated at reflux overnight. Then the solvent was removed in vacuo and the crude was purified by column chromatography. The compound eluted between a 1:1 ratio of hexanes:EtOAc to 100% EtOAc) to give 91 mg (79 % yield) of 12a. 1H-NMR (300 MHz, CDCl3) δ ppm 7.63 (dt, J = 1.44, 7.73 Hz, 1H), 7.54 (t, J = 1.57 Hz, 1H), 7.30 (d, J = 8.49 Hz, 2H), 7.20 (bs, 1H), 7.12 (dt, J = 1.41, 7.62 Hz, 1H), 7.09-7.00 (m, 3H), 3.42 (s, 3H), 3.27 (s, 3H), 2.64 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 163.09, 139.80, 138.67, 137.28, 135.82, 134.65, 132.38(2C), 131.16, 130.83, 130.06, 129.11, 129.03(2C), 119.86, 111.60, 94.91, 42.03, 32.10, 12.04. ESI-MS m/z 528.92 (M+H)+.
5-(4-Chlorophenyl)-1-ethyl-4-(3-iodophenyl)-2-methyl-N-(methylsulfonyl)-1H-pyrrole-3-carboxamide (31a)
Concentrated H2SO4 (2 mL) was added to a cooled (0 °C) solution of 5a (620 mg, 1.19 mmol) in a mixture of DCM (5 mL) and THF (2 mL). After 10 min, the reaction was slowly quenched with saturated NaHCO3 and extracted with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated in vacuo to produce 569 mg of its carboxylic acid 5b as a white solid. Oxalyl chloride (222 μL, 2.44 mmol) followed by DMF (catalytic amount, ~ 10 drops) were added to a solution of the carboxylic acid (569 mg, 1.22 mmol) in DCM (7 mL) and heated to reflux for 30 min. After cooling, DCM and excess oxalyl chloride were removed in vacuo to produce the corresponding acid chloride. The resulting solid was redissolved in 1,2-DCE (10 mL), then methanesulfonamide (580 mg, 6.1 mmol), DMAP (75 mg, 0.61 mmol) were added and the solution heated under reflux overnight. After this time, the solvent was removed in vacuo and the crude was purified by column chromatography (the compound eluted between a 1:1 ratio of hexanes:EtOAc to 100% EtOAc) to give 526 mg (82% yield, 3 steps) of 31a. 1H-NMR (300 MHz, CDCl3) δ ppm 7.60 (dt, J = 1.42, 7.82 Hz, 1H), 7.55 (t, J = 1.52 Hz, 1H), 7.31 (d, J = 8.44 Hz, 2H), 7.22 (bs, 1H), 7.14-6.99 (m, 4H), 3.83 (q, J = 7.18 Hz, 2H), 3.27 (s, 3H), 2.65 (s, 3H), 1.17 (t, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 163.17, 139.74, 137.64, 137.13, 135.74, 134.78, 132.59(2C), 130.72, 130.61, 129.96, 129.37, 128.99(2C), 120.20, 111.67, 94.81, 41.99, 39.44, 16.08, 11.77.
5-(4-Chlorophenyl)-4-(3-iodophenyl)-1-isopropyl-2-methyl-N-(methylsulfonyl)-1H-pyrrole-3-carboxamide (32a)
Starting with 28a, compound 32a was prepared according to the procedure described for the preparation of compound 31a. 1H-NMR (300 MHz, CDCl3) δ ppm 7.58 (d, J = 7.70 Hz, 1H), 7.52 (s, 1H), 7.29 (d, J = 8.37 Hz, 2H), 7.18 (bs, 1H), 7.11-7.04 (m, 3H), 7.01 (t, J = 7.68 Hz, 1H), 4.35 (sept, J = 7.02, 1H), 3.25 (s, 3H), 2.73 (s, 3H), 1.44 (d, J = 7.09 Hz, 6H); 13C-NMR (75 MHz, CDCl3) δ ppm 163.28, 139.66, 137.34, 136.93, 135.87, 134.76, 132.94(2C), 131.08, 130.62, 130.13, 129.83, 128.79(2C), 120.16, 112.50, 94.69, 49.26, 41.87, 22.33(2C), 13.29.
2-(4-Chlorophenyl)-3-(3-iodophenyl)-1,5-dimethyl-1H-pyrrole (45)
Trifluoroacetic acid (3 mL) was added to a solution of 4b (500 mg, 1.11 mmol) in DCM (2 mL). After standing overnight at room temperature, the reaction was slowly quenched with saturated NaHCO3 and extracted with EtOAc. The combined organic layers was dried over Na2SO4, filtered and concentrated in vacuo to give crude 45. Purification by column chromatography (using a gradient of hexanes: EtOAc) produced 223 mg (49% yield) of 45. 1H-NMR (300 MHz, CDCl3) δ ppm 7.56 (t, J = 1.6 Hz, 1H), 7.40-7.30 (m, 3H), 7.16 (d, J = 8.5 Hz, 2H), 6.99-6.93 (m, 1H), 6.82 (t, J = 7.8 Hz, 1H), 6.12 (s, 1H), 3.34 (s, 3H), 2.29 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 139.02, 136.75, 134.04, 133.76, 132.52(2C), 131.61, 130.18, 129.90, 129.05(2C), 127.02, 120.59, 106.85, 94.51, 31.60, 12.68.
3-Chloro-5-(4-chlorophenyl)-4-(3-iodophenyl)-1,2-dimethyl-1H-pyrrole (13a)
N-chlorosuccinamide (91 mg, 0.68 mmol) was added to a solution of 45 (213 mg, 0.52 mmol) in DMF (3 mL). After overnight at room temperature, water was added and the mixture was extracted with EtOAc. The combined EtOAc layer was washed with brine, dried over Na2SO4, filtered and the solvent was removed in vacuo to give crude 13a. Purification by column chromatography (using a gradient of hexanes: EtOAc) produced 145 mg (63% yield) of 13a. 1H-NMR (300 MHz, CDCl3) δ ppm 7.58 (s, 1H), 7.47 (d, J = 7.8 Hz, 1H), 7.29 (d, J = 8.5 Hz, 2H), 7.11-7.01 (m, 3H), 6.91 (t, J = 7.8 Hz, 1H), 3.39 (s, 3H), 2.31 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 139.05, 136.16, 135.09, 133.92, 132.41(2C), 130.28, 129.77, 129.47, 129.01, 128.96(2C), 126.35, 118.83, 108.99, 93.98, 32.35, 10.28.
3-(3-Bromophenyl)-2-(4-chlorophenyl)-1,5-dimethyl-1H-pyrrole (47)
Starting with 46 (300 mg, 0.69 mmol), 181 mg (72%) of compound 47 was obtained according to the procedure described for the preparation of 45. 1H-NMR (300 MHz, CDCl3) δ ppm 7.37-7.28 (m, 3H), 7.20-7.11 (m, 3H), 6.98-6.90 (m, 2H), 6.12 (s, 1H), 3.32 (s, 3H), 2.28 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 138.94, 133.75, 132.51(2C), 131.60, 130.64, 130.18, 129.72, 129.05(2C), 128.03, 126.38, 122.44, 120.69, 106.87, 31.56, 12.66.
3-(3-Bromophenyl)-2-(4-chlorophenyl)-4-iodo-1,5-dimethyl-1H-pyrrole (48)
Starting with NIS (129 mg, 0.58 mmol) and 47 (173 mg, 0.48 mmol), 174 mg (75% yield) of compound 48 was obtained according to the procedure described for the preparation of 13a. 1H-NMR (300 MHz, CDCl3) δ ppm 7.37-7.22 (m, 4H), 7.11-6.95 (m, 4H), 3.46 (s, 3H), 2.40 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 138.17, 133.83, 133.55, 132.27(2C), 132.02, 130.91, 130.49, 129.51, 129.39, 129.37, 128.85(2C), 124.02, 121.90, 33.27, 14.00.
3-(3-Bromophenyl)-2-(4-chlorophenyl)-1,5-dimethyl-4-(trifluoromethyl)-1H-pyrrole (14a)
Methyl 2,2-difluoro-2-(fluorosulfonyl)acetate (785 μL, 6.2 mmol) and copper (I) iodide (142 mg, 0.74 mmol) were added to a solution of 48 (300 mg, 0.62 mmol) in DMF (3 mL). The solution was placed under vacuum and flushed with nitrogen three times, then heated to 100 °C overnight. After cooling to room temperature, the reaction was slowly quenched with saturated ammonium chloride, then extracted with diethyl ether, and the extracted solution was washed with water, brine, dried over Na2SO4, filtered and concentrated to give crude 14a. Purification by column chromatography (using a gradient of hexanes:EtOAc) provided 152 mg (57% yield) of 14a. 1H-NMR (300 MHz, CDCl3) δ ppm 7.32-7.22 (m, 4H), 7.08-6.97(m, 4H), 3.40 (s, 3H), 2.43 (d, J = 1.4 Hz, 3H).
5-(4-Chlorophenyl)-4-(3-iodophenyl)-1,2-dimethyl-1H-pyrrole-3-carbonitrile (15a)
To a solution of 9a (86 mg, 0.19 mmol) and pyridine (31 μL, 0.38 mmol) in 1,4-dioxane (2 mL) at 0 °C was added trifluoroacetic anhydride (TFAA) (30 μL, 0.21 mmol) dropwise. After 3 h at room temperature, the reaction was quenched with water and extracted with EtOAc. The EtOAc solution was washed with brine, dried over Na2SO4 and concentrated in vacuo to provide crude 15a. Purification by column chromatography (gradient of hexanes:EtOAc) provided 65 mg (79%) of 15a. 1H-NMR (300 MHz, CDCl3) δ ppm 7.56-7.46 (m, 2H), 7.36 (d, J = 8.39 Hz, 2H), 7.17-7.07 (m, 3H), 6.95 (t, J = 7.91 Hz, 1H), 3.40 (s, 3H), 2.47 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 139.18, 137.82, 135.83, 135.21, 134.87, 132.43, 130.42, 130.22, 129.32, 129.20, 128.28, 122.29, 116.88, 94.36, 91.91, 32.43, 12.09.
4-Chloro-N-(3-iodobenzylidene)aniline (50)
A solution of 3-iodobenzaldehyde (4.1 g, 17.7 mmol), and 4-chloroaniline (2.25 g, 17.7 mmol) in toluene (70 mL) was heated to reflux in a Dean-Stark apparatus. After reaction overnight, the solvent was removed in vacuo and the crude product was used in the following reaction. 1H-NMR (300 MHz, CDCl3) δ ppm 8.31 (d, J = 16.03 Hz, 2H), 7.81 (d, J = 7.71 Hz, 2H), 7.36 (d, J = 7.08 Hz, 2H), 7.24-7.09 (m, 3H).
1-(4-Chlorophenyl)-5-(3-iodophenyl)-1H-imidazole (18a)
A solution of 50 (6.05 g, 17.7 mmol), toluenesulfonylmethyl isocyanide (5.2 g, 26.6 mmol), potassium carbonate (4.9 g, 35.4 mmol) in 120 mL of MeOH and 50 mL of DME was refluxed for two hours. After cooling to room temperature, the reaction was quenched with water and extracted with EtOAc. The EtOAc solution was washed with brine, dried over Na2SO4 and concentrated in vacuo to provide crude 18a. Purification by column chromatography (the compound eluted between 1:1 and 1:2 hexanes:EtOAc) provided 3.95 g (59%, 2 steps) of 18a. 1H-NMR (300 MHz, CDCl3) δ ppm 7.68 (d, J = 1.04 Hz, 1H), 7.63-7.58 (m, 2H), 7.40 (d, J = 8.80 Hz, 2H), 7.28 (d, J = 1.06 Hz, 1H), 7.13 (d, J = 8.80 Hz, 2H), 7.01-6.96 (m, 2H).
N-(4-Chlorophenyl)-3-iodobenzamide (52)
4-chloroaniline (2.06 g, 16.13 mmol) was added to a solution of 3-iodobenzoic acid 51 (2.0 g, 8.06 mmol), EDCI (2.32 g, 12.09 mmol), HOBt (1.56 g, 12.09 mmol), and DIEA (4.2 mL, 24.18 mmol) in 130 mL of DCM. After stirring overnight, the solvent was removed in vacuo and the crude was purified by column chromatography to give 2.4 g (83%) of 52. 1H-NMR (300 MHz, CDCl3) δ ppm 7.69 (s, 1H), 7.65 (d, J = 8.00 Hz, 1H), 7.40-7.24 (m, 5H), 7.07 (d, J = 7.74 Hz, 1H), 6.83 (s, 1H); 13C-NMR (75 MHz, CDCl3) δ ppm 206.32, 144.03, 137.70, 137.59, 135.22, 134.86, 130.52, 129.26, 128.13, 127.69, 94.74.
Ethyl 1-(4-chlorophenyl)-5-(3-iodophenyl)-1H-imidazole-4-carboxylate (23a)
Potassium tert-butoxide (0.516 g, 4.61 mmol) was added to a solution of 52 (1.5 g, 4.19 mmol), at 0 °C, in THF. After 20 min at 0 °C, the reaction was cooled to −35 °C and diethyl chlorophosphate (783 μL, 5.45 mmol) was added slowly. After 30 min at 0 °C, the reaction was cooled to −35 °C, then ethyl isocyanoacetate (504 μL, 4.61 mmol) and t-BuOK (0.516, 4.61 mmol) were added and the reaction was allowed to warm to room temperature. After 4 h, the reaction was quenched with saturated sodium bicarbonate and extracted with EtOAc. The EtOAc solution was washed with brine, dried over Na2SO4 and concentrated in vacuo to provide crude 23a. Purification by column chromatography (the compound eluted between 1:1 and 1:2 hexanes:EtOAc) provided 1.15 g (61%) of 23a. 1H-NMR (300 MHz, CDCl3) δ ppm 7.76 (s, 1H), 7.69-7.63 (m, 2H), 7.34 (d, J = 8.68 Hz, 2H) 7.15 (d, J = 7.83 Hz, 1H), 7.10-6.98 (m, 3H), 4.28 (q, J = 7.12 Hz, 2H), 1.28(t, J = 7.13 Hz, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 162.37, 139.53, 137.81, 137.73, 135.86, 134.77, 133.65, 131.09, 129.97, 129.80, 129.74(2C), 129.48, 126.98(2C), 93.33, 60.56, 14.17. ESI-MS m/z 453.50 (M+H)+.
1-(4-Chlorophenyl)-5-(3-iodophenyl)-N-(methylsulfonyl)-1H-imidazole-4-carboxamide (23b)
Compound 23a was converted to its corresponding carboxylic acid in the manner described for the preparation of 4b. A solution of this carboxylic acid (300 mg, 0.71 mmol) in 3 mL of thionyl chloride was heated at reflux for 2 h. After cooling, thionyl chloride was removed in vacuo to produce the corresponding acid chloride. The resulting solid was re-dissolved in 10 mL of 1,2-DCE, followed by the addition of methane sulfonamide (337 mg, 3.55 mmol), and DMAP (43 mg, 0.355 mmol), and the solution was heated at reflux overnight. After this time, the reaction was cooled, water was added and extracted with EtOAc. The EtOAc solution was washed with brine, dried over Na2SO4 and concentrated in vacuo to provide crude 23b. Purification by column chromatography provided 195 mg (55% yield) of 23b. 1H-NMR (300 MHz, CDCl3) δ ppm 9.57 (s, 1H), 7.69 (d, J = 7.92 Hz, 1H), 7.66 (s, 1H), 7.61 (s, 1H), 7.39 (d, J = 8.70 Hz, 2H), 7.11-6.99 (m, 3H), 3.35 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 160.19, 139.39, 138.67, 137.39, 136.27, 135.61, 133.53, 130.69, 130.23(2C), 130.17, 129.97, 128.81, 127.19(2C), 93.75, 42.00. ESI-MS m/z 502.67 (M+H)+.
1-(4-Chlorophenyl)-5-(3-iodophenyl)-1H-pyrazole (19a)
A solution of 3-iodoacetophenone 53a (5.0 g, 20.3 mmol), and N,N-dimethylformamide dimethylacetal (41 mL, 304.8 mmol) in 200 mL of toluene was refluxed with azeotropic removal of water using a Dean-Stark apparatus. After 3 h, the solvent was removed in vacuo to give 55a as an oil. A solution of crude 55a (20.3 mmol), and 4-chlorophenylhydrazine hydrochloride (56) (4.0 g, 22.3 mmol) in ethanol was heated to reflux for 4 h. After cooling, EtOH was removed and the residue re-dissolved in EtOAc. The EtOAc solution was washed with H2O, then brine, dried over Na2SO4, filtered and concentrated to give crude 19a. Purification by column chromatography (hexanes:EtOAc) provided 6.48 g (84%, 2 steps) of 19a. 1H-NMR (300 MHz, CDCl3) δ ppm 7.75-7.60 (m, 3H), 7.33 (d, J = 8.57 Hz, 2H), 7.23 (d, J = 8.61 Hz, 2H), 7.12-6.97 (m, 2H), 6.51 (d, J = 1.28 Hz, 1H).
Ethyl 5-(3-bromophenyl)-1-(4-chlorophenyl)-1H-pyrazole-4-carboxylate (24a)
Starting with ethyl 3-(3-bromophenyl)-3-oxopropanoate 53b (1.0 g, 3.69 mmol), 0.67 g (44%, 2 steps) of compound 24a was prepared according to the procedure described for the preparation of 19a. 1H-NMR (300 MHz, CDCl3) δ ppm 8.18 (s, 1H), 7.56-7.50 (m, 2H), 7.29 (d, J = 8.84 Hz, 2H), 7.22 (t, J = 7.68 Hz, 1H), 7.18-7.10 (m, 3H), 4.22 (q, J = 7.14 Hz, 2H), 1.23 (t, J = 7.14 Hz, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 162.70, 143.66, 142.89, 137.56, 134.18, 133.65, 132.56, 130.80, 129.82, 129.38, 129.19, 126.53, 122.21, 114.78, 60.51, 14.28.
5-(3-Bromophenyl)-1-(4-chlorophenyl)-N-(methylsulfonyl)-1H-pyrazole-4-carboxamide (24b)
Starting with 24a, compound 24b was obtained according to the procedure described for the preparation of 23b. 1H-NMR (300 MHz, CDCl3) δ ppm 8.17 (s, 1H), 7.64 (ddd, J = 1.24, 1.85, 7.91 Hz, 1H), 7.48 (t, J = 1.68 Hz, 1H), 7.38-7.27 (m, 4H), 7.14 (d, J = 8.88 Hz, 2H), 3.34 (s, 3H).
1-(4-Chlorophenyl)-2-(3-iodophenyl)ethanone (58)
A solution of 3-iodophenylacetic acid (57) (2.0 g, 7.63 mmol) and thionyl chloride (3 mL) was refluxed for 2 h. After cooling to room temperature, excess thionyl chloride was removed in vacuo, producing 3-iodophenylacetyl chloride as a brown solid. The crude 3-iodophenylacetyl chloride was dissolved in chlorobenzene (10 mL) and added to a 0 °C solution of aluminum chloride (1.73 g, 12.97 mmol) and chlorobenzene (50 mL). After 2 h at 0 °C, the reaction was poured into ice and extracted with DCM. The DCM solution was washed with brine, dried over sodium sulfate and concentrated in vacuo to provide crude 58. Purification by column chromatography (the compound eluted at 19:1 hexanes:EtOAc) provided 2.03 g (75%) of 58. 1H-NMR (300 MHz, CDCl3) δ ppm 7.93 (d, J = 8.68 Hz, 2H), 7.66-7.56 (m, 2H), 7.45 (d, J = 8.68 Hz, 2H), 7.21 (d, J = 7.82 Hz, 1H), 7.07 (t, J = 8.23 Hz, 1H), 4.20 (s, 2H); 13C-NMR (75 MHz, CDCl3) δ ppm 195.82, 140.14, 138.55, 136.57, 136.39, 134.81, 130.57, 130.15, 129.31, 128.99, 94.82, 44.94.
1-(4-Chlorophenyl)-3-(dimethylamino)-2-(3-iodophenyl)prop-2-en-1-one (59)
A solution of 58 (1.5 g, 4.21 mmol), and N,N-dimethylformamide dimethylacetal (54) (8.4 mL, 63.1 mmol) in 70 mL of toluene was heated to reflux with azeotropic removal of water using a Dean-Stark apparatus. After 3 h, the solvent was removed in vacuo to give 59 as an oil that was used without purification in the following reactions.
3-(4-Chlorophenyl)-4-(3-iodophenyl)-1-methyl-1H-pyrazole (21a) and 5-(4-chlorophenyl)-4-(3-iodophenyl)-1-methyl-1H-pyrazole (20a)
A solution of 59 (1.1 g, 2.67 mmol), methylhydrazine (210 μL, 4.01 mmol) and EtOH (80 mL) was refluxed for 4 h. After cooling, EtOH was removed and the residue redissolved in EtOAc. The EtOAc solution was washed with water, then brine, dried over Na2SO4, filtered and concentrated to give a crude mixture of 21a and 20a. Purification by column chromatography (the compounds eluted between a solvent mixture of 4:4:0.1 and 4:4:0.2 DCM:hexanes:ethyl ether) eluted 21a first, followed by 20a. 21a1H-NMR (300 MHz, CDCl3) δ ppm 7.66 (s, 1H), 7.59 (d, J = 7.87 Hz, 1H), 7.46 (s, 1H), 7.41 (d, J = 8.36 Hz, 2H), 7.29 (d, J = 8.48 Hz, 2H), 7.16 (d, J = 7.80 Hz, 1H), 7.01 (t, J = 7.74 Hz, 1H), 3.97 (s, 3H); 20a1H-NMR (300 MHz, CDCl3) δ ppm 7.70 (s, 1H), 7.58 (s, 1H), 7.49 (d, J = 7.74 Hz, 1H), 7.44 (d, J = 8.33 Hz, 2H), 7.23 (d, J = 8.44 Hz, 2H), 7.01 (d, J = 7.82 Hz, 1H), 6.92 (t, J = 7.75 Hz, 1H), 3.77 (s, 3H).
5-(4-Chlorophenyl)-4-(3-iodophenyl)isoxazole (22a)
A solution of 59 (300 mg, 0.73 mmol), hydroxylamine hydrochloride (56 mg, 0.801 mmol), sodium carbonate (108 mg, 1.02 mmol), MeOH (50 mL), H2O (25 mL) and AcOH (1 mL) was heated to reflux for 4 h. After cooling to room temperature, H2O was added and the mixture was extracted with EtOAc. The EtOAc solution was washed with sodium bicarbonate, then brine, dried over Na2SO4, filtered and concentrated to give crude 22a. Purification by column chromatography (hexanes:EtOAc) provided 247 mg (89%) of 22a. 1H-NMR (300 MHz, CDCl3) δ ppm 8.33 (s, 1H), 7.79-7.67 (m, 2H), 7.54 (d, J = 8.58 Hz, 2H), 7.36 (d, J = 8.59 Hz, 2H), 7.32 (d, J = 7.78 Hz, 1H), 7.13 (t, J = 7.77 Hz, 1H); 13C-NMR (75 MHz, CDCl3) δ ppm 163.43, 151.71, 137.44, 137.42, 136.59, 132.10, 130.84, 129.40, 128.63, 128.01, 125.75, 115.13, 94.98.
2-Bromo-1-(4-chlorophenyl)-2-(3-iodophenyl)ethanone (60)
A solution of bromine (86 μL, 1.68 mmol) in acetic acid (4 mL) was added to a solution of 58 (500 mg, 1.40 mmol) in DCM (4 mL). After 3 h at room temperature, H2O was added and the mixture was extracted with DCM. The extracted solution was washed sequentially with sodium bicarbonate, H2O, and brine, then dried over Na2SO4, filtered and concentrated to give crude 60. Purification by column chromatography (hexanes:EtOAc) provided 220 mg (36%) of 60. 1H-NMR (300 MHz, CDCl3) δ ppm 7.91 (d, J = 8.55 Hz, 2H), 7.87 (s, 1H), 7.63 (d, J = 7.91 Hz, 1H), 7.48 (d, J = 7.80 Hz, 1H), 7.40 (d, J = 8.53 Hz, 2H), 7.08 (t, J = 7.85 Hz, 1H), 6.21 (s, 1H); 13C-NMR (75 MHz, CDCl3) δ ppm 189.53, 140.55, 138.32, 137.96, 137.60, 132.17, 130.66, 130.61, 129.33, 128.66, 94.62, 48.74.
Ethyl 4-(4-chlorophenyl)-2-cyano-3-(3-iodophenyl)-4-oxobutanoate (61)
A mixture of potassium carbonate (636 mg, 4.60 mmol) and ethyl cyanoacetate (1.71 mL, 16.07 mmol) was heated at 45 °C. After 45 min, the mixture was allowed to cool to room temperature, then a solution of 60 (1.0g, 2.30 mmol) in 5 mL of Me2CO was added dropwise. After stirring overnight, H2O was added and the solution was extracted with EtOAc. The extracted solution was sequentially washed with H2O, and brine, then dried over Na2SO4, filtered and concentrated to give crude 61. Purification by column chromatography (hexanes:EtOAc) provided 1.01 g (94%) of 61.
Ethyl 2-chloro-5-(4-chlorophenyl)-4-(3-iodophenyl)-1H-pyrrole-3-carboxylate (62)
To 61 (500 mg, 1.07 mmol) was added 6 mL of 4M HCl in 1,4-dioxane. After four hours, the reaction was slowly quenched with saturated sodium bicarbonate and the solution extracted with EtOAc. The extracted solution was washed with brine, dried over Na2SO4, filtered and concentrated to give crude 62. Purification by column chromatography (the compound eluted with 100% DCM) provided 323 mg (62%) of 62. 1H-NMR (300 MHz, CDCl3) δ ppm 9.10 (s, 1H), 7.67-7.53 (m, 2H), 7.18 (d, J = 8.43 Hz, 2H), 7.13 (d, J = 7.67 Hz, 1H), 7.06-6.93 (m, 3H), 4.11 (q, 7.10 Hz, 2H), 1.08 (t, J = 7.11 Hz, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 163.72, 139.73, 136.97, 136.18, 133.57, 130.06, 129.72, 129.27, 129.09, 128.51, 127.64, 122.19, 121.89, 112.37, 93.73, 60.40, 14.11.
Ethyl 2-chloro-5-(4-chlorophenyl)-4-(3-iodophenyl)-1-methyl-1H-pyrrole-3-carboxylate (17a)
Potassium carbonate (274 mg, 1.98 mmol) was added to a solution of 62 (323 mg, 0.66 mmol) in DMF (3 mL). After 10 min at room temperature, iodomethane (86μL, 1.33 mmol) was added and the mixture stirred overnight. The reaction was slowly quenched with saturated ammonium chloride and extracted with EtOAc. The extracted solution was washed with brine, dried over Na2SO4, filtered and concentrated to give crude 17a. Purification by column chromatography (the compound eluted at a solvent mixture between 1:1 to 3:1 of DCM:hexanes) provided a quantitative yield of 17a. 1H-NMR (300 MHz, CDCl3) δ ppm 7.58-7.45 (m, 2H), 7.28 (d, J = 8.44 Hz, 2H), 7.05 (d, J = 8.42 Hz, 2H), 6.98 (d, J = 7.70 Hz, 1H), 6.88 (t, J = 7.67 Hz, 1H), 4.13 (q, J = 7.12 Hz, 2H), 3.49 (s, 3H), 1.08 (t, J = 7.13 Hz, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 163.43, 139.87, 136.91, 135.49, 134.53, 132.38, 130.84, 129.94, 129.20, 129.02, 128.93, 123.51, 122.76, 110.84, 93.24, 60.05, 32.64, 14.14.
Methyl 3-(4-chlorophenyl)-1-methyl-1H-pyrazole-5-carboxylate (64b) and methyl 5-(4-chlorophenyl)-1-methyl-1H-pyrazole-3-carboxylate (64a)
Methyl hydrazine (0.656 mL, 12.47 mmol) was added, dropwise, to a solution of methyl 4-(4-chlorophenyl)-2,4-dioxobutanoate 63 (2.0 g, 8.31 mmol), AcOH (2.4 mL), and EtOH (40 mL). After 3 h of stirring at room temperature, H2O was added and the mixture extracted with EtOAc. The extracted solution was washed with saturated sodium bicarbonate, then brine, dried over Na2SO4, filtered and concentrated to give a crude mixture of 64a and 64b. Purification by column chromatography (using a gradient of hexanes:EtOAc where 64b eluted at 9:1 and 64a eluted between 4:1 to 1:1) provided 0.824 g (40%) of 64b and 0.974 g (47%) of 64a. 64b: 1H-NMR (300 MHz, CDCl3) δ ppm 7.71 (d, J = 8.57 Hz, 2H), 7.36 (d, J = 8.56 Hz, 2H), 7.07 (s, 1H), 4.21 (s, 3H), 3.90 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 160.32, 148.88, 133.96, 133.77, 131.24, 129.06(2C), 126.91(2C), 108.03, 52.17, 39.83. 64a: 7.46 (d, J = 8.58 Hz, 2H), 7.37 (d, J = 8.59 Hz, 2H), 6.85 (s, 1H), 3.95 (s, 3H), 3.94 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 162.52, 143.87, 142.35, 135.23, 130.01(2C), 129.11(2C), 127.90, 108.92, 51.95, 38.27.
Methyl 3-(4-chlorophenyl)-4-iodo-1-methyl-1H-pyrazole-5-carboxylate (65b)
N-iodosuccinamide (903 mg, 4.01 mmol), followed by ceric ammonium nitrate (17 mg, 0.0308 mmol) was added to a solution of 64b (774 mg, 3.08 mmol) in MeCN (30 mL), and the mixture was heated at 70 °C overnight. After cooling to room temperature, the solvent was removed in vacuo and the residue re-dissolved in EtOAc. The EtOAc solution was washed with H2O, then brine, dried over Na2SO4, filtered and concentrated to give crude 65b. Purification by column chromatography (using a gradient of hexanes:EtOAc) provided 1.03 g (89% yield) of 65b. 1H-NMR (300 MHz, CDCl3) δ ppm 7.71 (d, J = 8.61 Hz, 2H), 7.41 (d, J = 8.61 Hz, 2H), 4.23 (s, 3H), 3.97 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 159.73, 151.89, 135.04, 134.73, 131.05, 130.23(2C), 128.64(2C), 64.17, 52.31, 41.57.
Methyl 5-(4-chlorophenyl)-4-iodo-1-methyl-1H-pyrazole-3-carboxylate (65a)
Starting with 64a (974 mg, 3.89 mmol), compound 65a (337 mg, 23% yield) was obtained according to the procedure described for the preparation of compound 65b. 1H-NMR (300 MHz, CDCl3) δ ppm 7.52 (d, J = 8.34 Hz, 2H), 7.33 (d, J = 8.39 Hz, 2H), 3.97 (s, 3H), 3.89 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 161.80, 146.46, 142.35, 136.30, 131.68(2C), 129.41(2C), 127.37, 63.65, 52.31, 39.28.
1-(4-Nitrophenyl)-4-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)piperazine (68)
Potassium carbonate (361 mg, 2.61 mmol) was added to a solution of 3-piperazinylphenylboronic acid pinacol ester 66 (500 mg, 1.74 mmol) and 1-fluoro-4-nitrobenzene 67 (269 mg, 1.91 mmol) in DMSO (5 mL). After stirring overnight, the reaction was quenched with 2N HCl and extracted with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated in vacuo to produce crude 68. Column chromatography, using a gradient of DCM:EtOAc, produced 238 mg (33% yield) of pure 68 as an orange oil. 1H-NMR (300 MHz, CDCl3) δ ppm 8.11 (d, J = 9.35 Hz, 2H), 7.45-7.27 (m, 3H), 7.09-7.02 (m, 1H), 6.82 (d, J = 9.41 Hz, 2H), 3.60-3.50 (m, 4H), 3.41-3.30 (m, 4H), 1.35 (s, 12H); 13C-NMR (75 MHz, CDCl3) δ ppm 154.75, 150.17, 138.54, 128.82, 126.98, 126.00(2C), 122.44, 119.45, 112.76(2C), 83.90(2C), 48.97(2C), 47.06(2C), 24.95(4C). ESI-MS m/z 410.42 (M+H)+.
5-(4-Chlorophenyl)-1-methyl-4-(3-(4-(4-nitrophenyl)piperazin-1-yl)phenyl)-1H-pyrazole-3-carboxylic acid (25a)
Pd(PPh3)4 (10 mg) was added to a solution of 65a (30 mg, 0.08 mmol), boronic ester 68 (65 mg, 0.16 mmol), and potassium carbonate (44 mg, 0.32 mmol) in DME/H2O (9 mL, 2:1). The mixture was placed under vacuum, flushed with nitrogen twice, and heated to 90 °C overnight. After cooling to room temperature, saturated NH4Cl was added and the mixture was extracted with EtOAc. The combined EtOAc layers were washed with brine, dried over Na2SO4, filtered through a plug of celite and the solvent was removed in vacuo to give crude 25a. Purification by reverse phase HPLC provided pure 25a. 1H-NMR (300 MHz, 10:1 CDCl3:CD3OD) δ ppm 8.15 (d, J = 9.17 Hz, 2H), 7.38 (d, J = 8.32 Hz, 2H), 7.22-7.12 (m, 3H), 6.96-6.82 (m, 4H), 6.78 (d, J = 7.54 Hz, 1H), 3.89 (s, 3H), 3.64-3.51 (m, 4H), 3.30-3.19 (m, 4H). ESI-MS m/z 518.50 (M+H)+.
3-(4-Chlorophenyl)-1-methyl-4-(3-(4-(4-nitrophenyl)piperazin-1-yl)phenyl)-1H-pyrazole-5-carboxylic acid (26a)
Starting with 65b, compound 26a was prepared according to the procedure described for the preparation of compound 25a. 1H-NMR (300 MHz, 10:1 CDCl3:CD3OD) δ ppm 8.14 (d, J = 9.35 Hz, 2H), 7.33-7.16 (m, 5H), 6.97-6.77 (m, 5H), 4.23 (s, 3H), 3.62-3.50 (m, 4H), 3.32-3.21 (m, 4H). ESI-MS m/z 518.33 (M+H)+.
5-(4-Chlorophenyl)-1,2-dimethyl-N-(methylsulfonyl)-4-(3-(4-(4-nitrophenyl)piperazin-1-yl)phenyl)-1H-pyrrole-3-carboxamide (12b)
A mixture of 12a (585 mg, 1.1 mmol), 1-(4-nitrophenyl)piperazine (916 mg, 4.43 mmol), copper (I) iodide (105 mg, 0.55 mmol), L-proline (127 mg, 1.1 mmol), and potassium carbonate (612 mg, 4.43 mmol) was placed under vacuum and then flushed with N2 three times. To this mixture was added DMSO (13 mL), the solution was placed under vacuum and flushed with N2 three times, then heated to 100 °C overnight. After cooling to room temperature, the reaction was slowly quenched with saturated ammonium chloride and stirred for 10 min. The solution was then extracted with DCM and the extracted solution was washed with saturated ammonium chloride, 2M hydrochloric acid, brine, dried over Na2SO4, filtered and concentrated to give crude 12b. Purification by column chromatography (using a gradient of DCM:EtOAc) provided 491 mg (74%) of 12b as a yellow solid. 1H-NMR (300 MHz, CDCl3) δ ppm 8.15 (d, J = 9.36 Hz, 2H), 7.53 (bs, 1H), 7.32-7.21 (m, 3H), 7.07 (d, J = 8.40 Hz, 2H), 6.93 (dd, J = 2.21, 8.16 Hz, 1H), 6.88-6.78 (m, 3H), 6.70 (d, J = 7.49 Hz, 1H), 3.62-3.52 (m, 4H), 3.45 (s, 3H), 3.39-3.29 (m, 4H), 3.24 (s, 3H), 2.67 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 163.39, 154.76, 150.58, 139.05, 139.02, 134.66, 134.36, 132.38(2C), 130.91, 130.42, 129.64, 128.91(2C), 126.19(2C), 123.20, 121.84, 119.16, 116.34, 113.05(2C), 111.38, 48.96(2C), 46.83(2C), 42.24, 32.13, 12.20. ESI-MS m/z 608.50 (M+H)+.
1-(3-(4-Chloro-2-(4-chlorophenyl)-1,5-dimethyl-1H-pyrrol-3-yl)phenyl)-4-(4-nitrophenyl)piperazine (13b)
Starting with 13a (123 mg, 0.28 mmol), 95 mg (65% yield) of compound 13b was obtained according to the procedure described for the preparation of 12b. 1H-NMR (300 MHz, CDCl3) δ ppm 8.11 (d, J = 9.3 Hz, 2H), 7.28 (d, J = 8.4 Hz, 2H), 7.18-7.07 (m, 3H), 6.81 (d, J = 9.4 Hz, 2H), 6.77-6.68 (m, 3H), 3.54-3.44 (m, 4H), 3.42 (s, 3H), 3.23-3.11 (m, 4H), 2.32 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 154.86, 150.18, 138.67, 134.69, 133.55, 132.52(2C), 130.92, 128.90, 128.80(2C), 128.70, 126.22, 126.10(2C), 122.32, 120.36, 118.60, 114.18, 112.81(2C), 108.96, 48.86(2C), 46.98(2C), 32.29, 10.25. ESI-MS m/z 521.67 (M+H)+.
1-(3-(2-(4-Chlorophenyl)-1,5-dimethyl-4-(trifluoromethyl)-1H-pyrrol-3-yl)phenyl)-4-(4-nitrophenyl)piperazine (14b)
Starting with 14a (152 mg, 0.36 mmol), 68 mg (34% yield) of compound 14b was obtained according to the procedure described for the preparation of 12b. 1H-NMR (300 MHz, CDCl3) δ ppm 8.14 (d, J = 9.3 Hz, 2H), 7.30-7.20 (m, 2H), 7.16-7.02 (m, 3H), 6.84 (d, J = 9.3 Hz, 2H), 6.76 (d, J = 8.4 Hz, 1H), 6.73-6.63 (m, 2H), 3.56-3.46 (m, 4H), 3.43 (s, 3H), 3.25-3.13 (m, 4H), 2.45 (s, 3H). ESI-MS m/z 555.17 (M+H)+.
5-(4-Chlorophenyl)-1,2-dimethyl-4-(3-(4-(4-nitrophenyl)piperazin-1-yl)phenyl)-1H-pyrrole-3-carbonitrile (15b)
Starting with 15a (216 mg, 0.50 mmol), 150 mg (59%) of compound 15b was obtained according to the procedure described for the preparation of 12b. 1H-NMR (300 MHz, CDCl3) δ ppm 8.14 (d, J = 9.36 Hz, 2H), 7.37 (d, J = 8.42 Hz, 2H), 7.22-7.08 (m, 3H), 6.91-6.79 (m, 3H), 7.76 (dd, J = 2.00, 8.14 Hz, 1H), 6.66 (d, J = 7.67 Hz, 1H), 3.58-3.47 (m, 4H), 3.42 (s, 3H), 3.29-3.17 (m, 4H), 2.49 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 154.89, 150.60, 139.03, 138.80, 134.67, 133.87, 132.67(2C), 130.07, 130.03, 129.45, 129.28(2C), 126.17(2C), 124.18, 120.96, 117.46, 117.23, 114.74, 112.88(2C), 91.98, 48.82(2C), 47.06(2C), 32.36, 12.08. ESI-MS m/z 512.33 (M+H)+.
5-(4-Chlorophenyl)-1-methyl-4-(3-(4-(4-nitrophenyl)piperazin-1-yl)phenyl)-2-(trifluoromethyl)-1H-pyrrole-3-carboxylic acid (16c)
Starting with 16b (484 mg, 0.96 mmol), 150 mg (27%) of compound 16c was obtained according to the procedure described for the preparation of 12b. 1H-NMR (300 MHz, CDCl3) δ ppm 8.10 (d, J = 9.36 Hz, 2H), 7.34 (d, J = 8.42 Hz, 2H), 7.13 (d, J = 8.43 Hz, 2H), 7.06 (t, J = 7.83 Hz, 1H), 6.88-6.68 (m, 4H), 6.55 (d, J = 7.65 Hz, 1H), 3.60 (s, 3H), 3.51-3.41 (m, 4H), 3.24-3.07 (m, 4H). ESI-MS m/z 585.42 (M+H)+.
Ethyl 2-chloro-5-(4-chlorophenyl)-1-methyl-4-(3-(4-(4-nitrophenyl)piperazin-1-yl)phenyl)-1H-pyrrole-3-carboxylate (17b)
Starting with 17a (353 mg, 0.71 mmol), 199 mg (51%, 2 steps) of compound 17b was obtained according to the procedure described for the preparation of 12b. 1H-NMR (300 MHz, CDCl3) δ ppm 8.12 (d, J = 9.33 Hz, 2H), 7.27 (d, J = 8.46 Hz, 2H), 7.16-7.04 (m, 3H), 6.84 (d, J = 9.41 Hz, 2H), 6.77 (dd, J = 1.76, 8.18 Hz, 1H), 6.71-6.60 (m, 2H), 4.13 (q, J = 7.17 Hz, 2H), 3.51 (s, 3H), 3.58-3.43 (m, 4H), 3.22-3.10 (m, 4H), 1.10 (t, J = 7.11 Hz, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 163.74, 154.81, 149.83, 138.62, 135.31, 134.17, 132.41(2C), 130.58, 129.65, 128.77(2C), 128.32, 126.06(2C), 124.57, 123.25, 122.91, 119.24, 114.72, 112.80(2C), 111.01, 59.98, 48.99(2C), 46.96(2C), 32.61, 14.13.
5-(4-Chlorophenyl)-4-(3-(4-(4-nitrophenyl)piperazin-1-yl)phenyl)isoxazole (22b)
Starting with 22a (100 mg, 0.24 mmol), 43 mg (39%) of compound 22b was obtained according to a modified procedure described for the preparation of 12b. For this scaffold the reaction is not heated; instead it is run at room temperature. 1H-NMR (300 MHz, CDCl3) δ ppm 8.35 (s, 1H), 8.15 (d, J = 9.33 Hz, 2H), 7.61 (d, J = 8.57 Hz, 2H), 7.41-7.29 (m, 3H), 7.03-6.80 (m, 5H), 3.65-3.52 (m, 4H), 3.41-3.30 (m, 4H); 13C-NMR (75 MHz, CDCl3) δ ppm 163.07, 154.79, 152.18, 151.36, 139.10, 136.36, 131.11, 130.33, 129.31(2C), 128.72(2C), 126.32, 126.19(2C), 120.63, 117.02, 116.29, 116.07, 113.05(2C), 48.67(2C), 47.16(2C). ESI-MS m/z 461.83 (M+H)+.
1-(4-Chlorophenyl)-N-(methylsulfonyl)-5-(3-(4-(4-nitrophenyl)piperazin-1-yl)phenyl)-1H-imidazole-4-carboxamide (23c)
Starting with 23b, compound 23c was obtained according to the procedure described for the preparation of 12b. 1H-NMR (300 MHz, CDCl3) δ ppm 8.15 (d, J = 9.28 Hz, 2H), 7.38 (d, J = 7.05 Hz, 2H), 7.22 (t, J = 7.63 Hz, 1H), 7.16-7.01 (m, 2H), 7.01-6.89 (m, 2H), 6.86 (d, J = 9.32 Hz, 2H), 6.70 (d, J = 7.04 Hz, 1H), 3.61-3.50 (m, 4H), 3.35 (s, 3H), 3.32-3.22 (m, 4H). ESI-MS m/z 581.92 (M+H)+.
1-(4-Chlorophenyl)-N-(methylsulfonyl)-5-(3-(4-(4-nitrophenyl)piperazin-1-yl)phenyl)-1H-pyrazole-4-carboxamide (24c)
Starting with 24b, compound 24c was obtained according to the procedure described for the preparation of 12b. 1H-NMR (300 MHz, CDCl3) δ ppm 8.26 (s, 1H), 8.13 (d, J = 9.19 Hz, 2H), 8.02 (bs, 1H), 7.37 (t, J = 7.94 Hz, 1H), 7.29 (d, J = 8.72 Hz, 2H), 7.18 (d, J = 8.68 Hz, 2H), 7.04 (d, J = 8.41 Hz, 1H), 6.93-6.78 (m, 3H), 6.74 (d, J = 7.39 Hz, 1H), 3.66-3.53 (m, 4H), 3.45-3.35 (m, 4H), 3.31 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 160.36, 154.62, 151.32, 143.95, 142.48, 138.93, 137.47, 134.41, 130.97, 129.40(2C), 128.34, 126.28(2C), 126.16(2C), 120.87, 117.81, 116.90, 115.42, 112.88(2C), 47.92(2C), 46.72(2C), 42.30. ESI-MS m/z 581.58 (M+H)+.
Ethyl 5-(4-chlorophenyl)-2-methyl-4-(3-(4-(4-nitrophenyl)piperazin-1-yl)phenyl)furan-3-carboxylate (27b)
Starting with 27a (400 mg, 0.95 mmol), 132 mg (27%, 2 steps) of compound 27b was obtained according to the procedure described for the preparation of 12b. 1H-NMR (300 MHz, CDCl3) δ ppm 8.10 (d, J = 9.38 Hz, 2H), 7.36-7.24 (m, 3H), 7.16 (d, J = 8.73 Hz, 2H), 6.98 (dd, J = 2.00, 8.19 Hz, 1H), 6.92-6.77 (m, 4H), 4.10 (q, J = 7.12 Hz, 2H), 3.63-3.48 (m, 4H), 3.39-3.26 (m, 4H), 2.68 (s, 3H), 1.07 (t, J = 7.13 Hz, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 163.86, 158.48, 154.74, 150.63, 146.43, 138.73, 134.53, 133.14, 129.34, 128.90, 128.63(2C), 126.70(2C), 126.05(2C), 123.11, 122.40, 118.17, 115.96, 115.67, 112.84(2C), 60.01, 49.02(2C), 47.02(2C), 14.39, 14.06. ESI-MS m/z 546.42 (M+H)+.
tert-Butyl 5-(4-chlorophenyl)-2-methyl-4-(3-(4-(4-nitrophenyl)piperazin-1-yl)phenyl)-1-propyl-1H-pyrrole-3-carboxylate (29b)
Starting with 29a (390 mg, 0.81 mmol), 242 mg (53%) of compound 29b was obtained according to the procedure described for the preparation of 12b. 1H-NMR (300 MHz, CDCl3) δ ppm 8.14 (d, J = 9.36 Hz, 2H), 7.24 (d, J = 8.42 Hz, 2H), 7.13-7.03 (m, 3H), 6.85 (d, J = 9.42 Hz, 2H), 6.77-6.60 (m, 3H), 3.79-3.68 (m, 2H), 3.55-3.43 (m, 4H), 3.22-3.09 (m, 4H), 2.59 (s, 3H), 1.53 (sex, J = 7.66 Hz, 2H), 1.28 (s, 9H), 0.76 (t, J =3H); 13C-NMR (75 MHz, CDCl3) δ ppm 165.53, 154.96, 149.82, 138.80, 137.20, 135.10, 133.66, 132.79(2C), 131.16, 129.77, 128.56(2C), 128.18, 126.14(2C), 124.35, 123.53, 119.27, 114.25, 113.16, 112.92(2C), 79.57, 49.25(2C), 47.11(2C), 45.99, 28.22(3C), 24.28, 11.70, 11.29. ESI-MS m/z 615.00 (M+H)+.
tert-Butyl 1-butyl-5-(4-chlorophenyl)-2-methyl-4-(3-(4-(4-nitrophenyl)piperazin-1-yl)phenyl)-1H-pyrrole-3-carboxylate (30b)
Starting with 30a (400 mg, 0.81 mmol), 227 mg (49%) of compound 30b was obtained according to the procedure described for the preparation of 12b. 1H-NMR (300 MHz, CDCl3) δ ppm 8.14 (d, J = 9.36 Hz, 2H), 7.24 (d, J = 8.42 Hz, 2H), 7.14-7.04 (m, 3H), 6.85 (d, J = 9.36 Hz, 2H), 6.76-6.58 (m, 3H), 3.83-3.70 (m, 2H), 3.55-3.44 (m, 4H), 3.23-3.12 (m, 4H), 2.59 (s, 3H), 1.49 (p, J = 7.94 Hz, 2H), 1.28 (s, 9H), 1.16 (sex, J = 7.45 Hz, 2H), 0.79 (t, J = 7.31 Hz, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 165.53, 154.96, 149.83, 138.82, 137.22, 135.06, 133.67, 132.82(2C), 131.16, 129.75, 128.55(2C), 128.18, 126.14(2C), 124.36, 123.55, 119.29, 114.25, 113.17, 112.93(2C), 79.57, 49.26(2C), 47.12(2C), 44.16, 33.04, 28.23(3C), 19.99, 13.72, 11.71. ESI-MS m/z 629.42 (M+H)+.
5-(4-Chlorophenyl)-1-ethyl-2-methyl-N-(methylsulfonyl)-4-(3-(4-(4-nitrophenyl)piperazin-1-yl)phenyl)-1H-pyrrole-3-carboxamide (31b)
Starting with 31a, compound 31b was prepared according to the procedure described for the preparation of compound 12b. 1H-NMR (300 MHz, CDCl3) δ ppm 8.14 (d, J = 9.34 Hz, 2H), 7.59 (bs, 1H), 7.32-7.18 (m, 3H), 7.11 (d, J = 8.40 Hz, 2H), 6.88-6.79 (m, 3H), 6.73 (s, 1H), 6.65 (d, J = 7.33 Hz, 1H), 3.86 (q, J = 7.02 Hz, 2H), 3.58-3.47 (m, 4H), 3.36-3.26 (m, 4H), 3.23 (s, 3H), 2.69 (s, 3H), 1.19 (t, J = 7.12 Hz, 3H); 13C-NMR (75 MHz, CDCl3) δ ppm 163.46, 154.54, 151.10, 138.84, 138.00, 134.46, 132.60(2C), 130.29, 130.21, 130.00, 128.86(2C), 126.17(2C), 122.40, 122.35, 118.75, 115.87, 112.89(2C), 111.44, 48.51, 46.90, 42.18, 39.42, 16.13, 11.99. ESI-MS m/z 622.17 (M+H)+.
5-(4-Chlorophenyl)-1-isopropyl-2-methyl-N-(methylsulfonyl)-4-(3-(4-(4-nitrophenyl)piperazin-1-yl)phenyl)-1H-pyrrole-3-carboxamide (32b)
Starting with 32a, compound 32b was prepared according to the procedure described for the preparation of compound 12b. 1H-NMR (300 MHz, CDCl3) δ ppm 8.12 (d, J = 9.34 Hz, 2H), 7.55 (bs, 1H), 7.30-7.14 (m, 3H), 7.10 (d, J = 8.38 Hz, 2H), 6.88-6.78 (m, 3H), 6.70 (s, 1H), 6.63 (d, J = 7.52 Hz, 1H), 4.39 (sept, J = 6.96 Hz, 1H), 3.60-3.47 (m, 4H), 3.33-3.24 (m, 4H), 3.23 (s, 3H), 2.77 (s, 3H), 1.46 (d, J = 7.08 Hz, 6H); 13C-NMR (75 MHz, CDCl3) δ ppm 163.63, 154.81, 150.99, 138.69, 137.87, 134.57, 134.44, 132.96(2C), 130.85, 130.71, 130.08, 128.66(2C), 126.14(2C), 122.31, 121.03, 118.68, 115.74, 112.82(2C), 112.08, 49.23, 48.45, 46.81, 42.05, 22.37, 13.54.
(R)-tert-Butyl 4-(4-(4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrophenylsulfonamido)phenyl)piperazine-1-carboxylate (37)
To a cooled (0 °C) solution of tert-butyl 4-(4-aminophenyl)piperazine-1-carboxylate 33 (2 g, 7.21 mmol) in pyridine (30 mL) was added 4-fluoro-3-nitrobenzene-1-sulfonyl chloride 34 (2.25 g, 9.37 mmol). After 30 minutes, pyridine was removed in vacuo and the oil was purified by column chromatography to give 1.82 g (53% yield) of intermediate 35. Intermediate 35 (1.82 g, 3.79 mmol) was re-dissolved in DMF (10 mL), then (R)-N1,N1-dimethyl-4-(phenylthio)butane-1,3-diamine 36 (1.02 g, 4.55 mmol) and DIEA (2 mL, 11.37 mmol) were added. After stirring overnight, the solvent was removed in vacuo and the crude was purified by column chromatography (using a gradient of DCM: EtOAc) to produce 2.14 g (82% yield) of 37 as a yellow solid. 1H-NMR (300 MHz, CDCl3) δ ppm 8.99 (d, J = 8.24 Hz, 1H), 8.49 (d, J = 2.21 Hz, 1H), 7.48 (dd, J = 1.83, 9.14 Hz, 1H), 7.35-7.28 (m, 2H), 7.25-7.16 (m, 3H), 7.02 (d, J = 8.82 Hz, 2H), 6.80 (d, J = 8.93 Hz, 2H), 6.61 (d, J = 9.34 Hz, 1H), 4.03-3.88 (m, 1H), 3.60-3.48 (m, 4H), 3.11 (d, J = 6.07 Hz, 2H), 3.09-3.01 (m, 4H), 2.53-2.40 (m, 1H), 2.35-2.24 (m, 1H), 2.20 (s, 6H), 2.11-1.94 (m, 1H), 1.90-1.74 (m, 1H), 1.47 (s, 9H); 13C-NMR (75 MHz, CDCl3) δ ppm 154.86, 149.70, 146.82, 134.95, 133.56, 131.11(2C), 130.93, 129.32(2C), 128.42, 127.84, 127.37, 125.34, 124.89(2C), 117.18(2C), 114.52, 80.18, 55.34, 51.55, 49.38(2C), 45.54(2C), 38.57, 30.59, 28.59(3C). ESI-MS m/z 685.33 (M+H)+.
(R)-4-(4-(Dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitro-N-(4-(piperazin-1-yl)phenyl)benzenesulfonamide (38)
Trifluoroacetic acid (5 mL) was added to a solution of 37 (2.14g, 3.12 mmol) in DCM (3 mL). After 5 h at room temperature, the reaction was slowly quenched with saturated NaHCO3 extracted with EtOAc, and solid NaCl was added to salt out the product. The combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo to give quantitative yield of 38 as an orange solid. 1H-NMR (300 MHz, CD3OD) δ ppm 8.30 (d, J = 2.11 Hz, 1H), 7.53 (dd, J = 2.09, 9.16 Hz, 1H), 7.24-7.15 (m, 2H), 7.10-6.97 (m, 5H), 6.92-6.82 (m, 3H), 4.13-3.99 (m, 1H), 3.37-3.31 (m, 1H), 3.18 (d, J = 5.87 Hz, 1H), 3.15-3.07 (m, 4H), 3.07-2.95 (m, 4H), 2.57-2.33 (m, 2H), 2.25 (s, 6H), 2.13-1.78 (m, 2H); 13C-NMR (75 MHz, CD3OD) δ ppm 150.43, 148.03, 136.52, 134.31, 131.70, 131.59(2C), 131.05, 130.04(2C), 128.04, 127.74, 126.90, 124.80(2C), 118.12(2C), 116.18, 56.62, 52.77, 50.11(2C), 45.90(2C), 45.32(2C), 39.47, 32.18. ESI-MS m/z 585.92 (M+H)+.
(R)-N-(4-(4-(3-(1-(4-Chlorophenyl)-1H-imidazol-5-yl)phenyl)piperazin-1-yl)phenyl)-4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrobenzenesulfonamide (18)
(Method A) A round bottom flask containing a solution of imidazole 18a (20 mg, 0.051 mmol), piperazine 38 (60 mg, 0.103 mmol), Pd(dba)2 (5.8 mg, 0.01 mmol), P(tBu)3 (50 μL), sodium tert-butoxide (10 mg, 0.103 mmol), toluene (2 mL) and DMF (1 mL) was placed under vacuum briefly, then flushed with N2 three times. The solution, under nitrogen, was heated to 90 °C overnight. After cooling, the reaction was filtered through celite, then through a plug of silica. The crude was dissolved in MeOH, 4.0 M HCl in dioxane was added and after 5 minutes the solvent was removed. The residue was re-dissolved in 3:1 MeOH/water and purified by reverse phase HPLC to give 12 mg (28% yield) of 18 as a yellow powder. 1H-NMR (300 MHz, CD3OD) δ ppm 9.23 (s, 1H), 8.33 (d, J = 2.0 Hz, 1H), 7.83 (s, 1H), 7.60 (dd, J = 2.0, 9.0 Hz, 1H), 7.55 (d, J = 8.8 Hz, 2H), 7.43 (d, J = 8.8 Hz, 2H), 7.30- 7.15 (m, 3H), 7.12-7.00 (m, 6H), 7.00-6.90 (m, 3H), 6.80-6.71 (m, 2H), 4.19-4.04 (m, 1H), 3.28-3.12 (m, 12H), 2.87 (s, 6H), 2.35-2.09 (m, 2H). ESI-MS m/z 837.50 (M+H)+.
(R)-5-(4-Chlorophenyl)-4-(3-(4-(4-(4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-1,2-dimethyl-1H-pyrrole-3-carboxamide (9)
Starting with 9a, compound 9 was obtained according to the procedure described for the preparation of 18. 1H-NMR (300 MHz, 10:1 CDCl3:CD3OD) δ ppm 8.45 (d, J = 2.2 Hz, 1H), 7.63 (dd, J = 2.3, 9.1 Hz, 1H), 7.29-7.21 (m, 4H), 7.20-7.11 (m, 4H), 7.10-7.03 (m, 4H), 6.97-6.85 (m, 3H), 6.80-6.74 (m, 3H), 4.13-4.05 (m, 1H), 3.43 (s, 3H), 3.31-3.20 (m, 9H), 3.19-3.09 (m, 3H), 2.82 (s, 6H), 2.62 (s, 3H), 2.40-2.05 (m, 2H). ESI-MS m/z 907.67 (M+H)+.
(R)-N-(4-(4-(3-(1-(4-Chlorophenyl)-1H-pyrazol-5-yl)phenyl)piperazin-1-yl)phenyl)-4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrobenzenesulfonamide (19)
Starting with 19a, compound 19 was obtained according to the procedure described for the preparation of 18. 1H-NMR (300 MHz, CD3OD) δ ppm 8.35 (d, J = 2.0 Hz, 1H), 7.73 (d, J = 1.8 Hz, 1H), 7.59 (dd, J = 2.0, 9.0 Hz, 1H), 7.40 (d, J = 8.8 Hz, 2H), 7.32-7.23 (m, 3H), 7.22-7.16 (m, 2H), 7.12-6.87 (m, 9H), 6.80-6.73 (m, 2H), 6.60 (d, J = 1.9 Hz, 1H), 4.16-4.02 (m, 1H), 3.29-3.12 (m, 11H), 2.87 (s, 6H), 2.31-2.09 (m, 2H). ESI-MS m/z 837.50 (M+H)+.
(R)-N-(4-(4-(3-(5-(4-Chlorophenyl)-1-methyl-1H-pyrazol-4-yl)phenyl)piperazin-1-yl)phenyl)-4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrobenzene-sulfonamide (20)
Starting with 20a, compound 20 was obtained according to the procedure described for the preparation of 18. 1H-NMR (300 MHz, 10:1 CDCl3:CD3OD) δ ppm 8.44 (d, J = 1.9 Hz, 1H), 7.74 (s, 1H), 7.64 (dd, J = 1.8, 9.1 Hz, 1H), 7.45 (d, J = 8.3 Hz, 2H), 7.31-7.23 (m, 4H), 7.23-7.09 (m, 6H), 7.08-7.00 (m, 2H), 6.94 (d, J = 8.30 Hz, 1H), 6.89-6.74 (m, 3H), 4.19-4.04 (m, 1H), 3.78 (s, 3H), 3.49-3.09 (m, 12H), 2.82 (s, 6H), 2.40-2.07 (m, 2H). ESI-MS m/z 851.33 (M+H)+.
(R)-N-(4-(4-(3-(3-(4-Chlorophenyl)-1-methyl-1H-pyrazol-4-yl)phenyl)piperazin-1-yl)phenyl)-4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrobenzenesulfonamide (21)
Starting with 21a, compound 21 was obtained according to the procedure described for the preparation of 18. 1H-NMR (300 MHz, 10:1 CDCl3:CD3OD) δ ppm 8.45 (d, J = 2.2 Hz, 1H), 7.62 (dd, J = 2.2, 9.1 Hz, 1H), 7.52 (s, 1H), 7.42 (d, J = 8.5 Hz, 2H), 7.31-7.19 (m, 5H), 7.18-7.10 (m, 3H), 7.07 (d, J = 8.9 Hz, 2H), 6.94-6.71 (m, 6H), 4.11-4.02 (m, 1H), 3.97 (s, 3H), 3.31-3.06 (m, 12H), 2.82 (s, 6H), 2.38-2.08 (m, 2H). ESI-MS m/z 851.58 (M+H)+.
(R)-5-(4-Chlorophenyl)-4-(3-(4-(4-(4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-N,1,2-trimethyl-1H-pyrrole-3-carboxamide (8)8
To a solution of 4 (50 mg, 0.055 mmol), EDCI (16 mg, 0.083 mmol), HOBt (11 mg, 0.083 mmol), and DIEA (28 μL, 0.165 mmol) in 5 mL of DCM, was added methyl amine (138 μL of 2M in THF, 0.275 mmol). After stirring overnight, the solvent was removed in vacuo and the crude was re-dissolved in MeOH, 4.0M HCl in dioxane was added and after 5 min the solvent was removed. The residue was re-dissolved in 3:1 MeOH/H2O and purified by reverse phase HPLC to give 34 mg (68%) of 8 as a yellow powder.
5-(4-Chlorophenyl)-4-(3-(4-(4-(4-((R)-4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-N-((1s,3s)-3-hydroxy-3-methylcyclobutyl)-1,2-dimethyl-1H-pyrrole-3-carboxamide (10)
Starting with (1s,3s)-3-amino-1-methylcyclobutanol and 4, compound 10 was obtained according to the procedure described for the preparation of 8. 1H-NMR (300 MHz, 10:1 CDCl3:CD3OD) δ ppm 8.37 (d, J = 2.1 Hz, 1H), 7.55 (dd, J = 2.1, 9.1 Hz, 1H), 7.21-7.15 (m, 4H), 7.14-7.05 (m, 4H), 7.03-6.95 (m, 4H), 6.86-6.77 (m, 3H), 6.71-6.59 (m, 3H), 4.06-3.96 (m, 1H), 3.80 (p, 7.9 Hz, 1H), 3.34 (s, 3H), 3.20-3.00 (m, 12H), 2.73 (s, 6H), 2.49 (s, 3H), 2.30-2.00 (m, 4H), 1.48-1.37 (m, 2H), 1.18 (s, 3H). ESI-MS m/z 991.42 (M+H)+.
(R)-N-(4-(4-(3-(2-(4-Chlorophenyl)-4-(3-hydroxy-3-methylazetidine-1-carbonyl)-1,5-dimethyl-1H-pyrrol-3-yl)phenyl)piperazin-1-yl)phenyl)-4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrobenzenesulfonamide (11)
Starting with 3-methylazetidin-3-ol and 4, compound 11 was obtained according to the procedure described for the preparation of 8. 1H-NMR (300 MHz, 10:1 CDCl3:CD3OD) δ ppm 8.45 (d, J =2.2 Hz, 1H), 7.63 (dd, J = 2.2, 9.1 Hz, 1H), 7.34-7.22 (m, 4H), 7.19-7.03 (m, 8H), 6.89 (d, J = 9.0 Hz, 2H), 6.83-6.73 (m, 2H), 6.67 (br.s., 1H), 6.61 (d, J = 7.6 Hz, 1H), 4.13-4.05 (m, 1H), 3.83 (q, 10Hz, 2H), 3.40 (s, 3H), 3.30-3.10 (m, 13H), 2.82 (s, 6H), 2.39 (s, 3H), 2.36-2.10 (m, 2H), 1.11 (s, 3H). ESI-MS m/z 977.17 (M+H)+.
(R)-5-(4-Chlorophenyl)-4-(3-(4-(4-(4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-1,2-dimethyl-N-(methylsulfonyl)-1H-pyrrole-3-carboxamide (12)
A round bottom flask containing a solution of compound 12b (100 mg, 0.16 mmol) in MeOH (10 mL) was placed under vacuum briefly, then flushed with N2 atmosphere three times. 10 % Pd-C (50 mg) was added to this solution, which was then purged again. The N2 was removed in vacuum and a balloon of H2 gas was connected to the flask. After 15 min, the reaction was filtered through celite, and the solvent was removed in vacuo to produce the aniline intermediate that was used without further purification. To a cooled (0 °C) solution of the aniline intermediate in pyridine (6 mL) was added 4-fluoro-3-nitrobenzene-1-sulfonyl chloride 34 (50 mg, 0.21 mmol). After 30 min, pyridine was removed in vacuo and the oil was purified by column chromatography to give 79 mg of an oily red intermediate. The resulting oil (79 mg, 0.10 mmol) was re-dissolved in DMF (2 mL), then (R)-N1,N1-dimethyl-4-(phenylthio)butane-1,3-diamine 36 (45 mg, 0.20 mmol) and DIEA (0.1 mL) were added. After standing overnight, the solvent was removed in vacuo and the crude was purified by column chromatography to produce 12 as a yellow solid. The solid was dissolved in MeOH, 4.0 M HCl in dioxane was added and after 5 minutes the solvent was removed. The residue was re-dissolved in 3:1 MeOH/H2O and purified by reverse phase HPLC to give 92 mg (58 %, 3 steps) of 12 as a yellow powder. 1H-NMR (300 MHz, 10:1 CDCl3:CD3OD) δ ppm 8.45 (d, J = 1.81 Hz, 1H), 7.61 (dd, J = 1.61, 8.84 Hz, 1H), 7.32-7.00(m, 13H), 6.94-6.82 (m, 3H), 6.79-6.63 (m, 3H), 4.14-4.00 (m, 1H), 3.45 (s, 3H), 3.31-3.07 (m, 15H), 2.82 (s, 6H), 2.65 (s, 3H), 2.37-2.05 (m, 2H). ESI-MS m/z 985.67 (M+H)+.
(R)-N-(4-(4-(3-(4-Chloro-2-(4-chlorophenyl)-1,5-dimethyl-1H-pyrrol-3-yl)phenyl)piperazin-1-yl)phenyl)-4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrobenzenesulfonamide (13)
Starting with 13b, compound 13 was obtained according to the procedure described for the preparation of 12. 1H-NMR (300 MHz, 10:1 CDCl3:CD3OD) δ ppm 8.45 (d, J = 2.2 Hz, 1H), 7.62 (dd, J = 2.3, 9.1 Hz, 1H), 7.31-7.23 (m, 4H), 7.18-7.02 (m, 8H), 6.90-6.72 (m, 6H), 4.17-4.07 (m, 1H), 3.43 (s, 3H), 3.29-3.06 (m, 12H), 2.81 (s, 6H), 2.33 (s, 3H), 2.40-2.10 (m, 2H). ESI-MS m/z 900.17 (M+H)+.
(R)-N-(4-(4-(3-(2-(4-Chlorophenyl)-1,5-dimethyl-4-(trifluoromethyl)-1H-pyrrol-3-yl)phenyl)piperazin-1-yl)phenyl)-4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrobenzenesulfonamide (14)
Starting with 14b, compound 14 was obtained according to the procedure described for the preparation of 12. 1H-NMR (300 MHz, 10:1 CDCl3:CD3OD) δ ppm 8.46 (d, J = 2.2 Hz, 1H), 7.61 (dd, J = 2.2, 9.1 Hz, 1H), 7.29-7.23 (m, 4H), 7.18-7.10 (m, 4H), 7.09-7.02 (m, 4H), 6.90-6.67 (m, 6H), 4.12-4.04 (m, 1H), 3.44 (s, 3H), 3.28-3.08 (m, 12H), 2.82 (s, 6H), 2.45 (d, J = 1.4 Hz, 3H), 2.37-2.08 (m, 2H). ESI-MS m/z 932.42 (M+H)+.
(R)-N-(4-(4-(3-(2-(4-Chlorophenyl)-4-cyano-1,5-dimethyl-1H-pyrrol-3-yl)phenyl)piperazin-1-yl)phenyl)-4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrobenzenesulfonamide (15)
Starting with 15b, compound 15 was obtained according to the procedure described for the preparation of 12. 1H-NMR (300 MHz, 10:1 CDCl3:CD3OD) δ ppm 8.46 (d, J = 2.2 Hz, 1H), 7.61 (dd, J = 2.2, 9.1 Hz, 1H), 7.36 (d, J = 9.0 Hz, 2H), 7.28-7.22 (m, 2H), 7.20-7.08 (m, 6H), 7.05 (d, H = 8.9 Hz, 2H), 6.87 (d, J = 9.0 Hz, 2H), 6.83-6.67 (m, 4H), 4.12-4.02 (m, 1H), 3.43 (s, 3H), 3.30-3.08 (m, 12H), 2.82 (s, 6H), 2.49 (s, 3H), 2.39-2.09 (m, 2H). ESI-MS m/z 889.33 (M+H)+.
(R)-5-(4-Chlorophenyl)-4-(3-(4-(4-(4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-1-methyl-2-(trifluoromethyl)-1H-pyrrole-3-carboxylic acid (16)
Starting with 16c, compound 16 was obtained according to the procedure described for the preparation of 12. 1H-NMR (300 MHz, 10:1 CDCl3:CD3OD) δ ppm 8.45 (d, J = 1.7 Hz, 1H), 7.61 (d, J = 9.1 Hz, 1H), 7.38-7.29 (m, 2H), 7.28-7.23 (m, 2H), 7.20-7.01 (m, 8H), 6.91-6.57 (m, 6H), 4.12-4.04 (m, 1H), 3.58 (s, 3H), 3.27-3.02 (m, 12H), 2.81 (s, 6H), 2.36-2.07 (m, 2H). ESI-MS m/z 962.58 (M+H)+.
(R)-N-(4-(4-(3-(5-(4-Chlorophenyl)isoxazol-4-yl)phenyl)piperazin-1-yl)phenyl)-4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrobenzenesulfonamide (22)
Starting with 22b, compound 22 was obtained according to the procedure described for the preparation of 12. 1H-NMR (300 MHz, 10:1 CDCl3:CD3OD) δ ppm 8.45 (d, J = 2.2Hz, 1H), 8.38 (s, 1H), 7.64 (dd, J = 2.3, 9.1 Hz, 1H), 7.61-7.58 (m, 2H), 7.39-7.31 (m, 3H), 7.28-7.23 (m, 2H), 7.17-7.09 (m, 5H), 7.52-7.00 (m, 3H), 6.99-6.90 (m, 2H), 6.78 (d, J = 9.4 Hz, 1H), 4.14-4.07 (m, 1H), 3.43-3.09 (m, 12H) 2.82 (s, 6H), 2.37-2.09 (m, 2H). ESI-MS m/z 838.33 (M+H)+.
(R)-1-(4-Chlorophenyl)-5-(3-(4-(4-(4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-N-(methylsulfonyl)-1H-imidazole-4-carboxamide (23)
Starting with 23c, compound 23 was obtained according to the procedure described for the preparation of 12. 1H-NMR (300 MHz, 10:1 CDCl3:CD3OD) δ ppm 8.45 (d, J = 2.2 Hz, 1H), 7.71 (s, 1H), 7.63 (dd, J = 2.1, 9.1 Hz, 1H), 7.36 (d, J = 9.0, 2H), 7.29-7.05 (m, 10H), 7.00-6.91 (m, 4H), 6.77 (d, J = 9.3 Hz, 1H), 6.71 (d, J = 7.6 Hz, 1H), 4.14-4.05 (m, 1H), 3.32 (s, 3H), 3.30-3.05 (m, 12H), 2.81 (s, 6H), 2.36-2.08 (m, 2H). ESI-MS m/z 959.42 (M+H)+.
(R)-1-(4-Chlorophenyl)-5-(3-(4-(4-(4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-N-(methylsulfonyl)-1H-pyrazole-4-carboxamide (24)
Starting with 24c, compound 24 was obtained according to the procedure described for the preparation of 12. 1H-NMR (300 MHz, 10:1 CDCl3:CD3OD) δ ppm 8.45 (d, J = 2.1 Hz, 1H), 8.24 (s, 1H), 7.63 (dd, J = 2.0, 9.1 Hz, 1H), 7.32-7.22 (m, 5H), 7.20-7.00 (m, 8H), 6.99-6.89 (m, 3H), 6.79-6.70 (m, 2H), 4.15-4.05 (m, 1H), 3.35-3.04 (m, 15H), 2.81 (s, 6H), 2.37-2.08 (m, 2H). ESI-MS m/z 958.42 (M+H)+.
(R)-5-(4-Chlorophenyl)-4-(3-(4-(4-(4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-1-methyl-1H-pyrazole-3-carboxylic acid (25)
Starting with 25a, compound 25 was obtained according to the procedure described for the preparation of 12. 1H-NMR (300 MHz, CD3OD) δ ppm 8.31 (d, J = 2.2 Hz, 1H), 7.60 (dd, J = 2.3, 9.2 Hz, 1H), 7.38-7.35 (m, 2H), 7.26-6.90 (m, 15H), 6.85 (d, J = 7.6 Hz, 1H), 4.11-4.07 (m, 1H), 3.82 (s, 3H), 3.45-3.33 (m, 9H), 3.21-3.14 (m, 3H), 2.84 (s, 6H), 2.25-2.15 (m, 2H). ESI-MS m/z 895.75 (M+H)+.
(R)-3-(4-Chlorophenyl)-4-(3-(4-(4-(4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-1-methyl-1H-pyrazole-5-carboxylic acid (26)
Starting with 26a, compound 26 was obtained according to the procedure described for the preparation of 12. 1H-NMR (300 MHz, CD3OD) δ ppm 8.32 (d, J = 2.3 Hz, 1H), 7.58 (dd, J = 2.3, 9.2 Hz, 1H), 7.30-7.25 (m, 3H), 7.20-7.14 (m, 4H), 7.09-6.96 (m, 9H), 6.90 (d, J = 10.2 Hz, 1H), 6.85 (d, J = 7.6 Hz, 1H), 4.18 (s, 3H), 4.10-4.06 (m, 1H), 3.37-3.31 (m, 9H), 3.21-3.15 (m, 3H), 2.84 (s, 6H), 2.24-2.13 (m, 2H). ESI-MS m/z 895.50 (M+H)+.
(R)-5-(4-Chlorophenyl)-4-(3-(4-(4-(4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-2-methylfuran-3-carboxylic acid (27)
To a solution of 27b (260 mg, 0.48 mmol) in 60 mL of 1:1:1 dioxane, EtOH, and H2O, was added NaOH (190 mg, 4.8 mmol), and the solution was refluxed until no compound 27b was observed by TLC. After cooling, the reaction was slowly neutralized with 1M HCl and the compound was extracted with EtOAc. The EtOAc solution was washed with brine, dried over Na2SO4 and concentrated in vacuo to produce 97 mg of the acid intermediate as a yellow solid. Starting with this acid, compound 27 was obtained according to the procedure described for the preparation of 12. 1H-NMR (300 MHz, CD3OD) δ ppm 8.32 (d, J = 2.2 Hz, 1H), 7.58 (dd, J = 2.3, 9.2 Hz, 1H), 7.35-6.97 (m, 16H), 6.91-6.88 (m, 2H), 4.09-4.07 (m, 1H), 3.34-3.31 (m, 9H), 3.21-3.15 (m, 3H), 2.84 (s, 6H), 2.65 (s, 3H), 2.25-2.15 (m, 2H). ESI-MS m/z 895.92 (M+H)+.
(R)-2-Chloro-5-(4-chlorophenyl)-4-(3-(4-(4-(4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-1-methyl-1H-pyrrole-3-carboxylic acid (17)
Starting with 17b, compound 17 was obtained according to the procedure described for the preparation of 27. 1H-NMR (300 MHz, 10:1 CDCl3:CD3OD) δ ppm 8.45 (d, J = 2.3 Hz, 1H), 7.62 (dd, J = 2.3, 9.1 Hz, 1H), 7.31-7.23 (m, 4H), 7.19-7.11 (m, 4H), 7.10-7.02 (m, 4H), 6.94-6.85 (m, 3H), 6.84-6.80 (m, 1H), 6.76 (d, J = 9.2 Hz, 2H), 4.13-4.04 (m, 1H), 3.51 (s, 3H), 3.31-3.07 (m, 12H), 2.81 (s, 6H), 2.35-2.08 (m, 2H). ESI-MS m/z 928.42 (M+H)+.
(R)-5-(4-Chlorophenyl)-4-(3-(4-(4-(4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-2-methyl-1-propyl-1H-pyrrole-3-carboxylic acid (29)
Concentrated H2SO4 (2 mL) was added to a cooled (0 °C) solution of 29b (92 mg, 0.15 mmol) in a mixture of DCM (2 mL) and THF (0.5 mL). After 10 minutes the reaction was slowly quenched with saturated NaHCO3 and extracted with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated in vacuo to produce 68 mg of its carboxylic acid as a yellow solid. Starting with this acid, compound 29 was obtained according to the procedure described for the preparation of 12. 1H-NMR (300 MHz, 10:1 CDCl3:CD3OD) δ ppm 8.45 (d, J = 2.17 Hz, 1H), 7.62 (dd, J = 2.16, 9.10 Hz, 1H), 7.30-7.22 (m, 4H), 7.18-7.10 (m, 4H), 7.10-7.03 (m, 4H), 6.94-6.73 (m, 6H), 4.14-4.02 (m, 1H), 3.79-3.69 (m, 2H), 3.34-3.19 (m, 9H), 3.19-3.07 (m, 3H), 2.81 (s, 6H), 2.62 (s, 3H), 2.37-2.06 (m, 2H), 1.54 (sex, J = 7.62 Hz, 2H), 0.77 (t, J = 7.39 Hz, 3H). ESI-MS m/z 936.83 (M+H)+.
(R)-1-Butyl-5-(4-chlorophenyl)-4-(3-(4-(4-(4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-2-methyl-1H-pyrrole-3-carboxylic acid (30)
Starting with 30b, compound 30 was obtained according to the procedure described for the preparation of 29. 1H-NMR (300 MHz, 10:1 CDCl3:CD3OD) δ ppm 8.45 (d, J = 2.11 Hz, 1H), 7.63 (dd, J = 1.92, 9.10 Hz, 1H), 7.31-7.20 (m, 4H), 7.20-7.04 (m, 8H), 7.04-6.85 (m, 5H), 6.78 (d, J = 9.24 Hz, 1H), 4.16-4.01 (m, 1H), 3.85-3.73 (m, 2H), 3.44-3.07 (m, 12H), 2.81 (s, 6H), 2.63 (s, 3H), 2.38-2.03 (m, 2H), 1.48 (sex, J = 6.87 Hz, 2H), 1.29-1.08 (m, 2H), 0.79 (t, J = 7.31 Hz, 3H). ESI-MS m/z 950.67 (M+H)+.
(R)-5-(4-Chlorophenyl)-4-(3-(4-(4-(4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-1-ethyl-2-methyl-N-(methylsulfonyl)-1H-pyrrole-3-carboxamide (31)
Starting with 31b, compound 31 was obtained according to the procedure described for the preparation of 12. 1H-NMR (300MHz, CD3OD) δ ppm 8.35 (d, J = 2.2 Hz, 1H), 7.62 (dd, J = 2.1, 9.1 Hz, 1H), 7.35 (d, J = 8.5 Hz, 2H), 7.23-6.93 (m, 15H), 6.80 (d, J = 7.6 Hz, 1H), 4.19-4.08 (m, 1H), 3.89 (q, J = 7.0 Hz, 2H), 3.46-3.33 (m, 9H), 3.26-3.16 (m, 7H), 2.87 (s, 6H), 2.70 (s, 1H), 2.55 (s, 3H), 2.34-2.11 (m, 2H), 1.13 (t, J = 7.1 Hz, 3H). ESI-MS m/z 999.83 (M+H)+.
(R)-5-(4-Chlorophenyl)-4-(3-(4-(4-(4-(4-(dimethylamino)-1-(phenylthio)butan-2-ylamino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-1-isopropyl-2-methyl-N-(methylsulfonyl)-1H-pyrrole-3-carboxamide (32)
Starting with 32b, compound 32 was obtained according to the procedure described for the preparation of 12. 1H-NMR (300MHz, CD3OD) δ ppm 8.34 (d, J = 2.2 Hz, 1H), 7.65 (dd, J = 2.1, 9.1 Hz, 1H), 7.35 (d, J = 8.4 Hz, 2H), 7.27-6.98 (m, 15H), 6.90 (d, J = 7.3 Hz, 1H), 4.40 (h, J = 7.0 Hz, 1H), 4.21-4.10 (m, 1H), 3.62-3.34 (m, 9H), 3.27-3.15 (m, 6H), 2.99 (s, 1H), 2.87 (s, 7H), 2.61 (s, 3H), 2.34-2.10 (m, 2H), 1.42 (d, J = 7.0 Hz, 6H). ESI-MS m/z 1013.83 (M+H)+.
Fluorescence Polarization based (FP) binding assays
Binding affinities of our synthesized compounds to Bcl-2, Bcl-xL and Mcl-1 were determined using an FP based competitive binding assay described previously.7 Briefly, pre-incubated protein/probe complex (1.5 and 1 nM for the Bcl-2 assay, 10 and 2 nM for the Bcl-xL assay, and 20 and 2 nM for the Mcl-1 assay, respectively) and serial dilutions of the inhibitors were incubated at room temperature for 2 h with gentle shaking. Millipolarization (mP) values were measured and plotted over total compound concentrations, nonlinear regression fitting to the binding curves generated the IC50 values, and the Ki values were calculated using the equation described previously.25
Cell growth inhibition assay
This assay was performed as previously reported.7 Human small cell lung cancer cell lines H146, H1963, H187, and H1417 were purchased from American Type Culture Collection (ATCC) and were maintained in RPMI-1640 medium containing 10% FBS. Various concentrations of compounds were incubated for 4 days in 96-well flat bottom cell culture plates that were seeded at a density of 1×1014 cells/well. At the end of incubation, the effect of compounds on cell growth was evaluated by WST-assay. In this assay, 20 μL of WST-8 dye was added to each well and incubated for an additional 1–2 h, and then the absorbance was measured, at 450 nm, in a microplate reader (Molecular Devices). Using the GraphPad Prism software (GraphPad Software, La Jolla), the IC50 values were calculated by comparing absorbance in the untreated cells and the cells treated with the compounds. The standard deviation for each compound was obtained by performing a minimum of three independent experiments in each cell line.
In Vivo Pharmacodynamic (PD) and Efficacy Studies in the H146 Xenograft Model
We employed a similar procedure to that used in our previous studies.7, 8 An H146 small-cell lung cancer xenograft model was used for our in vivo PD and efficacy studies. Xenograft tumors (one tumor per mouse) were developed by injection of 5 × 106 H146 cancer cells with Matrigel, subcutaneously, on the dorsal side of the SCID mice (from Charles River).
For the PD studies, mice bearing tumors approximately 100 mm3 in volume were given a single dose of 31, 32 (15 mg/kg) or vehicle. The tumor tissues were harvested at 3h, 6h or 24h, followed by Western blot analysis to determine levels of PARP and caspase-3, as well as cleaved PARP and caspase-3.
For the efficacy studies, mice with tumor volumes between 100 and 150 mm3 were randomized into different groups (8 mice per group) with mean tumor volume of 126 mm3. Each group of mice was treated intravenously with either vehicle control or 31 or 32 at 15 mg/kg, daily, 5 days/week for 2 weeks. Tumor sizes and animal weights were measured 3 times per week during the treatment and twice per week after the treatment. Data are presented as mean tumor volumes ± SEM. Statistical analyses were performed using two-way ANOVA and unpaired two-tailed t test, using Prism (version 4.0, GraphPad, La Jolla, CA). P < 0.05 was considered statistically significant. The efficacy experiment was performed under the guidelines of the University of Michigan Committee for Use and Care of Animals.
Scheme 1.

The convergent Method A and stepwise Method B for the preparation of 4, 5, and 7-32
Scheme 4.
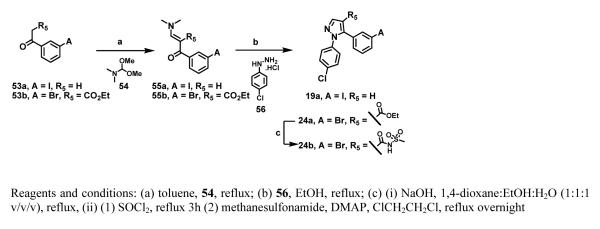
Synthesis of pyrazole scaffolds 19a and 24b
Scheme 5.

Synthesis of pyrazoles 20a and 21a, isoxazole 22a, and 2-chloropyrrole 17a
ACKNOWLEDGMENTS
This research was supported in part by a grant from the National Cancer Institute, National Institutes of Health (U19CA113317), the University of Michigan Cancer Center (Core Grant P30CA046592), and Ascentage Pharma. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817).
ABBREVIATIONS USED
- AcOH
acetic acid
- DIEA
N,N-diisopropylethylamine
- EDCI
1-ethyl-(3-dimethylaminopropyl)carbodiimide
- Et3N
triethylamine
- EtOAc
ethyl acetate
- EtOH
ethanol
- FBS
fetal bovine serum
- FP
fluorescence-polarization
- HOBt
hydroxybenzotriazole
- MeOH
methanol
- mP
millipolarization
- NIS
N-iodosuccinamide
- PARP
poly ADP ribose polymerase
- Pd(dba)2
Bis(dibenzylidene-acetone)palladium(0)
- SCID
Severe combined immunodeficient
- TosMIC
Toluenesulfonylmethyl isocyanide
Footnotes
Accession Codes: Coordinates for compound 4 complexed with Bcl-xL were deposited into the Protein Data Bank under Accession Number 3SP7.
References
- 1.Cory S, Adams JM. Killing cancer cells by flipping the Bcl-2/Bax switch. Cancer Cell. 2005;8:5–6. doi: 10.1016/j.ccr.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 2.Labi V, Erlacher M, Kiessling S, Villunger A. BH3-only proteins in cell death initiation, malignant disease and anticancer therapy. Cell Death Differ. 2006;13:1325–1338. doi: 10.1038/sj.cdd.4401940. [DOI] [PubMed] [Google Scholar]
- 3.Labi V, Grespi F, Baumgartner F, Villunger A. Targeting the Bcl-2-regulated apoptosis pathway by BH3 mimetics: a breakthrough in anticancer therapy? Cell Death Differ. 2008;15:977–987. doi: 10.1038/cdd.2008.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 5.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 6.Park CM, Bruncko M, Adickes J, Bauch J, Ding H, Kunzer A, Marsh KC, Nimmer P, Shoemaker AR, Song X, Tahir SK, Tse C, Wang X, Wendt MD, Yang X, Zhang H, Fesik SW, Rosenberg SH, Elmore SW. Discovery of an orally bioavailable small molecule inhibitor of prosurvival B-cell lymphoma 2 proteins. J Med Chem. 2008;51:6902–6915. doi: 10.1021/jm800669s. [DOI] [PubMed] [Google Scholar]
- 7.Zhou H, Chen J, Meagher JL, Yang CY, Aguilar A, Liu L, Bai L, Cong X, Cai Q, Fang X, Stuckey JA, Wang S. Design of Bcl-2 and Bcl-xL Inhibitors with Subnanomolar Binding Affinities Based upon a New Scaffold. J Med Chem. 2012;55:4664–4682. doi: 10.1021/jm300178u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou H, Aguilar A, Chen J, Bai L, Liu L, Meagher JL, Yang CY, McEachern D, Cong X, Stuckey JA, Wang S. Structure-based design of potent Bcl-2/Bcl-xL inhibitors with strong in vivo antitumor activity. J Med Chem. 2012;55:6149–6161. doi: 10.1021/jm300608w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J, Zhou H, Aguilar A, Liu L, Bai L, McEachern D, Yang CY, Meagher JL, Stuckey JA, Wang S. Structure-based discovery of BM-957 as a potent small-molecule inhibitor of Bcl-2 and Bcl-xL capable of achieving complete tumor regression. J Med Chem. 2012;55:8502–8514. doi: 10.1021/jm3010306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartwig JF, Kawatsura M, Hauck SI, Shaughnessy KH, Alcazar-Roman LM. Room-Temperature Palladium-Catalyzed Amination of Aryl Bromides and Chlorides and Extended Scope of Aromatic C-N Bond Formation with a Commercial Ligand. J Org Chem. 1999;64:5575–5580. doi: 10.1021/jo990408i. [DOI] [PubMed] [Google Scholar]
- 11.Zhang H, Cai Q, Ma D. Amino acid promoted CuI-catalyzed C-N bond formation between aryl halides and amines or N-containing heterocycles. J Org Chem. 2005;70:5164–5173. doi: 10.1021/jo0504464. [DOI] [PubMed] [Google Scholar]
- 12.Tararov V, Korostylev A, Boerner A, Bobal P, Frantisek J, Stohandl J, Denike K, Jeker N. Process for preparing C5 products and their use for atorvastatin synthesis. 2006 EP1705175A1. [Google Scholar]
- 13.Roth BD, Blankley CJ, Chucholowski AW, Ferguson E, Hoefle ML, Ortwine DF, Newton RS, Sekerke CS, Sliskovic DR, Stratton CD, Wilson MW. Inhibitors of cholesterol biosynthesis. 3. Tetrahydro-4-hydroxy-6-[2-(1H-pyrrol-1-yl)ethyl]-2H-pyran-2-one inhibitors of HMG-CoA reductase. 2. Effects of introducing substituents at positions three and four of the pyrrole nucleus. J Med Chem. 1991;34:357–366. doi: 10.1021/jm00105a056. [DOI] [PubMed] [Google Scholar]
- 14.Chen QY, Wu SW. Methyl Fluorosulfonyldifluoroacetate - a New Trifluoromethylating Agent. Journal of the Chemical Society-Chemical Communications. 1989:705–706. [Google Scholar]
- 15.Wendt MD, Rockway TW, Geyer A, McClellan W, Weitzberg M, Zhao X, Mantei R, Nienaber VL, Stewart K, Klinghofer V, Giranda VL. Identification of novel binding interactions in the development of potent, selective 2-naphthamidine inhibitors of urokinase. Synthesis, structural analysis, and SAR of N-phenyl amide 6-substitution. J Med Chem. 2004;47:303–324. doi: 10.1021/jm0300072. [DOI] [PubMed] [Google Scholar]
- 16.Almansa C, Alfon J, de Arriba AF, Cavalcanti FL, Escamilla I, Gomez LA, Miralles A, Soliva R, Bartroli J, Carceller E, Merlos M, Garcia-Rafanell J. Synthesis and structure-activity relationship of a new series of COX-2 selective inhibitors: 1,5-diarylimidazoles. J Med Chem. 2003;46:3463–3475. doi: 10.1021/jm030765s. [DOI] [PubMed] [Google Scholar]
- 17.Yang J, Teng Y, Ara S, Rallapalli S, Cook JM. An Improved Process for the Synthesis of 4H-Imidazo[1,5-a][1,4]benzodiazepines. Synthesis (Stuttg) 2009;40:nihpa145687. doi: 10.1055/s-0028-1083358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Menozzi G, Fossa P, Cichero E, Spallarossa A, Ranise A, Mosti L. Rational design, synthesis and biological evaluation of new 1,5-diarylpyrazole derivatives as CB1 receptor antagonists, structurally related to rimonabant. Eur J Med Chem. 2008;43:2627–2638. doi: 10.1016/j.ejmech.2008.01.043. [DOI] [PubMed] [Google Scholar]
- 19.Lange JH, van Stuivenberg HH, Coolen HK, Adolfs TJ, McCreary AC, Keizer HG, Wals HC, Veerman W, Borst AJ, de Looff W, Verveer PC, Kruse CG. Bioisosteric replacements of the pyrazole moiety of rimonabant: synthesis, biological properties, and molecular modeling investigations of thiazoles, triazoles, and imidazoles as potent and selective CB1 cannabinoid receptor antagonists. J Med Chem. 2005;48:1823–1838. doi: 10.1021/jm040843r. [DOI] [PubMed] [Google Scholar]
- 20.Olivera R, SanMartin R, Dominguez E. A combination of tandem amine-exchange/heterocyclization and biaryl coupling reactions for the straightforward preparation of phenanthro[9,10-d]pyrazoles. J Org Chem. 2000;65:7010–7019. doi: 10.1021/jo000609i. [DOI] [PubMed] [Google Scholar]
- 21.Habeeb AG, Rao PNP, Knaus EE. Design and syntheses of diarylisoxazoles: Novel inhibitors of cyclooxygenase-2 (COX-2) with analgesic-antiinflammatory activity. Drug Development Research. 2000;51:273–286. [Google Scholar]
- 22.Kajino M, Hasuoka A, Nishida H. Preparation of substituted 1-heterocyclylsulfonyl-2-aminomethyl-5-(hetero)aryl-1H-pyrrole derivatives as acid secretion inhibitors for treating ulcer and related disorders. 2007 WO2007026916A1. [Google Scholar]
- 23.Schmidt A, Habeck T, Kindermann MK, Nieger M. New pyrazolium-carboxylates as structural analogues of the pseudo-cross-conjugated betainic alkaloid Nigellicine. Journal of Organic Chemistry. 2003;68:5977–5982. doi: 10.1021/jo0344337. [DOI] [PubMed] [Google Scholar]
- 24.Brough PA, Aherne W, Barril X, Borgognoni J, Boxall K, Cansfield JE, Cheung KM, Collins I, Davies NG, Drysdale MJ, Dymock B, Eccles SA, Finch H, Fink A, Hayes A, Howes R, Hubbard RE, James K, Jordan AM, Lockie A, Martins V, Massey A, Matthews TP, McDonald E, Northfield CJ, Pearl LH, Prodromou C, Ray S, Raynaud FI, Roughley SD, Sharp SY, Surgenor A, Walmsley DL, Webb P, Wood M, Workman P, Wright L. 4,5-diarylisoxazole Hsp90 chaperone inhibitors: potential therapeutic agents for the treatment of cancer. J Med Chem. 2008;51:196–218. doi: 10.1021/jm701018h. [DOI] [PubMed] [Google Scholar]
- 25.Nikolovska-Coleska Z, Wang R, Fang X, Pan H, Tomita Y, Li P, Roller PP, Krajewski K, Saito NG, Stuckey JA, Wang S. Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal Biochem. 2004;332:261–273. doi: 10.1016/j.ab.2004.05.055. [DOI] [PubMed] [Google Scholar]